

 and
and - 1 Department of Experimental Oncology, European Institute of Oncology, Milan, Italy
- 2 IFOM, The FIRC Institute of Molecular Oncology Foundation, Milan, Italy
Acute myeloid leukemia (AML) is, as other types of cancer, a genetic disorder of somatic cells. The detection of somatic molecular abnormalities that may cause and maintain AML is crucial for patient stratification. The development of mutation-specific therapeutic interventions will hopefully increase cure rates and improve patients’ quality of life. This review illustrates how next generation sequencing technologies are changing the study of cancer genomics of adult AML patients.
Acute myeloid leukemia (AML) is the most frequent hematological malignancy in adults, with an estimated worldwide annual incidence of three to four cases per 100,000 people. Despite intensive research for new therapies and prognostic markers, it is still a disease with a highly variable prognosis among patients and a high mortality rate. Indeed, less than 50% of adult AML patients have a 5-year overall survival rate (OS), and, in the elderly, only 20% survive 2 years (Gregory et al., 2009).
In general, both prognosis and treatment choice for AML patients are based on the presence or absence of specific genetic alterations, which determine AML classification in three risk based-categories: favorable, intermediate, and unfavorable. This classification is usually based on cytogenetic information. AML with a favorable prognosis includes patients with inv(16) (that generates the CBFB–MYH11 fusion protein), t(15;17) (that generates the PML–RARA fusion protein), or t(8;21) (that generates the AML1–ETO fusion protein). The 5-year OS rate of patients in this category is 55%. The unfavorable subgroup includes patients with monosomy 5, monosomy 7, 11q23 (that generates MLL-highly variable breakpoints on the partner fusion protein), or complex cytogenetics, and the 5-year OS rate is reduced to 11%. Favorable prognosis AML patients are usually treated with primary chemotherapy, while high-risk patients are considered for allogenic stem cell transplantation in first remission if a suitable donor is found. The intermediate subgroup includes normal karyotype (NK) AML patients. Patients belonging to this group have a 5-year OS rate ranging between 24 and 42%, depending on the study, but it is still largely unclear what might be the best therapeutic strategy for them (Gregory et al., 2009; Tefferi et al., 2009).
More recently, other mutations associated to AML have been identified (FLT3, CEBP, NPM1, IDH1/2) and their prognostic power investigated particularly in the intermediate risk category. FLT3–ITD and CEBPA mutations seem to associate with a bad prognosis, while NPM1 and IDH1/2 are controversial. However, several challenges still lie ahead and markers are needed to predict prognosis and sensibility to treatment.
Understanding the genetic lesions associated to AML is also important in order to adjust for specific therapies. For example, Acute Promyelocytic Leukemia (APL, one of the AML subtypes) is treated with a combination of the differentiation-inducing agent ATRA (all-trans retinoic acid) and chemotherapy, which induces long-term remissions or cure in 75–85% of patients. Some of the newly described genetic lesions (e.g., FLT3) may be targeted by specific inhibitors which have shown anti-leukemic efficacy in preliminary studies, and are now currently being evaluated in phase III clinical trials.
The advent of second- (or next) generation sequencing technologies has dramatically accelerated biological and biomedical discoveries by enabling comprehensive analysis of genomes, transcriptomes, and DNA–protein interactions. These technologies allow the identification of cancer-associated mutations at a single-base resolution in an unbiased manner, and will likely revolutionize our understanding of cancer. A comprehensive description of somatic mutations in cancer is essential as it can (i) shed light on tumor initiation and progression mechanisms, (ii) assist patient stratification for prognosis and treatment choice, and (iii) allow the identification of new genes that can be specifically targeted by therapy.
Massive parallel sequencing is now discovering a growing number of submicroscopic somatic mutations with prognostic significance. These, together with the primary somatic genetic abnormalities already identified, are enabling the drawing of patient mutation profiles and will hopefully have a major impact on the clinical management of AML, not only as independent prognostic factors, but also as the foundation of genome-informed personalized cancer treatments.
In this review, we will examine the somatic mutations recently identified using next generation sequencing (NGS). First, we will describe which types of mutations can be detected by sequencing and comment on the pros and cons of different technological approaches (synthesized in Table 3). Then, we will describe all the identified mutations and the subsets of recurring mutations according to sequencing technology and mutation type (cataloged in Tables 1 and 2). Finally, we will discuss future perspectives in the use of NGS technologies in the clinical setting and existing open challenges.
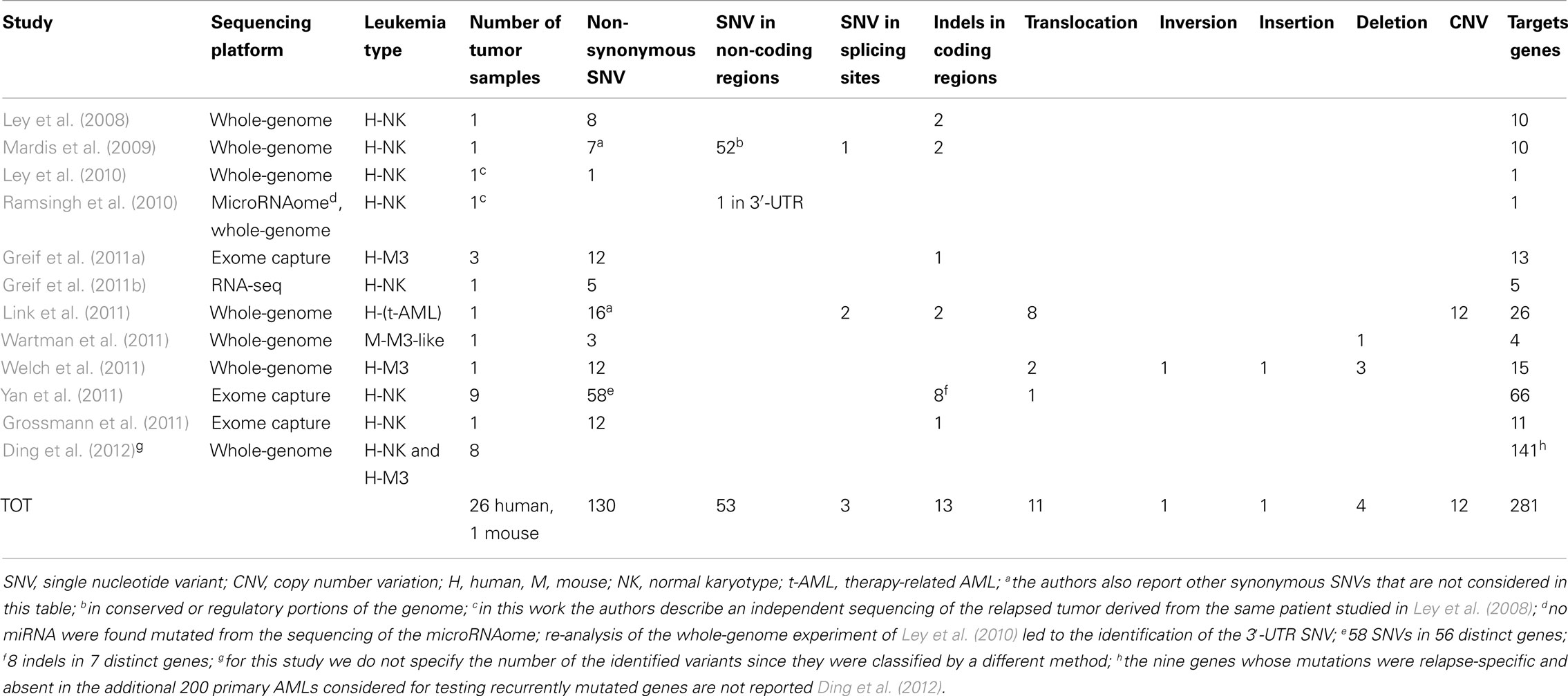
Table 1. Numerical summary of identified and validated variants found in adult AMLs by NGS technologies.
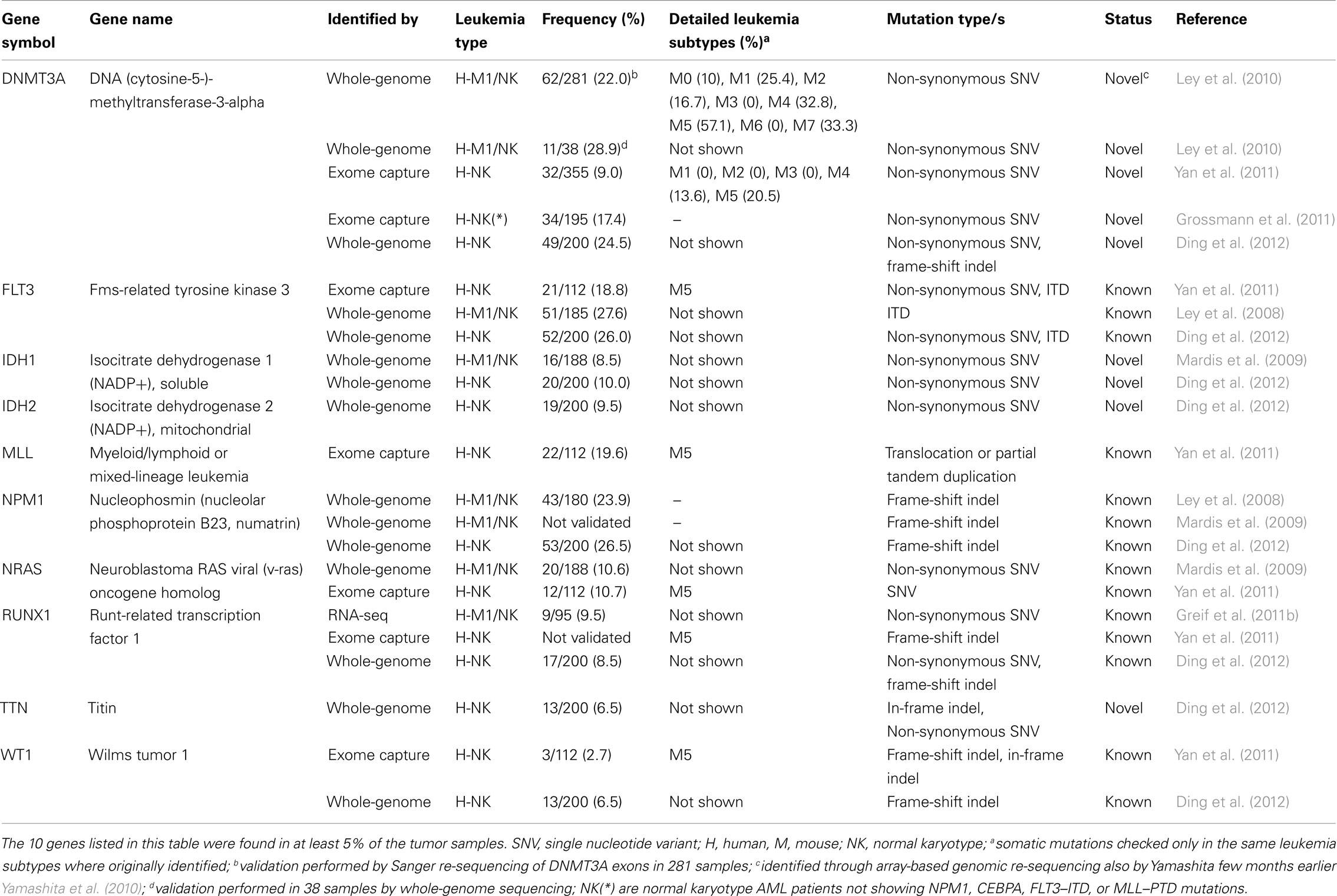
Table 2. Catalog of genes targeted by recurrent molecular genetic abnormalities in adult AMLs as detected by NGS technologies.
Massive Parallel Sequencing Approaches for Mutational Analysis in AML
To identify AML somatic mutations by NGS, sequencing is usually performed on DNA or RNA obtained from bone marrow samples (with high level of tumor cellularity) and normal tissues (skin biopsies or peripheral blood) from the same AML patient when in clinical remission. This approach aims to define somatic variants, including single nucleotide variants (SNVs), short deletions and insertions (indels), structural variants (SVs) such as translocations, long insertions or deletions, and copy number variations (CNVs), which are present in the tumor sample and absent in the matched control sample. Usually, the sequences from tumor and normal samples are mapped to the reference genome and the sequence changes (variants) that differ from the reference genome are identified. Variants present in both tumor and control samples (generally referred to as germline variants) and variants matching known single nucleotide polymorphisms (SNPs) are discarded.
All the identified variants are then validated by using an independent sequencing technology, for example DNA Sanger sequencing. Finally, the validated variants are usually tested on a large number of clinical samples, in order to determine their actual frequency and to identify recurrent mutations.
Currently, there are three experimental approaches, which are most frequently utilized to identify somatic mutations by NGS: whole-genome sequencing, exome-sequencing, and transcriptome-sequencing (also known as RNA-sequencing). Whole-genome sequencing allows the identification of the entire DNA sequence of a given sample, at single-base resolution level. Exome-sequencing, instead, is preceded by an exome capture step that selects the coding regions of the genome (representing ∼1% of the genome). RNA-sequencing measures the transcriptome.
Sequencing is performed using either single-end or paired-end tags (PET). In PET, short and paired reads are obtained from the ends of DNA fragments for sequencing. The use of PET in genome re-sequencing has advantages over the use of single tags, as it allows higher mapping specificity and the identification of small and large insertions, deletions, and translocations, which is not possible using single-end tags.
The two parameters to take into considerations to understand data analysis and interpretation are the “coverage” and the “read lengths.” Coverage is the number of tags aligned to each base of the reference genome. A high coverage is desired because it can overcome errors in base calling and assembly, and it can reduce false positives. Longer read lengths are more easily mapped to the reference genome, increasing the proportion of the genome that is mappable. Moreover, longer read lengths are essential for the detection of small indels.
Each of these techniques has pros and cons (see Table 3). Whole-genome sequencing allows identification of all the possible variants at once, and it is the best method to study chromosomal rearrangements; however, it is expensive ($5000–$15,000 per sample, depending on the sequencing services and coverage) and requires a high amount of starting material (usually 1 μg of genomic DNA). Exome-sequencing reduces costs ($1000–$2000 per sample), but not the amount of starting material (usually around 3 μg of genomic DNA), and allows high coverage in coding regions. Exome-sequencing relies on a capture step that may not have uniform efficiency, and the identification of chromosomal rearrangements is restricted to exonic regions. RNA-sequencing is capable of detecting variants present in the transcriptome and fusion genes of expressed genes (Maher et al., 2009). RNA-sequencing, which necessitates 0.1–4 μg of RNA as starting material, further reduces costs ($300–$500 per sample); importantly, while allowing identification of tumor-specific fusion transcripts or mRNA-splice variants, it also offers information on gene expression levels. There are three main disadvantages, however, in using RNA-sequencing to detect somatic variants. First, the identification of the corresponding normal sample is challenging and, even if one could successfully identify it, gene expression in cancer cells is altered from that of normal cells. Second, SNVs and indels within genes that are transcribed at very low levels or in those for which mutations may induce mRNA degradation may be missed. Finally, the chance of errors due to reverse transcriptase and the phenomenon of RNA editing (Li et al., 2011) can make these data difficult to interpret (Meyerson et al., 2010).

Table 3. Comparison of pros and cons of whole-genome sequencing, exome-sequencing, and RNA-sequencing.
Whole-Genome Sequencing
The first demonstration of the possibility to identify somatic mutations in cancer genomes using sequencing technologies was obtained in a patient with AML (NK, M1 subtype; Ley et al., 2008). The authors, using single-end whole-genome sequencing, identified mutations in the entire genome but decided to validate only those which (i) had occurred in coding sequences, (ii) were non-synonymous, or (iii) were predicted to alter splicing sites (all the 181 identified variants and 28 manually selected indels). In this first study, the percentage of computationally identified false positive variants was quite high, since only 5% of the identified mutations could be validated. The authors discovered 10 non-synonymous somatic mutations: eight novel SNVs and two previously described indels (i.e., in NPM1 and FLT3; Table 1). They sequenced the 8 novel SNVs in 187 additional AML cases but could not find any of these variants.
