


- 1Unit of Pathology, Institute for Cancer Research and Treatment, Candiolo, Torino, Italy
- 2Department of Biology and Biotechnology, University of Pavia, Pavia, Italy
In 2002, Al-Tassan and co-workers described for the first time a recessive form of inherited polyposis associated with germline mutations of MUTYH, a gene encoding a base excision repair (BER) protein that counteracts the DNA damage induced by the oxidative stress. MUTYH-associated polyposis (MAP) is now a well-defined cancer susceptibility syndrome, showing peculiar molecular features that characterize disease progression. However, some aspects of MAP, including diagnostic criteria, genotype-phenotype correlations, pathogenicity of variants, as well as relationships between BER and other DNA repair pathways, are still poorly understood. A deeper knowledge of the MUTYH expression pattern is likely to refine our understanding of the protein role and, finally, to improve guidances for identifying and handling MAP patients.
Introduction
About one-third of colorectal cancers (CRC) are ascribable to a genetic predisposition. In approximately 5% of cases, CRC occurs in the context of Mendelian syndromes and is associated with highly penetrant gene mutations; in 20–25% of cases it can be considered as “familial” and likely due to low/medium-penetrant genetic variants (reviewed by de la Chapelle, 2004). More generally, all non-Mendelian CRCs can be regarded as complex diseases, where multiple genetic variants and environmental factors can modulate the disease risk. Genome-wide association studies (GWAS) have recently identified over a dozen risk loci, statistically associated with CRC (Tomlinson, 2012). The present challenge is to assess their functional significance and to clarify their interaction with probable or well-established environmental risk factors (Dunlop et al., 2012).
Among CRC predisposing syndromes, Hereditary Non-Polyposis Colorectal Cancer (HNPCC) or Lynch syndrome (OMIM#114500; NCBI database of human genes and genetic phenotype: www.ncbi.nlm.nih.gov/omim) is the most common disease; this autosomal dominant disorder is characterized by early onset CRC and extracolonic manifestations. HNPCC is associated with inherited defects in DNA Mismatch Repair (MMR) genes, primarily MSH2 and MLH1 (reviewed by Lynch et al., 2009). A less common syndrome is Familial Adenomatous Polyposis (FAP; OMIM#175100); this autosomal dominant condition is characterized by the development of hundred to thousands of colorectal adenomas predisposing to CCR. Congenital retinal pigment epithelial hypertrophy (CHRPE), gastric and duodenal adenomas, desmoid tumors, and thyroid cancers, are the possible extracolonic manifestations. FAP is associated with germline mutations in APC that encodes a tumor suppressor protein acting as a regulator of WNT signal transduction pathway. A milder form of FAP, termed attenuated FAP (AFAP), is characterized by the development of less than 100 colorectal adenomas, a more limited expression of extracolonic features, and a delay in onset of CRC; germline mutations associated with AFAP have mainly been detected in the 5′ and 3′ end of APC, and in the alternatively spliced exon 9 of the gene (reviewed by Jasperson et al., 2010).
In 2002, Al-Tassan and coworkers described for the first time a recessive form of polyposis associated with biallelic mutations of MUTYH (Al-Tassan et al., 2002; Jones et al., 2002; Sieber et al., 2003); this gene encodes a base excision repair (BER) protein that counteracts DNA damage induced by oxidative stress. This disease is currently known as MAP for MUTYH-associated polyposis.
The MAP Patients: Phenotype, CRC Risk, and Surveillance
Presently, due to the variability of clinical features in MUTYH mutation carriers (Morak et al., 2010), the diagnostic criteria for MAP are not fully established and patients presenting FAP-like phenotypes are frequently difficult to classify. Usually, patients directed to MUTYH genetic testing have disease family history compatible with an autosomal recessive mode of inheritance, colorectal polyposis, and no identifiable mutations in APC (Sampson et al., 2003; Russell et al., 2006; reviewed by Aretz, 2010). In biallelic MUTYH mutation carriers the colonic phenotype resembles that of AFAP, with onset in the fourth-fifth decade and a limited number of adenomas (30-100) that increase susceptibility to CRC (Venesio et al., 2004; Filipe et al., 2009); however, unlike AFAP, hyperplastic and sessile serrated polyps can be found in MAP (Boparai et al., 2008; Zorcolo et al., 2011). Importantly, approximately 60% of MAP patients with polyposis have CRC at first presentation (mean age at diagnosis 48 years) (Nielsen et al., 2009). Moreover, a number of MAP patients with CRC and no polyps have been reported (reviewed by Nielsen et al., 2011). In a population-based screening, the penetrance of CRC has been estimated 20% and 43% at 50 and at 60 years of age, respectively (Lubbe et al., 2009). Since oxidative stress occurs in various cell types, MUTYH inactivation can be expected to predispose not only to intestinal, but also to extraintestinal lesions. Key extracolonic manifestations include predisposition to duodenal adenomas and cancer (Nielsen et al., 2006); in addition, constitutive gene mutations have been detected in patients with endometrial carcinoma (Barnetson et al., 2007; Tricarico et al., 2009). A recent multicenter study showed that the incidence of extraintestinal malignancies among MAP cases is almost twice that of the general population, with a significant increase in the incidence of ovarian, bladder, and skin cancers, and a trend of increased risk of breast tumors; interestingly, this cancer spectrum overlaps with HNPCC syndrome (Vogt et al., 2009). Thyroid carcinomas have been documented; however, in contrast to FAP, MAP does not appear to be associated with this cancer type (Ponti et al., 2005; Vogt et al., 2009; Pervaiz et al., 2010).
While the strong impact of biallelic MUTYH mutations on CRC risk has been demonstrated, the cancer risk associated with germline monoallelic mutations is still controversial. Initially, heterozygous germline mutations had been reported to increase the risk of CRC later in life without apparent excess risk of polyposis (Farrington et al., 2005), and both biallelic and monoallelic mutation carriers had been reported more likely to have first/second-degree relatives with CRC compared with non-carriers (Croitoru et al., 2004). However, subsequent meta-analyses have produced conflicting results, indicating both a non-significant and a significant increased risk of CRC for monoallelic mutation carriers (Peterlongo et al., 2006; Tenesa et al., 2006; Webb et al., 2006). More recently, the analysis of MAP family members confirmed previous data that monoallelic carriers have a two-fold increase of risk of CRC (Jones et al., 2009; Win et al., 2011); analogous results have been obtained through a systematic evaluation of clinicopathologic/epidemiologic and genetic data in a series of CRC cases and controls from a multisite CRC registry (Cleary et al., 2009).
