 Eva Rettinger1* Vida Meyer1 Hermann Kreyenberg1 Andreas Volk2
Eva Rettinger1* Vida Meyer1 Hermann Kreyenberg1 Andreas Volk2 Selim Kuçi1
Selim Kuçi1 Andre Willasch1 Ewa Koscielniak3
Andre Willasch1 Ewa Koscielniak3 Simone Fulda4 Winfried S. Wels2
Simone Fulda4 Winfried S. Wels2 Halvard Boenig5 Thomas Klingebiel1
Halvard Boenig5 Thomas Klingebiel1 Peter Bader1*
Peter Bader1*- 1 Department of Pediatric Hematology, Oncology and Hemostaseology, University Children’s Hospital of Frankfurt/Main, Goethe-University Frankfurt/Main, Frankfurt/Main, Germany
- 2 Chemotherapeutisches Forschungsinstitut, Georg-Speyer-Haus, Frankfurt/Main, Germany
- 3 Department of Pediatric Oncology and Hematology, Olgahospital Stuttgart, Stuttgart, Germany
- 4 Institute for Experimental Cancer Research in Pediatrics, Goethe-University Frankfurt/Main, Frankfurt/Main, Germany
- 5 Institute for Transfusion Medicine and Immunohematology, Goethe-University Frankfurt/Main, Division for Cell Processing, German Red Cross Blood Donor Service Baden-Württemberg-Hessen, Frankfurt/Main, Germany
Allogeneic stem cell transplantation (allo-SCT) has become an important treatment modality for patients with high-risk acute myeloid leukemia (AML) and is also under investigation for soft tissue sarcomas. The therapeutic success is still limited by minimal residual disease (MRD) status ultimately leading to patients’ relapse. Adoptive donor lymphocyte infusions based on MRD status using IL-15-expanded cytokine-induced killer (CIK) cells may prevent relapse without causing graft-versus-host-disease (GvHD). To generate preclinical data we developed mouse models to study anti-leukemic- and anti-tumor-potential of CIK cells in vivo. Immunodeficient mice (NOD/SCID/IL-2Rγc−, NSG) were injected intravenously with human leukemic cell lines THP-1, SH-2 and with human rhabdomyosarcoma (RMS) cell lines RH41 and RH30 at minimal doses required for leukemia or tumor engraftment. Mice transplanted with THP-1 or RH41 cells were randomly assigned for analysis of CIK cell treatment. Organs of mice were analyzed by flow cytometry as well as quantitative polymerase chain reaction for engraftment of malignant cells and CIK cells. Potential of CIK cells to induce GvHD was determined by histological analysis. Tissues of the highest degree of THP-1 cell expansion included bone marrow followed by liver, lung, spleen, peripheral blood (PB), and brain. RH30 and RH41 engraftment mainly took place in liver and lung, but was also detectable in spleen and PB. In spite of delayed CIK cell expansion compared with malignant cells, CIK cells injected at equal amounts were sufficient for significant reduction of RH41 cells, whereas against fast-expanding THP-1 cells 250 times more CIK than THP-1 cells were needed to achieve comparable results. Our preclinical in vivo mouse models showed a reliable 100% engraftment of malignant cells which is essential for analysis of anti-cancer therapy. Furthermore our data demonstrated that IL-15-activated CIK cells have potent cytotoxic capacity against AML and RMS cells without causing GvHD.
Introduction
Allogeneic stem cell transplantation (allo-SCT) is an established method in the treatment of high-risk acute leukemia in children (Bader et al., 2004; Rettinger et al., 2011), and may also play a role in the treatment of high-risk soft tissue sarcoma in children. Especially patients with primary alveolar rhabdomyosarcoma (RMS) above the age of 10 years with bone or bone marrow (BM) metastases have no realistic chance to survive their disease with conventional treatment (Koscielniak et al., 1997; Klingebiel et al., 2008; Perez et al., 2011). By evaluating the impact of allo-SCT in both, leukemia and soft tissue sarcoma patients, minimal residual disease (MRD) after transplantation is the reason for disease recurrence, and therefore is the strongest negative effector of the therapeutic success of allo-SCT. Once a stem cell recipient relapses, further therapeutic options are limited. For instance, donor lymphocyte infusion (DLI) is only efficacious for a limited number of diseases, and the high T cell doses required for DLI raise the risk for severe graft-versus-host-disease (GvHD; Rettinger et al., 2011).
Cytokine-induced killer (CIK) cells are known in principle to be capable of eradicating a variety of both hematological and solid malignancies in a non-MHC-restricted manner, without possessing significant alloreactive potential (Takayama et al., 2000; Baker et al., 2001; Edinger et al., 2003; Kornacker et al., 2006; Sangiolo et al., 2009; Kim et al., 2007a,b,c). In recent years, the application of CIK cells has evolved from experimental observations into early clinical studies (Introna et al., 2007), and CIK cells are now considered superior to lymphokine-activated killer (LAK) cells and tumor-infiltrating lymphocytes (TIL) for cancer immunotherapy. In this context, CIK cells exhibit a stronger anti-tumor effect and continue to differentiate and survive in vivo without maintenance of exogenous cytokines after injection (Olioso et al., 2009). Activated CIK cells represent a heterogeneous population of polyclonal T cells sharing both natural killer (NK) phenotype and functional properties of NK cells (Pievani et al., 2011). CIK cells can be efficiently expanded in vitro from peripheral blood (PB), BM mononuclear cells and umbilical cord blood by addition of interferon (IFN)γ, activating antibody directed against CD3 and interleukin (IL)-2 (Lu and Negrin, 1994; Thorne et al., 2006). We recently used IL-15 for further CIK cell activation and expansion (Rettinger et al., 2012). We could show that IL-15-activated CIK cells have an increased anti-leukemic potential in vitro compared to conventional IL-2-activated CIK cells. Furthermore, our modified protocol allowed us to shorten ex vivo expansion time of CIK cells. Therefore, in this study we used IL-15-activated CIK cells after 10 days of culture for in vitro and in vivo analyses.
NOD/SCID/IL-2Rγc− (NSG) mice have a phenotype of severe combined immunodeficiency lacking functional T, B, and NK lymphocytes and, therefore, permit establishment of human xenografts (Ishikawa et al., 2005; Shultz et al., 2005). Other than NSG mouse models in many cases lacked reliable engraftment of malignant cells. A reliable engraftment of malignant cells, best mimicking engraftment sites of human malignancies is essential for functional analysis of human cellular therapies in preclinical animal models.
In this study we focused on the principal biological characteristics and engraftment sites of human acute myeloid leukemia (AML) and RMS cells injected via the tail vein in sublethally irradiated NSG mice. In addition, IL-15-activated day 10 CIK cells were inoculated for functional analyses regarding anti-tumor, anti-leukemic and GvHD potential in NSG mice, which had received grafts of human AML and RMS cells.
Materials and Methods
AML and Soft Tissue Sarcoma Cells
M4 subtype AML cell line, THP-1 was obtained and cultured as previously described (Rettinger et al., 2012). M2 subtype AML cell line, SH-2 was purchased from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany) and was maintained in IMDM medium supplemented with 20% fetal calf serum (FCS), L-glutamine, and antibiotics (penicillin 100 U/mL, streptomycin 100 μg/mL) according to the manufacturer’s instructions. Alveolar RMS RH30, RH41, and embryonal RMS TE671 cell lines were obtained and cultured as described (Kuci et al., 2010b). After written informed consent, primary Ewing’s sarcoma cells were harvested from a resected thoracic tumor of a Ewing’s sarcoma patient. The Ewing’s sarcoma patient was diagnosed with the first relapse after allo-SCT. Ewing sarcoma samples were cryopreserved and subsequently thawed to be used in the experiments.
Generation of CIK Cells
The Ethical Review Board of the Medical Faculty of the University Hospital Frankfurt/Main, Germany approved the study protocol to take blood from healthy volunteers after written informed consent for the purpose of generating cellular therapies against leukemia and soft tissue sarcomas (Geschäfts-Nr. 298/07). CIK cells were generated from peripheral blood mononuclear cells (PBMC) after standard Ficoll separation as previously described (Rettinger et al., 2012). In brief, cells were resuspended at a density of 3 × 106 cells/mL in RPMI 1640, supplemented with 10% FCS, L-glutamine and antibiotics and primed by adding 1000 U/mL IFN-γ on day 0 and 100 ng/mL anti-CD3 antibody (MACS GMP CD3 pure, Miltenyi Biotech, Bergisch Gladbach, Germany) and 500 U/mL IL-2 within the following 24 h of culture. At day 4 of culture, cell density was adjusted to 1 × 106 cells/mL. About 500 U/mL IL-2 or 50 ng/mL IL-15 and culture medium were added every 3 days, respectively. CIK cells were expanded over 10 days. On day 10 of culture CIK cells were harvested and used for analysis.