In the following year, the same group sequenced another patient with cytogenetically normal AML-M1 (Mardis et al., 2009), using paired-end whole-genome sequencing. In this second attempt, it was decided to validate not only SNVs and indels present in coding regions and in consensus-splice site regions, but also those present in non-coding genes, in conserved regions, or in regions having regulatory potentials. Ultimately, they identified 7 non-synonymous SNVs, 1 splice site SNV, 2 indels in coding regions, and 52 somatic point mutations in conserved or regulatory portions of the genome (Table 1). They tested these mutations in additional 188 AML samples and found that the mutations on the IDH1 gene were also present in other AML samples at a frequency of ∼10% (Table 2). Furthermore, one of the 52 mutations found in conserved or regulatory portions of the genome was detected in one additional AML tumor. Previously identified mutations, such as NPM1 and NRAS, were also found amongst the mutations within coding regions.
One year later, the researchers re-sequenced the genome from the relapsed AML and control samples of the original patient reported in 2008 (Ley et al., 2008), using paired-end sequencing in order to obtain a higher depth of coverage (Ley et al., 2010). They found, among several other non-synonymous new mutations (not described) a 1-base pair (bp) deletion in the DNA methyltransferase-3-alpha (DNMT3A) gene (identified through array-based genomic re-sequencing just few months before; Yamashita et al., 2010; Table 1). To assess DNMT3A mutation frequency, the authors amplified and sequenced by Sanger technique the 24 exons of DNMT3A in 188 additional de novo AML samples (and their matched normal counterparts) and in other 93 AML samples (without corresponding normal controls). They ascertained that DNMT3A variants were present in 62 of the total 281 AML DNA samples examined (22%), definitely proving that DNMT3A is recurrently mutated in AML. All the variations identified in the 188 matched-sample validation set were confirmed to derive from somatic mutational events, since DNMT3A mutations were not found in the normal sample set. Two distinctive categories of DNMT3A mutations were found: highly frequent SNVs, producing variations in the R882 amino acid residue, and ∼20 other different widely distributed missense mutations.
From this study, a mutually exclusive relationship was found between DNMT3A mutations and the three classical AML translocations [t(15;17), t(8;21), and inv(16)], which correlate with low cytogenetic risk. The same had been already observed for mutations of NPM1, IDH1, and IDH2 that usually do not appear in AML cells when one of the above-mentioned chromosomal rearrangements is present. However, an association between the DNMT3A mutation and mutations of these genes, and also FLT3, was shown very clearly. Co-occurrence of DNMT3A mutations with MLL genomic variants, present in 11 of the 281 patients examined, was also never observed. Variations in the DNMT3A genomic sequence were frequently found enriched in NK samples (44/119 NK samples, 37%). Indeed, the presence of DNMT3A mutations, concomitantly with variations in FLT3, NPM1, IDH1, and IDH2, contributed to identify a group of patients that strictly associated with an intermediate cytogenetic risk, and to specifically exclude patients with an adverse prognosis. Finally, DNMT3A mutations were found associated with poor event-free and overall survival, regardless of NPM1 status, age, and cytogenetic risk; patients also carrying FLT3 tandem duplication had a significantly worse outcome. So far, the DNMT3A mutation is the most frequent novel genomic variation in AMLs identified and characterized thanks to the application of massive parallel sequencing technologies (Table 2).
Welch et al. (2011) have recently described a successful clinical application of whole genomic sequencing, presenting the case of a patient with a difficult diagnosis of AML: the patient appeared to have a hyper-granular APL-like leukemia, but it was impossible to detect the PML–RARA oncogene by routine cytogenetic profiling or FISH, and PCR was not done. The correct identification of an APL is a critical requirement since APLs are the only AMLs that can be cured without allogeneic stem cell transplantation. Given the complexity of this case, the authors decided to apply whole-genome sequencing to the patient’s leukemia cells (Table 1). This led to the identification of the insertion of a segment of chromosome 15 (containing the LOXL1 and PML genes) into the second intron of RARA on chromosome 17, generating the PML–RARA fusion gene and two other fusion genes: LOXL1–PML and RARA–LOXL1. In the end, the patient was correctly diagnosed with APL and got into remission after being treated with ATRA. Thus, whole-genome sequencing can detect translocations that may be missed by cytogenetic profiling. Indeed, by analyzing 11 other cases of AML with APL-resembling features, the authors also found that, in two of these, the PML–RARA fusion gene had derived from an insertional translocation instead of a translocation. In addition, Welch and colleagues identified, in the same tumor sample, the presence of 12 non-synonymous SNVs, 1 inversion, 2 additional translocations and 4 deletions. The frequencies of the 12 SNVs were consistent with the presence of two different leukemic clones.
Finally, Link et al. (2011) identified a novel cancer susceptibility gene by sequencing leukemic bone marrow and normal skin samples from a patient with therapy-related AML and multiple early onset primary tumors. They detected a germline deletion variant that had caused the elimination of exons 7–9 of the TP53 gene. Furthermore, the authors discovered 16 non-synonymous SNVs, 2 variants in splice sites, 2 indels in coding regions, 8 SVs, and 12 somatic copy number alterations (Table 1).
Whole-genome sequencing has been also used to find somatic mutations in mouse models of APL (Wartman et al., 2011). Wartman et al., in fact, identified three somatic non-synonymous SNVs in leukemia samples from a PML–RAR knock-in mouse (Table 1). One of the three mutations affected the Jak1 gene and recurred in 6 of the 89 additionally screened mice. An identical mutation in the human JAK1 gene had been already described in human APLs. Furthermore, the authors found a 150-kb somatic deletion on chromosome X affecting the Kdm6a gene. A similar mutation was also found in one of the 150 AML patients regarded as the human leukemia population of comparison.
Development of drug resistance has been linked to hundreds of gene mutations in experimental models, using in vitro cell lines or transgenic mice (e.g., MDR-1). There is no confirmation, however, of any of them having a specific role in acquired clinical resistance following anticancer therapy, or that they can be used as prognostic factors to predict treatment outcome. Thus, the molecular basis of chemoresistance in human tumors, including AMLs, remains largely unknown.
Recently, Ding et al. (2012) have reported the whole-genome analysis of primary/relapse tumor-pairs from 8 AML patients, using NGS technologies. This is the first report of an extensive search of tumor mutations in relapsing tumors. Initially, the authors analyzed each tumor pair using a sequence protocol that allows identification of high frequency mutations. They used a sequence coverage of ∼30×, corresponding to low cell detection sensitivity. With this approach, Ding and colleagues documented the existence of relapse-specific mutations in all the analyzed cases. The authors then looked for the presence of these relapse-specific mutations in the primary tumors of origin, using a sequence protocol that allows identification of low-frequency mutations (in this second phase the sequence coverage was ∼500×, which corresponds to a cell detection sensitivity of around 5%). Interestingly, under these experimental conditions, a few relapse-specific mutations could be also detected in the respective primary tumors. These data represent a direct demonstration that chemotherapy can induce the selection of rare tumor sub-populations harboring specific gene mutations (clonal selection). As clonal selection was not shown in three of the eight analyzed cases but some relapse-specific mutations were still found, alternative mechanisms of chemoresistance might have been present in these patients (the mutation could have been acquired during treatment). On the other hand, they might have been already present in the primary tumor, but had escaped identification due to the limited sensitivity of the detection assay (∼5%). Regrettably, the authors did not investigate whether the identified relapse-specific mutations were indeed responsible of the chemoresistance (i.e., whether they were chemoresistance-specific mutations). This study identified a total of 141 mutated genes present in primary AML, of which 129 were novel mutations in AML. Using 200 AML cases whose exomes were sequenced as part of the Cancer Genome Atlas AML project, Ding et al. identified 126 of the 129 novel mutations in other AML samples.
Exome-Sequencing
Most whole-genome sequencing analyses only focused on variants present in coding regions, as mutations in the coded portion of the genome are easier to interpret because of their putative impact on protein functions. This approach, although restrictive, has been nevertheless successful allowing the identification of many novel mutations. Since the publication of the first exome-sequencing study in 2009 (Ng et al., 2009), many groups have been reporting the use of exome-sequencing to identify mutations present in cancer or in other pathological conditions (Meyerson et al., 2010; Singleton, 2011 for reviews). Novel mutations identified by exome-sequencing in AML (Grossmann et al., 2011; Yan et al., 2011) and APL (Greif et al., 2011a) patients have been also recently published.
Yan et al. (2011) published exome-sequencing data from bone marrow and control tissues derived from nine patients with AML-M5. They validated 58 SNVs and 8 indels with Sanger sequencing, identifying 66 somatic mutations in 63 genes (Table 1). These somatic mutations included known variants (e.g., in NRAS and in FLT3) as well as the MLL–MLLT4 fusion gene. Other five AML-M5 cases without matched normal samples were sequenced and the authors focused on additional mutations occurring in the 63 identified genes. Furthermore, the authors checked all the sequence changes detected in the 63 genes in other 98 AML-M5 leukemia samples (94 newly diagnosed and 4 relapsed); these variants were not present in the control set, consisting of 509 normal samples from healthy donors, or in the matched control samples. In total 112 samples were tested and amongst these 14 genes were mutated, each in at least 2 of the 112 cases. Yan and colleagues selected 5 of these 14 genes (DNMT3A, ATP2A, C10orf2, CCND3, GATA2) plus a gene mutated only in one case (NSD1) and sequenced their entire coding regions in the 98 AML-M5 leukemia samples, discovering three different DNMT3A variants in ∼20% of the samples. Interestingly, they observed that individuals with DNMT3A mutations had a worse prognosis than those without and that these mutations were common in elderly patients.
To find cooperative mutations in APL, Greif et al. (2011a) examined the exome-sequencing data of three APL patients who did not have mutations in FLT3. After the exclusion of annotated polymorphisms, the authors confirmed a total of 12 non-synonymous SNVs and 1 indel in coding regions (Table 1). The identified mutations (including known mutations such as WT1 and NRAS) did not overlap in the three APL patients, suggesting that the spectrum of mutations that can cooperate with PML–RARA might be large and diverse.
NPM1 and CEBPA mutations are found in 60% of NK AML cases, but the remaining 40% are not well characterized. To better characterize this second group of AMLs, Grossmann et al. (2011) sequenced a NK AML case with no mutations of the NPM1, CEBPA, FLT3–ITD, or MLL gene and identified 12 non-synonymous SNVs and 1 frame-shift deletion, corresponding to 11 distinct genes (Table 1). All these mutations were found to be heterozygous. The authors selected 4 of these 11 genes (BCOR, YY2, SSRP1, and DNMT3A) and performed deep-sequencing analysis of all their exons in other AML patients who had a karyotype similar to their original AML case (i.e., a NK in the absence of NPM1, CEBPA, FLT3–ITD mutations, and MLL partial tandem duplication, PTD). They found that one case (1/16; 6.25%) carried a mutation in the SSRP1 gene, 4 (4/30; 13.3%) in DNMT3A and 5 (5/30; 16.6%) in BCOR. BCOR frequency was confirmed in a total of 82 NK cases with the above genetic features (14/82; 17%). In a second phase of the study, to assess the real frequency of BCOR mutations in unselected patients with NK AMLs, Grossmann et al. analyzed 262 unselected NK AML patients from an independent Italian cohort characterized for mutations in NPM1, FLT3–ITD, and DNMT3A. They found BCOR mutations in 10/262 (3.8%) cases; all these patients had a karyotype similar to their initial index patient. Thus BCOR mutations appear to be mostly enriched in the least characterized subgroup of NK AML, the subgroup with wild type NPM1, FLT3–ITD, IDH1, and MLL genes. The authors also studied the frequency of BCOR mutations in 131 AML patients with cytogenetic abnormalities but no mutation was found. Interestingly, BCOR mutations were usually associated with DMNT3A and only rarely with NPM1; finally, for NK leukemias, mutation of the BCOR gene appeared associated with a worse outcome.
Transcriptome-Sequencing
Greif et al. (2011b) had shown that transcriptome-sequencing by RNA-seq could also be used to identify recurrent or rare mutations in leukemia. A bone marrow sample (≥90% cellularity) from an NK AML patient and a normal sample from the peripheral blood of the same patient were compared by RNA-seq. Five tumor-specific SNVs (in RUNX1, TLE4, SHKBP1, XPO7, and RRP8 genes) were identified and validated (Table 1). Except for the mutation in the RUNX1 gene, a known recurrent mutation in AML, the other four were novel mutations. Variants in TLE4 and SHKBP1 were considered potentially relevant for further characterizations. TLE4, in fact, had been previously identified as a putative tumor suppressor and a possible cooperative gene of AML1–ETO in AML patients with chromosome 9q deletions (Dayyani et al., 2008). SHKBP1, on the other hand, is putatively linked to leukemia through the interaction with SETA which mediates its binding to CBL, an ubiquitin ligase involved in the degradation of FLT3. To evaluate the frequency of these mutations, the authors re-sequenced the coding sequence for both TLE4 and SHKBP1, as well as for RUNX1, in 95 additionally NK AML patients. The authors found two missense mutations (2%) for TLE4 and SHKBP1 and nine missense mutations (9.5%) for RUNX1. Notably, RUNX1, TLE4, and SHKBP1 mutations were mutually exclusive; moreover, TLE4 was found in samples carrying NPM1 and CEBPA variants, whereas SHKBP1 was found in combination with NMP1 and FLT3 mutations. To date, this is the only high-throughput experiment that has studied AML by RNA-seq.
Small non-coding RNAs play a key role in regulating a large variety of biological processes, including tumorigenesis. Thus, it is expected that they will be affected by mutations, like their cognate “coding genes.” In a recently published genome wide analysis of microRNAs (miRNAs; Ramsingh et al., 2010), the authors applied NGS technologies to the characterization of the microRNAome in a sample from the same AML patient previously studied in 2008 (Ley et al., 2008). They looked for miRNA mutations, aberrant expression, and miRNA binding-site mutations, detecting several new miRNAs (some of them expressed differently in the tumor and control samples), no somatic mutations of miRNA genes, and one somatic mutation in the 3′-UTR of the TNFAINP2 gene, which may result in the acquisition of a novel miRNA binding-site (Table 1). However, this gene was not mutated in 187 de novo AMLs, suggesting that this mutation is rare in primary AMLs. Likewise, no somatic mutations of miRNA genes were identified in this leukemic genome.
Genomics of Myelodysplastic Syndromes by NGS
Together with AMLs, myelodysplastic syndromes (MDSs), and myeloproliferative neoplasms (MPNs) include the majority of myeloid malignancies. Thus, it is worth mentioning some mutations recently identified with NGS technologies in these pathologies in relation to AML mutations.
Myelodysplastic syndromes represent a heterogeneous group of clonal hemopathies, characterized by bone marrow dysplasia, aberrant differentiation, peripheral cytopenia, increased incidence in old age and risk of progression to AML. At the end of 2011, four significant papers described specific mutations identified in MDSs by exome and whole-genome sequencing (Papaemmanuil et al., 2011; Visconte et al., 2011; Yoshida et al., 2011; Graubert et al., 2012). These recent publications, as well as corollary papers published soon after (Malcovati et al., 2011; Makishima et al., 2012) clearly indicate that, besides karyotypic abnormalities (i.e., 5q−, −7/7q−, trisomy 8, 20q−, and −Y) and “prototypic” gene mutations (e.g., TET2, RUNX1, TP53, ASXL1, NRAS/KRAS, EZH2, JAK2, and MPL), which had been linked to MDS for years, components of the splicing machinery are recurrent targets of mutations in MDSs and in myelodysplasia (e.g., U2AF1/U2AF35, SRSF2, ZRSR2, SF3B1, SF3A1). In particular, surprisingly high mutation frequencies (20–85%) were reported in the SF3B1 gene (Papaemmanuil et al., 2011; Visconte et al., 2011; Yoshida et al., 2011; Makishima et al., 2012); these were almost specific to the MDS subtypes refractory anemia with ring sideroblast (RARS) and RARS associated with marked Thrombocytosis (RARS-T), suggesting that they might be virtually pathognomonic to these MDS groups.
Little overlap was observed between SF3B1 and all the other mutations identified in genes of the spliceosome complex and those found so far in AML (Table S1 in Supplementary Material), suggesting that these splicing pattern mutations have a distinctive association with the pathogenesis of MDSs. Notably, 3 out of the 57 AML samples (5.3%) from a 2087 patient cohort screened for target re-sequencing were reported to contain SF3B1 mutations (Papaemmanuil et al., 2011); however, this is the first report of SF3B1 mutations in primary AML (even from larger cohorts), and it is possible that the AML in these three patients derives from the evolution of a preexisting MDS. This is indeed the case for the two AML patients (2/38) carrying a somatic SF3B1 mutation in the study of Malcovati et al. (2011).