A large population-based series of patients and controls has been screened for the presence of the two most common MUTYH mutations in MAP (i.e., Y179C and G396D): following genotype-phenotype correlation, the evaluation of genotype-specific CRC risk indicated that monoallelic mutation status is not clinically relevant (Lubbe et al., 2009). Accordingly, a marginal monoallelic effect has been reported in a recent study aimed at refining the estimates of CRC risk associated with mono- and bi-allelic MUTYH mutations (Theodoratou et al., 2010).
During a workshop held in Mallorca in 2006 and 2007, European experts on hereditary gastrointestinal cancer established recommendations for the clinical management of polyposis. According to these guidelines, patients with more than 10 adenomas should be referred for genetic counselling, and MUTYH mutation analysis should be performed. The screening should start between 18 and 20 years, the same age as recommended in AFAP. Since patients frequently develop only a few adenomas and CRC is often localized in the proximal colon, colonoscopy at 2-yearly intervals instead of sigmoidoscopy should be performed. Due to the relatively high risk for duodenal cancer, upper gastrointestinal endoscopy should start between 25 and 30 years of age. If surgery is required, IRA is sufficient in most cases to eliminate cancer risk (Vasen et al., 2008).
Recently, Nieuwenhuis and collaborators (2012) underlined the high risk in MAP patients to develop CRC even under surveillance, suggesting colonoscopy at short intervals (1–2-year intervals) starting from the age of 18–20 years, and recommending (sub)total colectomy when surgery for CRC is justified.
In the absence of clear-cut data on monoallelic MUTYH mutation carriers, CRC surveillance should follow guidelines proposed for subjects with a family history of CRC, without intensive screening, i.e., colonoscopy starting at age 40 and repeated every five years (Levin et al., 2008).
The lifetime risk for extracolonic cancers seems considerable; however, it is uncertain if more aggressive cancer surveillance for these lesions than is recommended for the general population would be valuable (Nielsen et al., 2009; Terdiman, 2009).
Molecular Mechanisms Underlying MAP
Al-Tassan and colleagues (2002) reported for the first time that predisposition to multiple colorectal adenomas and carcinomas can be inherited in an autosomal recessive manner. By analyzing tumor DNAs from three affected individuals of the same family, they found that APC tumor suppressor gene was somatically inactivated due to the frequent occurrence of G:C to T:A transversions. Such a pattern was suggestive of a defect in the repair system that prevents mutations caused by spontaneous oxidation of guanine to 8-oxoguanine, the most prevalent product of the oxidative stress (reviewed by David et al., 2007). Accordingly, the human genes encoding for the enzymes of the BER pathway were entirely sequenced and germline biallelic mutations in MUTYH were detected in the affected members of the family. Subsequent studies have unequivocally established the association between MUTYH germline mutations and predisposition to adenomas and CRCs, confirming the preponderance of G:C to T:A transversions as a molecular feature of MAP-associated tumors (reviewed by Lipton and Tomlinson, 2004, and by Sampson et al., 2005).
In addition to transversions along APC resulting in stop codon formation and gene inactivation, somatic GGT to TGT transversions, giving rise to the G12C KRAS activating-mutation, were frequently detected in MAP CRCs (Lipton et al., 2003; Jones et al., 2004; Nielsen et al., 2009). Boparai and collaborators (2008) reported that not only hyperplastic polyps (HPs) and sessile serrated adenomas (SSAs) can be a typical expression of MAP, but that they also have a characteristic molecular background. In particular, KRAS gene mutations were shown to be present in 70% of HP/SSA in MAP patients, compared with 17% of HP/SSA in sporadic cases. Of relevance, G:C to T:A transversions accounted for 94% of the mutations in MAP-HP/SSA, with respect to 29% in sporadic HPs/SSAs. Moreover, comparing adenomas and HPs/SSAs in MAP patients, APC mutations were only detectable in adenomas, indicating two possible tumor pathways, one leading to adenomas via APC mutations, and the other leading to HPs/SSAs via KRAS activation.
On the whole, MAP cancers appear to follow a distinct progression pathway compared to pathways occurring in CRCs, i.e., with either chromosomal instability (CIN) or with high-frequency microsatellite instability (MSI-H). However, some features overlap with CIN phenotype, including frequent APC/KRAS mutations, while others with MSI-H phenotype, including unfrequent LOH at APC locus and near-diploid karyotype (Lipton et al., 2003; Johnson et al., 2005). Tumors from both monoallelic and biallelic MUTYH mutations carriers often show low-frequency microsatellite instability (MSI-L), suggesting functional interactions between BER and MMR systems (Cleary et al., 2009). Interestingly, HLA class I expression loss has been reported in both MAP and MMR-deficient tumors (De Miranda et al., 2009), suggesting that the mutagenic background of these tumor types triggers the generation of aberrant peptides likely acting as tumor neo-antigens. The elicited immune reaction might then selectively favor the outgrowth of cancer cell clones that have lost HLA class I expression, avoiding cancer cell recognition and elimination by the immune system (De Miranda et al., 2009).
In sporadic carcinogenesis, the issue of a possible role of MUTYH has been addressed by Halford and collaborators (2003) by screening a large sample of sporadic CRCs for somatic alterations of MUTYH, as well as of MTH1 and OGG1 BER genes: somatic inactivation of these genes does not appear a frequent mechanism directly involved in colorectal tumorigenesis. On the contrary, a reduced expression of MUTYH has been reported in human gastric cancer, where reduction in protein amount proved to be associated with a poor prognosis (Shinmura et al., 2011).
The MUTYH Gene
The MUTYH gene (previously termed MYH or hMYH), the human ortholog of the Escherichia coli mutY, was first cloned by Slupska and coworkers in 1996; it is about 11,200 bp in length, is localized on the short arm of chromosome 1 (1p32.1-p34.3), and contains 16 exons (NCBI Genomic refseq ID seq ID: NG_008189.1).
Many MUTYH genetic variants have been reported, having or likely not having a phenotypic effect; in this regard, the Leiden Open Variation Database represents an extremely valuable tool to evaluate gene mutations that have been identified both in healthy and affected subjects (http://chromium.liacs.nl/LOVD2/colon_cancer/home.php?action=switch_db).
MUTYH encodes a DNA glycosylase that is expressed both in the nucleus and in the mitochondria. The coding sequence generates three classes of mRNAs (namely α, β, and γ) that correspond to a total of 10 possible mature transcripts; these transcripts are produced by three independent transcription initiation sites and by the occurrence of different exon 3 alternative splicing events, and correspond to seven protein isoforms (Ohtsubo et al., 2000; reviewed by Parker and Eshleman, 2003). In accordance with the Human Genome Variation Society nomenclature, the longest transcript (transcript <5: 1945 bp) is used as coding reference sequence (NCBI transcript refseq ID: NM_001128425.1). Transcripts starting from the first AUG retain the sequence that corresponds to the N-terminal aminoacids likely acting as mitochondrial targeting signal (MTS) (Ohtsubo et al., 2000). The relative amounts of each of the ten possible mRNAs in different tissues as well as their subcellular localization are only partially characterized. Among different human tissues, the largest total amount of mRNA has been observed in thymus, adult brain, testis, and kidney, whereas heart, salivary gland, liver, and pancreas show lower MUTYH expression levels compared to other tissues (Ohtsubo et al., 2000). Very recently, it has been reported that not only MUTYH expression is different in various organs and positively correlated with proliferative activity, but also the first exons of the gene are used in a tissue-specific manner. In addition, transcripts encoding mitochondrial proteins have been shown to predominate in muscle tissues, while the highest amount of transcripts encoding nuclear proteins have been detected in testes and colon (Plotz et al., 2012). As example of protein expression, Figure 1 shows the diffuse localization of MUTYH in normal cells of colon epithelium.
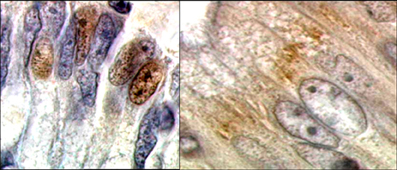
Figure 1. Diffuse localization of wild-type MUTYH protein in colonic epithelial cells, as evidenced with specific antibodies (Courtesy of Dr. M. Risio).
The BER System and the MUTYH Protein
A number of deleterious processes and pathological conditions may result from DNA oxidation, including initiation and progression of cancer. DNA oxidation arises from its interaction with exogenous molecules or from the action of reactive oxygen species (ROS); these can be generated by the metabolism of exogenous compounds as well as by cellular processes including respiration and inflammation.
The BER system is a cellular defense against the damaging effects of ROS. Generally, BER is initiated by damage-specific DNA glycosylases that recognize oxidized bases and excise the damaged nucleotides, allowing the restoration of the parental DNA sequence by further processing of endonucleases, polymerases, and ligases (reviewed by Fortini and Dogliotti, 2007; van Loon et al., 2010). A very well known oxidation product is the highly mutagenic 7,8-dihydro-8-oxoguanine (8-oxoguanine or 8-oxoG) which is able to form both 8-oxoG:C and 8-oxoG:A stable base pairs. Failure to remove the incorporated 8-oxoG before the next round of DNA replication results in G:C to T:A transversion mutations (reviewed by David et al., 2007).
The occurrence of 8-oxoG in the DNA arises from two pathways: incorporation into DNA of the oxidized precursor 8-oxo-dGTP during DNA synthesis and direct oxidation of guanine in DNA. The 8-oxo-dGTP can be removed from the nucleotide pool by the oxidized purine nucleoside triphosphatase MTH1, which prevents the selection of the oxidized guanine by polymerases during DNA replication. In addition to this sanitizing mechanism, two other BER enzymes act as second line of defense, namely OGG1 and MUTYH. OGG1 has a high specificity for the 8-oxoG:C and removes the oxidized guanine, allowing restoration of a G:C base pair, while MUTYH is a DNA glycosylase able to recognize and remove either adenine from a mismatch with 8-oxoG, or 2-OH-adenine from a mismatch with guanine (reviewed by Nakabeppu et al., 2004; Ushijima et al., 2005).
MUTYH is the functional counterpart of E. coli MutY and is a highly evolutionarily conserved protein; it belongs to a large superfamily of structurally related DNA glycosylases with a signature helix-hairpin-helix motif followed by a Gly/Pro-rich (denoted as HhH-GPD domain) region. This domain contains the active site pocket responsible for the 8-oxoG:A mispair recognition and for the glycolytic excision of the substrate adenine (Lee and Verdine, 2009). MUTYH shows an N-terminal domain containing, apart from the mitochondrial localization signal (MLS) and a putative nuclear localization signal (NLS), the replication protein A (RPA) interacting motif; the C-terminal domain contains a NLS and the proliferating cell nuclear antigen (PCNA) binding region. The interaction with PCNA is crucial in replication-coupled repair in order to increase and direct the glycosylase activity of MUTYH to the newly synthesized nascent DNA strand, thus preventing replication errors caused by a 8-oxoG template (Hayashi et al., 2002; reviewed by Parker and Eshleman, 2003; van Loon et al., 2010). In vitro experiments have shown that MUTYH physically interacts with the MSH2/MSH6 heterodimeric complex via the MSH6 subunit, demonstrating that this MMR protein complex stimulates the glycosylase activity of MUTYH by enhancing the protein affinity for the 8-oxoG:A mismatches (Gu et al., 2002; Bai et al., 2005). Besides multiple interactions with replication and repair proteins, MUTYH activity can also be modulated by post-translational modifications that regulate protein localization and level (Hirano et al., 2003).
The 8-oxoG accumulation in nuclear and in mitochondrial DNA triggers two distinct cell death pathways that, although independent of each other, are both associated with BER and initiated by MUTYH activity (Ichikawa et al., 2008; Oka et al., 2008). Oka and coworkers (2008) demonstrated that the knockdown of MUTYH results in escaping from both types of pathways and proposed a tumor suppressor role of MUTYH due to its capability to induce death of pre-cancerous cells that have accumulated high levels of 8-oxoG in nuclear or in mitochondrial DNA.
ROS can be produced by the cellular components of the inflammation; oxidative stress and consequent DNA damage play a key role in the pathogenesis of ulcerative colitis (UC), a form of inflammatory bowel disease characterized by an increased risk of CRC. In particular, the UC-associated tumors display both an accumulation of 8-oxoG and an altered expression of MUTYH protein in cancer cells (Gushima et al., 2009). Accordingly, in a mouse model of UC, Mutyh has recently been shown to play a major role in maintaining intestinal integrity by influencing the inflammatory response (Casorelli et al., 2010).
Germline Mutations of MUTYH in MAP Patients
Approximately 30% of APC mutation-negative polyposis cases can be attributed to MUTYH biallelic mutations; on the whole, these account for less than 1% of all CRC cases (reviewed by Cheadle and Sampson, 2007; Cleary et al., 2009). To date, nearly 300 variants have been identified at the MUTYH locus, including about 80 pathogenic mutations distributed throughout the gene and located at positions corresponding to different functional domains of the protein (see: Leiden Open Variation Database). Although various types of alterations have been reported in MAP patients, including nonsense, small insertion/deletion, and splicing variants, missense mutations represent the great majority of the detected changes. A number of variants appear recurrent in different populations, with Y179C (previously annotated as Y165C) and G396D (previously annotated as G382D) missense mutations accounting together for about 70% of germline alterations found in European patients (reviewed by Cheadle and Sampson, 2007); however, in Asian populations, Y179C and G396D must be rare, since neither mutation has been found in MAP patients. On the other hand, other mutations have proven to be recurrent in patients from particular populations (reviewed by Poulsen and Bisgaard, 2008). Taken together, these findings indicate that sequencing of the entire MUTYH open reading frame has to be performed for the genetic testing, especially in populations of mixed ethnicity.
Recently, a large gene deletion spanning exons 4–16 has been found in two unrelated patients showing an attenuated phenotype; due to this observation, appropriate methods to detect gene rearrangements should be considered, at least for patients carrying either a single heterozygous mutation or a (apparently) homozygous disease-causing mutation (Rouleau et al., 2011; Torrezan et al., 2011). Besides rare mutations, a polymorphic allele (SNP rs3219468: G>C) associated with a significant reduction of a MUTYH transcription product has recently been implicated in CRC risk (Plotz et al., 2012).
The identification of germline mutations in patients with inherited CRC syndromes, including MAP, is extremely important to allow mutation carriers to be included in cancer surveillance programs which have been proven to save lives. Many of the identified disease-gene mutations result in loss-of-function of the encoded protein, indicating a clear pathogenic significance. However, significance remains uncertain for a large proportion of the identified variants, some of which may contribute to increase the risk of cancer. For MAP, the major problem is represented by missense mutations, the pathogenetic role of which cannot easily be assessed; this leaves open questions about their diagnostic interpretation and about counseling management of the patients in whom they are detected.
Since MUTYH mitigates the mutagenic potential of 8-oxoG by preventing accumulation of transversion mutations, mutY-deficient E. coli cells have been used to study if the human heterologous mutated proteins have the ability to complement the mutator phenotype in these cells (Chmiel et al., 2003; Bai et al., 2005, 2007). Analogously, to characterize the functional effect of some missense mutations, human proteins have been expressed in Mutyh-defective mouse fibroblasts and their biological activity has been related to the 8-oxoG level in the genome and to the response of cells following oxidative stress induction (Molatore et al., 2010). Further, lymphoblastoid cell lines derived from MAP patients have been established and immunoprecipitated proteins from whole cell extracts have been tested in vitro (Alhopuro et al., 2005; Parker et al., 2005). Owing to the capacity of MUTYH to recognize and excise adenine from DNA duplexes containing either 8-oxoG:A or G:2-OH-A mismatches, in vitro tests with purified proteins have been performed. By using different oligonucleotides containing specific mismatches as substrate, the enzymatic activities of bacterial (Al-Tassan et al., 2002; Chmiel et al., 2003; Livingston et al., 2005), mouse (Hirano et al., 2003; Pope et al., 2005; Ushijima et al., 2005; Yanaru-Fujisawa et al., 2008), and human (Bai et al., 2005, 2007; Ali et al., 2008; Kundu et al., 2009; D'Agostino et al., 2010) purified proteins have been investigated. The glycosylase activity has been evaluated by measuring the amount of cleavage fragments derived from DNA duplexes, while the DNA-binding capacity has mainly been investigated through the formation of DNA-protein complexes in electrophoresis mobility shift assay (EMSA). Since surface plasmon resonance (SPR) technology is extremely sensitive to detect subtle and time-dependent changes in DNA-protein interaction and to investigate the enzymatic activity on different DNA substrates, this method has successfully been used to evaluate the binding of some mutated human MUTYH proteins to an 8-oxoG:A-containing DNA substrate (D'Agostino et al., 2010). Recently, computational methods based on amino acids substitutions within homologous proteins with known 3D structures have been applied to predict changes of different proteins, thus correlating amino acids substitutions with functional effects possibly resulting in phenotype changes (Worth et al., 2007). As for other proteins, the understanding of the mechanisms by which mutations affect MUTYH protein structure can provide new insights into its activity and, ultimately, on the pathogenicity of gene mutations. Figure 2 shows an example of the structure-based analysis of MUTYH mutated proteins.
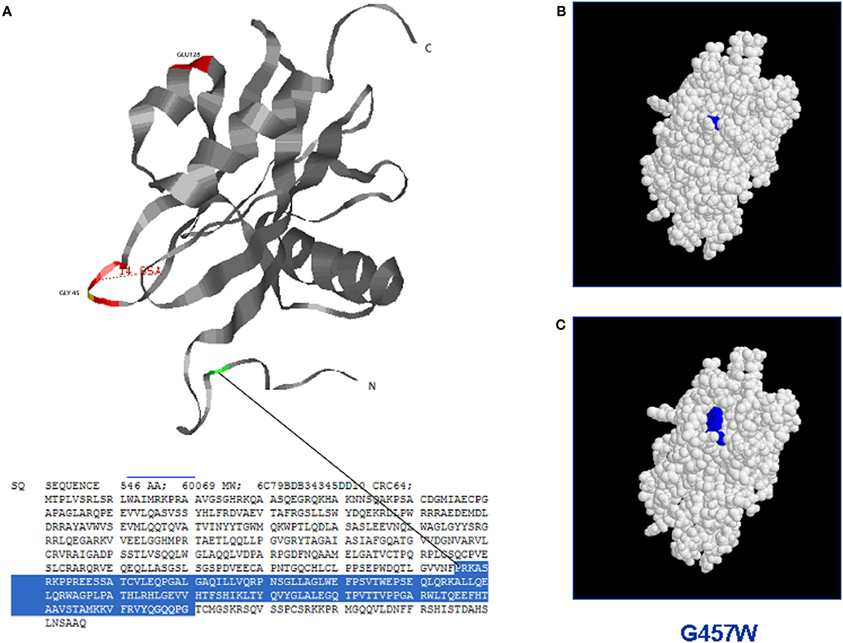
Figure 2. Structure-based analysis of mutant MUTYH protein by homology modelling (Site Directed Mutator): the figure shows the possible switch of the MUTYH protein folding when a single aminoacid residue is mutated in the NUDIX domain. (A) Three dimensional structure of the NUDIX domain of MUTYH protein (PDB database); (B) Protein folding of the wild-type NUDIX domain. (C) Protein switch of the NUDIX domain containing the substitution G457W.
On the whole, the majority of MUTYH tested variants have been found to be totally or partially devoid of DNA-glycosylase, with variable adenine-removal residual capability. As an example, functional assays demonstrated that G396D capability is far more similar to the wild-type protein compared to Y179C missense mutation. Accordingly, clinical data indicate a milder phenotypic effect of G396D compared to Y179C. Indeed, Nielsen and collaborators (2009) reported that MAP patients with homozygous G396D mutations or with compound heterozygous G396D/Y179C mutations show a milder phenotype when compared to homozygous carriers of Y179C mutations.
