
- Center for Gene Regulation in Health and Disease, Department of Biological, Geological, and Environmental Sciences, Cleveland State University, Cleveland, OH, USA
In a number of microbial pathogens that undergoes antigenic variation to evade the host’s immune attack, genes encoding surface antigens are located at subtelomeric loci, and recent studies have revealed that telomere components play important roles in regulation of surface antigen expression in several of these pathogens, indicating that telomeres play critical roles in microbial pathogen virulence regulation. Importantly, although telomere protein components and their functions are largely conserved from protozoa to mammals, telomere protein homologs in microbial pathogens and humans have low sequence homology. Therefore, pathogen telomere components are potential drug targets for therapeutic approaches because first, most telomere proteins are essential for pathogens’ survival, and second, disruption of pathogens’ antigenic variation mechanism would facilitate host’s immune system to clear the infection.
Introduction
Telomeres are nucleoprotein complexes located at the ends of linear chromosomes. In most eukaryotic cells, telomere DNA consists of simple repetitive TG-rich sequences and is maintained by telomerase, a ribonucleoprotein that contains both a catalytic protein subunit and an RNA component providing the template for de novo telomere DNA synthesis (Greider and Blackburn, 1987).
A number of proteins have been identified that specifically associate with the telomere DNA. In mammalian cells, the core telomere protein complex termed “Shelterin” (de Lange, 2005) contains two duplex TTAGGG repeat binding factors, TRF1 and TRF2 (Chong et al., 1995; Bilaud et al., 1997; Broccoli et al., 1997), a single-stranded telomere DNA binding protein, POT1 (Baumann and Cech, 2001), and RAP1 (Li et al., 2000), TIN2 (Kim et al., 1999), and TPP1 (Houghtaling et al., 2004; Liu et al., 2004; Ye et al., 2004) that interact with TRFs or POT1. In addition, a trimeric CST complex containing CTC1, STN1, and TEN1 has also been identified to bind the single-stranded telomere DNA (Miyake et al., 2009; Wan et al., 2009). Fission yeast has a very similar telomere protein complex (Miyoshi et al., 2008), and many telomere protein homologs have been identified in budding yeast, too (Lewis and Wuttke, 2012). Recent studies have led to the identification of TRF (Li et al., 2005) and RAP1 (Yang et al., 2009) homologs in Trypanosoma brucei, a protozoan parasite belongs to the kinetoplastids group, suggesting that the telomere complex is largely conserved from protozoan to mammalian cells. Telomeres, together with their associated protein components, form a specialized structure so that the natural chromosome ends are properly protected (Stewart et al., 2011), while maintenance of a stable length of telomere DNA provides adequate docking sites for telomere binding proteins. Therefore, telomeres are essential for genome stability and sustained cell proliferation.
Although telomeres are predominantly maintained by telomerase in most eukaryotes, DNA homologous recombination can also serve as an important means for telomere maintenance (McEachern and Haber, 2006; Nabetani and Ishikawa, 2011). In addition, subtelomeric DNA recombination appears to be a major factor for genome plasticity, which may help to diversify the sequences of subtelomeric genes (Corcoran et al., 1988; Pologe and Ravetch, 1988; De Bruin et al., 1994; Louis, 1995). For several microbial pathogens whose virulence genes are located next to telomeres, this can also be an important pathogenesis mechanism to enhance their virulence (see below).
A telomere position effect (TPE) phenomenon has been observed in a number of organisms, where the expression of genes located at subtelomeres is suppressed by the nearby telomere chromatin structure (Gottschling et al., 1990; Baur et al., 2001; Koering et al., 2002; Park et al., 2002; Pedram et al., 2006). TPE is well studied in S. cerevisiae, where ScRap1 binds the duplex telomere DNA (Longtine et al., 1989). Both ScRap1 and yKu (a heterodimer complex that binds DNA ends in a sequence-independent manner; Riha et al., 2006) can recruit the Sir4 silencer to the telomere, and ScRap1 can also recruit Sir3 (Moretti et al., 1994; Martin et al., 1999; Moretti and Shore, 2001; Luo et al., 2002). Together, Sir3 and Sir4 recruit Sir2 (Moazed et al., 1997; Strahl-Bolsinger et al., 1997; Buchberger et al., 2008; Martino et al., 2009), which is an NAD+-dependent histone deacetylase (Tanny et al., 1999; Landry et al., 2000) and can remove the acetyl group from histone H3 at K9 and K14 residues and from histone H4K16 (Imai et al., 2000). Sir2 activity and the interaction between Sir3/4 and histone tails are necessary for establishing and propagating of the heterochromatic structure from telomere to chromosome internal regions (Hoppe et al., 2002). Similarly, TPE in human cells appears to be mediated by the heterochromatic chromatin structure, as treatment with Trichostatin A, an inhibitor of class I and II histone deacetylases, led to decreased TPE (Koering et al., 2002).
Antigenic Variation and Phenotypic Switch in Microbial Pathogens
Many microbial pathogens that infect mammals have adopted antigenic variation to avoid eradication by the host immune system so that they can maintain persistent infections and enhance the chances of being transmitted to new hosts. Antigenic variation is the phenomenon that a pathogen changes its surface antigen presented to the host immune system regularly and much more frequently than spontaneous gene mutation. The term of antigenic variation usually encompasses both phase variation and true antigenic variation. In phase variation (such as phenotypic switching), the expression of an individual antigen switches between “on” or “off” states. Multiple genes from the same gene family can be expressed at the same time, and each gene’s expression state is relatively independent to that of other genes in the same family. Phenotypic switching can contribute to the virulence of the pathogen because expressing different types or various number of surface molecules may enhance or weaken adhesion of the pathogen to the host. In true antigenic variation, a certain antigen switches among different forms. The antigen is usually expressed in a mutually exclusive manner – a single gene from a multi-copy gene family is expressed at any time. In general, both antigenic variation and phenotypic switching can occur through two general types of mechanisms: genetic and epigenetic (Deitsch et al., 2009). A genetic event involves changes in DNA sequences of an antigen encoding gene or its regulatory elements so that either its expression level or its gene product is changed. An epigenetic event only affects a gene expression level but does not change its DNA sequences. However, recent studies suggest that epigenetic changes such as chromatin remodeling may also influence genetic events such as DNA recombination (Benetti et al., 2007; Bisht et al., 2008). Common mechanisms of antigenic variation have evolved in different pathogens, including bacteria, fungi, and parasites, possibly due to similar selection pressure exerted from the mammalian immune responses. However, in this chapter, we will focus on those mechanisms that are influenced or likely to be affected by the telomere structure.
TPE Participates in the Regulation of EPA Expression in C. glabrata
Candida glabrata is part of the normal human mucosal flora and usually commensal, but it can cause opportunistic mucosal and bloodstream infections in immunocompromised individuals. During infection, binding of the pathogen to host cell proteins or microbial competitors would help to reduce the chance of clearance by the host. Therefore, the adherence of C. glabrata to host cells has been proposed to play an important role in its virulence (Kaur et al., 2005).
When cultured human epithelial cells are used, 95% of in vitro C. glabrata adherence depends on an adhesin molecule that binds the host N-acetyl lactosamine-containing glycoconjugates (Castano et al., 2005) and is encoded by the EPA1 gene (Kapteyn et al., 1999), which belongs to the EPA gene family. So far, a total of 23 putative EPA genes and pseudogenes have been identified in C.glabrata strain BG2 based on their sequence similarity (Kaur et al., 2005). Seven EPA genes encode full-length GPI-anchored proteins, among which Epa1 is a lectin (Cormack et al., 1999), Epa6 and Epa7 are confirmed to be adhesins (Castano et al., 2005), and Epa2 and Epa3 are predicted to be cell wall proteins (De Las Penas et al., 2003). All seven EPA genes located at subtelomeric regions (Figure 1; De Las Penas et al., 2003; Castano et al., 2005; Iraqui et al., 2005).
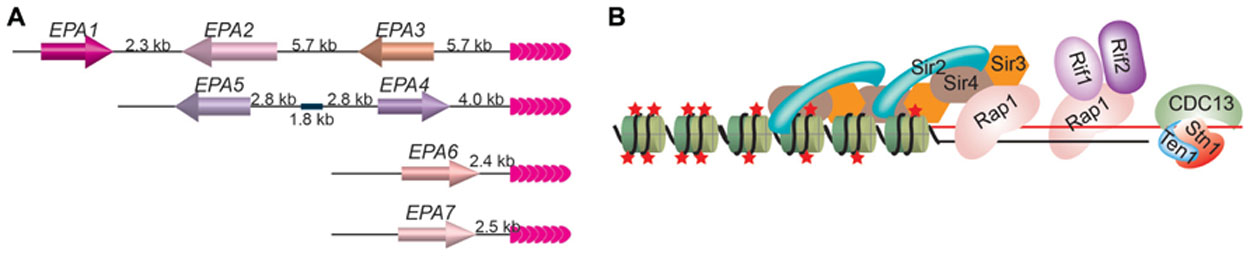
FIGURE 1. (A) EPA1–7 are located at subtelomeric loci in C. glabrata. The positions of seven EPA genes at their respective chromosome end loci are shown. EPA1 is furthest away from the telomere and is the only one that is expressed normally, while EPA 2–7 are usually silenced by TPE. Pink arrowheads, telomere repeats. (B) The telomere protein complex in budding yeast. Rap1 is the duplex telomere DNA binding factor, while Cdc13/Stn1/Ten1 binds to single-stranded telomere DNA. Rap1 recruits Sir3 and Sir4, which in turn recruits Sir2. Sir3 and Sir4 can also interact with histones directly. Sir2’s deacetylase activity maintains the hypoacetylated state of histones. Rap1 also recruits Rif1 and Rif2. Red stars, histone acetylation groups; green cylinders, nucleosomes.
Normally, only EPA1 gene is active, while EPA2–7 genes are silenced by TPE, which depends on telomere protein Rap1. Deletion of the C-terminal 28 amino acids of Rap1 led to derepression of EPA4–7 and sometimes also EPA2 and EPA3 (De Las Penas et al., 2003). Silencing of subtelomeric EPA genes also depends on Sir proteins (De Las Penas et al., 2003; Castano et al., 2005). Deletion of SIR3 led to hyper expression of EPA1 and derepression of EPA2–7, although the derepression of EPA2, EPA3, and EPA4/5 is mild. Deletion of SIR4 also led to derepression of EPA6. In the case of deletion of SIR3, expression of EPA6 and EPA7 appears to contribute to the hyper-adherent phenotype, indicating that TPE can be directly involved in regulation of pathogen virulence (Castano et al., 2005). Interestingly, Epa6 expression is associated with the ability of C. glabrata cells to form biofilm on plastic surface (Iraqui et al., 2005). Biofilm formed by microbial pathogens can increase infection probability and is of great clinical importance because microorganisms adopting this life form is more tolerant or resistant to host defense machinery and anti-microbial agents than free cells.
This TPE regulated adhesin expression is well exploited by C. glabrata to adapt to the host environment. C. glabrata is an nicotinic acid (NA or vitamin niacin) auxotroph, as it lost all the BNA genes involved in the NA synthesis except BNA5 (Domergue et al., 2005). When growing in urine, where NA is limited, the activity of Sir2, an NAD+-dependent histone deacetylase, decreases correspondingly since NA is the precursor of NAD+. As a consequence, TPE level decreases, and EPA1, 6, and 7 genes are highly expressed (Domergue et al., 2005). This effect can be reverted by adding NA or a related compound nicotinamide (NAM). Most importantly, when using an established murine model of urinary track infection, transurethrally inoculated C. glabrata has an elevated colonization frequency in bladder and kidney, which is dependent on EPA1, 6, and 7 gene expression, and mice fed with high-NA diet are no longer susceptible to high rate of colonization of C. glabrata (Domergue et al., 2005). Therefore, in C. glabrata, TPE plays an important role in regulation of virulence gene expression.
Sir2-Mediated TPE Plays an Essential Role in Manoallelic Expression of var Genes in P. falciparum
Plasmodium falciparum is a protozoan parasite in the Apicomplexa phylum that causes the most severe form of malaria, which is a debilitating and sometimes fatal disease mostly found in tropical and subtropical regions of the world. During P. falciparum infection in a human host, the parasite invades first hepatocytes then erythrocytes. One major reason why it is very difficult to eliminate these parasites once an infection is established is that P. falciparum undergoes antigenic variation at the erythrocyte stage (Dzikowski and Deitsch, 2009). At this stage, P. falciparum cells produce erythrocyte membrane protein 1 (PfEMP1), which is encoded by var genes and is transported to the infected erythrocyte membrane (Baruch et al., 1995; Smith et al., 1995; Su et al., 1995). Expression of PfEMP1 on the infected cell surface allows the infected erythrocyte to adhere to the endothelium of the post-capillary venules and avoid circulation through the spleen, where the infected cells will be destroyed (Baruch, 1999). Therefore, expression of PfEMP1 on host cell surface is critical for prolonged parasite infection. However, PfEMP1 is also susceptible to host antibody recognition and subsequent immune attack. As an important virulence mechanism, P. falciparum regularly switches the expressed PfEMP1, therefore effectively evading the host immune attack (Roberts et al., 1992).
There are ~60 var genes in the P. falciparum genome (Gardner et al., 2002). However, only one var gene is expressed at any moment (Roberts et al., 1992). Based on its upstream regulatory elements, var genes can be classified into three groups (Figure2; Kraemer and Smith, 2003; Lavstsen et al., 2003). Those with UpsA and transcribed toward the telomere and those with UpsB and transcribed away from the telomere are located at subtelomeric loci (Figure 2A), while the ones with UpsC are located at chromosome internal loci (Figure 2B; Voss et al., 2000; Gardner et al., 2002; Kraemer and Smith, 2003; Lavstsen et al., 2003). Monoallelic expression of var gene is regulated at multiple levels, and telomeres appear to play an important role (Dzikowski and Deitsch, 2009).
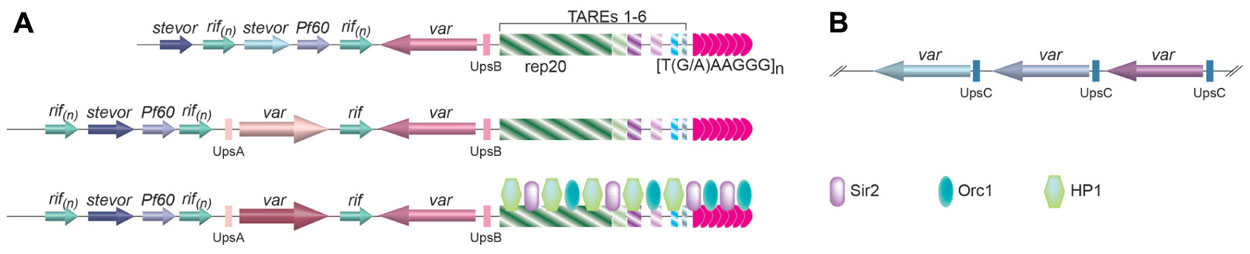
FIGURE 2. (A) The organization of subtelomere elements in P. falciparum. Immediately internal to the telomere tract are six telomere-associated repeat elements (TAREs 1–6), with the largest one, rep20, located furthest away from the telomere repeats. One or two var genes are usually found immediately upstream of rep20, followed by the rifin, stevor, and Pf60 gene families. Depending on the upstream flanking sequences, three classes of var genes have been identified. The ones with associated UpsB and UpsA are located at subtelomeric regions and transcribed in opposite directions as drawn, while the ones with associated UpsC are located as gene arrays inchromosome-internal loci (B). Little is known about the telomere proteins in Plasmodium, except that Sir2 and Orc1 is located at the telomere vicinity (Mancio-Silva et al., 2008), and HP1 is associated with subtelomeric TAREs (Perez-Toledo et al., 2009), which are shown in the bottom diagram in(A).
Telomere position effect in P. falciparum spreads ~55kb along the chromosome from telomeres and was first observed by targeting a reporter gene to the rep20 repeats located at the subtelomeric regions (Figure 2A; Duraisingh et al., 2005). Rep20 is the most telomere-distal telomere associated repetitive element (TARE) and is usually adjacent to the subtelomeric var gene promoter. TPE in P. falciparum depends on Sir2 (Duraisingh et al., 2005; Freitas-Junior et al., 2005; Tonkin et al., 2009), which is both a histone deacetylase and an ADP-ribosyltransferase (Merrick and Duraisingh, 2007; Chakrabarty et al., 2008) and is localized at the telomeres, where histones H4 acetylation is absent (Freitas-Junior et al., 2005). By examining subnuclear localization of a number of genetic markers along chromosome 2 by FISH, it is also inferred that chromatin structure is more condensed for telomere-proximal regions than telomere-distal ones (Freitas-Junior et al., 2005). The direct evidence of involving TPE in var gene regulation came from the observation that deletion of PfSir2 led to a significant increase in transcription of a subset of var genes, particularly the var genes with UpsA and at the subtelomere regions (Duraisingh et al., 2005).
RAP1-Mediated Silencing is Essential for Monoallelic Expression of VSG in T. brucei
The kinetoplastids are a group of flagellated protozoa. Three members of kinetoplastids are of great clinical importance because they cause human diseases: Trypanosoma brucei causes human African trypanosomiasis or sleeping sickness, Trypanosoma cruzi causes South America trypanosomiasis or Chagas disease, and several Leishmania species cause leishmaniasis. Of these three trypanosomatids (the kinetoplastid organisms that only have a single flagellum), only T. brucei undergoes antigenic variation, which is an important mechanism of its pathogenesis and one of its most interesting biological aspects (Barry and McCulloch, 2001).
Trypanosoma brucei is transmitted between mammalian hosts by an insect vector, tsetse (Glossina spp.). While inside the mid-gut of a tsetse fly, T. brucei cells are in the non-virulent proliferative stage, procyclic form (PF), and several procyclic acidic repetitive proteins (PARPs, or procyclins) are expressed on its surface. After T. brucei cells migrate into the salivary gland of the tsetse fly, they differentiate into the metacyclic form, stop proliferating, and acquire virulence. When a tsetse fly takes a blood meal, T. brucei cells can be injected into a mammalian host. They stay in the bloodstream or extracellular spaces in the host and quickly differentiate into bloodstream form (BF). The slender BF is proliferative, while the stumpy form is quiescent and non-proliferative. The metacyclic form and BF cells express variant surface glycoproteins (VSGs) as their major surface glycoprotein (Mehlert et al., 1998). When a tsetse fly bites the infected mammalian host, stumpy bloodstream form T. brucei cells taken into the midgut of a tsetse can quickly differentiate into the PF, ending the life cycle (Matthews, 2005).
Trypanosoma brucei is exposed to the host’s immune system and is vulnerable to both the innate (inflammations, complements, etc.) and adaptive immune responses (antibody, killer T cells, etc.). However, T. brucei has evolved a sophisticated antigenic variation mechanism and regularly switches its surface VSG coat, thus effectively evading the host’s immune response (Barry and McCulloch, 2001).
Antigenic variation in T. brucei has two essential aspects: switch to express a different VSG gene (VSG switching) and monoallelic expression of VSG. Although there are >1,500 VSG genes and pseudogenes in the T. brucei genome (Berriman et al., 2005), only one type of VSG is expressed at any time. After a new VSG gene is turned on, it is essential to turn off the previously active VSG so that the old surface antigen is no longer presented to the host immune system. In addition, expressing only one VSG gene at a time would allow the VSG gene pool to be used for a maximum period of time, enabling a persistent infection. Therefore, both VSG switching and monoallelic expression of VSG are critical for antigenic variation and have been the focus of intensive research for several decades.
There are 11 pairs of megabase chromosomes (0.9–5.7Mb), several intermediate chromosomes (300–900kb), and ~100 copies of minichromosomes (50–100kb) in T. brucei genome (Melville et al., 2000; Alsford et al., 2001; Berriman et al., 2005). The majority of VSG genes are found in long tandem arrays of repeated genes at subtelomeric locations on megabase chromosomes (Figure 3C). Approximately 200 copies of VSG genes are found immediately upstream of telomeres of the minichromosomes, which carry besides the VSG genes, only repetitive sequences, including 177bp repeats in the chromosome internal region and telomere repeats at the chromosome ends (Figure 3D; Alsford et al., 2001). The VSG genes in subtelomeric gene arrays and on minichromosome are often referred to as basic VSG copies because they are transcriptional silent. The rest of VSGs are found in VSG expression sites. In BF T. brucei cells, VSGs are expressed exclusively from bloodstream form VSG expression sites (B-ESs) which are RNA polymerase I (RNAP I)-transcribed, polycistronic transcription units located at subtelomere loci (Figure 3A; de Lange and Borst, 1982; Gunzl et al., 2003). VSG is the last gene in any B-ES and is usually within 1.5kb from the telomere repeats, while the promoter is often 40–60kb upstream of the VSG (Hertz-Fowler et al., 2008). In contrast, at the metacyclic stage, VSGs are expressed from metacyclic VSG expression sites (M-ESs), which are monocistronic transcription units located at the subtelomeric regions (Figure 3B), with the promoter located only ~5kb from the telomere (Lenardo et al., 1984; Cornelissen et al., 1985). Although the M-ESs have much simpler organizations than the B-ESs, much less is understood about metacyclic than bloodstream VSG expression regulation. T. brucei has multiple B-ESs (e.g., Lister 427 has 15 different B-ESs), usually carrying different VSGs, but all B-ESs have very similar genomic organization with ~90% sequence identity (Hertz-Fowler et al., 2008). Earlier studies focused on B-ES promoters also showed that they are almost always identical (Zomerdijk et al., 1990, 1991; Pham et al., 1996). Therefore, how T. brucei manages to fully express only one B-ES and VSG had been a great puzzle for more than a couple of decades.
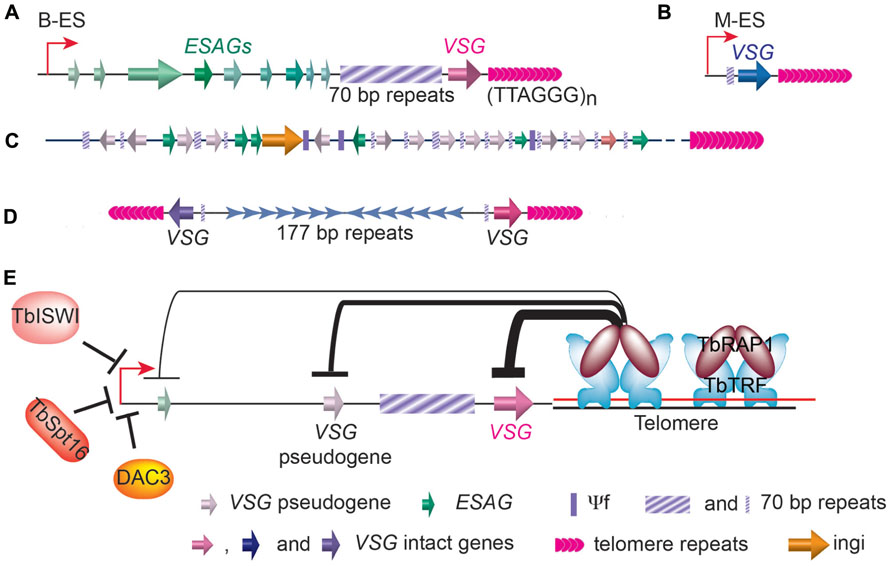
FIGURE 3. Distribution of VSG genes in T. brucei genome. (A) In a bloodstream form VSG expression site (B-ES), the VSG gene is the last one in the large polycistronic transcription unit and is located within 2kb of the telomere repeats. A stretch of 70bp repeats with various length is located upstream of the VSG gene followed by a number of ES associated genes (ESAGs). (B) The metacyclic VSG expression site (M-ES) is a monocistronic transcription unit also located at subtelomeric region. (C) Most VSG genes and pseudogenes (and some ESAG genes) are found in gene arrays located at subtelomeric regions on megabase chromosomes. Short stretches of 70bp repeats are found upstream of each gene. (D) On minichromosomes, single VSG genes and upstream 70bp repeats are also found at subtelomeric regions. (E) The telomere protein, TbRAP1, has been shown to play an important role in silencing subtelomeric VSG genes. TbTRF and TbRAP1 are two known T. brucei telomere proteins. TbRAP1-mediated silencing is stronger (thick line) at telomere-proximal VSG locus and weaker (thin line) at telomere-distal ES promoter region. Several factors important for ES promoter silencing are also shown.
A number of studies in the last decade have shown that VSG expression is regulated at multiple levels. First, transcription elongation from B-ES promoter appears to be regulated. Silent B-ES promoters are actually mildly active (Vanhamme et al., 2000), but transcription elongation is quickly attenuated after a few kilobases, effectively stopping transcription long before the VSG genes. Second, chromatin structure of the active B-ES is very different from the silent B-ESs. The active B-ES has very few nucleosomes while silent ESs are packed with nucleosomes (Figueiredo and Cross, 2010; Stanne and Rudenko, 2010). Chromatin remodeling also plays an important role in regulating the B-ES expression: Deletion of the histone H3K79 methyltransferase TbDot1b led to a 10-fold increase in transcription throughout the silent B-ESs (Janzen et al., 2006). Additional chromatin remodelers have been shown to affect B-ES promoter but not downstream VSG expression (Figure 3E): Depletion of a Swi/Snf homolog, TbISWI, led to an elevated transcription from the silent B-ES promoters (Hughes et al., 2007; Stanne et al., 2011); DAC3, a histone deacetylase homologs, is required for B-ES promoter silencing (Wang et al., 2010); and depletion of TbSpt16, a subunit of the FACT chromatin remodeling complex, also led to an ~20-fold increase in silent B-ES promoter transcription (Denninger et al., 2010). Third, ever since the discovery that VSGs are exclusively expressed from subtelomeric regions (de Lange and Borst, 1982), it has been proposed that telomeres may play an important role in VSG expression regulation (Dreesen et al., 2007). This hypothesis was supported by the fact that TPE has been observed in T. brucei (Horn and Cross, 1997a; Glover and Horn, 2006).
Although the earlier studies provided promising evidence for TPE, direct evidence linking TPE and VSG silencing was lacking for a long time. In addition, although the T. brucei Sir2 homolog plays an essential role in TPE at reporter marked telomeres without native B-ESs, its deletion does not affect VSG silencing at all (Alsford et al., 2007). Furthermore, Glover et al. (2007) was able to target an I-Sce I digestion site together with a neo reporter gene downstream of the VSG gene and immediately upstream of the telomere in a telomerase null background. Induction of ectopic I-SceI expression led to immediate cleavage and loss of the marked telomere. Within 9h, degradation of the reporter gene and the subtelomeric VSG gene was also observed. Although a mild derepression of the reporter gene was observed shortly before it was degraded, the VSG gene was not derepressed at all (Glover et al., 2007). These observations raised a great deal of doubts whether telomeres are indeed necessary for proper VSG silencing.
It was difficult to examine the roles of the telomere in antigenic variation directly without identifying any telomere specific proteins. Earlier attempts to identify telomere DNA binding factors in T. brucei using biochemical approaches led to the identification of a couple of telomere DNA binding activities without identification of the responsible proteins (Eid and Sollner-Webb, 1995, 1997).
The first T. brucei Shelterin homolog, TbTRF, was identified using an in silico approach (Li et al., 2005), and a yeast 2-hybrid screen using TbTRF as bait led to the identification of T.brucei RAP1 (Yang et al., 2009), another integral component of the T.brucei telomere complex (Figure 3E). When TbRAP1 was depleted by RNAi (Shi et al., 2000), a derepression of silent B-ES-linked VSGs can be detected (Yang et al., 2009). Using quantitative RT-PCR analysis, it was shown that all B-ES-linked silent VSGs had an elevated expression level upon depletion of TbRAP1, although the level of derepression varies among different VSGs, ranging from 8- to 56-fold. Subsequently, it was confirmed by IF that multiple VSGs are expressed simultaneously in individual cells on cell surface (Yang et al., 2009). Importantly, the TbRAP1-mediated silencing is position dependent. First, only subtelomeric B-ES-linked VSGs were affected. Genes located in chromosome internal regions including RNAP I transcribed rDNA and RNAP II transcribed telomerase protein gene, a ribosomal protein gene, and a glycolytic protein gene were not affected. Second, within an individual B-ES, the telomere-adjacent VSG gene is derepressed at the highest level, a VSG pseudogene located 7–20kb away from the telomere is derepressed at an intermediate level, and a reporter gene targeted immediately downstream of the B-ES promoter located 40–60kb away from the telomere is derepressed at the lowest level (Figure 3E). It is therefore convinced that the TbRAP1-mediated silencing originates from the telomere, demonstrating for the first time that the telomere structure indeed plays an essential role in VSG expression regulation (Yang et al., 2009).
However, the involvement of telomere in VSG expression regulation does not necessarily exclude other mechanisms mentioned above. In fact, TbRAP1-mediated silencing appears to block the elongation of the basal level transcription from the silent B-ES promoters, because in TbRAP1 deficient cells, derepressed VSGs are expressed at a level that is still ~100 fold lower than when the same VSG is in a fully active B-ES (Yang et al., 2009). Therefore, the observed quick attenuation of transcription elongation along silent B-ESs may well be the combined effect of a basal level transcription initiated from silent B-ES promoters and a TbRAP1-mediated TPE. The fact that derepressed VSGs are not expressed at its fullest potential also suggests that B-ES promoters are regulated by additional factors other than TPE. This is consistent with the observations that a number of chromatin remodeling factors are involved in B-ES promoter regulation as mentioned above.
Recent studies have made great contributions to our understanding of how VSG expression is silenced. However, how is allelic-exclusive expression of VSG achieved is not fully understood. It has been proposed that sufficient amount of RNAP I machinery, which is responsible for high level VSG transcription, may be accessible to only one B-ES, which would effectively ensure its monoallelic expression (Horn and McCulloch, 2010). In an IF analysis, Navarro and Gull (2001) found that in BF T. brucei cells, transcriptionally active RNAP I forms a small nuclear focus in addition to the large focus inside the nucleolus, where it transcribes rRNA. In addition, only the active B-ES but not the silent ones is co-localized with RNAP I in this ES body (ESB), which only exists in BF but not PF cells (Navarro and Gull, 2001). It is therefore hypothesized that ESB, enriched with RNAP I, can only accommodate one B-ES, which would effectively limit the number of active B-ES to one. In support of this view, when two different B-ESs were tagged with selective markers immediately downstream of their respective promoters and forced to be active simultaneously, the two B-ESs appear to switch back and forth rapidly and locate next to each other in the nucleus, presumably competing for available RNAP I at ESB (Chaves et al., 1999).
Telomere Length Affects VSG Switching Frequency and Mechanism in T. brucei
VSG switching can occur through several different pathways (Figure 4; Barry and McCulloch, 2001). In the so-called in situ switch, a silent B-ES promoter is turned fully active while the originally active B-ES promoter is turned off without any DNA rearrangements. There are 15 B-ESs carrying distinctive VSGs in the T. brucei Lister 427 cells, providing a small number of possible in situ switch opportunities (Hertz-Fowler et al., 2008). However, in situ VSG switching is usually a rare event, and VSG switching involving DNA recombination events are much more prevalent (Robinson et al., 1999). The large VSG gene pool, therefore, provides essentially endless possibilities for VSG switching.
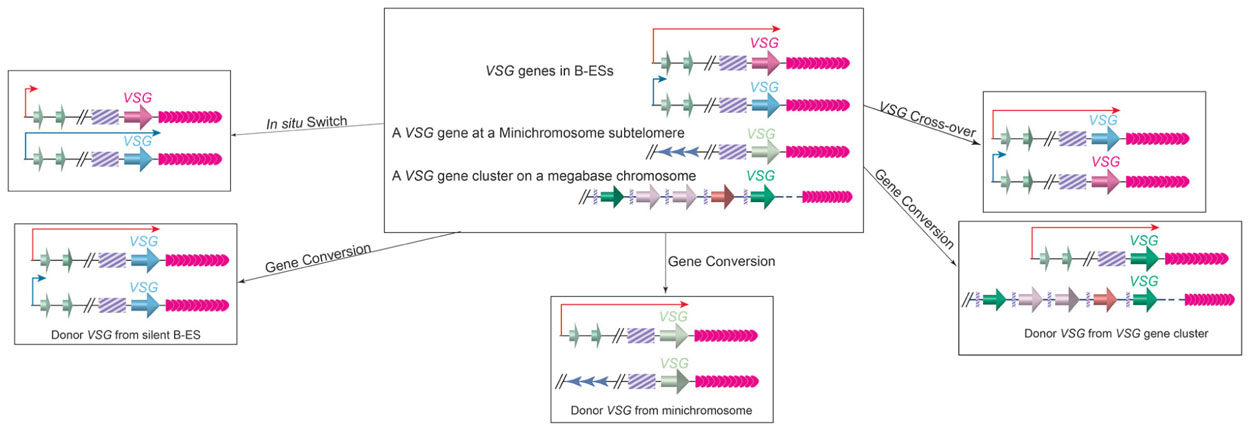
FIGURE 4. VSG switching can occur through in situ switch, gene conversion, or crossover. Top Middle, before switching, an active B-ES (long red arrow), a silent B-ES (short blue arrow), a VSG gene at a minichromosome subtelomere, and an array of VSG genes and pseudogenes on a megabase chromosome are shown. In situ switch (top left) results from turning on (long blue arrow) of the silent B-ES and turning off (short red arrow) of theactive B-ES simultaneously without any DNA rearrangements. In gene conversion, a silent VSG gene is duplicated into the active B-ES, and the originally active VSG gene is lost. The VSG donor can come from a silent B-ES (bottom left), a minichromosome subtelomere (bottom middle), or a VSG gene array (bottom-right). In VSG cross-over (top right), the active VSG and a silent VSG (most often from a silent B-ES) exchange their loci reciprocally, resulting in a new VSG gene in the active B-ES without losing any genetic information. The cross-over site is often found within the 70bp repeats upstream of the VSG genes, although it can locate more upstream, and all B-ESs have high sequence homology.
In gene conversion events, a silent VSG is copied into the active B-ES while the originally active VSG is lost. In this event, the donor can be any functional VSG gene in the genome. There is almost always a stretch of 70bp repeats upstream of a VSG gene, in which homologous recombination can initiate as DNA double-strand breaks (Boothroyd et al., 2009). In rare occasions, several VSG donors have been identified in a single VSG switching event, where each donor contributes only a fragment of the gene, generating a new mosaic VSG gene product (Marcello and Barry, 2007). Such mechanism has been proposed to be useful in late stage of persistent infection. More often, a silent B-ES is used as a donor possibly because long stretch of 70bp repeats (2 to >14kb) and telomere repeats (3–20kb) flank the VSG gene in any B-ES, and efficient homologous recombination can initiate from these sites. In fact, all B-ESs have very similar genome organization and are ~90% identical in sequences, so gene conversion event can initiate at places upstream of 70bp repeats and often a whole silent B-ES can be copied to replace the active B-ES (Pays et al., 1983b; Hertz-Fowler et al., 2008). Therefore, the terms of VSG gene conversion and ES gene conversion are used to differentiate different types of gene conversion events (Kim and Cross, 2010). In addition to gene conversion, reciprocal crossover event can occur in a VSG switching (Rudenko et al., 1996). In this case, the crossover usually occurs at the 70bp repeats, and the silent and active VSGs (often together with their respective downstream telomeres) simply trade places without deletion of large fragments of genetic information. It is worth to note that in a crossover switching, the originally silent VSG often comes from a silent B-ES, but it can also be from a minichromosome subtelomere. Finally, more complicated switching events involving loss of the active B-ES or VSG associated with an in situ switch have also been observed (Kim and Cross, 2011).
It has been shown that homologous DNA recombination is important for VSG switching in T. brucei (McCulloch and Barry, 1999). In homologous recombination, searching for DNA sequence homology and subsequent strand-invasion is a key step, at which RAD51 polymerizes around ssDNA to assemble a nucleoprotein helical filament and, with the help of ATP, extends the DNA structure and carries out the strand exchange process (Holloman, 2011). When ssDNA is coated with RPA (a single-strand-specific DNA binding protein), it will not be accessible by RAD51 without the help of a mediator, such as BRCA2 (Holloman, 2011). In T.brucei, six RAD51 related proteins have been identified: RAD51, DMC1, RAD51-3, RAD51-4, RAD51-5, and Rad51-6 (Proudfoot and McCulloch, 2005). Among these, deletion of TbRAD51 and TbRAD51-3 led to a decrease in VSG switching rate while deletion of TbRAD51-5 did not have any effect, and deletion of TbBRCA2 also led to a similar decreased VSG switching rate (Hartley and McCulloch, 2008). In addition, TbTOPO3α and TbRMI1, whose homologs in mammalian cells form a so-called RTR complex with The RecQ helicase BLM and suppress aberrant and inappropriate homologous recombination, were recently shown to be involved in regulation of VSG switching (Kim and Cross, 2010, 2011).
Apparently, homologous recombination is a major pathway for VSG switching. However, exactly how VSG switching is regulated is less clear. Several recent studies now indicate that the telomere structure can influence VSG switching greatly.
It has been shown that the active VSG-marked telomere is less stable than the silent telomeres (Bernards et al., 1983; Pays et al., 1983a; van der Ploeg et al., 1984; Myler et al., 1988; Horn and Cross, 1997b). Rapidly shortened active telomere arises frequently, which is quite similar to the TRD observed in yeast cells carrying abnormally long telomeres (Li and Lustig, 1996). Presumably the active transcription of the telomere is a major cause for the brittle active telomere (Rudenko and Van der Ploeg, 1989). With the presence of telomerase, shortened telomeres are elongated quickly (Horn et al., 2000). With frequent truncation and elongation, telomere length at the active chromosome end is often much more heterogeneous than those at silent telomeres (Bernards et al., 1983). However, in the absence of telomerase, the truncated active telomere remains short, allowing the isolation of clones baring extremely short active telomere in a relatively short culturing period (Dreesen and Cross, 2006). Interestingly, when such telomerase negative clones were obtained that carry extremely short active telomere, these clones tend to switch to express a new VSG (Dreesen and Cross, 2006). This observation led to the hypothesis that shorter telomeres may cause higher VSG switching rate (Dreesen et al., 2007). It is speculated that all active telomeres are prone to large telomere fragment deletions due to its active transcription state, but shorter telomeres are more likely to have a deletion landed in the subtelomeric region and to cause damage in the active VSG gene, which will force the parasite to go through VSG switching. Introducing a break at the I-SceI site targeted immediately upstream of the active VSG gene led to a 250-fold increase in VSG switching frequency, confirming part of this theory that damage to the active VSG gene will force the parasite to switch (Boothroyd et al., 2009).
Importantly, a recent study showed that cells carrying short active telomeres (~1.5kb) has an ~6.3-fold higher VSG switching frequency than cells carrying long telomeres (>10kb; Hovel-Miner et al., 2012). In addition, cells with short active telomere also have more gene conversion and much fewer telomere crossover events as VSG switching mechanism (Hovel-Miner et al., 2012). Therefore, telomere length indeed affects subtelomeric VSG switching. At least two Shelterin homologs have been identified in T. brucei (Li et al., 2005; Yang et al., 2009), which enabled further investigation of the telomere structure in VSG switching regulation. It is speculated that disruption of the heterochromatic telomere structure, especially in the case of depletion of TbRAP1 (Yang et al., 2009), may also lead to higher VSG switching rate, similar to what was observed in S. pombe (Bisht et al., 2008).
Does Telomere Affect Switching of Subtelomere-Located Surface Antigen in P. carinii and B. burgdorferi?
Pneumocystis carinii is a fungus that solely dwells in the lung tissue of mammals. Normally, P. carinii infection does not cause any symptom, but in immunocompromised individuals it can cause pneumonia. The complete life cycle of P. carinii is still not very well defined, mainly because of the lack of a continuous cultivation system. However, it is obvious that P. carinii can survive in the lower respiratory tract where strong and effective defense systems normally work to eliminate invaders, and the reason for persistent and effective P. carinii infection is that it undergoesantigenic variation at a high frequency (Cushion and Stringer, 2010).
The Major Surface Glycoprotein (MSG) is one of the major surface molecules of P. carinii that is involved in antigenic variation (Stringer, 2005). MSG is encoded by the MSG gene family. So far 73 MSG genes have been identified, all are located at the subtelomeric loci (Figure 5; Keely and Stringer, 2009). There are 17 chromosomes in P. carinii (Hong et al., 1990), indicating that on average at least two MSG genes are at each telomere, which is often the case in cloned terminal fragments from various chromosomes (Wada and Nakamura, 1996; Keely et al., 2005). Similar to the situation in T. brucei, only one MSG gene is transcribed at any time. Transcribed MSG messengers always contained an upstream conserved sequence (UCS; Wada et al., 1995; Edman et al., 1996; Wada and Nakamura, 1996; Sunkin and Stringer, 1997), which has only one copy in the P. carinii genome (Wada et al., 1995; Edman et al., 1996), suggesting that MSG is transcribed from a specific expression site marked with the unique UCS element. In addition, translation initiation codon on an MSG mRNA is located in the sequence transcribed from the UCS (Wada et al., 1995; Edman et al., 1996). Therefore, transcribing MSG from UCS-containing expression site is essential for proper MSG translation. Furthermore, the UCS encoded peptide contains a signal sequence that targets the pre-MSG protein into the endoplasmic reticulum, where it can be cleaved and glycosylated, then deposited on the cell surface (Sunkin et al., 1998). Hence the UCS peptide is also essential for MSG function, although it is not present on MSG found on the cell surface because it is likely removed in the endoplasmic reticulum.
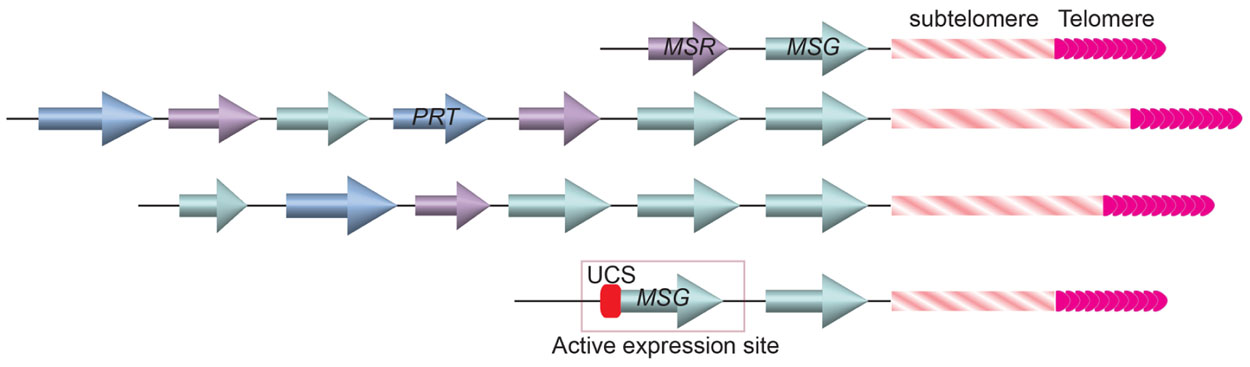
FIGURE 5. Gene arrays at the ends of three Pneumocystis carinii chromosomes. MSG genes (cyan colored arrows) are located closest to the telomere and subtelomeric repetitive sequences. A single copy UCS is found in the active MSG expression site immediately upstream of the MSG gene.
If P. carinii contains only one UCS-containing MSG expression site, how does it achieve antigenic variation? Computational analysis of MSG gene sequences suggested that these genes commonly undergo recombination (Wada and Nakamura, 1996; Keely et al., 2005; Keely and Stringer, 2009), which is not unlike the VSG switching in T. brucei. Similar to VSG, MSG is also the last transcribed gene on the chromosome (Wada and Nakamura, 1996; Keely et al., 2005). The proximity of MSG genes to telomeres suggests that the MSG switching events might also be regulated by the telomere structure, although this has not be investigated at all.
In a different microbial pathogen Borrelia burgdorferi, the spirochete that causes the Lyme disease, the gene encoding variant surface antigen is found at a subtelomere region on a linear plasmid (Zhang et al., 1997). B. burgdorferi also undergoes antigenic variation, and the lipoprotein VlsE is the variant surface protein (Schwan et al., 1991; Zhang et al., 1997; Zhang and Norris, 1998b; Steere et al., 2004; Norris, 2006). VlsE is encoded by the vls gene family located on the linear plasmid lp28-1 (Figure 6). Immediately next to the telomere is the active vlsE expression site. More upstream is the silent vls gene cluster (Zhang et al., 1997). Bacteria lost the lp28-1 exhibit an intermediate infectivity phenotype where it is hard to establish a persistent infection in the mouse model (Bankhead and Chaconas, 2007). Deletion of vlsE and silent vls cassettes also led to reduced persistent infection, indicating that antigenic variation through vls switching is an important virulence mechanism in B. burgdorferi (Zhang et al., 1997; Purser and Norris, 2000; Labandeira-Rey and Skare, 2001; Bankhead and Chaconas, 2007). The vlsE and the silent vls genes are highly homologous at the sequence level, and most of the sequence differences within the cassette regions are concentrated in six variable regions, VR1–VR6 (Zhang and Norris, 1998a). Segmental gene conversion between the silent cassettes and the vlsE cassette region occurs as early as 4days after infection in mice, and appears to continue throughout the course of infection (Zhang and Norris, 1998b). Because these recombination events appear to involve random segments of any silent cassette and occur continuously during infection, an almost unlimited number of VlsE amino acid sequence permutations are theoretically possible (Zhang and Norris, 1998a). Apparently, vls switching is not so unlike the VSG switching in T. brucei or MSG switching in P. carinii. However, nothing is known about the telomere structure at the ends of lp28 or any protein(s) associated with it. Therefore, it is unclear whether the nearby telomere structure might exhibit any influence to vls switching.
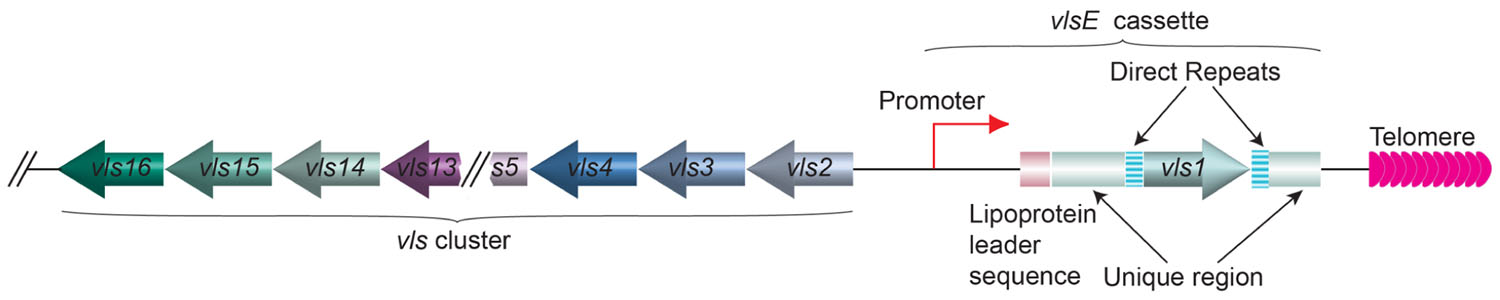
FIGURE 6. The organization of vlsE and the array of silent vls genes on lp28 linear plasmid of B. burgdorferi. The vls1 gene (moss colored arrow) is expressed from the vlsE expression site next to the telomere (pink arrows). The direct repeats (barred boxes) and the unique regions (green boxes) flanking the vls1 gene and the lipoprotein leader sequence (rouge box) upstream of vls1 are marked. The silent array includes vls2–16 genes (various colored arrows) going to the opposite direction from vls1 are located at the internal region of the linear plasmid.
Telomere Components as Potential Targets of Anti-Pathogen Agents
In this review, I have provided a detailed treatise of the telomere region, the adjoining genes and sites, and the regulatory elements and proteins in several microbial pathogens that undergo antigenic variation. As discussed, telomere forms a specialized heterochromatic structure that can influence the expression of genes located nearby. It appears that several microbial pathogens have conveniently taken advantage of this TPE to regulate expression of surface antigen-encoding gene families at subtelomeric regions. Further studies of the telomere structure and telomere-specific proteins in these microbial pathogens should provide more insight about the allelic exclusion expression of surface antigen genes. In addition, the subtelomeric region in many eukaryotic cells appears to be a DNA recombination hot spot, presumably contributing to gene diversity. This could be one of the reasons why many gene families encoding virulence factors are located at subtelomeric loci in microbial pathogens. One cannot help to speculate that the intrinsic plastic nature of the subtelomeres might facilitate antigenic variation. On the other hand, unchecked homologous recombination could cause hazardous genome instability, and the telomere structure with telomere-specific proteins is hypothesized to suppress subtelomeric recombination to maintain a relatively stable genome organization.
The essential functions of telomeres in maintaining genome integrity are conserved for all eukaryotic cells, and homologs of many telomere-specific proteins have been identified from protozoa to mammals. However, telomere homologs from mammals and those from the above-mentioned microbial pathogens have very low sequence homology. As the telomere components play important functions in regulation of virulence in several microbial pathogens, they are attractive drug targets for treatment of diseases caused by these pathogens. For example, TbRAP1 and hRAP1 has very limited sequence homology. It should be feasible to identify or develop agents that specifically target TbRAP1 but not hRAP1. These agents are expected to act as a double-edged sword. First, TbRAP1 is essential for trypanosome cell growth and dysfunctional TbRAP1 leads to cell growth arrest. Second, TbRAP1 is essential for complete subtelomeric VSG silencing, and lack of TbRAP1 leads to expression of multiple VSG proteins on trypanosome cell surface, which will facilitate the host immune system to eliminate the parasite efficiently.
Because most microbial pathogens grow much faster than their mammalian host, they are also more susceptible to agents that disrupt the telomere structure or cause telomere length attrition, which is more detrimental to fast growing cells. Because T. brucei has a 3′ single-stranded G-rich overhang at the end of the telomeres, compounds that target the G-quadruplex such as Imetelstat would be a good choice to inhibit T. brucei growth preferentially. Imetelstat is a lipid-conjugated oligonucleotide (previously known as GRN163), with excellent tissue penetration, bioavailability, and efficacy that has been used to against a variety of cancers (Dikmen et al., 2005; Herbert et al., 2005; Gellert et al., 2006). Other small molecule inhibitors that interact with the human telomeric DNA are available (Sun et al., 1997), but they may not inhibit parasites other than T. brucei due to telomere sequence dissimilarity. Another potential target would be telomerase that synthesize the telomere DNA in most eukaryotic cells. Although knockout telomerase is not expected to cause immediate deleterious effects in parasites such as T. brucei that carry long telomeres, changing the telomere sequence by incorporation of mutations into the telomerase RNA template may lead to more acute cell growth arrest due to disrupted binding of the mutant telomere DNA by normal telomere binding proteins, such as TRF.
We have just begun to understand the functions of telomeres in antigenic variation. New telomere components are continuously investigated for their potential roles in this important mechanism of pathogenesis. As we gain more knowledge, we expect to identify more suitable telomere components as good anti-pathogen targets.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alsford, S., Kawahara, T., Isamah, C., and Horn, D. (2007). A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol. Microbiol. 63, 724–736.
Alsford, S., Wickstead, B., Ersfeld, K., and Gull, K. (2001). Diversity and dynamics of the minichromosomal karyotype in Trypanosoma brucei. Mol. Biochem. Parasitol. 113, 79–88.
Bankhead, T., and Chaconas, G. (2007). The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 65, 1547–1558.
Barry, J. D., and McCulloch, R. (2001). Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite. Adv. Parasitol. 49, 1–70.
Baruch, D. I., Pasloske, B. L., Singh, H. B., Bi, X., Ma, X. C., Feldman, M., et al. (1995). Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 82, 77–87.
Baruch, D. I. (1999). Adhesive receptors on malaria-parasitized red cells. Baillieres Best Pract. Res. Clin. Haematol. 12, 747–761.
Baumann, P., and Cech, T. R. (2001). Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292, 1171–1175.
Baur, J. A., Zou, Y., Shay, J. W., and Wright, W. E. (2001). Telomere position effect in human cells. Science 292, 2075–2077.
Benetti, R., Gonzalo, S., Jaco, I., Schotta, G., Klatt, P., Jenuwein, T., et al. (2007). Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J. Cell Biol. 178, 925–936.
Bernards, A., Michels, P. A. M., Lincke, C. R., and Borst, P. (1983). Growth of chromosome ends in multiplying trypanosomes. Nature 303, 592–597.
Berriman, M., Ghedin, E., Hertz-Fowler, C., Blandin, G., Renauld, H., Bartholomeu, D. C., et al. (2005). The genome of the African trypanosome Trypanosoma brucei. Science 309, 416–422.
Bilaud, T., Brun, C., Ancelin, K., Koering, C. E., Laroche, T., and Gilson, E. (1997). Telomeric localization of TRF2, a novel human telobox protein. Nat. Genet. 17, 236–239.
Bisht, K. K., Arora, S., Ahmed, S., and Singh, J. (2008). Role of heterochromatin in suppressing subtelomeric recombination in fission yeast. Yeast 25, 537–548.
Boothroyd, C. E., Dreesen, O., Leonova, T., Ly, K. I., Figueiredo, L. M., Cross, G. A. M., et al. (2009). A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature 459, 278–281.
Broccoli, D., Smogorzewska, A., Chong, L., and de Lange, T. (1997). Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 17, 231–235.
Buchberger, J. R., Onishi, M., Li, G., Seebacher, J., Rudner, A. D., Gygi, S. P., et al. (2008). Sir3-nucleosome interactions in spreading of silent chromatin in Saccharomyces cerevisiae. Mol. Cell. Biol. 28, 6903–6918.
Castano, I., Pan, S. J., Zupancic, M., Hennequin, C., Dujon, B., and Cormack, B. P. (2005). Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 55, 1246–1258.
Chakrabarty, S. P., Saikumari, Y. K., Bopanna, M. P., and Balaram, H. (2008). Biochemical characterization of Plasmodium falciparum Sir2, a NAD+-dependent deacetylase. Mol. Biochem. Parasitol. 158, 139–151.
Chaves, I., Rudenko, G., Dirks-Mulder, A., Cross, M., and Borst, P. (1999). Control of variant surface glycoprotein gene-expression sites in Trypanosoma brucei. EMBO J. 18, 4846–4855.
Chong, L., van Steensel, B., Broccoli, D., Erdjument-Bromage, H., