 Heinrich Kovar1,26*
Heinrich Kovar1,26* Javier Alonso2 Pierre Aman3 Dave N. T. Aryee1,26 Jozef Ban1 Sue A. Burchill4 Stefan Burdach5 Enrique De Alava6 Olivier Delattre7 Uta Dirksen8 Argyro Fourtouna1 Simone Fulda9 Lee J. Helman10 David Herrero-Martin1 Pancras C. W. Hogendoorn11 Udo Kontny12 Elizabeth R. Lawlor13,14 Stephen L. Lessnick15 Antonio Llombart-Bosch16 Markus Metzler17 Richard Moriggl18 Stephan Niedan1 Jenny Potratz8 Françoise Redini19 Günther H. S. Richter5 Lucia T. Riedmann1 Claudia Rossig8 Beat W. Schäfer20 Raphaela Schwentner1 Katia Scotlandi21 Poul H. Sorensen22 Martin S. Staege23 Franck Tirode7 Jeffrey Toretsky24 Selena Ventura20 Angelika Eggert25 and Ruth Ladenstein1,26
Javier Alonso2 Pierre Aman3 Dave N. T. Aryee1,26 Jozef Ban1 Sue A. Burchill4 Stefan Burdach5 Enrique De Alava6 Olivier Delattre7 Uta Dirksen8 Argyro Fourtouna1 Simone Fulda9 Lee J. Helman10 David Herrero-Martin1 Pancras C. W. Hogendoorn11 Udo Kontny12 Elizabeth R. Lawlor13,14 Stephen L. Lessnick15 Antonio Llombart-Bosch16 Markus Metzler17 Richard Moriggl18 Stephan Niedan1 Jenny Potratz8 Françoise Redini19 Günther H. S. Richter5 Lucia T. Riedmann1 Claudia Rossig8 Beat W. Schäfer20 Raphaela Schwentner1 Katia Scotlandi21 Poul H. Sorensen22 Martin S. Staege23 Franck Tirode7 Jeffrey Toretsky24 Selena Ventura20 Angelika Eggert25 and Ruth Ladenstein1,26
- 1 Children’s Cancer Research Institute, St. Anna Kinderkrebsforschung, Vienna, Austria
- 2 Unidad de Tumores Sólidos Infantiles, Centro Nacional de Microbiología, Instituto de Salud Carlos III, Majadahonda, Spain
- 3 Department of Pathology, Sahlgrenska Cancer Center, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden
- 4 Leeds Institute of Molecular Medicine, Leeds, UK
- 5 Children’s Cancer Research Center and Roman Herzog Comprehensive Cancer Center, Klinikum rechts der Isar, Technical University, Munich, Germany
- 6 Department of Pathology, University Hospital of Salamanca, Cancer Research Center-IBMCC, University of Salamanca-CSIC, Salamanca, Spain
- 7 INSERM, U830 Génétique et Biologie des Cancers, Institut Curie, Paris, France
- 8 Pediatric Hematology and Oncology, University Children’s Hospital Münster, Münster, Germany
- 9 Institute for Experimental Cancer Research in Pediatrics, Goethe-University Frankfurt, Frankfurt am Main, Germany
- 10 Molecular Oncology Section, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
- 11 Department of Pathology, Leiden University Medical Centre, Leiden, Netherlands
- 12 Division of Pediatric Hematology and Oncology, University Children’s Hospital, Freiburg, Germany
- 13 Department of Pediatrics, University of Michigan, Ann Arbor, MI, USA
- 14 Department of Pathology, University of Michigan, Ann Arbor, MI, USA
- 15 Division of Pediatric Hematology and Oncology, Department of Oncological Sciences, Center for Children’s Cancer Research at Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT, USA
- 16 Department of Pathology, University of Valencia, Valencia, Spain
- 17 University Children’s Hospital, Erlangen, Germany
- 18 Ludwig Boltzmann Institute for Cancer Research, Vienna, Austria
- 19 INSERM, UMR 957, LUNAM Université, Laboratoire de Physiopathologie de la Résorption Osseuse et Thérapie des Tumeurs Osseuses Primitives EA3822, Nantes, France
- 20 Department of Oncology, University Children’s Hospital, Zurich, Switzerland
- 21 CRS Development of Biomolecular Therapies, Laboratory of Experimental Oncology, Rizzoli Institute, Bologna, Italy
- 22 Department of Molecular Oncology, British Columbia Cancer Research Centre, Vancouver, BC, Canada
- 23 Department of Pediatrics, Children’s Cancer Research Centre, Martin-Luther-University Halle-Wittenberg, Halle, Germany
- 24 Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC, USA
- 25 Department of Pediatric Oncology and Hematology, University Children’s Hospital, Essen, Germany
- 26 Department of Pediatrics, Medical University, Vienna, Austria
The European Network for Cancer Research in Children and Adolescents (ENCCA) provides an interaction platform for stakeholders in research and care of children with cancer. Among ENCCA objectives is the establishment of biology-based prioritization mechanisms for the selection of innovative targets, drugs, and prognostic markers for validation in clinical trials. Specifically for sarcomas, there is a burning need for novel treatment options, since current chemotherapeutic treatment protocols have met their limits. This is most obvious for metastatic Ewing sarcoma (ES), where long term survival rates are still below 20%. Despite significant progress in our understanding of ES biology, clinical translation of promising laboratory results has not yet taken place due to fragmentation of research and lack of an institutionalized discussion forum. To fill this gap, ENCCA assembled 30 European expert scientists and five North American opinion leaders in December 2011 to exchange thoughts and discuss the state of the art in ES research and latest results from the bench, and to propose biological studies and novel promising therapeutics for the upcoming European EWING2008 and EWING2012 clinical trials.
The European Network for Cancer Research in Children and Adolescents
Biomedical laboratory research into the origins and specifics of cancer has led to an almost logarithmic increment of biological knowledge over the last decade, clinical translation of this knowledge, however, is lagging far behind. Owing to multimodal treatment concepts in (international) multicenter trials, the prognosis has markedly improved in pediatric cancers within the past decades. Yet for more than 20 years now a plateau has been reached with regard to standard chemotherapeutic regimens. Although the clinical trials have clearly been of enormous benefit to patients, the rigid structure, and long duration of the trials restrict options for the clinical development of new therapeutics and the prospective testing of novel prognostic markers. One of the aims of the European Network for Cancer Research in Children and Adolescents (ENCCA), supported by the Framework Program 7 of the European Commission, is therefore the integration of biology into pediatric clinical trials. ENCCA aims at establishing mechanisms of prioritization for marker/target/drug selection to be incorporated into European clinical trials. Specifically in sarcoma, there is a high need for early detection of treatment resistant, metastatic, and progressive disease, and for novel therapeutic options for high risk patients whose survival chances are otherwise still below 30%. Unlike in pediatric leukemia and neuroblastoma, the international Ewing sarcoma (ES) research community had, so far, not established a platform for regular meetings and scientific exchange that would allow involvement of the whole expert community into the prioritization process. In the past, the Charles and Dana Nearburg Foundation kindly sponsored a few valuable ES expert meetings with limited European participation on the American side of the Atlantic in irregular, every 3 or 4 years, intervals. Then, from December 19 to 21, 2011, the first ENCCA-supported European Interdisciplinary ES Research Summit was held in Vienna, Austria. Thirty European and five North American expert scientists met to exchange and discuss their most recent, largely unpublished results, and to propose biological studies and novel promising therapeutics for the upcoming European EWING2008 and EWING2012 studies. Topics ranged from factors affecting the susceptibility to and pathomechanisms of the disease, tumor stem cells, and tumor host interactions, over models for the study of the tissue of origin, mechanisms of metastasis, and drug screening, to novel diagnostic/prognostic markers and approaches. In the discussion, particular emphasis was laid on overcoming bottlenecks and the translatability of research results into clinical practice.
EWS-FLI1 Signature Based Identification of Therapeutic Compounds and Drug Targets
The major driver of ES, the EWS-FLI1 gene fusion, was discovered exactly 20 years ago (Delattre et al., 1992), and there is ample experimental evidence that inhibition of EWS-FLI1 interferes with tumor growth. EWS-FLI1 is an aberrant transcription factor, and as such it deregulates the expression of a multitude of genes. Several labs use EWS-FLI1 signature based approaches to screen for potential therapeutic drugs which mimic the effects of EWS-FLI1 knockdown, as pioneered by Stegmaier et al. (2007). Starting from meta analysis of EWS-FLI1 dependent gene expression data sets, luciferase reporter constructs controlled by EWS-FLI1 regulated promoters are used for read out in high-throughput drug screens. The group of Beat Schäfer (University Children’s Hospital Zurich) monitored EWS-FLI1 activity using reporters for EWS-FLI1 repressed PHLDA1 and EWS-FLI1 activated IL1RAP while Lee Helman’s lab (NCI, NIH, Bethesda) used the NR0B1 promoter for that purpose. Schäfer screened a LOPAC library (Library of Pharmacologically Active Compounds) of 1280 known compounds and identified several drugs already known to be effective against ES including Etoposide and Doxorubicin but also Idarubicin, 5-Bromo-2-deoxyuridine, and Fenretinide. Schäfer reported on a particularly active novel drug, PKC412 (Midostaurin, previously shown to modify phosphorylation of PAX-FKHR in alveolar rhabdomyosarcoma), and presented data on in vitro apoptosis induction and in vivo growth reduction of ES cell lines in xenografts (Boro et al., 2012) (Table 1).
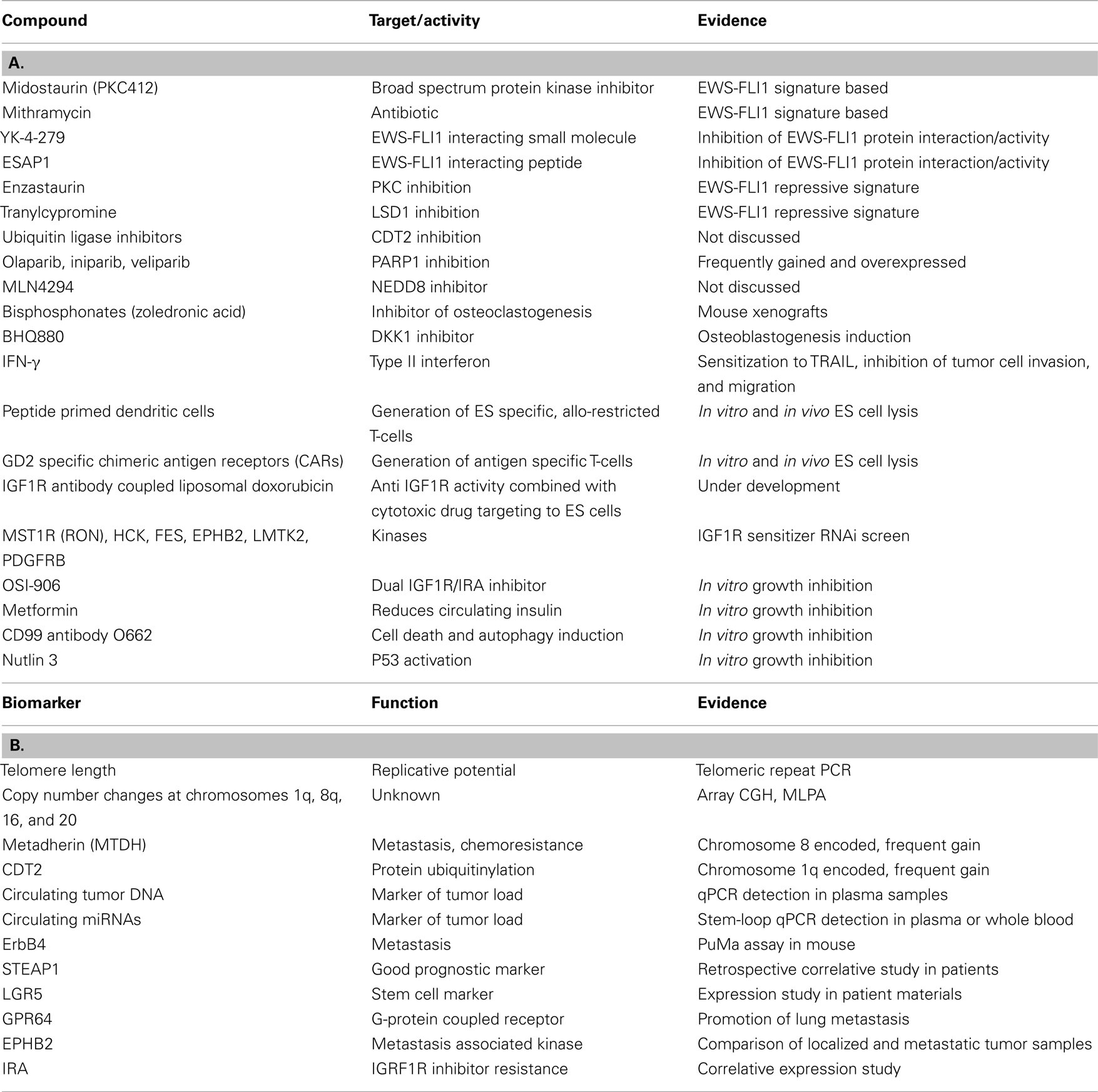
Table 1. Candidate compounds (A) and biomarkers (B) discussed at the Ewing sarcoma summit.
Helman described their preclinical results on mithramycin, an old antibiotic from streptomyces, which they have identified in a 56,000 natural product EWS-FLI1 signature based screen (Grohar et al., 2011) (Table 1). He reviewed published molecular (inhibition of EWS-FLI1 cooperating transcription factors SP1, E2F, and ETS) and first clinical results in a patient with ES (Kofman et al., 1973), and reported on their own findings of tumor regression in TC32 xenografts. Consistent with the modulation of EWS-FLI1 signature genes, they found that mithramycin reduces EWS-FLI1 expression. A phase II clinical trial with 10 patients is about to commence at the NCI and Vanderbilt University using mithramycin at a dose half of what has been previously used, hopefully reducing the risk of bleeding and other negative side effects. Particularly the risk of bleeding has been attributed to mitramycin treatment due to an effect of the drug on platelet function associated with thrombocytopenia. As a next step, Helman plans a combination screen of mithramycin with kinase specific siRNAs to identify potential drug sensitizers. But also other, more potent (active at concentrations of 0.3 nM), compounds from the initial screen are currently under investigation in 22 ES cell lines. Since the first EWS-FLI1 signature modulating drug, cytarabine, has failed to induce objective responses in a phase II clinical trial (DuBois et al., 2009), it is hoped that these newly identified compounds will show good clinical activity and ultimately lead to improved survival rates of ES patients.
While the mechanisms by which these compounds affect EWS-FLI1 and/or its downstream signature are still unknown, Jeff Toretsky (Georgetown University, Washington, DC, USA) provided an update on their studies of agents directly targeting EWS-FLI1, the small molecule YK-4-279 and the peptide ESAP1 (Erkizan et al., 2009, 2011) (Table 1). He reported that different enantiomers of YK-4-279 have different biological activity, offering exciting new opportunities to increase therapeutic efficacy by decreasing IC50 values (i.e., to <1 μM for YK-4-279 enantiomer 1).
During the discussion of EWS-FLI1 and signature directed agents, the question was raised – and left unanswered – as to which surrogate markers might be used to monitor EWS-FLI1 expression/activity in a clinical setting? This may become a central issue when following drug response in therapeutic trials.
EWS-FLI1 Mediated Gene Repression and Epigenetics
EWS-FLI1 specific expression signatures have also been the basis for integrated genomics approaches to identify key downstream pathways for potential therapeutic intervention. Heinrich Kovar (Children’s Cancer Research Institute, Vienna, Austria) reported on blocked forkhead box (FOX) transcription factor and NOTCH signaling pathways (Ban et al., 2008; Bennani-Baiti et al., 2011) in ES, on how NOTCH impacts mechanistically on p53, and on the in vitro activity of drugs reactivating these pathways. While these represent examples of indirect EWS/FLI repressed pathways, Stephen Lessnick (Huntsman Cancer Institute, Salt Lake City, UT, USA) reported evidence for direct gene repression by EWS/FLI. Lessnick provided evidence for the involvement of proteins that modulate epigenetic alterations in EWS/FLI mediated direct repression of critical target genes in Ewing’s sarcoma. Franck Tirode (Institut Curie, Paris, France) highlighted another EWS-FLI1 directly regulated epigenetic mechanism of gene repression, which involves the direct EWS-FLI1 target gene PRKCbeta. This kinase has been demonstrated to phosphorylate histone H3T6 in prostate cancer cells controlling demethylation at histone H3K4 (Metzger et al., 2010). Strikingly, Tirode found a strong overlap between EWS-FLI1 and PRKCB regulated genes in ES and outlined a few examples. Thus, epigenetic mechanisms appear to play a central role in ES pathogenesis (Figure 1).