The expression pattern of MUTYH mutated proteins has been evaluated on CRC tissue sections to test whether immunohistochemistry can be used in clinical practice to identify carriers of germline mutations with functional effects. However, the specificity of the expression pattern has proven to be uncertain and immunostaining does not seem an appropriate tool to discriminate CRC tissues from patients with or without biallelic germline mutations (Di Gregorio et al., 2006; van der Post et al., 2009).
Concluding Remarks
During the last 10 years MAP has clearly become a distinct cancer-susceptibility syndrome with respect to both FAP and AFAP. However, in order to refine clinical guidances, some aspects still poorly understood that concern the function of MUTYH protein deserve to be elucidated. Unsolved problems deal with genotype–phenotype correlations, pathogenicity of unclassified variants, cross-talk between BER and other DNA repair pathways including MMR, overlapping between MAP and HNPCC syndromes, and last, but not least, tumor suppressor role of MUTYH in pre-cancerous cells subjected to oxidative stress. A better understanding of the regulation of MUTYH isoforms, of their relative expression in different tissues, and of their subcellular fate, will likely allow us to clarify some of the above issues. By taking into account the complexity of the expression pattern, the effect on splicing efficiency and accuracy should be evaluated for a number of MUTYH variants that have been identified in patients without clear disease-causing mutations. Indeed, a fraction of gene mutations, including synonymous nucleotide changes, have been shown to have roles in various diseases or to modify disease severity through splicing perturbation rather than other molecular mechanisms (reviewed by Cartegni et al., 2002; Wang and Cooper, 2007).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partly supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC).
Abbreviations
MAP, MUTYH-associated polyposis; CRC, colorectal cancer; HNPCC, hereditary non-polyposis colorectal cancer;, FAP, familial adenomatous polyposis; BER, base excision repair; MMR, mismatch repair; OMIM, Online Mendeleian Inheritance in Man.
References
Alhopuro, P., Parker, A. R., Lehtonen, R., Enholm, S., Jarvinen, H. J., Mecklin, J. P., Karhu, A., Eshleman, J. R., and Aaltonen, L. A. (2005). A novel functionally deficient MYH variant in individuals with colorectal adenomatous polyposis. Hum. Mutat. 26, 393–401.
Ali, M., Kim, H., Cleary, S., Cupples, C., Gallinger, S., and Bristow, R. (2008). Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology 2, 499–507.
Al-Tassan, N., Chmiel, N. H., Maynard, J., Fleming, N., Livingston, A. L., Williams, G. T., Hodges, A. K., Davies, D. R., David, S. S., Sampson, J. R., and Cheadle, J. P. (2002). Inherited variants of MYH associated with somatic G:C>T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232.
Aretz, S. (2010). The differential diagnosis and surveillance of hereditary gastrointestinal polyposis syndromes. Dtsch. Arztebl. Int. 107, 163–173.
Bai, H., Jones, S., Guan, X., Wilson, T. M., Sampson, J. R., Cheadle, J. P., and Lu, A. L. (2005). Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 33, 597–604.
Bai, H., Grist, S., Gardner, J., Suthers, G., Wilson, T. M., and Lu, A. L. (2007). Functional characterization of human MutY homolog (hMYH) missense mutation (R231L) that is linked with hMYH-associated polyposis. Cancer Lett. 250, 74–81.
Barnetson, R. A., Devlin, L., Miller, J., Farrington, S. M., Slater, S., Drake, A. C., Campbell, H., Dunlop, M. G., and Porteous, M. E. (2007). Germline mutation prevalence in the base excision repair gene, MYH, in patients with endometrial cancer. Clin. Genet. 72, 551–555.
Boparai, K. S., Dekker, E., Van Eeden, S., Polak, M. M., Bartelsman, J. F., Mathus-Vliegen, E. M., Keller, J. J., and van Noesel, C. J. (2008). Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology 135, 2014–2018.
Cartegni, L., Chew, S. L., and Krainer, A. R. (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 3, 285–298.
Casorelli, I., Pannellini, T., De Luca, G., Degan, P., Chiera, F., Iavarone, I., Giuliani, A., Butera, A., Boirivant, M., Musiani, P., and Bignami, M. (2010). The Mutyh base excision repair gene influences the inflammatory response in a mouse model of ulcerative colitis. PLoS ONE 5:e12070. doi: 10.1371/journal.pone.0012070
Cheadle, J. P., and Sampson, J. R. (2007). MUTYH-associated polyposis–from defect in base excision repair to clinical genetic testing. DNA Repair 63, 274–279.
Chmiel, N. H., Livingston, A. L., and David, S. S. (2003). Insight into the functional consequences of inherited variants of the hMYH adenine glycosylase associated with colorectal cancer: complementation assays with hMYH variants and pre-steady-state kinetics of the corresponding mutated E. coli enzymes. J. Mol. Biol. 327, 431–443.
Cleary, S. P., Cotterchio, M., Jenkins, M. A., Kim, H., Bristow, R., Green, R., Haile, R., Hopper, J. L., LeMarchand, L., Lindor, N., Parfrey, P., Potter, J., Younghusband, B., and Gallinger, S. (2009). Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 136, 1251–1260.
Croitoru, M. E., Cleary, S. P., Di Nicola, N., Manno, M., Selander, T., Aronson, M., Redston, M., Cotterchio, M., Knight, J., Gryfe, R., and Gallinger, S. (2004). Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J. Natl. Cancer Inst. 96, 1631–1634.
D'Agostino, V. G., Minoprio, A., Torreri, P., Marinoni, I., Bossa, C., Petrucci, T. C., Albertini, A. M., Ranzani, G. N., Bignami, M., and Mazzei, F. (2010). Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA Repair 9, 700–707.
David, S. S., O'Shea, V. L., and Kundu, S. (2007). Base-excision repair of oxidative DNA damage. Nature 447, 941–950.
de la Chapelle, A. (2004). Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 4, 769–780.
De Miranda, N. F., Nielsen, M., Pereira, D., van Puijenbroek, M., Vasen, H. F., Hes, F. J., van Wezel, T., and Morreau, H. (2009). MUTYH-associated polyposis carcinomas frequently lose HLA class I expression - a common event amongst DNA-repair-deficient colorectal cancers. J. Pathol. 219, 69–76.
Di Gregorio, C., Frattini, M., Maffei, S., Ponti, G., Losi, L., Pedroni, M., Venesio, T., Bertario, L., Varesco, L., Risio, M., and Ponz de Leon, M. (2006). Immunohistochemical expression of MYH protein can be used to identify patients with MYH-associated polyposis. Gastroenterology 131, 439–444.