In vitro Cytotoxicity Analysis by Europium Release Assays
Europium release assay was used for in vitro cytotoxicity analysis as previously described (Rettinger et al., 2012). In brief, target cells were labeled with BATDA (Perkin Elmer, Boston, USA) washed and co-cultured with CIK cells in duplicates or triplicates at an effector to target cell ratio (E:T ratio) of 20:1 in U-bottom-96-well culture plates (NUNC, Roskilde, Denmark). Supernatant was collected from each well and co-incubated with europium solution (Europium, Perkin Elmer, Turku, Finland) on flat-bottom-96-well plates (NUNC, Roskilde, Denmark). Fluorescence data were recorded using a time resolved fluorometer (1420-018 Victor, Perkin Elmer, Waltham, MA, USA). The percentage of specific cytolysis was calculated for each well as described previously (Rettinger et al., 2012), and means with SD were calculated from each duplicate or triplicate.
Establishment of Mouse Models for Human AML and RMS
NSG mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and were maintained in the animal facilities of the University of Frankfurt/Main, Germany under specific pathogen-free conditions. The described research was approved by the Animal Care Committee of Frankfurt am Main University and the Regierungspräsidium Darmstadt, Germany (Gen. Nr. F. 133/08).
For the establishment of human leukemia and RMS models in mice, 6- to 8-week old NSG mice were sublethally irradiated with 200 cGy 24 h prior to intravenous (i.v.) injection of AML or RMS cells (Biobeam 2000, Eckert and Ziegler, Bebig, Germany). Subsequently, mice received tail vein injections of either 1 × 104 or 1 × 103 THP-1 cells. In parallel, mice were transplanted with 5 × 106, 2.5 × 106, 1 × 106, or 0.5 × 106 SH-2 cells. For RMS engraftment, mice were inoculated with 1 × 106 RH41 cells or 1 × 105 RH30 cells, respectively.
Animals were euthanized on days 5, 15, 25, 30, 32, and 45 after transplantation of malignant cells, or monitored for survival. Mice showing visible signs of poor health or physical abnormalities were sacrificed. Mice were sacrificed by carbon dioxide asphyxiation followed by cervical dislocation.
CIK Cell Treatment of AML or RMS Mice
In the first series of experiments, mice were inoculated i.v. with the minimal cell dose of 1 × 104 THP-1 cells and 1 × 106 RH41 cells sufficient to induce cancer. Randomly selected mice were injected with 1 × 106 IL-15-activated day 10 CIK cells 1 day after transplantation of malignant cells.
In the second series of experiments CIK cell dose was increased to 2.5 × 106 cells and CIK cells were inoculated 24 h after injection of 1 × 104 THP-1 cells into randomly selected mice. THP-1 and RH41 injected mice were sacrificed 25 and 45 days after transplantation of malignant cells and organs were analyzed for engraftment of malignant cells.
Harvest of Human Cells from Organs of NSG Mice
Cell suspensions of liver, spleen, lung, brain, PB, and BM were prepared. Briefly, BM cells were collected from each tibia and femur by flushing bones with culture medium. Mouse erythrocytes within BM and PB samples were lysed with lysing buffer (Mouse Erythrocyte Lysing Kit, R&D Systems, Wiesbaden, Germany) and washed once with washing buffer (Mouse Erythrocyte Lysing Kit, R&D Systems, Wiesbaden, Germany) according to the manufacturer’s instructions. Other cell suspensions were prepared from mouse organs digested by collagenase, filtered through a 100-μM cell strainer and washed with phosphate buffered saline (PBS). Aliquots of cell suspensions were analyzed by four-color flow cytometry analysis (FACS) and quantitative polymerase chain reaction (qPCR).
Quantification and Characterization of Human Cells in the Organs of NSG Mice by FACS
Leukemia burden in CIK cell treated and untreated mice were quantified by FACS. In addition, CIK cells were characterized by FACS. Cells were washed once in PBS resuspended in 100 μL PBS, and incubated with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, phycoerythrin–cyanin (Cy) 5-, or allophycocyanin (APC)-conjugated-anti human monoclonal antibodies (mAbs) for 20 min at 4°C, washed twice in PBS and resuspended in 200 μL of PBS. The frequency of human CD45 (FITC, PerCP, APC, BD Biosciences, Heidelberg, Germany; PE, BD Pharmingen, Heidelberg, Germany), CD3 (PE, PerCP, BD Biosciences; APC, BD Pharmingen), CD4 (FITC, APC, BD Pharmingen), CD8 (FITC, PE, BD Biosciences; APC, BD Pharmingen), CD25 (PE, BD Pharmingen), CD56 (FITC, PE, BD Biosciences; APC, BD Pharmingen), TCRαβ (PE, Miltenyi Biotec, Bergisch Gladbach Germany), TCRγδ (FITC, BD Biosciences), and CD33 (PerCP, BD Biosciences) expressing cells was analyzed. Isotype-matched fluorochrome-conjugated IgGs were used as controls. After gating on viable cells, data on 5 × 105 events were acquired by flow cytometer BD FACSCalibur (BD Biosciences) using Cell Quest software (BD Biosciences).
Quantification and Discrimination of Human Cells in the Organs of NSG Mice by PCR
DNA PCR was performed from cell suspensions as previously described (Kuci et al., 2010a). In brief, genomic DNA was extracted using the QIAamp blood and tissue kit (Qiagen, Hilden, Germany). As a first step a quantitative real time PCR approach was used to assess the amount of human cells of each tissue sample by specific amplification of the human albumin gene (Pongers-Willemse et al., 1999). As a standard was used the serial dilution preparation of human DNA in mouse DNA. For each reaction 50 ng DNA were processed and the assay was able to detect one human cell beneath 1000 murine cells. As a second step within the human cell fraction proportion of CIK and tumor cells were discriminated by human specific STR-genotyping similar to chimerism analyses (Scharf et al., 1995). Informative loci for the discrimination of THP-1 and CIK cells were D7S820 and for the discrimination of RH41 and CIK cells D3S1358, D13S317, and D18S51, respectively. Leukemia or tumor burden of each mouse was obtained by adding leukemia or tumor signals per analyzed organ. Primers and probes were achieved from Eurofins (Eurofins MWG GmbH, Ebersberg, Germany) and genotyping was performed using the STR multiplex PCR system Powerplex16 (Promega GmbH, Mannheim, Germany).
Histology
Histological analysis was performed by mfd diagnostics (mfd diagnostics GmbH, Wendelsheim, Germany). Tissue was fixed in 100% buffered formalin, paraffin embedded, sectioned, and stained with hematoxylin–eosin (HE). Microscopy analysis was performed by mfd diagnostics using Zeiss AXIO Imager A1/M1.
Statistical Analysis
Results were compared using the paired Student’s two tailed t-test. A p-value <0.05 was considered to be significant.
Results
In vitro Cytotoxicity of CIK Cells
Our previous studies demonstrated that IL-15-activated CIK cells in part were superior in killing of leukemia cells compared with IL-2-stimulated CIK cells (Rettinger et al., 2012). This study was performed, to confirm these findings against a variety of sarcoma cells. After 10 days of in vitro culture IL-15-stimulated CIK cells and conventional IL-2-stimulated CIK cells were added to target cells at an E:T ratio of 20:1 and tested in europium release assays. Target cells used were TE671, RH30, primary Ewing’s sarcoma and RH41 cells. Interestingly, IL-15 activation resulted in significantly increased cytotoxicity against TE671 (p < 0.0237, Figure 1A), RH30 (p < 0.0123, Figure 1B), and primary Ewing’s sarcoma cells (p < 0.0154, Figure 1C) compared to IL-2-activated CIK cells. In vitro cytotoxicity of IL-15-stimulated CIK cells was not increased against RH41 cells (Figure 1D). Altogether, IL-15-stimulation was vastly different among the individual cell lines and was equivalent of IL-2-stimulation in RH41 cells in vitro.
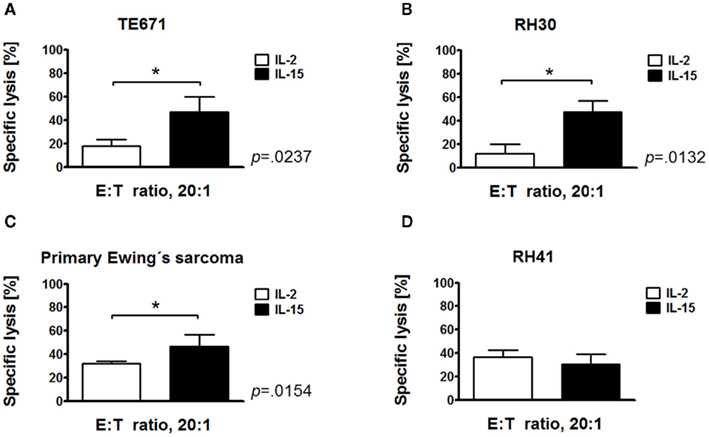
Figure 1. Increased cytotoxic potential of IL-15-stimulated cytokine-induced killer cells against sarcoma cells. Cytotoxicity of IL-15-stimulated and conventional IL-2-stimulated CIK cells after 10 days of culture against embryonal rhabdomyosarcoma (RMS) cell line TE671 (A), alveolar RMS cell lines RH30 (B) and RH41 (D), and primary Ewing’s sarcoma cells (C) was analyzed by europium release assay in vitro. Mean values from three independent experiments are shown. IL-15-activated CIK cells exhibited a significantly increased cytotoxicity against TE671 (p < 0.0237), RH30 (p < 0.0123) and primary Ewing’s sarcoma cells (p < 0.0154) compared to IL-2-activated CIK cells. Cytotoxicity of IL-15-activated CIK cells was not increased against RH41 cells (D).
Establishment of Human AML and RMS Models in NSG Mice
We suggested that IL-15-activated CIK cells may have differential effects in vivo compared with our in vitro findings, and therefore may also be effective against RH41 and THP-1 cells in vivo, which both were equivalent killed by IL-2- and IL-15-activated CIK cells (Rettinger et al., 2012). Besides RH41 and THP-1 models, we established additional xenotransplant models of both tumor entities, AML and RMS, in immunodeficient NSG mice. Taking into consideration the excellent proliferative capacity of AML cells, we performed dose-finding experiments to determine the minimal AML dose required for a delayed, but reliable engraftment of human AML cells. The determined cell dosage differed markedly between cell lines, and was 1 × 104 in case of fast-expanding THP-1 cells (manufacturer’s instructions: doubling time, 35–50 h; Figure 2A) and 0.5 × 106 in case of slowly expanding SH-2 cells (manufacturer’s instructions: doubling time, 80 h; Figure 2B). However, despite injection of a minimum of THP-1 cells, mice showed physical abnormalities within 24–34 days after injection, whereas mice injected with a minimum of SH-2 cells developed symptoms within a period of 82–103 days post-transplant. Further analyses of organs of affected mice showed that most of the THP-1 engraftment took place in the BM, but was also decreasingly detectable in lung, spleen, liver, PB, and brain (Figure 2C). The earliest time point for PCR-based detection of THP-1 engraftment within analyzed organs (BM, PB, liver, spleen, lung, and brain) taken together was 15 days after transplantation. From then, cells expanded dramatically 665-fold (day +15, 0.20; day +32, 132.95) within a period of 17 days (Figure 2D).
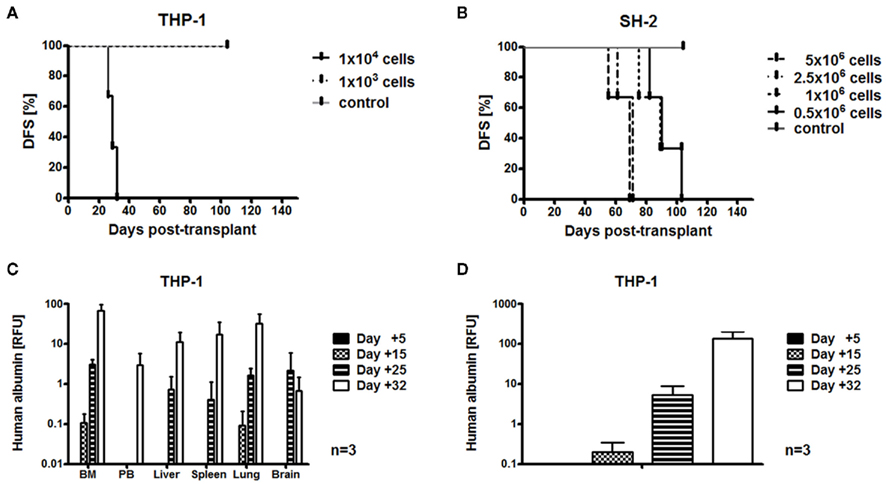
Figure 2. Establishment of humanized acute myeloid leukemia mouse models. Preclinical AML models were established in immunodeficient NSG mice. Results from three independent experiments are shown. Minimal cell doses required for AML engraftment were 1 × 104 in case of THP-1 cells (A) and 0.5 × 106 in case of SH-2 cells (B). Mice injected with a minimum of THP-1 cells showed disease development within 24–34 days (A). Mice injected with a minimum of SH-2 cells developed disease within 82–103 days (B). PCR-based detection of human albumin showed that most of the THP-1 engraftment occurred in the bone marrow (BM), but was also detectable at decreasing amounts in lung, spleen, liver peripheral blood (PB), and brain (C). THP-1 engraftment within bone marrow (BM), peripheral blood (PB), liver, spleen, lung, and brain taken together increased by 665-fold (day +15, 0.20; day +32, 132.95) within 17 days (D).
According to their proliferative capacity, 1 × 105 of RH30 cells (manufacturer’s instructions: doubling time, 35 h) and 1 × 106 of RH41 cells (manufacturer’s instructions: doubling time, 24–36 h) were found to be sufficient for tumorigenesis. RMS engraftment of RH30 and RH41 cells was monitored at days +15, +30 and >+45 post-transplant. Engraftment of RH30 cells in BM, PB, liver, spleen, and lung taken together was detectable 15 days after transplantation (Figure 3A), and RH30 injected mice, which were observed for survival, became ill 48–69 days after injection. Most of the RMS engraftment took place within the reticuloendothelial system (RES) of the liver (Figures 3B,C), but was also detectable to a decreasing extent in lung, BM, spleen, and PB (Figure 3D). PCR results demonstrated that RH30 cells expanded 627-fold (day +15, 0.21; >day +45, 131.71; Figure 3A). RH41 signals within analyzed compartments (BM, PB, liver, spleen, and lung) taken together were detected at day +15 (Figure 3E), and RH41 mice, which were observed for survival, showed signs of discomfort 45–51 days after transplantation. Expansion rate of RH41 cells, which were injected at higher cell doses compared with RH30 cells was 18020-fold (day +15, 0.01; >day +45, 180.20) within 30 days after RMS engraftment (Figure 3E). Most of the RH41 engraftment took place in liver and lung, but was also detectable to a decreasing extent in BM, PB, and spleen (Figures 3F,G).
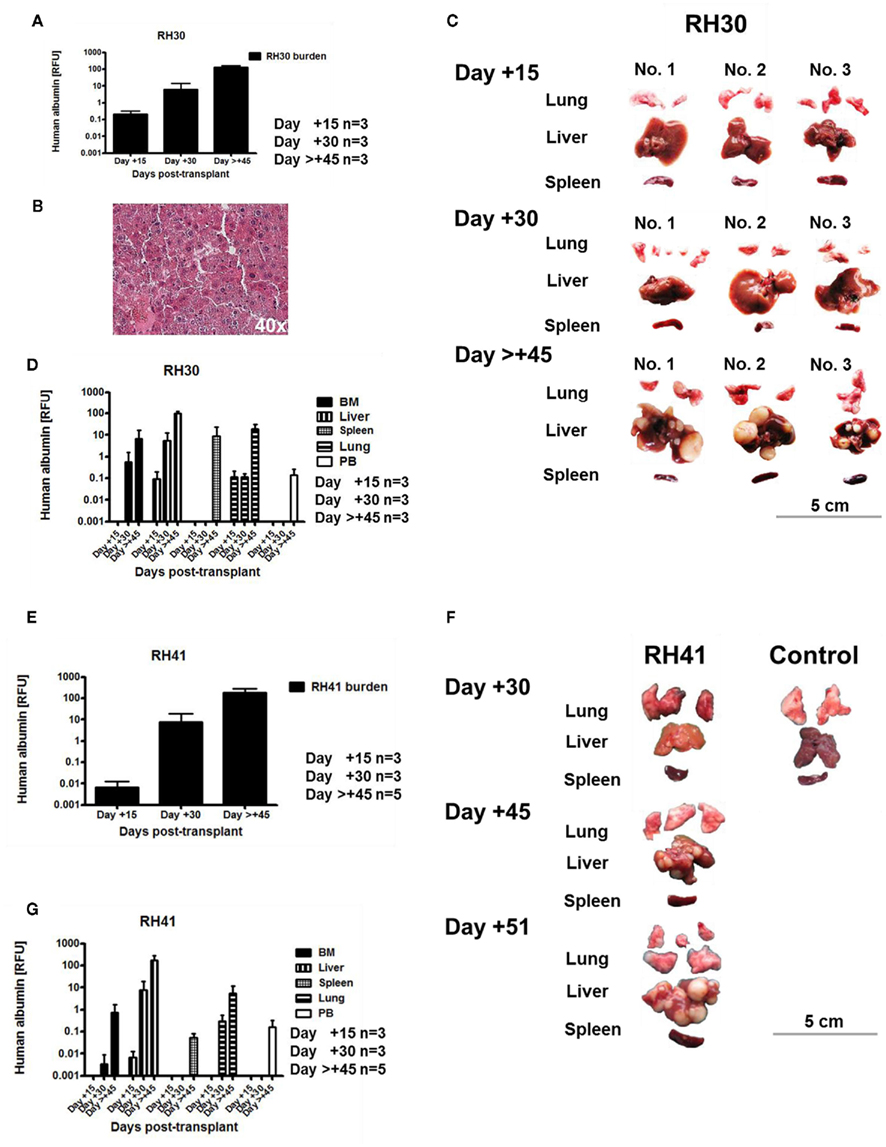
Figure 3. Establishment of humanized rhabdomyosarcoma mouse models. Results from three independent experiments demonstrated that intravenous injection of 1 × 105 RH30 cells was sufficient for tumorigenesis. PCR-based detection of human albumin in bone marrow (BM), peripheral blood (PB), liver, spleen, and lung taken together showed that RH30 cells engrafted by day +15 and expanded 627-fold within 30 days (A). Most of the RH30 engraftment occurred within the reticuloendothelial system (RES) of the liver as shown by HE-staining of one representative histological analysis (B), but was also detectable to a decreasing extent in lung, bone marrow (BM), spleen, and peripheral blood (PB) (C,D). In parallel, injection of 1 × 106 RH41 cells was sufficient for engraftment of tumor cells. RH41 signals within analyzed compartments (BM, PB, liver, spleen, and lung) were detectable at day +15 and increased by 18020-fold within 30 days (E). Most of RH41 engraftment occurred in liver and lung like shown by one representative example (F), but was also detectable in bone marrow, blood, and spleen (G).