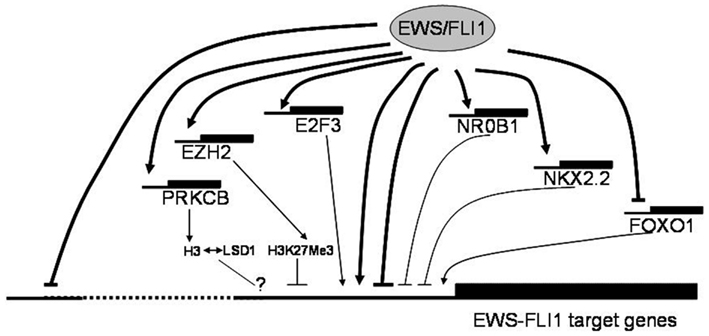
Figure 1. Putative mechanisms of EWS-FLI1 mediated gene regulation discussed at the Ewing sarcoma summit. EWS-FLI1 serves as a direct and indirect transcriptional regulator of a multitude of genes. It preferentially binds to proximal promoter regions directly activating genes, in part in synergy with other transcription factors such as E2Fs, which are themselves direct EWS-FLI1 targets. Transcriptional repression downstream of EWS-FLI1 likely comprises several components. The mechanism of direct repression (which is frequently associated with distal binding) remains largely unknown. However, several transcription factors, among them NR0B1, NKX2.2 and, most recently, repression of FOXO1 have been reported as mediators of indirect transcriptional repression by EWS-FLI1. There is also accumulating evidence for the involvement of epigenetic modifiers (EZH2, HDACs, PRKCB, LSD1) in negative transcriptional regulation by the chimeric ETS oncogene. The consequences of PRKCB mediated histone H3T6 phosphorylation in ES, previously reported to redirect the demethylating activity of LSD1 from H3K4Me2 to H3K9Me2 thus favoring gene activation over repression in androgen dependent prostate cancer (Metzger et al., 2010), remain to be defined.
Susceptibility to ES and Disease Progression
Chromatin accessibility regulated by epigenetic mechanisms may also be responsible for variations in susceptibility to the development and progression of the disease. Poul Sorensen (University of British Columbia Vancouver, BC, Canada) discussed epigenetic regulation of genes as a source of gene expression differences between primary ES and matched metastasis derived cell lines.
Under the leadership of Olivier Delattre (Institut Curie, Paris, France), the question of susceptibility to ES has been addressed in an international genome-wide association study (GWAS) on approximately 1500 ES cases and more than 4000 Caucasian controls (Postel-Vinay et al., 2012). Candidate loci have been detected on chromosomes 1, 10, and 15, and for each of these loci SNPs in the vicinity of three genes, TARDBP, EGR2, and BMP, have been discovered independently associating with ES at very high odds ratios. Also, the SNPs on chromosomes 1 and 10 are very rare in Africans, consistent with the rarity of ES in this population. For EGR2, the risk allele has been found to be associated with increased, for TARDBP with decreased, expression. TARDBP on chromosomal region 1p36.22 is of particular interest since its product, TDP-43, associates with FET (FUS, EWS, TAF15) proteins, and is frequently mutated in amyotrophic lateral sclerosis (as is FUS, for review Ince et al., 2011).
Although at this point the role of genes in the vicinity of the enriched SNPs for the development of ES remains unknown, the finding of TARDBP as a candidate ES predisposition gene moves the question of a potential functional interaction of EWS-FLI1 with EWS-containing protein complexes into focus. Pierre Aman (University of Gothenburg, Sweden) demonstrated coordinate but heterogeneous expression of all three FET proteins within a tissue as monitored by single cell qPCR and immunohistochemistry. Similarly, FET fusion product levels show variation. Based on results on the FUS-DDIT3 gene fusion in myxoid liposarcoma, Aman hypothesized that there might exist a critical intermediate FET fusion protein expression level defining a transformation window, while high expression results in senescence and/or apoptosis. Protein interaction studies for EWS and EWS-FLI1 by co-immunoprecipitation or affinity chromatography are ongoing in the Aman and Toretsky labs which will help elucidate the functional relationship between FET and associated proteins and their oncogenic fusions. Preliminary results suggest that one-third of proteins associated with EWS-FLI1 are spliceosomal while only about 8% are transcription proteins.
An intriguing new aspect with regard to how gene expression is regulated in ES and may contribute to disease susceptibility or progression was provided by Martin Staege (Martin-Luther-University Halle-Wittenberg, Halle, Germany). He explored the role of human endogenous retrovirus (HERV) like sequences which constitute up to 10% of the genome and which are epigenetically regulated. Importantly, HERVs do not share homology with other species. Staege reported on a screen for association between DNA microarray data and HERV loci (16 Mio putative HERVs) asking which HERVs are expressed in ES. For that purpose, his group has established a MySQL data base of HERVs and a PEARL script calculating association between DNA microarray data and HERV loci. He has found a correlation between the expression of HERVs and flanking genes (as exemplified for MYC regulated genes). Based on this approach, two types of ES with different HERV association can be distinguished, but biological correlations cannot be drawn yet. In the next step, the group will address the question, which proteins are encoded by the expressed HERVs in ES (envelope proteins, superantigens). They have cloned HERV-env proteins into expression vectors for conditional expression of these proteins in cell lines. Ultimately, they will investigate if HERV sequences have an impact on patient survival or immune response, or may be used as markers for minimal residual disease detection. So far, validation suffers from the lack of sufficient clinically annotated materials.
Factors associated with disease progression are of particular interest as putative prognostic markers. Such biomarkers are urgently needed for early response prediction particularly in localized disease. Sue Burchill (Leeds Institute of Molecular Medicine, Leeds, UK) presented preliminary data on telomere length measurements (by PCR of telomeric repeats) in primary ES suggestive of shorter telomeres being associated with a worse overall (but not event free) survival (Table 1). Besides the difficulty of defining a standardized cut-off value for telomere lengths, the lack of complete clinical data has, so far, not allowed determination of a possible relationship of this result to patients′ age, which is known to be of prognostic relevance. Enrique De Alava (Salamanca University Hospital, Salamanca, Spain), Javier Alonso (Instituto de Salud Carlos III, Madrid, Spain) and Olivier Delattre reported on array comparative genome hybridization (aCGH) and Multiplex Ligation-dependent Probe Amplification analyses (MLPA) to quantify chromosomal and gene copy number variations in ES. These studies confirm earlier observations by conventional cytogenetic and fluorescence in situ hybridization (FISH) methods (Hattinger et al., 2002; Roberts et al., 2008; Savola et al., 2009) that the most recurrent copy number variations of prognostic relevance are gains of chromosomes 1q, 8q, and 20, and loss of chromosome 16q (Table 1). Delattre briefly discussed chromosome 8 encoded MTDH (metadherin, AEG-1), a gene whose overexpression associates with metastasis and chemoresistance in several solid tumors (for review Meng et al., 2011), as a putative drug target in ES. De Alava reported on two genes on chromosome 1q gained in 31% of cases, CDT2 and PARP1 (MacKintosh et al., 2011) as potential therapeutic targets (Table 1). For many years, PARP1 has been known to be overexpressed in ES (Prasad et al., 1990). It is involved in DNA damage repair, and according to De Alava’s presentation there have been speculations on whether PARP inhibitors (i.e., Olaparib, Iniparib, Veliparib) may act in synergy with DNA damage inducing drugs, or whether increased PARP expression is already a consequence of persistent EWS-FLI1 induced DNA damage in ES. Early phase clinical trials in ES combining PARP inhibitors with DNA damaging agents are ongoing (Table 1).