Dunlop, M. G., Tenesa, A., Farrington, S. M., Ballereau, S., Brewster, D. H., Kossler, T., Pharoah, P., Schafmayer, C., Hampe, J., Völzke, H., Chang-Claude, J., Hoffmeister, M., Brenner, H., von Holst, S., Picelli, S., Lindblom, A., Jenkins, M. A., Hopper, J. L., Casey, G., Duggan, D., Newcomb, P. A., Abulí, A., Bessa, X., Ruiz-Ponte, C., Castellví-Bel, S., Niittymäki, I., Tuupanen, S., Karhu, A., Aaltonen, L., Zanke, B., Hudson, T., Gallinger, S., Barclay, E., Martin, L., Gorman, M., Carvajal-Carmona, L., Walther, A., Kerr, D., Lubbe, S., Broderick, P., Chandler, I., Pittman, A., Penegar, S., Campbell, H., Tomlinson, I., and Houlston, R. S. (2012). Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42 103 individuals. Gut. doi: 10.1136/gutjnl-2011-300537. [Epub ahead of print].
Farrington, S. M., Tenesa, A., Barnetson, R., Wiltshire, A., Pren-dergast, J., Porteous, M., Campbell, H., and Dunlop, M. G. (2005). Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am. J. Hum. Genet. 77, 112–119.
Filipe, B., Baltazar, C., Albuquerque, C., Fragoso, S., Lage, P., Vitoriano, I., Mão de Ferro, S., Claro, I., Rodrigues, P., Fidalgo, P., Chaves, P., Cravo, M., and Nobre Leitão, C. (2009). APC or MUTYH mutations account for the majority of clinically well-characterized families with FAP and AFAP phenotype and patients with more than 30 adenomas. Clin. Genet. 76, 242–255.
Fortini, P., and Dogliotti, E. (2007). Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 6, 398–409.
Gu, Y., Parker, A., Wilson, T. M., Bai, H., Chang, D. Y., and Lu, A. L. (2002). Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 277, 11135–11142.
Gushima, M., Hirahashi, M., Matsumoto, T., Fujita, K., Fujisawa, R., Mizumoto, K., Nakabeppu, Y., Iida, M., Yao, T., and Tsuneyoshi, M. (2009). Altered expression of MUTYH and an increase in 8-hydroxydeoxyguanosine are early events in ulcerative colitis-associated carcinogenesis. J. Pathol. 219, 77–86.
Halford, S. E., Rowan, A. J., Lipton, L., Sieber, O. M., Pack, K., Thomas, H. J., Hodgson, S. V., Bodmer, W. F., and Tomlinson, I. P. (2003). Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am. J. Pathol. 162, 1545–1548.
Hayashi, H., Tominaga, Y., Hirano, S., McKenna, A. E., Nakabeppu, Y., and Matsumoto, Y. (2002). Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr. Biol. 12, 335–339.
Hirano, S., Tominaga, Y., Ichinoe, A., Ushijima, Y., Tsuchimoto, D., Honda-Ohnishi, Y., Ohtsubo, T., Sakumi, K., and Nakabeppu, Y. (2003). Mutator phenotype of MUTYH-null mouse embryonic stem cells. J. Biol. Chem. 278, 38121–38124.
Ichikawa, J., Tsuchimoto, D., Oka, S., Ohno, M., Furuichi, M., Sakumi, K., and Nakabeppu, Y. (2008). Oxidation of mitochondrial deoxynucleotide pools by exposure to sodium nitroprusside induces cell death. DNA Repair 7, 418–430.
Jasperson, K. W., Tuohy, T. M., Neklason, D. W., and Burt, R. W. (2010). Hereditary and familial colon cancer. Gastroenterology 138, 2044–2058.
Johnson, V., Lipton, L. R., Cummings, C., Eftekhar Sadat, A. T., Izatt, L., Hodgson, S. V., Talbot, I. C., Thomas, H. J., Silver, A. J., and Tomlinson, I. P. (2005). Analysis of somatic molecular changes, clinicopathological features, family history, and germline mutations in colorectal cancer families: evidence for efficient diagnosis of HNPCC and for the existence of distinct groups of non-HNPCC families. J. Med. Genet. 42, 756–762.
Jones, S., Emmerson, P., Maynard, J., Best, J. M., Jordan, S., Williams, G. T., Sampson, J. R., and Cheadle, J. P. (2002). Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C–>T:A mutations. Hum. Mol. Genet. 11, 2961–2967.
Jones, S., Lambert, S., Williams, G. T., Best, J. M., Sampson, J. R., and Cheadle, J. P. (2004). Increased frequency of the k-ras G12C mutation in MYH polyposis colorectal adenomas. Br. J. Cancer 90, 1591–1593.
Jones, N., Vogt, S., Nielsen, M., Christian, D., Wark, P. A., Eccles, D., Edwards, E., Evans, D. G., Maher, E. R., Vasen, H. F., Hes, F. J., Aretz, S., and Sampson, J. R. (2009). Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology 137, 489–494.
Kundu, S., Brinkmeyer, M. K., Livingston, A. L., and David, S. S. (2009). Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair 8, 1400–1410.
Lee, S., and Verdine, G. L. (2009). Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 106, 18497–18502.
Levin, B., Lieberman, D. A., McFarland, B., Andrews, K. S., Brooks, D., Bond, J., Dash, C., Giardiello, F. M., Glick, S., Johnson, D., Johnson, C. D., Levin, T. R., Pickhardt, P. J., Rex, D. K., Smith, R. A., Thorson, A., and Winawer, S. J., American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force, American College of Radiology Colon Cancer Committee. (2008). Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American cancer society, the US multi-society task force on colorectal cancer, and the american college of radiology. Gastroenterology 134, 1570–1595.
Lipton, L., Halford, S. E., Johnson, V., Novelli, M. R., Jones, A., Cummings, C., Barclay, E., Sieber, O., Sadat, A., Bisgaard, M. L., Hodgson, S. V., Aaltonen, L. A., Thomas, H. J., and Tomlinson, I. P. (2003). Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 63, 7595–7599.
Lipton, L., and Tomlinson, I. (2004). The multiple colorectal adenoma phenotype and MYH, a base excision repair gene. Clin. Gastroenterol. Hepatol. 2, 633–638.
Livingston, A. L., Kundu, S., Henderson Pozzi, M., Anderson, D. W., and David, S. S. (2005). Insight into the roles of tyrosine 82 and glycine 253 in the Escherichia coli adenine glycosylase MutY. Biochemistry 44, 14179–14190.
Lubbe, S. J., Di Bernardo, M. C., Chandler, I. P., and Houlston, R. S. (2009). Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J. Clin. Oncol. 27, 3975–3980.
Lynch, H. T., Lynch, P. M., Lanspa, S. J., Snyder, C. L., Lynch, J. F., and Boland, C. R. (2009). Review of the lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 76, 1–18.
Molatore, S., Russo, M. T., D'Agostino, V. G., Barone, F., Matsumoto, Y., Albertini, A. M., Minoprio, A., Degan, P., Mazzei, F., Bignami, M., and Ranzani, G. N. (2010). MUTYH mutations associated with familial adenomatous polyposis: functional characterization by a mammalian cell-based assay. Hum. Mutat. 31, 159–166.
Morak, M., Laner, A., Bacher, U., Keiling, C., and Holinski-Feder, E. (2010). MUTYH-associated polyposis - variability of the clinical phenotype in patients with biallelic and monoallelic MUTYH mutations and report on novel mutations. Clin. Genet. 78, 353–363.
Nakabeppu, Y., Tsuchimoto, D., Furuichi, M., and Sakumi, K. (2004). The defense mechanisms in mammalian cells against oxidative damage in nucleic acids and their involvement in the suppression of mutagenesis and cell death. Free Radic. Res. 38, 423–429.
Nielsen, M., Poley, J. W., Verhoef, S., van Puijenbroek, M., Weiss, M. M., Burger, G. T., Dommering, C. J., Vasen, H. F., Kuipers, E. J., Wagner, A., Morreau, H., and Hes, F. J. (2006). Duodenal carcinoma in MUTYH-associated polyposis. J. Clin. Pathol. 59, 1212–1215.
Nielsen, M., Joerink-van de Beld, M. C., Jones, N., Vogt, S., Tops, C. M., Vasen, H. F., Sampson, J. R., Aretz, S., and Hes, F. J. (2009). Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology 136, 471–476.
Nielsen, M., Morreau, H., Vasen, H. F., and Hes, F. J. (2011). MUTYH-associated polyposis (MAP). Crit. Rev. Oncol. Hematol. 79, 1–16.
Nieuwenhuis, M. H., Vogt, S., Jones, N., Nielsen, M., Hes, F. J., Sampson, J. R., Aretz, S., and Vasen, H. F. (2012). Evidence for accelerated colorectal adenoma-carcinoma progression in MUTYH-associated polyposis? Gut 61, 734–738.
Ohtsubo, T., Nishioka, K., Imaiso, Y., Iwai, S., Shimokawa, H., Oda, H., Fujiwara, T., and Nakabeppu, Y. (2000). Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 28, 1355–1364.
Oka, S., Ohno, M., Tsuchimoto, D., Sakumi, K., Furuichi, M., and Nakabeppu, Y. (2008). Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 27, 421–432.
Parker, A. R., and Eshleman, J. R. (2003). Human MutY: gene structure, protein functions and interactions, and role in carcinogenesis. Cell Mol. Life Sci. 60, 2064–2083.
Parker, A. R., Sieber, O. M., Shi, C., Hua, L., Takao, M., Tomlinson, I. P., and Eshleman, J. R. (2005). Cells with pathogenic biallelic mutations in the human MUTYH gene are defective in DNA damage binding and repair. Carcinogenesis 26, 2010–2018.
Pervaiz, M. A., Eppolito, A., and Schmidt, K. (2010). Papillary thyroid cancer in a patient with MUTYH-associated polyposis (MAP). Fam. Cancer 9, 595–597.
Peterlongo, P., Mitra, N., Sanchez de Abajo, A., de la Hoya, M., Bassi, C., Bertario, L., Radice, P., Glogowski, E., Nafa, K., Caldes, T., Offit, K., and Ellis, N. A. (2006). Increased frequency of disease-causing MYH mutations in colon cancer families. Carcinogenesis 27, 2243–2249.
Plotz, G., Casper, M., Raedle, J., Hinrichsen, I., Heckel, V., Brieger, A., Trojan, J., and Zeuzem, S. (2012). MUTYH gene expression and alternative splicing in controls and polyposis patients. Hum. Mutat. 33, 1067–1074.
Ponti, G., Ponz de Leon, M., Maffei, S., Pedroni, M., Losi, L., Di Gregorio, C., Gismondi, V., Scarselli, A., Benatti, P., Roncari, B., Seidenari, S., Pellacani, G., Varotti, C., Prete, E., Varesco, L., and Roncucci, L. (2005). Attenuated familial adenomatous polyposis and Muir-Torre syndrome linked to compound biallelic constitutional MYH gene mutations. Clin. Genet. 68, 442–447.
Pope, M. A., Chmiel, N. H., and David, S. S. (2005). Insight into the functional consequences of hMYH variants associated with colorectal cancer: distinct differences in the adenine glycosylase activity and the response to AP endonucleases of Y150C and G365D murine MYH. DNA Repair 4, 315–325.
Poulsen, M. L., and Bisgaard, M. L. (2008). MUTYH Associated Polyposis (MAP). Curr. Genomics 9, 420–435.
Rouleau, E., Zattara, H., Lefol, C., Noguchi, T., Briaux, A., Buecher, B., Bourdon, V., Sobol, H., Lidereau, R., and Olschwang, S. (2011). First large rearrangement in the MUTYH gene and attenuated familial adenomatous polyposis syndrome. Clin. Genet. 80, 301–303.
Russell, A. M., Zhang, J., Luz, J., Hutter, P., Chappuis, P. O., Berthod, C. R., Maillet, P., Mueller, H., and Heinimann, K. (2006). Prevalence of MYH germline mutations in Swiss APC mutation-negative polyposis patients. Int. J. Cancer 118, 1937–1940.
Sampson, J. R., Dolwani, S., Jones, S., Eccles, D., Ellis, A., Evans, D. G., Frayling, I., Jordan, S., Maher, E. R., Mak, T., Maynard, J., Pigatto, F., Shaw, J., and Cheadle, J. P. (2003). Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 362, 39–41.
Sampson, J. R., Jones, S., Dolwani, S., and Cheadle, J. P. (2005). MutYH (MYH) and colorectal cancer. Biochem. Soc. Trans. 33, 679–683.
Shinmura, K., Goto, M., Suzuki, M., Tao, H., Yamada, H., Igarashi, H., Matsuura, S., Maeda, M., Konno, H., Matsuda, T., and Sugimura, H. (2011). Reduced expression of MUTYH with suppressive activity against mutations caused by 8-hydroxyguanine is a novel predictor of a poor prognosis in human gastric cancer. J. Pathol. 225, 414–423.
Sieber, O. M., Lipton, L., Crabtree, M., Heinimann, K., Fidalgo, P., Phillips, R. K., Bisgaard, M. L., Orntoft, T. F., Aaltonen, L. A., Hodgson, S. V., Thomas, H. J., and Tomlinson, I. P. (2003). Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N. Engl. J. Med. 348, 791–799.
Slupska, M. M., Baikalov, C., Luther, W. M., Chiang, J. H., Wei, Y. F., and Miller, J. H. (1996). Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol. 178, 3885–3892.
Tenesa, A., Campbell, H., Barnetson, R., Porteous, M., Dunlop, M., and Farrington, S. M. (2006). Association of MUTYH and colorectal cancer. Br. J. Cancer 95, 239–242.
Terdiman, J. P. (2009). MYH-associated disease: attenuated adenomatous polyposis of the colon is only part of the story. Gastroenterology 137, 1883–1886.
Theodoratou, E., Campbell, H., Tenesa, A., Houlston, R., Webb, E., Lubbe, S., Broderick, P., Gallinger, S., Croitoru, E. M., Jenkins, M. A., Win, A. K., Cleary, S. P., Koessler, T., Pharoah, P. D., Küry, S., Bézieau, S., Buecher, B., Ellis, N. A., Peterlongo, P., Offit, K., Aaltonen, L. A., Enholm, S., Lindblom, A., Zhou, X. L., Tomlinson, I. P., Moreno, V., Blanco, I., Capellà, G., Barnetson, R., Porteous, M. E., Dunlop, M. G., and Farrington, S. M. (2010). A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. Br. J. Cancer 103, 1875–1884.
Tomlinson, I. (2012). Colorectal cancer genetics: from candidate genes to GWAS and back again. Mutagenesis 27, 141–142.
Torrezan, G. T., da Silva, F. C., Krepischi, A. C., Santos, É. M., de O Ferreira, F., Rossi, B. M., and Carraro, D. M. (2011). Breakpoint characterization of a novel large intragenic deletion of MUTYH detected in a MAP patient: case report. BMC Med. Genet. 12, 128.
Tricarico, R., Bet, P., Ciambotti, B., Di Gregorio, C., Gatteschi, B., Gismondi, V., Toschi, B., Tonelli, F., Varesco, L., and Genuardi, M. (2009). Endometrial cancer and somatic G>T KRAS transversion in patients with constitutional MUTYH biallelic mutations. Cancer Lett. 274, 266–270.
Ushijima, Y., Tominaga, Y., Miura, T., Tsuchimoto, D., Sakumi, K., and Nakabeppu, Y. (2005). A functional analysis of the DNA glycosylase activity of mouse MUTYH protein excising 2-hydroxyadenine opposite guanine in DNA. Nucleic Acids Res. 33, 672–682.
van der Post, R. S., Kets, C. M., Ligtenberg, M. J., van Krieken, J. H., and Hoogerbrugge, N. (2009). Immunohistochemistry is not an accurate first step towards the molecular diagnosis of MUTYH-associated polyposis. Virchows Arch. 454, 25–29.
van Loon, B., Markkanen, E., and Hübscher, U. (2010). Oxygen as a friend and enemy: how to combat the mutational potential of 8-oxo-guanine. DNA Repair 9, 604–616.
Vasen, H. F., Möslein, G., Alonso, A., Aretz, S., Bernstein, I., Bertario, L., Blanco, I., Bülow, S., Burn, J., Capella, G., Colas, C., Engel, C., Frayling, I., Friedl, W., Hes, F. J., Hodgson, S., Järvinen, H., Mecklin, J. P., Møller, P., Myrhøi, T., Nagengast, F. M., Parc, Y., Phillips, R., Clark, S. K., de Leon, M. P., Renkonen-Sinisalo, L., Sampson, J. R., Stormorken, A., Tejpar, S., Thomas, H. J., and Wijnen, J. (2008). Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 57, 704–713.
Venesio, T., Molatore, S., Cattaneo, F., Arrigoni, A., Risio, M., and Ranzani, G. N. (2004). High frequency of MYH gene mutations in a subset of patients with familial adenomatous polyposis. Gastroenterology 126, 1681–1685.
Vogt, S., Jones, N., Christian, D., Engel, C., Nielsen, M., Kaufmann, A., Steinke, V., Vasen, H. F., Propping, P., Sampson, J. R., Hes, F. J., and Aretz, S. (2009). Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 137, 1976–1985.
Wang, G. S., and Cooper, T. A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 8, 749–761.
Webb, E. L., Rudd, M. F., and Houlston, R. S. (2006). Colorectal cancer risk in monoallelic carriers of MYH variants. Am. J. Hum. Genet. 79, 768–771.
Win, A. K., Cleary, S. P., Dowty, J. G., Baron, J. A., Young, J. P., Buchanan, D. D., Southey, M. C., Burnett, T., Parfrey, P. S., Green, R. C., Le Marchand, L., Newcomb, P. A., Haile, R. W., Lindor, N. M., Hopper, J. L., Gallinger, S., and Jenkins, M. A. (2011). Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int. J. Cancer 129, 2256–2262.
Worth, C. L., Bickerton, G. R., Schreyer, A., Forman, J. R., Cheng, T. M., Lee, S., Gong, S., Burke, D. F., and Blundell, T. L. (2007). A structural bioinformatics approach to the analysis of nonsynonymous single nucleotide polymorphisms (nsSNPs) and their relation to disease. J. Bioinform. Comput. Biol. 5, 1297–1318.
Yanaru-Fujisawa, R., Matsumoto, T., Ushijima, Y., Esaki, M., Hirahashi, M., Gushima, M., Yao, T., Nakabeppu, Y., and Iida, M. (2008). Genomic and functional analyses of MUTYH in Japanese patients with adenomatous polyposis. Clin. Genet. 73, 545–553.
Keywords: base excision repair (BER), colorectal cancer (CRC), familial adenomatous polyposis (FAP), hereditary non-polyposis colorectal cancer (HNPCC), mismatch repair (MMR), MUTYH-associated polyposis (MAP)
Citation: Venesio T, Balsamo A, D'Agostino VG and Ranzani GN (2012) MUTYH-associated polyposis (MAP), the syndrome implicating base excision repair in inherited predisposition to colorectal tumors. Front. Oncol. 2:83. doi: 10.3389/fonc.2012.00083
Received: 11 May 2012; Paper pending published: 07 June 2012;
Accepted: 16 July 2012; Published online: 02 August 2012.
Edited by:
Tomáš Skricka, Rašín School, Czech RepublicReviewed by:
Zilvinas Saladzinskas, Lithuanian University of Health Sciences, LithuaniaZdenek Kala, Masaryk University Brno, Czech Republic
Copyright © 2012 Venesio, Balsamo, D'Agostino and Ranzani. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Tiziana Venesio, Institute for Cancer Research and Treatment, Strada Provinciale 142, 10060 Candiolo, Torino, Italy. e-mail:dGl6aWFuYS52ZW5lc2lvQGlyY2MuaXQ=