In vivo Cytotoxicity of IL-15-Stimulated CIK Cells Against RMS
Having established the xenotransplant tumor models, we assessed the cytotoxicity of IL-15-activated CIK cells after 10 days of ex vivo culture against RH41 cells in vivo. With regard to clinical translatability, RH41 cells were used best mimicking resistant tumor cells. NSG mice were sublethally irradiated and transplanted with 1 × 106 RH41 cells. About 1 × 106 CIK cells were injected in one part of RH41-transplanted mice 24 h after tumor cell injection. CIK cell treated mice and untreated controls were analyzed for RMS engraftment 45 days after injection of RH41 cells. Gross examination of organs was equivalent in CIK cell treated and untreated mice. PCR-based results confirmed that small numbers of injected CIK cells were able to migrate to sites where tumor was found (Figure 4A). Furthermore, these CIK cells were capable of significantly reducing RH41 burden by comparing compartments of CIK cell treated mice (Figure 4A) with untreated controls (Figure 4B) where RH41 cells were detectable. Results of analyzed compartments taken together confirmed that CIK cell treatment (n = 3) resulted in a significantly reduced tumor burden compared with the untreated controls (n = 5; control group, 180 ± 104; treatment group, 67 ± 6, p < 0.001; Figure 4C).
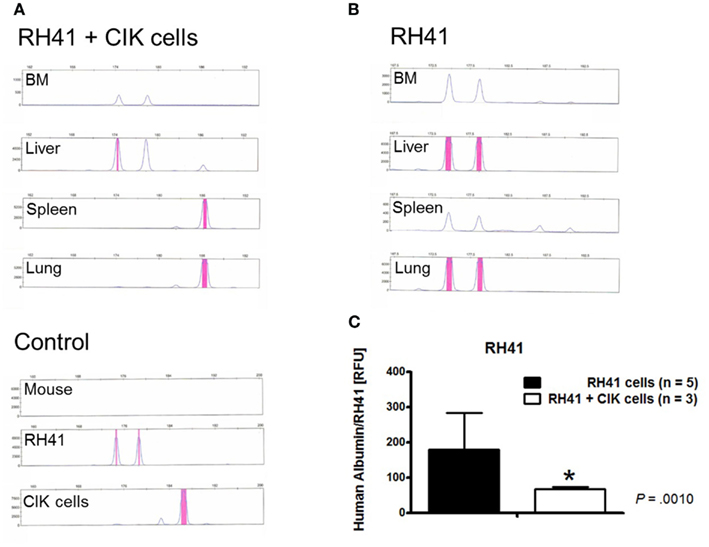
Figure 4. In vivo cytotoxicity of IL-15-stimulated cytokine-induced killer cells against rhabdomyosarcoma. CIK cell treated (n = 3) and untreated controls (n = 5) among RH41-transplanted mice were analyzed 45 days after transplantation of malignant cells. Chimerism analyses of one representative example within the treatment group showed that CIK cells were detectable in the liver spleen and lung (A). RH41 engraftment was most pronounced in liver and lung, but was also seen to a lesser extent in the bone marrow (BM) and spleen like shown by one representative untreated control (B). PCR results from all analyzed organs of the treatment and control group (untreated mice, n = 5; CIK cell treated mice, n = 3) taken together showed, that CIK cells were able to significantly reduce tumor growth when compared to untreated mice [p < 0.001, (C)].
In vivo Cytotoxicity of IL-15-Stimulated CIK Cells Against AML
Cytotoxicity of IL-15-stimulated day 10 CIK cells was determined in THP-1-injected mice 25 days after transplantation. Previously analyzed, fast-expanding THP-1 cells (Rettinger et al., 2012) were used in this study best representing AML engraftment in patients. Groups of mice inoculated with 1 × 104 THP-1 cells were injected with different doses of CIK cells. To this end, 1 × 106 or 2.5 × 106 CIK cells were transplanted via the tail vein in THP-1-injected mice. Despite aggressive expansion of THP-1 cells, PCR-based results demonstrated that CIK cells were capable of reducing THP-1 burden in all analyzed compartments (BM, PB, liver, spleen, lung, and brain) taken together. Furthermore, our results demonstrated that cytotoxicity of CIK cells was dose-dependent (untreated control group, 161 ± 56; treatment groups, 1 × 106 CIK cells, 74 ± 56; 2.5 × 106 CIK cells, 43 ± 35, p < 0.0001, Figure 5A). In this series of experiments, chimerism analysis was used to determine THP-1 and CIK cell signals in BM, liver, spleen, lung, brain, and PB of mice (Figure 5B). Comparable results were obtained by FACS analysis of BM samples of CIK cell treated mice and untreated controls (Figure 5C). Here, THP-1 cells were quantified by staining with human anti-CD45 and anti-CD33 antibodies. Engraftment of THP-1 cells (CD45+CD33+) and CIK cells (CD45+CD33−) was negatively correlated and THP-1 engraftment was even inhibited in two out of seven mice injected with 2.5 × 106 CIK cells (Figure 5C). THP-1 engraftment was markedly reduced but not inhibited after injection of 1 × 106 CIK cells. This might be due to the fact that compared to THP-1 expansion, CIK cell expansion was delayed and CIK cells (CD45+) were not detectable within 30 days post-transplant like shown by anti-CD45 staining within organs of representative examples after injection of 1 × 106 CIK cells only (Figure 5D). FACS analysis of organs of THP-1-injected mice treated with 2.5 × 106 CIK cells (CD3+) confirmed that CIK cells infiltrated leukemia sites, resulting in a reduction or even elimination of CD33+ expressing THP-1 cells (Figure 5E). In this analysis CIK cells were detected by staining them with a human anti-CD3 antibody whereas THP-1 cells were quantified by staining with a human anti-CD33 antibody.
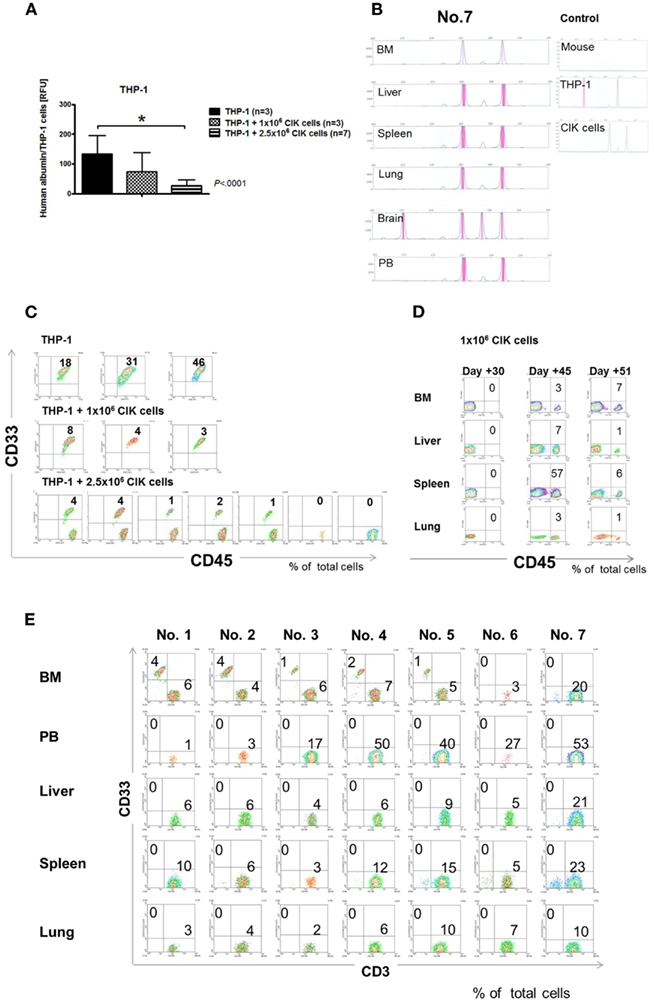
Figure 5. Anti-leukemic activity of IL-15-stimulated cytokine-induced killer cells. Mice were inoculated i.v. with the minimal leukemic dose of 1 × 104 THP-1 cells. About 1 × 106 and 2.5 × 106 CIK cells were transplanted via the tail vein into one part of mice injected with THP-1 cells. Bone marrow (BM), peripheral blood (PB), liver, spleen, lung, and brain were analyzed for THP-1 and CIK cell engraftment by PCR 25 days after transplantation of malignant cells. Results of analyzed compartments taken together showed that CIK cell treatment resulted in a markedly decreased THP-1 engraftment compared with the untreated controls (A). Reduction of leukemia burden was dose-dependent. Chimerism analysis was used to distinguish between AML and CIK cells. Chimerism results like shown here by one representative example [mouse no. 7, 1 × 104 THP-1 cells and 2.5 × 106 CIK cells, (B)] demonstrated that CIK cells were detectable in analyzed organs. Except the brain, THP-1 signals were not detectable in CIK cell infiltrated organs. Bone marrow results obtained by flow cytometry, confirmed that CIK cells (CD45+CD33−) were capable of reducing AML (CD45+CD33+) engraftment in a dose-dependent manner (C). Representative examples of mice injected with 1 × 106 CIK cells (CD45+) only, demonstrated that CIK cells expanded as early as 30 days post-transplant (D). Therefore, expansion of 1 × 106 injected CIK cells was delayed compared with THP-1 engraftment. Flow cytometry analysis from seven independent experiments within the treatment group which were performed 25 days after THP-1 injection confirmed that CIK cells (CD3+) injected at higher doses (2.5 × 106 CIK cells) were capable of reducing or even eliminating AML (CD33+) engraftment (E).