The discussed genes on chromosomes 1 and 8 are examples for candidate markers of disease progression. However, it is unlikely that either a single gene or a single chromosome aberration will ever enable reliable prediction of prognosis as a basis for therapy stratification. Whole genome targeted methods will be required to monitor combinations of copy number and sequence alterations. There was broad agreement that although MLPA and aCGH may be versatile and comparably easy to establish diagnostic tools in the context of a clinical trial (as pioneered in the European neuroblastoma study), next generation sequencing-based methods would be optimal for that purpose. Delattre reported that the French National Cancer Institute (INCa) is willing to fund a pilot exome and RNA sequencing study of about 100 clinically well annotated localized and metastatic ES as part of the International Cancer Genome Consortium, and invited European centers to submit diagnostic tumor and matched blood/bone marrow (for germline control) samples. Poul Sorensen reported that the comparison (whole exome and RNAseq) of matched normal, primary, and metastasis samples from three metastatic patients revealed that only 40–70% of SNVs were found to be shared between primary tumors and metastases. These results are consistent with massive genetic changes during tumor evolution. Therefore, if one wants to adopt therapy to progression-associated mutations it is essential to collect tumor material at a time right before any change in therapy is initiated, as Lee Helman pointed out. However, Uta Dirksen (University Hospital, Munster, Germany) mentioned that in most European pediatric cancer centers – with a few exceptions (i.e., Department of Pediatrics, Technical University, Munich, Germany, as reported by Stefan Burdach) – follow-up tumor material from metastatic lesions is rarely available, since, mainly for ethical reasons, protocols simply do not allow for further biopsies to be taken after initial diagnosis. In addition, diagnosis is usually done on fine needle or trephine biopsies critically restricting the amount of tumor material available for biological studies. Whole exome sequencing by NGS, however, requires a minimum of 5 μg DNA from fresh tumor tissue, since prior amplification is not recommended. This bottleneck in accessibility of follow-up tumor material calls for non-invasive approaches to follow therapy response in ES patients.
Biomarkers from Blood and Bone Marrow
Markus Metzler (University Hospital Erlangen, Germany) presented results from a pilot monitoring study of circulating tumor DNA (ctDNA) in the plasma of ES patients during therapy (Table 1). After characterization of genomic breakpoints of the EWS-FLI1 gene fusion on the initial biopsy material by long-range PCR, ctDNA isolated from plasma samples during therapy is quantified by real-time quantitative PCR. This method allows assessing tumor burden and response to chemotherapy in a minimally invasive way. Preliminary results on 27 patients reveal peaks after each treatment cycle and Metzler reported on the initiation of a pilot study in collaboration with the University Hospital Munster.
Tumor specific microRNA signatures are an area of great diagnostic potential. The labs of Heinrich Kovar (Children’s Cancer Research Institute, Vienna, Austria) and Katia Scotlandi (Rizzoli Institute, Bologna, Italy) have previously reported on EWS-FLI1 and ES associated miRNA patterns, respectively (Ban et al., 2010; Nakatani et al., 2011). Kovar’s group found that EWS-FLI1 knockdown decreases AGO2 expression in ES cells, and Günther Richter (Department of Pediatrics, Technical University, Munich, Germany) reported that EWS-FLI1 induced EZH2 up-regulates AGO2, and experimental AGO2 suppression results in decreased ES tumor growth and metastasis in mice, while AGO1 knockdown enhances tumorigenicity. Based on specific ES miRNA signatures, Sue Burchill suggested the monitoring of circulating microRNAs (miR) in plasma or whole blood of ES patients as a surrogate marker for tumor burden (Table 1). She presented preliminary data on the use of a 7900 miR high-throughput stem-loop RT-PCR platform for this purpose. The subsequent discussion focused on the question of tumor specificity of circulating miR spectra in the light of chemotherapy induced metabolic and consequently miR expression changes in many tissues that remain to be defined.
An experimental approach to non-invasive diagnosis of ES tumor cells was presented by Udo Kontny: the use of CD99 targeted supramagnetic particles, which is currently developed at the University Hospital Freiburg, Germany.
Models for Ewing Sarcoma Research
Testing and optimization of appropriate animal models for new diagnostic and therapeutic approaches require tumor models with a close histo-pathology match and the right sarcoma microenvironment. ES cell lines are amply available to the research community, but in vitro cultivation, even under non-adherent conditions as spheroids or in soft agar, does not allow for tumor-microenvironment interactions. Subcutaneous injection of tumor cells into immuno compromised mice is most frequently used to investigate ES growth in vivo. Even though in humans ES never grow under the skin, this has been a useful research tool for simple growth studies. Antonio Llombart-Bosch (University of Valencia, Spain) reported on successful transplantation of 514 primary sarcoma pieces without dissociation in Nu/Nu BALBc mice, 16 of which were ES. The avoidance of stress associated with in vitro manipulation before transplantation resulted in >80% tumor xenograft take. This model can be used to study early stages of angiogenesis, and for preclinical therapy testing. Llombart-Bosch summarized his results on the immunohistochemical characterization of xenotransplanted sarcomas with respect to epithelial markers, CD99, Ki67, caveolin, p16, and p53. A major limitation of the method is that it does not work with core biopsies, and tumors have to be transplanted immediately after biopsy. However, this may in fact more accurately reflect ES growth than subcutaneously grown tumors following injection of mice with cell line suspensions.
Richard Moriggl (Ludwig Boltzmann Institute for Cancer Research, Vienna, Austria) reported that subcutaneous xenograft tumor growth from ES cell lines activates JAK/STAT signaling (which has also been observed in 70% of patient derived tumor materials), presumably via activation through the oncostatin M receptor, which is also prominently expressed on mesenchymal stem cells (MSCs). He provided in vitro evidence that treatment with oncostatin M results in decreased cell adhesion and the activation of several stemness transcription factors.
As an alternative to subcutaneous injections, Udo Kontny (University Hospital Freiburg, Germany) reported on the use of orthotopic xenotransplantation of ES cell lines into the mouse gastrocnemius muscle in his study of invasive growth into bone and for perfusion based imaging for early chemotherapy response detection. Intravenous tumor cell injection followed by in vitro cultivation of mouse lung slices is a novel potent method to investigate pulmonary metastasis ex vivo (PuMA assay, Mendoza et al., 2010). Poul Sorensen adopted this approach for his study of ES and described the specific growth conditions and their application to the investigation of ErbB4 in promoting migration, metastasis, and chemoresistance of ES (Table 1).
Subcutaneous and intravenous injections of ES cell lines have also been used by Günter Richter (Technical University, Munich, Germany) to study the role of two EWS-FLI1 induced ES specific genes, GPR64 and STEAP1 (Grunewald et al., 2012a) (Table 1). Comparing clinical data in patients with in vivo data in the xenograft models demonstrated the limitations of mouse models: in patients, high STEAP1 expression is associated with better survival (Grunewald et al., 2012b) while in the mouse it promotes tumor cell invasion.
Richard Moriggl gave an update on the development of a genetic EWS-FLI1 mouse model. Using a similar approach as Lin et al. (2008), he described the use of a Prx1 Cre recombinase driven conditional EWS-FLI1 knock-in to target the fusion protein to mesenchymal progenitor cells. He provided a detailed description of developmental changes that result in arrest of endochondral bone formation at an early stage (day 12.5) of chondroblastic development and the consequences for embryonal limb and craniofacial malformations. He also reported on preliminary results using a tamoxifen inducible modification of this model to turn-on EWS-FLI1 in the mesenchymal lineage after birth. So far, however, there is no indication of successful sarcomagenesis induction in the mice and further genetic modifications are necessary to develop a robust model.