Interestingly, Graubert et al. (2012) work examined directly the genetics of MDS when it evolves into secondary AML (sAML), studying, by whole-genome sequencing, a sAML patient sample and then genotyping the identified mutations in the matched MDS sample. The authors identified, among others, a missense mutation in the U2AF1/U2AF35 gene, an auxiliary factor of the U2 splicing complex; in 150 additional MDS de novo samples, this mutation had a frequency of 8.7%. In contrast to SF3B1 mutations that were associated with a relatively benign prognosis, mutations of the U2AF1/U2AF35 gene were associated with shorter survival and with an increased risk of developing sAML.
Further studies are needed; however, these results seem to suggest that even if AML and MDS mutation patterns overall share only few common mutated genes (16/290 AML mutated targets, Table S1 in Supplementary Material), this number is not expected to occur simply by chance (Fisher’s exact test P-value = 0.0045). Even more interesting, 6 of those 16 mutated genes belong to a group of 10 recurrent mutated genes found in AML (Fisher’s exact test P-value = 1.3e−09), suggesting that a selected fraction of recurrent mutations are involved in both AML and MDS pathogenesis. Thus genome sequencing of larger collections of samples may provide new insights into the molecular basis of MDS clinical heterogeneity and lead to the identification of syndrome subtypes with similar outcomes, e.g., AML progression and/or responses to therapy.
Recurring Somatic Mutations in AML: The State of the Art
The NGS studies described so far, led to the identification of 281 mutated genes in AML. Among them, 164 have been found in at least 2 AML patients (Table S1 in Supplementary Material), and only 10 are recurrent, i.e., they have a frequency higher than 5% and are found in more than 100 patients (Table 2). Notably, only 16 (∼6%) of the mutated genes were previously known, demonstrating how powerful NGS technologies can be for the discovery of AML-associated mutations.
Analysis of the prevalence of these mutations, however, reveals that 153 of the 265 novel mutations (∼58%) are found in at least two AML patients (Table S1 in Supplementary Material). Notably, most of them (149/153, 97%) have a frequency lower than 5% in AMLs. Thus, these data suggest the existence of two classes of mutated genes in AMLs: one comprising few (10/281, 3.6%) and frequently mutated genes, and the other comprising a larger set of genes with very low mutation frequencies. Although these are partial data, as these mutations need to be confirmed in a larger number of samples, known recurrent mutations appear to be over-represented in the data-set of AML-associated mutations (Fisher’s exact test P-value = 2.3e−06), suggesting that NGS major contribution to AML cancer genomics will probably be the detection of rare mutations (with a frequency lower than 5%). Yet, this might turn out to be a critical step for the identification of novel prognostic or therapeutic targets in AMLs.
In AMLs, much evidence suggests that primary translocations [inv(16); t(15;17); t(8;21); and 11q23 translocations] are sufficient to initiate leukemogenesis (initiating mutations), yet other genetic alterations are needed for the selection of the full leukemia-phenotype (cooperating mutations). In fact: (i) these primary translocations are frequently found as the only cytogenetic abnormality in AML blasts; (ii) the expression of the associated fusion proteins induces a pre-leukemic state in mice; (iii) the murine leukemias that eventually develop have morphological and clinical properties that are near-identical to those of the corresponding human leukemias. Thus, in AMLs with primary translocations, NGS might allow identification of mutations that cooperate with fusion proteins to determine the leukemia-phenotype.
Genomic analyses are available for six AML cases with primary translocations (five human APLs and one mouse APL; Table 1). Notably, the frequency of recurrent mutations in these cases is also extremely low (in total, 42 novel mutations were identified but none had a frequency higher than 5%), suggesting that myeloid leukemogenesis may initiate from the alteration of a few genetic pathways to then proceed through the alterations of many.
A similar scenario might apply to AMLs with a NK (78% of all sequenced cases). Mutations of NPM1 are found in ∼25% NK AMLs, are frequently associated with mutations of other recurrently mutated genes, such as FLT3, and never found together with primary translocations. Notably, as for the AML-associated fusion proteins, expression of mutant NPM1 in mice induces either a pre-leukemic state (Cheng et al., 2010, our unpublished data) or the occurrence of a frank leukemia, after a long (if expressed alone) or short (if co-expressed with others cooperative mutations) latency (Vassiliou et al., 2011, our unpublished data). Similarly to AMLs with primary translocations, AMLs with mutated NPM1 were found associated with 34 novel non-recurrent mutated genes by NGS. Thus, NGS might contribute to identify cooperating mutations in AMLs. Functional analyses of these mutations might then lead to the identification of cellular pathways that are critical for the selection of the leukemia-phenotype, providing a biological classification of leukemias, regardless of the initiating genetic event.
Molecular and Functional Consequences of Mutations in Recurrently Targeted Genes in AML
To derive information about the molecular and patho-functional impact of mutations directly from the type of mutation and from their location is always a not-trivial mission. In general, it might be true that when a genetic variant is found persistently located at a single amino acid position, the lesion may trigger a gain-of-function deleterious mechanism, as already established for known oncogenic mutations (e.g., RAS, NPM1). Loss-of-function is instead suggested by the finding of widely distributed divergent mutations along the structure of the gene, as often observed for several classical tumor suppressor genes (e.g., BRCA1 and TP53). Actually, often, “hot spot” and dispersed mutations can be both found in the same gene, making a prediction more difficult. This is the case of DNMT3A, the DNA (cytosine-5-)-methyltransferase-3-alpha, one of the most interesting newly identified recurrent targets of mutations in AML.
DNMT3A is an epigenetic modifying-enzyme known to be essential, together with DNMT3B, for the proper de novo methylation of DNA. It is one of the novel, most frequently mutated genes found in AML patients (DNMT3A mutation frequency: ∼20%) and it is one of those also discovered to be recurrently mutated in MDS (about 8%; Walter et al., 2011). Its mutated form in AML (i) is associated with mutations of NPM1, FLT3, IDH1, and CBPA, (ii) never appears in AML characterized by translocation events, (iii) is prevalent in AML with NK, and (iv) is associated with poor survival.
Nearly half of the mutations in the DNMT3A gene are concentrated in positions affecting arginine 882 (R882), a conserved residue of the methyltransferase (MT) domain. The remaining variations are more largely distributed along the length of the gene, although preferentially targeting the MT domain, as well. This structural observation suggests a loss-of-function mechanism. In support of this hypothesis, in vitro experiments showed that mutations in the DNMT3A MT domain decrease the methyltransferase activity of DNMT3A. In contrast, overexpression of DNMT3A in PML–RARA expressing mice recently demonstrated the potential cooperative nature of DNMT3A to induce APL (Subramanyam et al., 2010). Indeed, transplantation into irradiated mice of PML–RARA+/DNMT3A+ bone marrow cells induced leukemia with shorter latency and higher penetrance than transplantation of cells only expressing the initiating protein PML–RARA, thus suggesting a gain-on-function mechanism, possibly combined with a dominant negative effect on the wild type proteins. Interestingly DNMT3A mutations, although not dramatically altering global DNA methylation levels in AML genomes, tend to produce modified methylation patterns in the proximity of specific DNA regions and genes (Ley et al., 2010). Further experiments are required to completely clarify mechanisms and roles of DNMT3A and its association with co-occurring recurrent and rare genomic alterations.
Future Prospective and Open Challenges
So far, tumor and control samples from 26 AML patients have been sequenced but larger numbers of samples are expected to be sequenced in the near future. These data will be crucial to dissect the complexity of somatic mutations, which contribute to AML pathogenesis. No doubt, NGS platforms will be also further employed for the discovery of mutations in mouse models of AML.
The last 3 years, characterized by increased NSG applications, have seen a dramatic reduction in the costs of data generation, an increase in coverage, and improvements in computational data analysis. Indeed, the number of validated mutations of the computationally predicted variants ranges from 5% (Ley et al., 2008) to 98% (Yan et al., 2011), thus reducing validation costs and increasing the automation of the identification processes. Cost reduction, increase in automation, availability of NGS in medium and small centers, and the possibility to simultaneously detect all the genetic variants existing in a cancer genome, have opened new opportunities for the employment of NGS in clinical settings.
Three main challenges remain to be addressed. First, while these technologies can detect many somatic mutations in each patient, only a subset of them is probably involved in cancer initiation and progression. Thus, it is essential to develop methods to distinguish between passenger and driver mutations (Stratton et al., 2009). Second, an increasing number of mutations have been identified in AML. What links these genetic alterations to cancer progression? What complex interactions underlie AML pathogenesis? Third, the use of massive parallel sequencing has also found a rewarding application in the identification of chromatin features; the next challenge will be to integrate AML genomic information with AML epigenomic profiles.
Help will come from the genome sequencing of 500 de novo AML cases by the TCGA (http://tcga-data.nci.nih.gov/tcga), an NIH consortium which aims to contribute to the understanding of the molecular basis of cancer through the gathering and analysis of different high-throughput data, such as DNA-sequencing, methylation, gene expression and miRNA expression data. We foresee different ways of interpreting the huge amount of information generated by cancer re-sequencing projects in order to link mutations identified by NGS technologies to leukemia progression.
To correlate cancer mutated genes to cancer behavior we will need to discriminate, within all the variants found in the sequencing projects, between passenger and driver mutations. Currently, the definition of driver mutations is usually based on mutation frequency (Wood et al., 2007), and mutations are defined as drivers when found in a larger number of AML genomes. Since many driver mutations may be infrequent and contributing to cancer development only in few tumors, we will need to test a large number of tumor samples in order to discriminate between rare driver mutations and passenger mutations. Anyway, there are other purely computational ways to identify driver mutations, independent of the evaluation of mutation frequency. These methods can identify driver mutations among those that cause changing in the amino acid sequence of the associated protein. Methods as SIFT (Kumar et al., 2009) and PolyPhen-2 (Adzhubei et al., 2010) can predict for each non-synonymous mutation the impact of the amino acid substitution on protein structure and functions, using different features such as sequence homology, amino acid physicochemical properties and protein structure-based features. Recently, new methods that use network-based approaches (Torkamani and Schork, 2009; Cerami et al., 2010; Vandin et al., 2011) or machine learning algorithms (Carter et al., 2009) have been developed to identify driver mutations.
However, since the vast majority of somatic mutations are shared only between few patients, it might be more important to identify driver pathways rather than driver mutations. Indeed, driver pathways can be reconstructed using network models containing driver mutations and other genes that may link them (Vandin et al., 2012). The identification of driver pathways is important to rationalize targets for therapeutic intervention. Many recent works identify driver pathways by integrating different types of high-throughput data, such as copy number variant data and mRNA expression data, to identify driver CNVs, to stratify patients and to obtain mechanistic and prognostic insights (Akavia et al., 2010; Vaske et al., 2010; Jörnsten et al., 2011).
Lastly, it is possible to correlate specific mutations found in very large collections of patient samples with clinical outcomes, using unsupervised and supervised machine learning methods. With the unsupervised methods it is possible to cluster the mutational profiles of different patients to identify common alterations. With the supervised methods we can identify the features (or biomarkers) that can better classify specific subgroups of AML.
In order to determine what most likely drives AML, in addition to the development of computational methods that can prioritize candidate mutations, we will need to functionally characterize these mutated genes, using in vitro and/or in vivo experiments.
The possibility of recognizing a subset of genetic variations with predictive and prognostic value will pave the way to a mutation-specific, “personalized,” therapy choice. The molecular classification of AML patients will improve clinical outcome and be essential for disease monitoring. We expect that in a not so distant future, testing the presence of mutated genes in biopsies before treatment will become clinical routine practice.
So far studies have focused on the identification of mutations that are present in the majority of tumor cells. Indeed, these works have identified mutations with a frequency usually higher than 25%. If we increase the coverage, and we improve bioinformatics pipelines, we could aim to identify small sub-clones (<1%). Such an achievement could help addressing many important open questions such as the clonal evolution of tumors and, of clinical interest, the prediction of resistance to anti-tumoral treatments. Indeed, Ding et al. (2012) observed changes in mutant allele frequencies between the primary tumor and the relapse tumor, as well as clonal evolution in 5/8 patients, suggesting that a population with potentially chemo-resistant mutations might pre-exist and expand after treatment.
“Mutations” Glossary Box
Genomic mutations, genetic variants, genomic alterations, or simply mutations or variants: they are all synonyms indicating variations found in the DNA sequence derived from an individual with respect to the “Reference genome sequence.”
Mutations can be germline or somatic. A “germline mutation” gives rise to a mutation in the offspring; it is present in every cell. SNPs belong to this class. A “somatic mutation” or “somatic variant” is a mutation acquired during the life span of an individual in a specific area of the body (e.g., bone marrow); the cell where the somatic mutation occurs, may give rise to a clonal proliferation event. A somatic variant can be easily distinguished from a germline one by comparing the region of the mutated DNA sequence with a corresponding sequence obtained from another tissue of the same individual: in the first case the sequences will be different, in the second identical. Both germline and somatic mutations can be neutral (i.e., do not produce an observable pathological phenotype) or deleterious (i.e., are directly responsible or contribute to establish a perturbed unhealthy condition). Neutrality and deleteriousness are not always obvious, but can be predicted based on the features of the specific areas of genomic DNA, such as coding and regulatory potential or involvement in splicing mechanisms.
Recurrent mutation: it generally indicates that the same somatic mutation is found in different individuals, usually carrying a tumor of the same type. Herein, a recurrent mutation is defined as found in “at least 5% of the tested samples.” Since the chance of finding a recurrent event is very low, it likely reflects the importance that a somatic mutation may have on a tumorigenic or disease predisposing phenotype.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from AIRC and Italian Ministry of Health. LR is supported by a Reintegration AIRC/Marie Curie International Fellowship in Cancer Research. We thank C. Ronchini and E. Colombo for helpful discussions; P. Dalton and R. Aina for scientific editing.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Molecular_and_Cellular_Oncology/abstract/21275
Table S1. Catalog of all the genes with molecular genetic abnormalities in adult AML as detected by NGS technologies. The 290 genes listed in this table were found in one or more tumor samples. This table includes also the nine genes whose mutations were relapse-specific and absent in the additional 200 primary AMLs considered for testing recurrently mutated genes (Ding et al., 2012).
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., Kondrashov, A. S., and Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249.
Akavia, U. D., Litvin, O., Kim, J., Sanchez-Garcia, F., Kotliar, D., Causton, H. C., Pochanard, P., Mozes, E., Garraway, L. A., and Pe’er, D. (2010). An integrated approach to uncover drivers of cancer. Cell 143, 1005–1017.
Carter, H., Chen, S., Isik, L., Tyekucheva, S., Velculescu, V. E., Kinzler, K. W., Vogelstein, B., and Karchin, R. (2009). Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 69, 6660–6667.
Cerami, E., Demir, E., Schultz, N., Taylor, B. S., and Sander, C. (2010). Automated network analysis identifies core pathways in glioblastoma. PLoS ONE 5, e8918. doi:10.1371/journal.pone.0008918
Cheng, K., Sportoletti, P., Ito, K., Clohessy, J. G., Teruya-Feldstein, J., Kutok, J. L., and Pandolfi, P. P. (2010). The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 115, 3341–3345.
Dayyani, F., Wang, J., Yeh, J.-R. J., Ahn, E.-Y., Tobey, E., Zhang, D.-E., Bernstein, I. D., Peterson, R. T., and Sweetser, D. A. (2008). Loss of TLE1 and TLE4 from the del(9q) commonly deleted region in AML cooperates with AML1-ETO to affect myeloid cell proliferation and survival. Blood 111, 4338–4347.
Ding, L., Ley, T. J., Larson, D. E., Miller, C. A., Koboldt, D. C., Welch, J. S., Ritchey, J. K., Young, M. A., Lamprecht, T., McLellan, M. D., McMichael, J. F., Wallis, J. W., Lu, C., Shen, D., Harris, C. C., Dooling, D. J., Fulton, R. S., Fulton, L. L., Chen, K., Schmidt, H., Kalicki-Veizer, J., Magrini, V. J., Cook, L., McGrath, S. D., Vickery, T. L., Wendl, M. C., Heath, S., Watson, M. A., Link, D. C., Tomasson, M. H., Shannon, W. D., Payton, J. E., Kulkarni, S., Westervelt, P., Walter, M. J., Graubert, T. A., Mardis, E. R., Wilson, R. K., and DiPersio, J. F. (2012). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510.
Graubert, T. A., Shen, D., Ding, L., Okeyo-Owuor, T., Lunn, C. L., Shao, J., Krysiak, K., Harris, C. C., Koboldt, D. C., Larson, D. E., McLellan, M. D., Dooling, D. J., Abbott, R. M., Fulton, R. S., Schmidt, H., Kalicki-Veizer, J., O’Laughlin, M., Grillot, M., Baty, J., Heath, S., Frater, J. L., Nasim, T., Link, D. C., Tomasson, M. H., Westervelt, P., DiPersio, J. F., Mardis, E. R., Ley, T. J., Wilson, R. K., and Walter, M. J. (2012). Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 44, 53–57.
Gregory, T. K., Wald, D., Chen, Y., Vermaat, J. M., Xiong, Y., and Tse, W. (2009). Molecular prognostic markers for adult acute myeloid leukemia with normal cytogenetics. J. Hematol. Oncol. 2, 23.
Greif, P. A., Yaghmaie, M., Konstandin, N. P., Ksienzyk, B., Alimoghaddam, K., Ghavamzadeh, A., Hauser, A., Graf, A., Krebs, S., Blum, H., and Bohlander, S. K. (2011a). Somatic mutations in acute promyelocytic leukemia (APL) identified by exome sequencing. Leukemia 25, 1519–1522.
Greif, P. A., Eck, S. H., Konstandin, N. P., Benet-Pagès, A., Ksienzyk, B., Dufour, A., Vetter, A. T., Popp, H. D., Lorenz-Depiereux, B., Meitinger, T., Bohlander, S. K., and Strom, T. M. (2011b). Identification of recurring tumor-specific somatic mutations in acute myeloid leukemia by transcriptome sequencing. Leukemia 25, 821–827.
Grossmann, V., Tiacci, E., Holmes, A. B., Kohlmann, A., Martelli, M. P., Kern, W., Spanhol-Rosseto, A., Klein, H.-U., Dugas, M., Schindela, S., Trifonov, V., Schnittger, S., Haferlach, C., Bassan, R., Wells, V. A., Spinelli, O., Chan, J., Rossi, R., Baldoni, S., De Carolis, L., Goetze, K., Serve, H., Peceny, R., Kreuzer, K. A., Oruzio, D., Specchia, G., Di Raimondo, F., Fabbiano, F., Sborgia, M., Liso, A., Farinelli, L., Rambaldi, A., Pasqualucci, L., Rabadan, R., Haferlach, T., and Falini, B. (2011). Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 118, 6153–6163.
Jörnsten, R., Abenius, T., Kling, T., Schmidt, L., Johansson, E., Nordling, T. E. M., Nordlander, B., Sander, C., Gennemark, P., Funa, K., Nilsson, B., Lindahl, L., and Nelander, S. (2011). Network modeling of the transcriptional effects of copy number aberrations in glioblastoma. Mol. Syst. Biol. 7, 486.
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081.
Ley, T. J., Ding, L., Walter, M. J., McLellan, M. D., Lamprecht, T., Larson, D. E., Kandoth, C., Payton, J. E., Baty, J., Welch, J., Harris, C. C., Lichti, C. F., Townsend, R. R., Fulton, R. S., Dooling, D. J., Koboldt, D. C., Schmidt, H., Zhang, Q., Osborne, J. R., Lin, L., O’Laughlin, M., McMichael, J. F., Delehaunty, K. D., McGrath, S. D., Fulton, L. A., Magrini, V. J., Vickery, T. L., Hundal, J., Cook, L. L., Conyers, J. J., Swift, G. W., Reed, J. P., Alldredge, P. A., Wylie, T., Walker, J., Kalicki, J., Watson, M. A., Heath, S., Shannon, W. D., Varghese, N., Nagarajan, R., Westervelt, P., Tomasson, M. H., Link, D. C., Graubert, T. A., DiPersio, J. F., Mardis, E. R., and Wilson, R. K. (2010). DNMT3A Mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433.
Ley, T. J., Mardis, E. R., Ding, L., Fulton, B., McLellan, M. D., Chen, K., Dooling, D., Dunford-Shore, B. H., McGrath, S., Hickenbotham, M., Cook, L., Abbott, R., Larson, D. E., Koboldt, D. C., Pohl, C., Smith, S., Hawkins, A., Abbott, S., Locke, D., Hillier, L. W., Miner, T., Fulton, L., Magrini, V., Wylie, T., Glasscock, J., Conyers, J., Sander, N., Shi, X., Osborne, J. R., Minx, P., Gordon, D., Chinwalla, A., Zhao, Y., Ries, R. E., Payton, J. E., Westervelt, P., Tomasson, M. H., Watson, M., Baty, J., Ivanovich, J., Heath, S., Shannon, W. D., Nagarajan, R., Walter, M. J., Link, D. C., Graubert, T. A., DiPersio, J. F., and Wilson, R. K. (2008). DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 4567218, 66–72.
Li, M., Wang, I. X., Li, Y., Bruzel, A., Richards, A. L., Toung, J. M., and Cheung, V. G. (2011). Widespread RNA and DNA sequence differences in the human transcriptome. Science 333, 53–58.
Link, D. C., Schuettpelz, L. G., Shen, D., Wang, J., Walter, M. J., Kulkarni, S., Payton, J. E., Ivanovich, J., Goodfellow, P. J., Le Beau, M., Koboldt, D. C., Dooling, D. J., Fulton, R. S., Bender, R. H., Fulton, L. L., Delehaunty, K. D., Fronick, C. C., Appelbaum, E. L., Schmidt, H., Abbott, R., O’Laughlin, M., Chen, K., McLellan, M. D., Varghese, N., Nagarajan, R., Heath, S., Graubert, T. A., Ding, L., Ley, T. J., Zambetti, G. P., Wilson, R. K., and Mardis, E. R. (2011). The identification of a novel TP53 cancer susceptibility mutation through whole genome sequencing of a patient with therapy-related AML. JAMA 305, 1568–1576.
Maher, C. A., Kumar-Sinha, C., Cao, X., Kalyana-Sundaram, S., Han, B., Jing, X., Sam, L., Barrette, T., Palanisamy, N., and Chinnaiyan, A. M. (2009). Transcriptome sequencing to detect gene fusions in cancer. Nature 458, 97–101.
Makishima, H., Visconte, V., Sakaguchi, H., Jankowska, A. M., Abu Kar, S., Jerez, A., Przychodzen, B., Bupathi, M., Guinta, K., Afable, M. G., Sekeres, M. A., Padgett, R. A., Tiu, R. V., and Maciejewski, J. P. (2012). Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210.
Malcovati, L., Papaemmanuil, E., Bowen, D. T., Boultwood, J., Della Porta, M. G., Pascutto, C., Travaglino, E., Groves, M. J., Godfrey, A. L., Ambaglio, I., Gallí, A., Da Vià, M. C., Conte, S., Tauro, S., Keenan, N., Hyslop, A., Hinton, J., Mudie, L. J., Wainscoat, J. S., Futreal, P. A., Stratton, M. R., Campbell, P. J., Hellström-Lindberg, E., Cazzola, M., and Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium and of the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative. (2011). Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118, 6239–6246.
Mardis, E. R., Ding, L., Dooling, D. J., Larson, D. E., McLellan, M. D., Chen, K., Koboldt, D. C., Fulton, R. S., Delehaunty, K. D., McGrath, S. D., Fulton, L. A., Locke, D. P., Magrini, V. J., Abbott, R. M., Vickery, T. L., Reed, J. S., Robinson, J. S., Wylie, T., Smith, S. M., Carmichael, L., Eldred, J. M., Harris, C. C., Walker, J., Peck, J. B., Du, F., Dukes, A. F., Sanderson, G. E., Brummett, A. M., Clark, E., McMichael, J. F., Meyer, R. J., Schindler, J. K., Pohl, C. S., Wallis, J. W., Shi, X., Lin, L., Schmidt, H., Tang, Y., Haipek, C., Wiechert, M. E., Ivy, J. V., Kalicki, J., Elliott, G., Ries, R. E., Payton, J. E., Westervelt, P., Tomasson, M. H., Watson, M. A., Baty, J., Heath, S., Shannon, W. D., Nagarajan, R., Link, D. C., Walter, M. J., Graubert, T. A., DiPersio, J. F., Wilson, R. K., and Ley, T. J. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066.
Meyerson, M., Gabriel, S., and Getz, G. (2010). Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 11, 685–696.
Ng, S. B., Turner, E. H., Robertson, P. D., Flygare, S. D., Bigham, A. W., Lee, C., Shaffer, T., Wong, M., Bhattacharjee, A., Eichler, E. E., Bamshad, M., Nickerson, D. A., and Shendure, J. (2009). Targeted capture and massively parallel sequencing of twelve human exomes. Nature 461, 272–276.
Papaemmanuil, E., Cazzola, M., Boultwood, J., Malcovati, L., Vyas, P., Bowen, D., Pellagatti, A., Wainscoat, J. S., Hellstrom-Lindberg, E., Gambacorti-Passerini, C., Godfrey, A. L., Rapado, I., Cvejic, A., Rance, R., McGee, C., Ellis, P., Mudie, L. J., Stephens, P. J., McLaren, S., Massie, C. E., Tarpey, P. S., Varela, I., Nik-Zainal, S., Davies, H. R., Shlien, A., Jones, D., Raine, K., Hinton, J., Butler, A. P., Teague, J. W., Baxter, E. J., Score, J., Galli, A., Della Porta, M. G., Travaglino, E., Groves, M., Tauro, S., Munshi, N. C., Anderson, K. C., El-Naggar, A., Fischer, A., Mustonen, V., Warren, A. J., Cross, N. C. P., Green, A. R., Futreal, P. A., Stratton, M. R., and Campbell, P. J. (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 365, 1384–1395.
Ramsingh, G., Koboldt, D. C., Trissal, M., Chiappinelli, K. B., Wylie, T., Koul, S., Chang, L.-W., Nagarajan, R., Fehniger, T. A., Goodfellow, P., Magrini, V., Wilson, R. K., Ding, L., Ley, T. J., Mardis, E. R., and Link, D. C. (2010). Complete characterization of the microRNAome in a patient with acute myeloid leukemia. Blood 116, 5316–5326.
Subramanyam, D., Belair, C. D., Barry-Holson, K. Q., Lin, H., Kogan, S. C., Passegué, E., and Blelloch, R. (2010). PML-RARα and Dnmt3a1 cooperate in vivo to promote acute promyelocytic leukemia. Cancer Res. 70, 8792–8801.
Tefferi, A., Thiele, J., and Vardiman, J. W. (2009). The 2008 World Health Organization classification system for myeloproliferative neoplasms. Cancer 115, 3842–3847.
Torkamani, A., and Schork, N. J. (2009). Identification of rare cancer driver mutations by network reconstruction. Genome Res. 19, 1570–1578.
Vandin, F., Upfal, E., and Raphael, B. J. (2011). Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol. 18, 507–522.
Vandin, F., Upfal, E., and Raphael, B. J. (2012). De novo discovery of mutated driver pathways in cancer. Genome Res. 22, 375–385.
Vaske, C. J., Benz, S. C., Sanborn, J. Z., Earl, D., Szeto, C., Zhu, J., Haussler, D., and Stuart, J. M. (2010). Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26, i237–i245.
Vassiliou, G. S., Cooper, J. L., Rad, R., Li, J., Rice, S., Uren, A., Rad, L., Ellis, P., Andrews, R., Banerjee, R., Grove, C., Wang, W., Liu, P., Wright, P., Arends, M., and Bradley, A. (2011). Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 43, 470–475.
Visconte, V., Makishima, H., Jankowska, A., Szpurka, H., Traina, F., Jerez, A., O’Keefe, C., Rogers, H. J., Sekeres, M. A., Maciejewski, J. P., and Tiu, R. V. (2011). SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26, 542–545.
Walter, M. J., Ding, L., Shen, D., Shao, J., Grillot, M., McLellan, M., Fulton, R., Schmidt, H., Kalicki-Veizer, J., O’Laughlin, M., Kandoth, C., Baty, J., Westervelt, P., DiPersio, J. F., Mardis, E. R., Wilson, R. K., Ley, T. J., and Graubert, T. A. (2011). Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25, 1153–1158.
Wartman, L. D., Larson, D. E., Xiang, Z., Ding, L., Chen, K., Lin, L., Cahan, P., Klco, J. M., Welch, J. S., Li, C., Payton, J. E., Uy, G. L., Varghese, N., Ries, R. E., Hoock, M., Koboldt, D. C., McLellan, M. D., Schmidt, H., Fulton, R. S., Abbott, R. M., Cook, L., McGrath, S. D., Fan, X., Dukes, A. F., Vickery, T., Kalicki, J., Lamprecht, T. L., Graubert, T. A., Tomasson, M. H., Mardis, E. R., Wilson, R. K., and Ley, T. J. (2011). Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J. Clin. Invest. 121, 1445–1455.
Welch, J. S., Westervelt, P., Ding, L., Larson, D. E., Klco, J. M., Kulkarni, S., Wallis, J., Chen, K., Payton, J. E., Fulton, R. S., Veizer, J., Schmidt, H., Vickery, T. L., Heath, S., Watson, M. A., Tomasson, M. H., Link, D. C., Graubert, T. A., DiPersio, J. F., Mardis, E. R., Ley, T. J., and Wilson, R. K. (2011). Use of whole genome sequencing to diagnose a cryptic fusion oncogene. JAMA 305, 1577–1584.
Wood, L. D., Parsons, D. W., Jones, S., Lin, J., Sjöblom, T., Leary, R. J., Shen, D., Boca, S. M., Barber, T., Ptak, J., Silliman, N., Szabo, S., Dezso, Z., Ustyanksky, V., Nikolskaya, T., Nikolsky, Y., Karchin, R., Wilson, P. A., Kaminker, J. S., Zhang, Z., Croshaw, R., Willis, J., Dawson, D., Shipitsin, M., Willson, J. K., Sukumar, S., Polyak, K., Park, B. H., Pethiyagoda, C. L., Pant, P. V., Ballinger, D. G., Sparks, A. B., Hartigan, J., Smith, D. R., Suh, E., Papadopoulos, N., Buckhaults, P., Markowitz, S. D., Parmigiani, G., Kinzler, K. W., Velculescu, V. E., and Vogelstein, B. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113.
Yamashita, Y., Yuan, J., Suetake, I., Suzuki, H., Ishikawa, Y., Choi, Y. L., Ueno, T., Soda, M., Hamada, T., Haruta, H., Akada, S., Miyazaki, Y., Kiyoi, H., Ito, E., Naoe, T., Tomonaga, M., Toyota, M., Tajima, S., Iwama, A., and Mano, H. (2010). Array-based genomic resequencing of human leukemia. Oncogene 29, 3723–3731.
Yan, X.-J., Xu, J., Gu, Z.-H., Pan, C.-M., Lu, G., Shen, Y., Shi, J.-Y., Zhu, Y.-M., Tang, L., Zhang, X.-W., Liang, W. X., Mi, J. Q., Song, H. D., Li, K. Q., Chen, Z., and Chen, S. J. (2011). Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 43, 309–315.
Yoshida, K., Sanada, M., Shiraishi, Y., Nowak, D., Nagata, Y., Yamamoto, R., Sato, Y., Sato-Otsubo, A., Kon, A., Nagasaki, M., Chalkidis, G., Suzuki, Y., Shiosaka, M., Kawahata, R., Yamaguchi, T., Otsu, M., Obara, N., Sakata-Yanagimoto, M., Ishiyama, K., Mori, H., Nolte, F., Hofmann, W. K., Miyawaki, S., Sugano, S., Haferlach, C., Koeffler, H. P., Shih, L. Y., Haferlach, T., Chiba, S., Nakauchi, H., Miyano, S., and Ogawa, S. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69.
Keywords: acute myeloid leukemia, next generation sequencing, somatic mutations, recurrent mutations
Citation: Riva L, Luzi L and Pelicci PG (2012) Genomics of acute myeloid leukemia: the next generation. Front. Oncol. 2:40. doi: 10.3389/fonc.2012.00040
Received: 22 December 2011; Paper pending published: 24 January 2012;
Accepted: 27 February 2012; Published online: 01 May 2012.
Edited by:
Napoleone Ferrara, Genentech, USAReviewed by:
Keisuke Ito, Beth Israel Deaconess Medical Center, USAShridar Ganesan, University of Medicine and Dentistry of New Jersey, USA
Copyright: © 2012 Riva, Luzi and Pelicci. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Pier Giuseppe Pelicci, Department of Experimental Oncology, European Institute of Oncology, Via Adamello, 16, 20139 Milan, Italy. e-mail:cGllcmdpdXNlcHBlLnBlbGljY2lAaWZvbS1pZW8tY2FtcHVzLml0
†Present address: Laura Riva, Center for Genomic Science of IIT@SEMM, Istituto Italiano di Tecnologia at the IFOM-IEO Campus, Milan, Italy.
‡Laura Riva and Lucilla Luzi have contributed equally to this work.