CIK Cell Phenotype and Potential to Induce GvHD
Flow cytometric analysis revealed that CIK cells at the time of injection displayed a CD3+CD56− rather than CD3+CD56+ or CD3−CD56+ phenotype (Figure 6A). These T cells expressed CD8 and CD25 rather than CD4 molecules (Figure 6A). BM analyses of cancer-infiltrating CIK cells in vivo exclusively demonstrated a CD3+CD56− T cell phenotype (Figure 6B). In addition, most of these T cells expressed CD4 rather than CD8 antigens (Figure 6B).
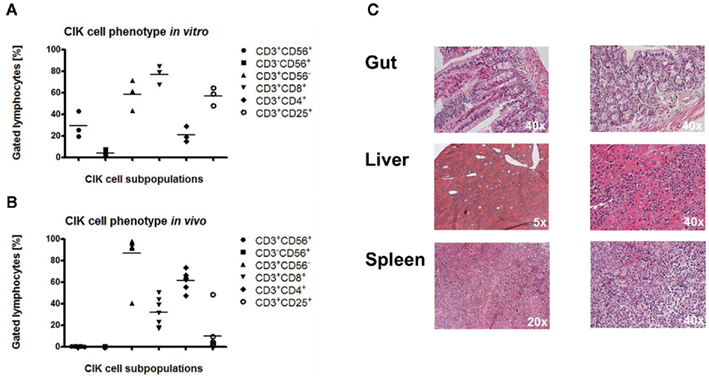
Figure 6. Phenotype and alloreactive potential of cytokine-induced killer cells. Before injection, CIK cells were CD3+CD56− or CD3+CD56+ rather than CD3+CD56+ like shown by FACS analyses of three independent examples (A). Most of these T cells expressed CD8 and CD25 rather than CD4 antigens (A). Cytometric analysis of injected CIK cells from seven independent bone marrow samples within the treatment group demonstrated predominantly a CD3+CD56− T cell phenotype (B). Most of these T cells were CD4+ rather than CD8+(B). Histological analysis from one representative CIK cell treated mouse with the best CIK cell engraftment (mouse no. 7, Figures 5B,E, 1 × 104 THP-1 cells + 2.5 × 106 CIK cells) showed no xenogenic GvHD in classical GvHD targeted tissues like gut and liver as well as in spleen, the preferred homing sites of CIK cells (C).
Graft-versus-host-disease targeted tissues like the gastrointestinal tract, liver, and spleen were analyzed in terms of xenogenic GvHD mediated by CIK cells. Histological analyses of gut, liver, and spleen of one representative mouse, with the highest degree of CIK cell expansion (mouse no. 7, Figures 5B,E) demonstrated no or mild xenogenic GvHD potential of IL-15-stimulated CIK cells (Figure 6C).
Discussion
The aim of this study was to evaluate the efficacy of ex vivo expanded, IL-15-activated day 10 CIK cells against AML and sarcoma cells in vivo in an optimized preclinical mouse model with the goal of clinical translatability. We therefore established a disease model for AML and sarcoma cells in NSG mice injected via the tail vein with a minimal leukemic or tumorigenic dose. The feasibility and efficacy of infused CIK cells in AML and RMS bearing mice was then analyzed in terms of regression of malignant cells or alloreactivity of CIK cells.
Transplantation of human AML cells into different strains of immunodeficient mice has led to preclinical models used to investigate AML and sarcoma biology, and efficacy of immunotherapy (Bonnet and Dick, 1997; Hudson et al., 1998; Vormoor et al., 2001; Ito et al., 2002; Feuring-Buske et al., 2003; Meyerrose et al., 2003; Ishikawa et al., 2005; Kawano et al., 2005; Shultz et al., 2005, 2007; Siegler et al., 2005). NSG mice have been investigated as models to generate human hematopoiesis in a murine host (Ito et al., 2002; Shultz et al., 2005). The IL-2Rγc-chain deficiency in NSG mice, also lacking functional B cells and T cells, impairs signaling through multiple cytokine receptors, which block NK cell development resulting in additional defects in innate immunity (Shultz et al., 2005, 2007). Agliano et al. (2008) observed a fast development of leukemia-related symptoms and a high percentage of leukemia cells in blood, BM, and spleen of intraperitoneally (i.p.) injected non-irradiated NSG mice. Total body irradiation in SCID mutant mice was reported to increase radiosensitivity, associated with some unpredictable mouse mortality (Shultz et al., 2007). In addition, recent reports showed a significant engraftment of human cells and a prolonged life span in non-irradiated NSG mice (Nakamura et al., 2005; Watanabe et al., 2007). Despite these observations, to best mimic conditioning prior to transplantation in humans, we decided to irradiate the animals before injection of human cells. Our results showed that tail vein injection of human AML and RMS cells resulted in a 100% engraftment of transplanted cells. We found that doses of 1 × 104–1 × 106 cells per mouse were sufficient to obtain with certainty leukemia or sarcoma engraftment within 3–8 weeks. We suggested that the log difference in cell requirement may depend on the proliferative capacity and migration characteristics of injected cells. As one reason, cell requirement was less in fast-expanding AML subtype M4 cells, and more in slowly expanding AML subtype M2 or RMS cells. We observed most of the leukemia engraftment in the BM, and most of the RMS engraftment in liver and lung. The RES of liver and lung usually represents with plenty of microvasculature, and velocity of blood flow inside is slow. Hence, it appears likely that infused RMS cells, which are larger than AML cells, were retained at these sites. In contrast to our finding, that 0.5 × 106 i.v. injected SH-2 cells were sufficient for leukemic engraftment in NSG mice, other groups reported that 1 × 107 subcutaneously (s.c.) or i.v. injected SH-2 cells were needed for engraftment in 5/8 BALB/c nude mice and 4/10 SCID mice 8 weeks after injection (Qiu et al., 2008). We suggest that a preclinical model with reliable engraftment rates is essential for analysis of cellular therapy.
Adoptive cellular immunotherapy is an important treatment to eliminate residual leukemia or tumor cells after allo-SCT. Over the past 20 years the development of CIK cell immunotherapy for the treatment of hematological and solid malignancies has received considerable attention. CIK cells are capable of a broad MHC-unrestricted anti-leukemic and anti-tumor activity as documented both in vitro and in vivo in murine models (Schmidtwolf et al., 1991, 1994; Lu and Negrin, 1994; Hoyle et al., 1998; Sweeney et al., 1999; Alvarnas et al., 2001; Baker et al., 2001; Verneris et al., 2001; Edinger et al., 2003). Limited data are available on the efficacy of CIK cells against solid tumors (Scheffold et al., 2002; Wang et al., 2002; Helms et al., 2010; Sangiolo, 2011). In previous work we modified CIK cell generation by using IL-15 for CIK cell activation (Rettinger et al., 2012). IL-15 activation significantly enhanced CIK cell-mediated cytotoxicity against leukemia cells in vitro, whereas the alloreactive potential of IL-15-stimulated CIK cells remained low. Furthermore, we could show that IL-15 activation can minimize the culture period prior to adoptive transfer without affecting the in vitro efficacy of CIK cells. Hence, IL-15-stimulation may change the outcome of CIK cell generation from terminally differentiated and potentially exhausted day 21 CIK cells to day 10 CIK cells with a more naïve phenotype and potent proliferative capacity.
In our experiments, mice received grafts of human AML and RMS cells. In order to demonstrate that IL-15-activated CIK cells have differential effects in vivo, rather than in vitro, and to best mimic clinical situations of high-risk patients, RH41 cells resistant to FAS- and TRAIL-induced apoptosis (Petak et al., 2000, 2003) and fast-expanding THP-1 cells, which were previously tested (Rettinger et al., 2012) were used as targets for in vivo analysis. TRAIL is important for CIK cell-mediated induction of apoptosis (Kuci et al., 2010b). Therefore, TRAIL-resistance of RH41 cells may have influenced in vitro cytotoxicity of IL-15-activated CIK cells in this study.
Experimental mice were then assigned randomly to different treatment groups, and were infused with equal amounts of CIK cells in the RMS setting, and 100–250 times increased amounts of CIK cells compared with injected malignant cells in the leukemia setting. To avoid non-engraftment of leukemia and tumor cells by co-transplantation of CIK cells, CIK cells were injected at least 24 h after administration of malignant cells. We observed that i.v. injection of 2.5 × 106 or 1 × 106 CIK cells resulted in a strong inhibition of AML and sarcoma growth. Hereby CIK cells were capable of reducing AML engraftment in a dose-dependent manner after being injected within 24 h after administration of malignant cells. This time point might represent MRD status, and might therefore be optimal for MRD-directed preemptive immunotherapy. An excess of leukemia/tumor cells will likely not be overcome by immunological measures. Furthermore, our study results demonstrated, that CIK cells widely distributed to many organs through the blood stream. It was previously reported that CIK cells migrated to tumor sites within 7 h after injection, and remained detectable at these sites for an additional 9 days (Verneris et al., 2000; Edinger et al., 2003; Skitzki et al., 2004; Kornacker et al., 2006). Our results revealed the presence of considerable numbers of CIK cells in different compartments up to 51 days post-transplant. But, CIK cell movement via the blood circulation and CIK cell expansion at tumor sites depended on the number of CIK cells infused. Some reports showed that CIK cells first entered into the lung, after inoculation of nude mice (Hazelrigg et al., 2002; Kim et al., 2009), peaked there within 2–6 h after injection, then declined, and re-distributed to organs such as liver, spleen, and kidney within 24 h. Another report demonstrated that CIK cells infused via the tail vein homed directly to the RES (Li et al., 2011). In contrast, in our study aggregation and expansion of infused CIK cells mostly occurred in BM and spleen at the time of analysis.
Cytokine-induced killer cell populations undergo a maturation process during ex vivo expansion. During this process we could show in previous studies that CIK cell subpopulations arise with a maximum of anti-tumor activity and proliferative capacity within day 7–14 of culture (Kuci et al., 2010b; Rettinger et al., 2012). Terminally differentiated effector CD3+CD56+ T cell subpopulations are known to possess potent cytotoxicity, but have low proliferative capacity (Wajchman et al., 2004; Powell et al., 2005). On the other hand, CD3+CD56− cells, which represent early effector T cells, exhibit reduced cytotoxicity but a higher capacity for proliferation, home to lymphoid tissues, and persist in vivo (Gattinoni et al., 2005). Therefore the entire expanded CIK cell population may represent an ideal cell population for adoptive immunotherapy. The CD3+CD56+ subset could deliver potent cytotoxicity for the immediate destruction of malignant cells while the less potent CD3+CD56− cell subset could proliferate and persist in vivo for a longer duration and therefore provide a continuous source of cells with long term cytotoxic activity. In addition, the small CD3−CD56+ NK cell subset has been shown to efficiently lyse MHC class I-deficient tumor targets that escape T cell recognition. The spleen is known to be the best organ to detect T cells, whereas NK cells are best detectable in BM (Varga et al., 2010). At the time of inoculation most of CIK cells showed CD3+CD56− followed by CD3+CD56+ and CD3+CD56− phenotype. Most of CD3+CD56− CIK cells expressed CD8 and CD25 rather than CD4 molecules. After injection we detected CD3+CD56− but no CD3+CD56+ or CD3−CD56+ cells at the time of analysis. In addition we demonstrated that persisting CD3+CD56− cells were CD4+ rather than CD8+, suggesting that the phenotype of CIK cells at the time of adoptive transfer reverted without cytokine stimulation in vivo and resulted in an expansion of CD3+CD4+ CIK cells in vivo.
It was shown that adoptive transfer of allogeneic CIK cells in a murine model caused minimal GvHD-like symptoms as CIK cells infiltrated GvHD targeted tissues to a lesser degree and more transiently than conventional T cells (Baker et al., 2001; Verneris et al., 2001; Beilhack et al., 2005; Nishimura et al., 2008). Irradiated Balb/c mice tolerated up to 20 × 106 day 14 CIK cells which caused minimal GvHD whereas as few as 2.5 × 106 splenocytes, as a T cell equivalent in mice, induced acute lethal GvHD. As one reason, CIK cells showed a slower division rate, higher susceptibility to apoptosis, increased IFNγ release, and reduced expression of homing molecules (Duffner et al., 2003; Welniak et al., 2004; Nishimura et al., 2008). IFNγ has a protective effect against GvHD at early time points after transplantation. Homing molecules and chemokine receptors direct alloreactive cells toward GvHD targeted inflamed tissues. Irradiation induced tissue inflammation in our experimental setting. However, we observed only mild tissue damage in mice when infused CIK cells persisted and expanded at these sites. Affected mice showed minimal weight loss, and were somewhat hunched and scrubby.
In conclusion, human CIK cells traffic and survive in a murine recipient for prolonged periods of time with minimal xenogenic GvHD. CIK cells retain strong anti-leukemia and anti-tumor activity and homing capacity to leukemia and tumor sites. Therefore, CIK cells may be an effective alternative to prevent or treat leukemia or sarcoma relapse after allo-SCT. From our findings, we expect that the MRD state will be most susceptible to immunotherapy. Therefore diagnostics for MRD and preemptive immunotherapy using CIK cells to prevent relapse in patients with leukemia or sarcoma should be considered early after allo-SCT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the “Förderung Nachwuchforscher 2009 Frankfurt (Beschluss F 13/09, R88/2009),” the Edith von Heyden-Vermächtnis 2007 (Eva Rettinger), the subproject (TP 2.1) of the “Translational Sarcoma Research Network” of the Bundesministerium für Bildung und Forschung (No: 01GM0872), Germany, the Else Kröner-Fresenius-Stiftung (P65/09//A112/09; Peter Bader; Eva Rettinger), the Ernst-Schering-Foundation (Andreas Volk), and the LOEWE Center for Cell and Gene Therapy Frankfurt/Main funded by: Hessisches Ministerium für Wissenschaft und Kunst (HMWK) funding reference number: III L 4- 518/17.004 (2010; Peter Bader; Thomas Klingebiel) for funding of this study.
Authorship and Disclosures
Eva Rettinger, Vida Meyer, Hermann Kreyenberg, Peter Bader acquired, analyzed, and helped interpret data. Andreas Volk, Selim Kuçi, Andre Willasch, Ewa Koscielniak, Simone Fulda, Winfried S. Wels, Halvard Boenig, Thomas Klingebiel, participated/contributed in/to the acquisition and interpretation of data. All authors reviewed and approved the final version of the manuscript.
References
Agliano, A., Martin-Padura, I., Mancuso, P., Marighetti, P., Rabascio, C., Pruneri, G., Shultz, L. D., and Bertolini, F. (2008). Human acute leukemia cells injected in NOD/LtSz-scid/IL-2R gamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int. J. Cancer 123, 2222–2227.
Alvarnas, J. C., Linn, Y. C., Hope, E. G., and Negrin, R. S. (2001). Expansion of cytotoxic CD3(+)CD56(+) cells from peripheral blood progenitor cells of patients undergoing autologous hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 7, 216–222.
Bader, P., Kreyenberg, H., Hoelle, W., Dueckers, G., Handgretinger, R., Lang, P., Kremens, B., Dilloo, D., Sykora, K. W., Schrappe, M., Niemeyer, C., von Stackelberg, A., Gruhn, B., Henze, G., Greil, J., Niethammer, D., Dietz, K., Beck, J. F., and Klingebiel, T. (2004). Increasing mixed chimerism is an important prognostic factor for unfavorable outcome in children with acute lymphoblastic leukemia after allogeneic stem-cell transplantation: possible role for pre-emptive immunotherapy? J. Clin. Oncol. 22, 1696–1705.
Baker, J., Verneris, M. R., Ito, M., Shizuru, J. A., and Negrin, R. S. (2001). Expansion of cytolytic CD8(+) natural killer T cells with limited capacity for graft-versus-host disease induction due to interferon gamma production. Blood 97, 2923–2931.
Beilhack, A., Schulz, S., Baker, J., Beilhack, G. F., Wieland, C. B., Herman, E. I., Baker, E. M., Cao, Y. A., Contag, C. H., and Negrin, R. S. (2005). In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood 106, 1113–1122.
Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737.
Duffner, U., Lu, B., Hildebrandt, G. C., Teshima, T., Williams, D. L., Reddy, P., Ordemann, R., Clouthier, S. G., Lowler, K., Liu, C., Gerard, C., Cooke, K. R., and Ferrara, J. L. M. (2003). Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp. Hematol. 31, 897–902.
Edinger, M., Cao, Y. A., Verneris, M. R., Bachmann, M. H., Contag, C. H., and Negrin, R. S. (2003). Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood 101, 640–648.
Feuring-Buske, M., Gerhard, B., Cashman, J., Humphries, R. K., Eaves, C. J., and Hogge, D. E. (2003). Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia 17, 760–763.
Gattinoni, L., Klebanoff, C. A., Palmer, D. C., Wrzesinski, C., Kerstann, K., Yu, Z. Y., Finkelstein, S. E., Theoret, M. R., Rosenberg, S. A., and Restifo, N. P. (2005). Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8(+) T cells. J. Clin. Invest. 115, 1616–1626.
Hazelrigg, M. R., Hirsch, J. I., and Merchant, R. E. (2002). Distribution of adoptively transferred, tumor-sensitized lymphocytes in the glioma-bearing rat. J. Neurooncol. 60, 143–150.
Helms, M. W., Prescher, J. A., Cao, Y. A., Schaffert, S., and Contag, C. H. (2010). IL-12 enhances efficacy and shortens enrichment time in cytokine-induced killer cell immunotherapy. Cancer Immunol. Immunother. 59, 1325–1334.
Hoyle, C., Bangs, C. D., Chang, P., Kamel, O., Mehta, B., and Negrin, R. S. (1998). Expansion of Philadelphia chromosome-negative CD3(+)CD56(+) cytotoxic cells from chronic myeloid leukemia patients: in vitro and in vivo efficacy in severe combined immunodeficiency disease mice. Blood 92, 3318–3327.
Hudson, W. A., Li, Q., Le, C., and Kersey, J. H. (1998). Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia 12, 2029–2033.
Introna, M., Borleri, G., Conti, E., Franceschetti, M., Barbui, A. M., Broady, R., Dander, E., Gaipa, G., d’Amico, G., Biagi, E., Parma, M., Pogliani, E. M., Spinelli, O., Baronciani, D., Grassi, A., Golay, J., Barbui, T., Biondi, A., and Rambaldi, A. (2007). Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: a phase I study. Haematologica 92, 952–959.
Ishikawa, F., Yasukawa, M., Lyons, B., Yoshida, S., Miyamoto, T., Yoshimoto, G., Watanabe, T., Akashi, K., Shultz, L. D., and Harada, M. (2005). Development of functional human blood and immune systems in NOD/SCID/IL2 receptor gamma chain(null) mice. Blood 106, 1565–1573.
Ito, M., Hiramatsu, H., Kobayashi, K., Suzue, K., Kawahata, M., Hioki, K., Ueyama, Y., Koyanagi, Y., Sugamura, K., Tsuji, K., Heike, T., and Nakahata, T. (2002). NOD/SCID/gamma(null)(c) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100, 3175–3182.
Kawano, N., Ishikawa, F., Shimoda, K., Yasukawa, M., Nagafuji, K., Miyamoto, T., Baba, E., Tanaka, T., Yamasaki, S., Gondo, H., Otsuka, T., Ohshima, K., Shultz, L. D., Akashi, K., and Harada, M. (2005). Efficient engraftment of primary adult T-cell leukemia cells in newborn NOD/SCID/beta 2-microglobulin(null) mice. Leukemia 19, 1384–1390.
Kim, H. M., Kang, J. S., Lim, J., Kim, J. Y., Kim, Y. J., Lee, S. J., Song, S., Hong, J. T., Kim, Y., and Han, S. B. (2009). Antitumor activity of cytokine-induced killer cells in nude mouse xenograft model. Arch. Pharm. Res. 32, 781–787.
Kim, H. M., Kang, J. S., Lim, J., Park, S. K., Lee, K., Yoon, Y. D., Lee, C. W., Lee, K. H., Han, G., Yang, K. H., Kim, Y. J., Kim, Y., and Han, S. B. (2007a). Inhibition of human ovarian tumor growth by cytokine-induced killer cells. Arch. Pharm. Res. 30, 1464–1470.
Kim, H. M., Lim, J., Park, S. K., Kang, J. S., Lee, K., Lee, C. W., Lee, K. H., Yun, M. J., Yang, K. H., Han, G., Kwon, S. W., Kim, Y., and Han, S. B. (2007b). Antitumor activity of cytokine-induced killer cells against human lung cancer. Int. Immunopharmacol. 7, 1802–1807.
Kim, H. M., Lim, J., Yoon, Y. D., Ahn, J. M., Kang, J. S., Lee, K., Park, S. K., Jeong, Y. J., Kim, J. M., Han, G., Yang, K. H., Kim, Y. J., Kim, Y., and Han, S. B. (2007c). Anti-tumor activity of ex vivo expanded cytokine-induced killer cells against human hepatocellular carcinoma. Int. Immunopharmacol. 7, 1793–1801.
Klingebiel, T., Boos, J., Beske, F., Hallmen, E., Int-Veen, C., Dantonello, T., Treuner, J., Gadner, H., Marky, I., Kazanowska, B., and Koscielniak, E. (2008). Treatment of children with metastatic soft tissue sarcoma with oral maintenance compared to high dose chemotherapy: report of the HD CWS-96 trial. Pediatr. Blood Cancer 50, 739–745.
Kornacker, M., Moldenhauer, G., Herbst, M., Weilguni, E., Tita-Nwa, F., Harter, C., Hensel, M., and Ho, A. D. (2006). Cytokine-induced killer cells against autologous CLL: direct cytotoxic effects and induction of immune accessory molecules by interferon-gamma. Int. J. Cancer 119, 1377–1382.
Koscielniak, E., Klingebiel, T. H., Peters, C., Hermann, J., Burdach, S. T., BenderGotze, C., MullerWeihrich, S. T., and Treuner, J. (1997). Do patients with metastatic and recurrent rhabdomyosarcoma benefit from high-dose therapy with hematopoietic rescue? Report of the German/Austrian pediatric bone marrow transplantation group. Bone Marrow Transplant. 19, 227–231.
Kuci, S., Kuci, Z., Kreyenberg, H., Deak, E., Putsch, K., Huenecke, S., Amara, C., Koller, S., Rettinger, E., Grez, M., Koehl, U., Latifi-Pupovci, H., Henschler, R., Tonn, T., von Laer, D., Klingebiel, T., and Bader, P. (2010a). CD271 antigen defines a subset of multipotent stromal cells with immunosuppressive and lymphohematopoietic engraftment-promoting properties. Haematologica 95, 651–659.
Kuci, S., Rettinger, E., Voss, B., Weber, G., Stais, M., Kreyenberg, H., Willasch, A., Kuci, Z., Koscielniak, E., Kloss, S., von Laer, D., Klingebiel, T., and Bader, P. (2010b). Efficient lysis of rhabdomyosarcoma cells by cytokine-induced killer cells: implications for adoptive immunotherapy after allogeneic stem cell transplantation. Haematologica 95, 1579–1586.
Li, Y., Qu, Y. H., Wu, Y. F., Wang, X. P., Wei, J., Huang, W. G., Zhou, D. H., Fang, J. P., Huang, K., and Huang, S. L. (2011). Bone marrow mesenchymal stem cells reduce the antitumor activity of cytokine-induced killer/natural killer cells in K562 NOD/SCID mice. Ann. Hematol. 90, 873–885.
Lu, P. H., and Negrin, R. S. (1994). A novel population of expanded human Cd3+Cd56+ cells derived from T-cells with potent in-vivo antitumor-activity in mice with severe combined immunodeficiency. J. Immunol. 153, 1687–1696.
Meyerrose, T. E., Herrbrich, P., Hess, D. A., and Nolta, J. A. (2003). Immune-deficient mouse models for analysis of human stem cells. Biotechniques 35, 1262–1272.
Nakamura, Y., Ito, M., Yamamoto, T., Yan, X. Y., Yagasaki, H., Kamachi, Y., Kudo, K., and Kojima, S. (2005). Engraftment of NOD/SCID/gamma c(null) mice with multilineage neoplastic cells from patients with juvenile myelomonocytic leukaemia. Br. J. Haematol. 130, 51–57.
Nishimura, R., Baker, J., Beilhack, A., Zeiser, R., Olson, J. A., Sega, E. I., Karimi, M., and Negrin, R. S. (2008). In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood 112, 2563–2574.
Olioso, P., Giancola, R., Di Riti, M., Contento, A., Accorsi, P., and Iacone, A. (2009). Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: a pilot clinical trial. Hematol. Oncol. 27, 130–139.
Perez, E. A., Kassira, N., Cheung, M. C., Koniaris, L. G., Neville, H. L., and Sola, J. E. (2011). Rhabdomyosarcoma in children: a SEER population based study. J. Surg. Res. 170, E243–E251.
Petak, I., Douglas, L., Tillman, D. M., Vernes, R., and Houghton, J. A. (2000). Pediatric rhabdomyosarcoma cell lines are resistant to Fas-induced apoptosis and highly sensitive to TRAIL-induced apoptosis. Clin. Cancer Res. 6, 4119–4127.
Petak, I., Vernes, R., Szucs, K. S., Anozie, M., Izeradjene, K., Douglas, L., Tillman, D. M., Phillips, D. C., and Houghton, J. A. (2003). A caspase-8-independent component in TRAIL/Apo2L-induced cell death in human rhabdomyosarcoma cells. Cell Death Differ. 10, 729–739.
Pievani, A., Borleri, G., Pende, D., Moretta, L., Rambaldi, A., Golay, J., and Introna, M. (2011). Dual-functional capability of CD3(+)CD56(+) CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood 118, 3301–3310.
Pongers-Willemse, M. J., Seriu, T., Stolz, F., d’Aniello, E., Gameiro, P., Pisa, P., Gonzalez, M., Bartram, C. R., Panzer-Grumayer, E. R., Biondi, A., San Miguel, J. F., and van Dongen, J. J. M. (1999). Primers and protocols for standardized detection of minimal residual disease in acute lymphoblastic leukemia using immunoglobulin and T cell receptor gene rearrangements and TAL1 deletions as PCR targets – report of the BIOMED-1 CONCERTED ACTION: investigation of minimal residual disease in acute leukemia. Leukemia 13, 110–118.
Powell, D. J., Dudley, M. E., Robbins, P. F., and Rosenberg, S. A. (2005). Transition of late-stage effector T cells to CD27(+) CD28(+) tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood 105, 241–250.
Qiu, H. Y., Xue, Y. Q., Zhang, J., Pan, J. L., Dai, H. P., Wu, Y. F., Wang, Y., Chen, S. N., and Wu, D. P. (2008). Establishment and characterization of a new human acute myelocytic leukemia cell line SH-2 with a loss of Y chromosome, a derivative chromosome 16 resulting from an unbalanced translocation between chromosomes 16 and 17, monosomy 17, trisomy 19, and p53 alteration. Exp. Hematol. 36, 1487–1495.
Rettinger, E., Kuci, S., Naumann, I., Becker, P., Kreyenberg, H., Anzaghe, M., Willasch, A., Koehl, U., Bug, G., Ruthardt, M., Klingebiel, T., Fulda, S., and Bader, P. (2012). The cytotoxic potential of interleukin-15 stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy 14, 91–103.
Rettinger, E., Willasch, A. M., Kreyenberg, H., Borkhardt, A., Holter, W., Kremens, B., Strahm, B., Woessmann, W., Mauz-Koerholz, C., Gruhn, B., Burdach, S., Albert, M. H., Schlegel, P. G., Klingebiel, T., and Bader, P. (2011). Preemptive immunotherapy in childhood acute myeloid leukemia for patients showing evidence of mixed chimerism after allogeneic stem cell transplantation. Blood 118, 5681–5688.
Sangiolo, D. (2011). Cytokine induced killer cells as promising immunotherapy for solid tumors. J. Cancer 2, 363–368.
Sangiolo, D., Mesiano, G., Carnevale-Schianca, F., Piacibello, W., Aglietta, M., and Cignetti, A. (2009). Cytokine induced killer cells as adoptive immunotherapy strategy to augment graft versus tumor after hematopoietic cell transplantation. Expert Opin. Biol. Ther. 9, 831–840.
Scharf, S. J., Smith, A. G., Hansen, J. A., Mcfarland, C., and Erlich, H. A. (1995). Quantitative-determination of bone-marrow transplant engraftment using fluorescent polymerase chain-reaction primers for human identity markers. Blood 85, 1954–1963.
Scheffold, C., Kornacker, M., Scheffold, Y. C., Contag, C. H., and Negrin, R. S. (2002). Visualization of effective tumor targeting by CD8+natural killer T cells redirected with bispecific antibody F(ab’)(2)HER2xCD3. Cancer Res. 62, 5785–5791.
Schmidtwolf, I. G. H., Lefterova, P., Johnston, V., Huhn, D., Blume, K. G., and Negrin, R. S. (1994). Propagation of large numbers of T-cells with natural-killer-cell markers. Br. J. Haematol. 87, 453–458.
Schmidtwolf, I. G. H., Negrin, R. S., Kiem, H. P., Blume, K. G., and Weissman, I. L. (1991). Use of a SCID mouse human lymphoma model to evaluate cytokine-induced killer-cells with potent antitumor cell-activity. J. Exp. Med. 174, 139–149.
Shultz, L. D., Ishikawa, F., and Greiner, D. L. (2007). Humanized mice in translational biomedical research. Nat. Rev. Immunol. 7, 118–130.
Shultz, L. D., Lyons, B. L., Burzenski, L. M., Gott, B., Chen, X. H., Chaleff, S., Kotb, M., Gillies, S. D., King, M., Mangada, J., Greiner, D. L., and Handgretinger, R. (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma(null) mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489.
Siegler, U., Kalberer, C. P., Nowbakht, P., Sendelov, S., Meyer-Monard, S., and Wodnar-Filipowicz, A. (2005). Activated natural killer cells from patients with acute myeloid leukemia are cytotoxic against autologous leukemic blasts in NOD/SCID mice. Leukemia 19, 2215–2222.
Skitzki, J., Craig, R. A., Okuyama, R., Knibbs, R. N., McDonagh, K., Chang, A. E., and Stoolman, L. M. (2004). Donor cell cycling, trafficking, and accumulation during adoptive immunotherapy for murine lung metastases. Cancer Res. 64, 2183–2191.
Sweeney, T. J., Mailander, V., Tucker, A. A., Olomu, A. B., Zhang, W. S., Cao, Y. A., Negrin, R. S., and Contag, C. H. (1999). Visualizing the kinetics of tumor-cell clearance in living animals. Proc. Natl. Acad. Sci. U.S.A. 96, 12044–12049.
Takayama, T., Sekine, T., Makuuchi, M., Yamasaki, S., Kosuge, T., Yamamoto, J., Shimada, K., Sakamoto, M., Hirohashi, S., Ohashi, Y., and Kakizoe, T. (2000). Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet 356, 802–807.
Thorne, S. H., Negrin, R. S., and Contag, C. H. (2006). Synergistic antitumor effects of immune cell-viral biotherapy. Science 311, 1780–1784.
Varga, N. L., Bárcena, A., Fomin, M. E., and Muench, M. O. (2010). Detection of human hematopoietic stem cell engraftment in the livers of adult immunodeficient mice by an optimized flow cytometric method. Stem Cell Stud. 1, e5.
Verneris, M. R., Ito, M., Baker, E., Arshi, A., Negrin, R. S., and Shizuru, J. A. (2001). Engineering hematopoietic grafts: purified allogeneic hematopoietic stem cells plus expanded CD8(+) NK-T cells in the treatment of lymphoma. Biol. Blood Marrow Transplant. 7, 532–542.
Verneris, M. R., Kornacker, M., Mailander, V., and Negrin, R. S. (2000). Resistance of ex vivo expanded CD3(+)CD56(+) T cells to Fas-mediated apoptosis. Cancer Immunol. Immunother. 49, 335–345.
Vormoor, J., Baersch, G., Decker, S., Hotfilder, M., Schafer, K. L., Pelken, L., Rube, C., Van Valen, F., Jurgens, H., and Dockhorn-Dworniczak, B. (2001). Establishment of an in vivo model for pediatric Ewing tumors by transplantation into NOD/scid mice. Pediatr. Res. 49, 332–341.
Wajchman, H. J., Pierce, C. W., Varma, V. A., Issa, M. M., Petros, J., and Dombrowski, K. E. (2004). Ex vivo expansion of CD8(+)CD56(+) and CD8(+)CD56(-) natural killer T cells specific for MUC1 mucin. Cancer Res. 64, 1171–1180.
Wang, F. S., Liu, M. X., Zhang, B., Shi, M., Lei, Z. Y., Sun, W. B., Du, Q. Y., and Chen, J. M. (2002). Antitumor activities of human autologous cytokine-induced killer (CIK) cells against hepatocellular carcinoma cells in vitro and in vivo. World J. Gastroenterol. 8, 464–468.
Watanabe, S., Ohta, S., Yajima, M., Terashima, K., Ito, M., Mugishima, H., Fujiwara, S., Shimizu, K., Honda, M., Shimizu, N., and Yamamoto, N. (2007). Humanized NOWSCID/IL2R gamma(null) mice transplanted with hematopoietic stem cells under nonmyeloablative conditions show prolonged life spans and allow detailed analysis of human immunodeficiency virus type 1 pathogenesis. J. Virol. 81, 13259–13264.
Keywords: preclinical, NSG mice, CIK cells, rhabdomyosarcoma, leukemia, immunotherapy
Citation: Rettinger E, Meyer V, Kreyenberg H, Volk A, Kuçi S, Willasch A, Koscielniak E, Fulda S, Wels WS, Boenig H, Klingebiel T and Bader P (2012) Cytotoxic capacity of IL-15-stimulated cytokine-induced killer cells against human acute myeloid leukemia and rhabdomyosarcoma in humanized preclinical mouse models. Front. Oncol. 2:32. doi: 10.3389/fonc.2012.00032
Received: 19 January 2012; Paper pending published: 11 February 2012;
Accepted: 19 March 2012; Published online: 09 April 2012.
Edited by:
Charles G. Mullighan, St. Jude Children’s Research Hospital, USAReviewed by:
David Shook, St. Jude Children’s Research Hospital, USAMichael R. Verneris, University of Minnesota, USA
Copyright: © 2012 Rettinger, Meyer, Kreyenberg, Volk, Kuçi, Willasch, Koscielniak, Fulda, Wels, Boenig, Klingebiel and Bader. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Eva Rettinger and Peter Bader, Department of Pediatric Hematology, Oncology and Hemostaseology, University Children’s Hospital of Frankfurt/Main, Goethe-University Frankfurt/Main, Theodor-Stern-Kai 7, 60590 Frankfurt/Main, Germany. e-mail:ZXZhLnJldHRpbmdlckBrZ3UuZGU=;cGV0ZXIuYmFkZXJAa2d1LmRl