Zebrafish models are increasingly used as an alternative to mice. There is a plethora of genetic variants available, they are easy to manipulate and breed, and, because of their transparency, simple to monitor. Pancras Hogendoorn (Leiden University Medical Center, Leiden, Netherlands) pointed out that bone development and the involved cytokines in zebrafish are highly homologous to mammals and they could, therefore, be particularly useful for studying bone sarcomas. He gave examples of research projects studying invasion, angiogenesis, and metastasis after yolk sac injection of ES cells. The beauty of this model is that invasion, angiogenesis, and tumor cell spread can be followed in real-time under the confocal microscope. Tumor host interactions can be easily studied by discrimination of human and fish genes. Preclinical lead development can profit from high-throughput screening of thousands of fish in a simple 96 well format in a single 1 week experiment under standardized experimental conditions. Poul Sorensen, with his collaborator Jason Berman of Dalhousie University in Halifax, used the zebrafish model to study the interaction of the splicing factor YB1 with EWS-FLI1. They found that hypoxia is inducing YB1, which increases polysomal (translated) HIF1α mRNA while decreasing translation of proliferation genes like CCND1 (Evdokimova et al., 2009), potentially affecting angiogenesis. Hogendoorn mentioned that zebrafish can be bred at temperatures as high as 34° to approach the human situation, and it was discussed whether these conditions could result in a kind of permanent heat shock. In addition, Hogendoorn reported on an in vitro 3D matrix model, in which tumor cells are inoculated into agar in 96 well format by robotic spotting with subsequent automated monitoring of outgrowth by a digital camera.
Tumor Initiating Cells
The efficiency of engraftment of solid tumor cells in mice and fish depends on the content of tumor initiating cells which are known to be particularly chemo resistant. Previously, such properties have been assigned to a small subpopulation of CD133 positive ES cells (Suva et al., 2009), which has subsequently been challenged by other researchers (Jiang et al., 2010). Uta Dirksen described the properties of a side population of ES cells as candidate tumor initiating cells. She found that this population of small cells retained pluripotency in differentiation assays, and led to increased soft agar colony growth associated with high tumor induction in mouse xenografts at subcutaneous and intravenous injection of only a few thousand cells of this particular side population of ES cells. The, presumably, tumor initiating sarcoma cells displayed increased chemoresistance to Doxorubicin and Etoposide. Importantly, these cells did neither express consistent stem cell marker (i.e., CD133 negative), nor increased levels of ABC transporters, and no stem cell transcription factors NANOG, OCT4, or SOX2.
Elizabeth Lawlor (University of Michigan) talked about expression and putative role in ES stem cells of the transmembrane protein LGR5 (GPR49). LGR5 is a well established stem cell marker highly expressed during embryonic development and post-natally in normal hair follicle and intestinal stem cells. High-levels of LGR5 are also associated with tumor initiation, cancer stem cells, and early relapse in colorectal cancer (Merlos-Suarez et al., 2011; Walker et al., 2011). LGR5 is a canonical Wnt target gene and recent data show that activation of LGR5 by its ligands, the R-spondin family of matricellular proteins, also potentiates canonical β catenin/Wnt signaling and activates stem cell proliferation (de Lau et al., 2011). R-spondins are expressed in developing bone and Lawlor has found that human neural crest stem cells and subpopulations of ES cells express LGR5. Ongoing studies are determining if LGR5 is just a marker or whether it is functionally involved in tumor initiation, stemness, and chemoresistance of ES cells (Table 1).
Targeting Tumor/Microenvironment Interactions and the Immune System
Besides the need for defining the subpopulation of chemoresistant tumor (stem) cells responsible for disease recurrence, tumor-microenvironmental interactions may offer entry points for supportive therapies. Françoise Redini (INSERM U957, Nantes, France) investigated the activity of bisphosphonates (i.e., Zoledronic acid) to inhibit osteoclastogenesis (Table 1). Osteolysis is an important mechanism of many bone targeting neoplasms to generate space for tumor growth. In turn, osteoclasts release factors promoting tumor cell growth. ES express RANKL which is known to stimulate osteoclastogenesis. In addition, primary ES contain high-levels of M-CSF and TNF-α, which might be secreted from tumor infiltrating macrophages. Since osteoclasts are hardly detectable in ES, macrophages may take their role in osteolysis and tumor stimulation. Redini reported that Zoledronic acid (Zometa, Novartis) induced caspase 3 dependent apoptosis of tumor cells in vitro and bone tumor regression in mice (Odri et al., 2010). In addition she demonstrated that ectopic soluble RANKL receptor osteoprotegerin (OPG), soluble TNF receptor, and DKK1 inhibitors (BHQ880, Novartis) inhibit ES growth by stimulating osteoblastogenesis (as monitored by increased bone marker expression) (Table 1).
Influencing the interaction between the tumor and the immune system is a further area of hope in novel ES therapy approaches. About 50% of ES are resistant to the apoptotic function of TRAIL. Udo Kontny and others had previously demonstrated that despite expression of TRAIL receptors DR4 and DR5, about 25% of tumors have low expression of caspase 8 and do therefore not respond to TRAIL treatment. IFN-γ had been shown to sensitize TRAIL-mediated apoptosis by upregulation of Caspase 8 in neuroblastoma and ES (Yang et al., 2003; Lissat et al., 2007) and to inhibit the formation of metastases in mice (Merchant et al., 2004) (Table 1). Kontny now reported that they found IFNgamma to inhibit invasion and migration of ES cells in vitro. Redini described a novel TRAIL receptor isoform on ES cells, a truncated DR4. It remains to be established, how this isoform may be involved in TRAIL resistance of tumor cells (Picarda et al., 2012). Günter Richter provided evidence that the EWS-FLI1 induced protein GPR64, which regulates expression of the metalloprotease MMP1, critically promotes lung metastasis. Moreover, experimental suppression of GPR64 expression rendered tumor cells more sensitive to TRAIL and chemotherapy causing decreased local tumor growth (Table 1).
Richter also described the experimental approach that Stefan Burdach′s group at the Technical University of Munich is taking to generate ES specific T-cell responses by in vitro priming (peptide non-amers) of dendritic cells. After selection of T-cells with peptide-MHC-pentamer-fluorophores, ES specific allo-restricted T-cells were shown to specifically kill tumor cells in vitro and upon tail vein injection in immunocompromised mice they delayed ES growth (Thiel et al., 2011). Based on this approach, a clinical trial is planned to be initiated in 2013 (Table 1).
Claudia Rössig (University Children’s Hospital Münster, Germany) explored the use of chimeric antigen receptors (CARs), that is T-cell receptor signaling domains linked to a tumor antigen specific antibody chain, to generate antigen specific T-cells for ES immunotherapy (Park et al., 2011; Kalos, 2012). She suggested using CARs for the carbohydrate GD2 that have recently been successfully applied successfully to the treatment of neuroblastoma (Louis et al., 2011) (Table 1). She found surface GD2 in 11/11 ES cell lines and 3/3 primary cell cultures (Kailayangiri et al., 2012). Moreover, diagnostic ES biopsies from 12 of 14 patients had uniform GD2 expression. Anti-GD2 CARs lyse ES cells under adherent growth conditions and in spheres (six cell lines) and have anti-tumor activity in an ES xenograft model. These CARs also work in an autologous setting. The theoretical potential of autoimmune reactions against other GD2 expressing cell types, such as mesenchymal and neural crest derived stem cells, limiting the clinical application of this approach, remains to be established. To test if a patient may be eligible for a GD2-directed immunotherapy, GD2 expression has to be assessed on frozen sections.
Rössig further discussed the pros and cons of using natural killer (NK) cells as alternative innate immune effector cells to antigen specific T-cells. While NK cells are more potent in direct tumor cytolysis than T-cells, they do not persist in vivo for extended periods. She suggested to expand NK cells from ES patients and combine the signaling lymphocyte activation molecule-related receptor 2B4 (CD244) with an anti-GD2 antibody chain to produce CARs that overcome NK cell resistance of ES, as she had previously demonstrated in neuroblastoma (Altvater et al., 2009) (Table 1).
The IGF Pathway and Beyond
A well known pathway of tumor host interaction in ES patients is IGF1R signaling. Initial enthusiasm for IGF1R directed antibodies and small molecules to treat ES patients with refractory or relapsed disease has decreased, reflecting limited effectiveness in vivo (Ho and Schwartz, 2011). To improve efficacy, Udo Kontny reported on the development of an experimental therapy using IGF1R antibody coupled to liposomal doxorubicin (Table 1). Jenny Potratz (University Children’s Hospital Muenster, Germany) reported on the results of an RNAi screen for sensitizers to IGF1R inhibitors and described a number of promising kinases including HCK, FES, EPHB2, LMTK2, PDGFRB). By comparing 15 localized tumors to six metastases and four MSC samples, they filtered out EPHB2 which was differentially expressed and thus defines a candidate target for treatment of aggressive disease. The top candidate kinase sensitizing to IGF1R inhibitors was found to be MST1R (RON), a cMET related receptor tyrosine kinase (RTK; Potratz et al., 2010) (Table 1).
Katia Scotlandi identified a mechanism of resistance to IGF1R targeting compounds. She found that ES cells upregulate Insulin Receptor A (IRA) which binds to IGF2 (Garofalo et al., 2011). She therefore suggested to co-target IGF1R and IRA. The EU-FP7 funded project EUROSARC will initiate a phase I trial for co-targeting using the dual inhibitor OSI-906 (Table 1). Alternatively, the antidiabetic drug Metformin, which decreases circulating insulin, may hold promise for the treatment of ES (Table 1). It has been demonstrated to display anticancer activity by depriving cells of insulin, inhibiting mTOR, and decreasing AMPK involved in anoikis resistance (Del Barco et al., 2011; Ng et al., 2012). So far, all tested ES cell lines, even IGF1R resistant ones, have proved to be sensitive to metformin, though very high concentrations of metformin (10 mM) were required. Under these conditions, Scotlandi also observed Metformin to reactivate p53.
Scotlandi also discussed several other p53 reactivating drugs (i.e., Nutlin 3) and addressed the role of CD99 targeting antibody 0662 for the induction of caspase independent cell death and autophagy in ES (Table 1). Simone Fulda (Goethe-University, Frankfurt, Germany) discussed the role and therapeutic potential of the Hedgehog pathway comparing fusion negative alveolar rhabdomyosarcoma to ES. Other drugs and pathways of relevance to ES that were addressed during the meeting but not mentioned above comprise the PKC inhibitor Enzastaurin (F. Tirode), the NEDD8 inhibitor MLN4294, and protein ubiquitin ligase inhibitors (E. De Alava) (Table 1).
Conclusion and Next Steps
In order to move the huge amount of new biological knowledge presented at the meeting forward to the clinics, feasibility, and quality criteria were discussed. Experience from the EuroE.W.I.N.G.99 study (Le Deley et al., 2010; Le Deley et al., unpublished) has taught us to keep prospective marker studies in the context of multicentric clinical trials as simple as possible. Enrique DeAlava suggested to first perform a meta analysis of existing retrospective data for a candidate marker before proceeding to prospective evaluation – even though the validity of retrospective studies may be doubted due to the frequent incompleteness of clinical data. Such retrospective tumor collections exist in Valencia, where Antonio Llombart-Bosch has built an impressive tumor bank with samples from 841 ES cases, and 24 tissue arrays with 604 cases. This ES biobank has been entered into the virtual tumor bank of the European network of excellence EuroBoNet. Limited clinical data exist for about 270 cases which have been used recently to establish high desmoplakin expression as a good prognostic indicator. As a reference lab for the Spanish SEHOP study, 141 ES (120 frozen samples and tissue arrays) have been collected by Javier Alonso in Madrid. Clinically well annotated retrospective ES tumor banks, partly with genomic data (gene expression), already exist in Münster, Leeds, and Paris with samples from the European ES studies EICESS92 and EuroE.W.I.N.G.99. In addition, the German TranSaRNet supports a tumor bank on relapsed sarcoma, SAREZ.
Limitations in the availability of unfixed biopsy materials are commonly recognized as the most severe bottleneck for marker studies. Poul Sorensen pointed to tumor heterogeneity as a problem that does not allow detection of rare tumor driving cells when grinding the whole tumor. Assay development on fixed material is therefore urgently needed, and pathologists should routinely produce tissue micro arrays (TMAs) from the biopsies of all patients entered prospectively into a clinical trial.
Yet, since the causes of metastasis are still unknown, it remains doubtful whether the analysis of diagnostic samples from the primary tumor of patients with localized disease before therapy will allow prediction of chemotherapy response and relapse. Therefore, we should focus on collecting tumor material from metastatic patients and during disease progression. In order to guarantee the same quality of samples and avoid bias in an international multi centric setting, emphasis should be placed on (i) the number of samples, (ii) the stability of targets, (iii) the availability of standard operating procedures (SOPs) for sample collection, processing, and diagnostic assays, (iv) a low inter- and intra assay variability, (v) a high degree of practicability, (vi) built-in quality controls, (vii) guaranteed availability and quality of clinical data, and lastly (viii) funding. EuroBoNet has already established SOPs for sample preparation and processing; these are available through the EuroBoNet website. Apart from putative prognostic markers discussed at the meeting, participants considered the prospective evaluation of already established copy number variations by aCGH or MLPA, complemented by exploratory exome sequencing, to be the highest priority. In addition, the investigation of ctDNA in blood and bone marrow was viewed as an area of great promise for non-invasive assessment of therapy response. Considering novel therapies, participants pleaded for the introduction of more experimental therapies into the treatment of metastatic patients. After 2 days of discussions at this meeting, participants considered PARP inhibitors and mitramycin as the most promising agents for this purpose. In addition, a need for more pharmacogenetic and pharmacogenomic studies was expressed to assess factors of resistance/sensitivity to conventional chemotherapies.
Through the Biology and Sarcoma Workpackages (chaired by Angelika Eggert, University Clinics, Essen, Germany, and Stefan Bielak, Olga Hospital, Stuttgart, Germany) these suggestions will be presented to the European Ewing sarcoma study groups for consideration in the design of Ewing2008 and Ewing2012 trials.
Author Contributions
Javier Alonso, Pierre Aman, Dave N. T. Aryee1, Jozef Ban, Sue A. Burchill, Stefan Burdach, Enrique De Alava, Olivier Delattre, Uta Dirksen, Argyro Fourtouna, Simone Fulda, Lee J. Helman, David Herrero-Martin, Pancras C. W. Hogendoorn, Udo Kontny, Elizabeth R. Lawlor, Stephen L. Lessnick, Antonio Llombart-Bosch, Markus Metzler, Richard Moriggl, Stephan Niedan, Jenny Potratz, Françoise Redini, Günther H. S. Richter, Lucia T. Riedmann, Claudia Rossig, Beat W. Schäfer, Raphaela Schwentner, Katia Scotlandi, Poul H. Sorensen, Martin S. Staege, Franck Tirode, Jeffrey Toretsky, and Selena Ventura contributed equally and are listed in alphabetical order.
Angelika Eggert – ENCCA Biology work package leader
Ruth Ladenstein – project coordinator of ENCCA
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Altvater, B., Landmeier, S., Pscherer, S., Temme, J., Schweer, K., Kailayangiri, S., Campana, D., Juergens, H., Pule, M., and Rossig, C. (2009). 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 15, 4857–4866.
Ban, J., Bennani-Baiti, I., Kauer, M., Schaefer, K. L., Poremba, C., Jug, G., Schwentner, R., Smrzka, O., Muehlbacher, K., Aryee, D., and Kovar, H. (2008). EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res. 68, 7100–7109.
Ban, J., Jug, G., Mestdagh, P., Schwentner, R., Kauer, M., Aryee, D. N. T., Schaefer, K. L., Nakatani, F., Scotlandi, K., Reiter, M., Strunk, D., Speleman, F., Vandesompele, J., and Kovar, H. (2010). Hsa-mir-145 is the top EWS-FLI1 repressed microRNA involved in a positive feed-back loop in Ewing’s sarcoma. Oncogene 30, 2173–2180.
Bennani-Baiti, I., Aryee, D., Ban, J., MacHado, I., Kauer, M., Mühlbacher, K., Amann, G., Llombart-Bosch, A., and Kovar, H. (2011). Notch signaling is off and is uncoupled from HES1 expression in Ewing’s sarcoma. J. Pathol. 225, 353–363.
Boro, A., Pretre, K., Rechfeld, F., Thalhammer, V., Oesch, S., Wachtel, M., Schafer, B. W., and Niggli, F. K. (2012). Small-molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing’s sarcoma. Int. J. Cancer. doi:10.1002/ijc.27472 [Epub ahead of print].
de Lau, W., Barker, N., Low, T. Y., Koo, B. K., Li, V. S., Teunissen, H., Kujala, P., Haegebarth, A., Peters, P. J., van de Wetering, M., Stange, D. E., van Es, J. E., Guardavaccaro, D., Schasfoort, R. B., Mohri, Y., Nishimori, K., Mohammed, S., Heck, A. J., and Clevers, H. (2011). Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297.
Del Barco, S., Vazquez-Martin, A., Cufi, S., Oliveras-Ferraros, C., Bosch-Barrera, J., Joven, J., Martin-Castillo, B., and Menendez, J. A. (2011). Metformin: multi-faceted protection against cancer. Oncotarget 2, 896–917.
Delattre, O., Zucman, J., Plougastel, B., Desmaze, C., Melot, T., Peter, M., Kovar, H., Joubert, I., De Jong, P., and Rouleau, G. (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359, 162–165.
DuBois, S. G., Krailo, M. D., Lessnick, S. L., Smith, R., Chen, Z., Marina, N., Grier, H. E., and Stegmaier, K. (2009). Phase II study of intermediate-dose cytarabine in patients with relapsed or refractory Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr. Blood Cancer 52, 324–327.
Erkizan, H. V., Kong, Y., Merchant, M., Schlottmann, S., Barber-Rotenberg, J. S., Yuan, L., Abaan, O. D., Chou, T. H., Dakshanamurthy, S., Brown, M. L., Uren, A., and Toretsky, J. A. (2009). A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 15, 750–756.
Erkizan, H. V., Scher, L. J., Gamble, S. E., Barber-Rotenberg, J. S., Sajwan, K. P., Uren, A., and Toretsky, J. A. (2011). Novel peptide binds EWS-FLI1 and reduces the oncogenic potential in Ewing tumors. Cell Cycle 10, 3397–3408.
Evdokimova, V., Tognon, C., Ng, T., Ruzanov, P., Melnyk, N., Fink, D., Sorokin, A., Ovchinnikov, L. P., Davicioni, E., Triche, T. J., and Sorensen, P. H. (2009). Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 15, 402–415.
Garofalo, C., Manara, M. C., Nicoletti, G., Marino, M. T., Lollini, P. L., Astolfi, A., Pandini, G., Lopez-Guerrero, J. A., Schaefer, K. L., Belfiore, A., Picci, P., and Scotlandi, K. (2011). Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene 30, 2730–2740.
Grohar, P. J., Woldemichael, G. M., Griffin, L. B., Mendoza, A., Chen, Q. R., Yeung, C., Currier, D. G., Davis, S., Khanna, C., Khan, J., McMahon, J. B., and Helman, L. J. (2011). Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 103, 962–978.
Grunewald, T. G., Diebold, I., Esposito, I., Plehm, S., Hauer, K., Thiel, U., da Silva-Buttkus, P., Neff, F., Unland, R., Muller-Tidow, C., Zobywalski, C., Lohrig, K., Lewandrowski, U., Sickmann, A., da Costa, O. P., Görlach, A., Cossarizza, A., Butt, E., Richter, G. H., and Burdach, S. (2012a). STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 10, 52–65.
Grunewald, T. G., Ranft, A., Esposito, I., da Silva-Buttkus, P., Aichler, M., Baumhoer, D., Schaefer, K. L., Ottaviano, L., Poremba, C., Jundt, G., Jürgens, H., Dirksen, U., Richter, G. H., and Burdach, S. (2012b). High STEAP1 expression is associated with improved outcome of Ewing’s sarcoma patients. Ann. Oncol. doi:10.1093/annonc/mdr605. [Epub ahead of print].
Hattinger, C. M., Potschger, U., Tarkkanen, M., Squire, J., Zielenska, M., Kiuru-Kuhlefelt, S., Kager, L., Thorner, P., Knuutila, S., Niggli, F. K., Ambros, P. F., Gadner, H., and Betts, D. R. (2002). Prognostic impact of chromosomal aberrations in Ewing tumours. Br. J. Cancer 86, 1763–1769.
Ho, A. L., and Schwartz, G. K. (2011). Targeting of insulin-like growth factor type 1 receptor in Ewing sarcoma: unfulfilled promise or a promising beginning? J. Clin. Oncol. 29, 4581–4583.
Ince, P. G., Highley, J. R., Kirby, J., Wharton, S. B., Takahashi, H., Strong, M. J., and Shaw, P. J. (2011). Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 122, 657–671.
Jiang, X., Gwye, Y., Russell, D., Cao, C., Douglas, D., Hung, L., Kovar, H., Triche, T. J., and Lawlor, E. R. (2010). CD133 expression in chemo-resistant Ewing sarcoma cells. BMC Cancer 10, 116. doi:10.1186/1471-2407-10-116
Kailayangiri, S., Altvater, B., Meltzer, J., Pscherer, S., Luecke, A., Titze, U., Dierkes, C., Leuchte, K., Landmeier, S., Hotfilder, M., Dirksen, U., Hardes, J., Gosheger, G., Juergens, H., and Rossig, C. (2012). The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 106, 1123–1133.
Kalos, M. (2012). Muscle CARs and TcRs: turbo-charged technologies for the (T cell) masses. Cancer Immunol. Immunother. 61, 127–135.
Kofman, S., Perlia, C. P., and Economou, S. G. (1973). Mithramycin in the treatment of metastatic Ewing’s sarcoma. Cancer 31, 889–893.
Le Deley, M. C., Delattre, O., Schaefer, K. L., Burchill, S. A., Koehler, G., Hogendoorn, P. C., Lion, T., Poremba, C., Marandet, J., Ballet, S., Pierron, G., Brownhill, S. C., Nesslböck, M., Ranft, A., Dirksen, U., Oberlin, O., Lewis, I. J., Craft, A. W., Jürgens, H., and Kovar, H. (2010). Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-EWING 99 Trial. J. Clin. Oncol. 28, 1982–1988.
Lin, P. P., Pandey, M. K., Jin, F., Xiong, S., Deavers, M., Parant, J. M., and Lozano, G. (2008). EWS-FLI1 induces developmental abnormalities and accelerates sarcoma formation in a transgenic mouse model. Cancer Res. 68, 8968–8975.
Lissat, A., Vraetz, T., Tsokos, M., Klein, R., Braun, M., Koutelia, N., Fisch, P., Romero, M. E., Long, L., Noellke, P., MacKall, C. L., Niemeyer, C. M., and Kontny, U. (2007). Interferon-gamma sensitizes resistant Ewing’s sarcoma cells to tumor necrosis factor apoptosis-inducing ligand-induced apoptosis by up-regulation of caspase-8 without altering chemosensitivity. Am. J. Pathol. 170, 1917–1930.
Louis, C. U., Savoldo, B., Dotti, G., Pule, M., Yvon, E., Myers, G. D., Rossig, C., Russell, H. V., Diouf, O., Liu, E., Liu, H., Wu, M. F., Gee, A. P., Mei, Z., Rooney, C. M., Heslop, H. E., and Brenner, M. K. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056.
MacKintosh, C., Ordonez, J. L., Garcia-Dominguez, D. J., Sevillano, V., Llombart-Bosch, A., Szuhai, K., Scotlandi, K., Alberghini, M., Sciot, R., Sinnaeve, F., Hogendoorn, P. C., Picci, P., Knuutila, S., Dirksen, U., Debiec-Rychter, M., Schaefer, K. L., and de Álava, E. (2011). 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31, 1287–1298.
Mendoza, A., Hong, S. H., Osborne, T., Khan, M. A., Campbell, K., Briggs, J., Eleswarapu, A., Buquo, L., Ren, L., Hewitt, S. M., Dakir el, H., Garfield, S., Walker, R., Merlino, G., Green, J. E., Hunter, K. W., Wakefield, L. M., and Khanna, C. (2010). Modeling metastasis biology and therapy in real time in the mouse lung. J. Clin. Invest. 120, 2979–2988.
Meng, X., Zhu, D., Yang, S., Wang, X., Xiong, Z., Zhang, Y., Brachova, P., and Leslie, K. K. (2011). Cytoplasmic MTDH provides a survival advantage under conditions of stress by acting as an RNA binding protein. J. Biol. Chem. 287, 4485–4491.
Merchant, M. S., Yang, X., Melchionda, F., Romero, M., Klein, R., Thiele, C. J., Tsokos, M., Kontny, H. U., and MacKall, C. L. (2004). Interferon gamma enhances the effectiveness of tumor necrosis factor-related apoptosis-inducing ligand receptor agonists in a xenograft model of Ewing’s sarcoma. Cancer Res. 64, 8349–8356.
Merlos-Suarez, A., Barriga, F. M., Jung, P., Iglesias, M., Cespedes, M. V., Rossell, D., Sevillano, M., Hernando-Momblona, X., da Silva-Diz, V., Munoz, P., Clevers, H., Sancho, E., Mangues, R., and Batlle, E. (2011). The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8, 511–524.
Metzger, E., Imhof, A., Patel, D., Kahl, P., Hoffmeyer, K., Friedrichs, N., Muller, J. M., Greschik, H., Kirfel, J., Ji, S., Kunowska, N., Beisenherz-Huss, C., Günther, T., Buettner, R., and Schüle, R. (2010). Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature 464, 792–796.
Nakatani, F., Ferracin, M., Manara, M. C., Ventura, S., Del Monaco, V., Ferrari, S., Alberghini, M., Grilli, A., Knuutila, S., Schaefer, K. L., Mattia, G., Negrini, M., Picci, P., Serra, M., and Scotlandi, K. (2011). miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemosensitivity and malignancy. J. Pathol. 226, 796–805.
Ng, T. L., Leprivier, G., Robertson, M. D., Chow, C., Martin, M. J., Laderoute, K. R., Davicioni, E., Triche, T. J., and Sorensen, P. H. (2012). The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 19, 501–510.
Odri, G. A., Dumoucel, S., Picarda, G., Battaglia, S., Lamoureux, F., Corradini, N., Rousseau, J., Tirode, F., Laud, K., Delattre, O., Gouin, F., Heymann, D., and Redini, F. (2010). Zoledronic acid as a new adjuvant therapeutic strategy for Ewing’s sarcoma patients. Cancer Res. 70, 7610–7619.
Park, T. S., Rosenberg, S. A., and Morgan, R. A. (2011). Treating cancer with genetically engineered T cells. Trends Biotechnol. 29, 550–557.
Picarda, G., Surget, S., Guiho, R., Teletchea, S., Berreur, M., Tirode, F., Pellat-Deceunynck, C., Heymann, D., Trichet, V., and Redini, F. (2012). A functional, new short isoform of death receptor 4 in Ewing’s sarcoma cell lines may be involved in TRAIL sensitivity/resistance mechanisms. Mol. Cancer Res. 10, 336–346.
Postel-Vinay, S., Veron, A. S., Tirode, F., Pierron, G., Reynaud, S., Kovar, H., Oberlin, O., Lapouble, E., Ballet, S., Lucchesi, C., Kontny, U., González-Neira, A., Picci, P., Alonso, J., Patino-Garcia, A., de Paillerets, B. B., Laud, K., Dina, C., Froguel, P., Clavel-Chapelon, F., Doz, F., Michon, J., Chanock, S. J., Thomas, G., Cox, D. G., and Delattre, O. (2012). Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat. Genet. 44, 323–327.
Potratz, J. C., Saunders, D. N., Wai, D. H., Ng, T. L., Kinney, S. E., Carboni, J. M., Gottardis, M. M., Triche, T. J., Jurgens, H., Pollak, M. N., Aparicio, S. A., and Sorensen, P. H. (2010). Synthetic lethality screens reveal RPS6 and MST1R as modifiers of insulin-like growth factor-1 receptor inhibitor activity in childhood sarcomas. Cancer Res. 70, 8770–8781.
Prasad, S. C., Thraves, P. J., Bhatia, K. G., Smulson, M. E., and Dritschilo, A. (1990). Enhanced poly (adenosine diphosphate ribose) polymerase activity and gene expression in Ewing’s sarcoma cells. Cancer Res. 50, 38–43.
Roberts, P., Burchill, S. A., Brownhill, S., Cullinane, C. J., Johnston, C., Griffiths, M. J., McMullan, D. J., Bown, N. P., Morris, S. P., and Lewis, I. J. (2008). Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing’s sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children’s Cancer and Leukaemia Group. Genes Chromosomes Cancer 47, 207–220.
Savola, S., Klami, A., Tripathi, A., Niini, T., Serra, M., Picci, P., Kaski, S., Zambelli, D., Scotlandi, K., and Knuutila, S. (2009). Combined use of expression and CGH arrays pinpoints novel candidate genes in Ewing sarcoma family of tumors. BMC Cancer 9, 17. doi:10.1186/1471-2407-9-17
Stegmaier, K., Wong, J. S., Ross, K. N., Chow, K. T., Peck, D., Wright, R. D., Lessnick, S. L., Kung, A. L., and Golub, T. R. (2007). Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS Med. 4, e122. doi:10.1371/journal.pmed.0040122
Suva, M. L., Riggi, N., Stehle, J. C., Baumer, K., Tercier, S., Joseph, J. M., Suva, D., Clement, V., Provero, P., Cironi, L., Osterheld, M. C., Guillou, L., and Stamenkovic, I. (2009). Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 69, 1776–1781.
Thiel, U., Pirson, S., Muller-Spahn, C., Conrad, H., Busch, D. H., Bernhard, H., Burdach, S., and Richter, G. H. (2011). Specific recognition and inhibition of Ewing tumour growth by antigen-specific allo-restricted cytotoxic T cells. Br. J. Cancer 104, 948–956.
Walker, F., Zhang, H. H., Odorizzi, A., and Burgess, A. W. (2011). LGR5 is a negative regulator of tumourigenicity, antagonizes Wnt signalling and regulates cell adhesion in colorectal cancer cell lines. PLoS ONE 6, e22733. doi:10.1371/journal.pone.0022733
Yang, X., Merchant, M. S., Romero, M. E., Tsokos, M., Wexler, L. H., Kontny, U., MacKall, C. L., and Thiele, C. J. (2003). Induction of caspase 8 by interferon gamma renders some neuroblastoma (NB) cells sensitive to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but reveals that a lack of membrane TR1/TR2 also contributes to TRAIL resistance in NB. Cancer Res. 63, 1122–1129.
Keywords: Ewing sarcoma, animal models, sarcomagenesis, genomics, epigenetics, biomarkers, drug screen, prognosis
Citation: Kovar H, Alonso J, Aman P, Aryee DNT, Ban J, Burchill SA, Burdach S, De Alava E, Delattre O, Dirksen U, Fourtouna A, Fulda S, Helman LJ, Herrero-Martin D, Hogendoorn PCW, Kontny U, Lawlor ER, Lessnick SL, Llombart-Bosch A, Metzler M, Moriggl R, Niedan S, Potratz J, Redini F, Richter GHS, Riedmann LT, Rossig C, Schäfer BW, Schwentner R, Scotlandi K, Sorensen PH, Staege MS, Tirode F, Toretsky J, Ventura S, Eggert A and Ladenstein R (2012) The first European interdisciplinary Ewing sarcoma research summit. Front. Oncol. 2:54. doi: 10.3389/fonc.2012.00054
Received: 15 March 2012; Paper pending published: 17 April 2012;
Accepted: 10 May 2012; Published online: 29 May 2012.
Edited by:
Brigitte Charolotte Widemann, National Cancer Institute, USACopyright: © 2012 Kovar, Alonso, Aman, Aryee, Ban, Burchill, Burdach, De Alava, Delattre, Dirksen, Fourtouna, Fulda, Helman, Herrero-Martin, Hogendoorn, Kontny, Lawlor, Lessnick, Llombart-Bosch, Metzler, Moriggl, Niedan, Potratz, Redini, Richter, Riedmann, Rossig, Schäfer, Schwentner, Scotlandi, Sorensen, Staege, Tirode, Toretsky, Ventura, Eggert and Ladenstein. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Heinrich Kovar, Children’s Cancer Research Institute, St. Anna Kinderkrebsforschung and Medical University, Zimmermannplatz 10, 1090 Vienna, Austria. e-mail:aGVpbnJpY2gua292YXJAY2NyaS5hdA==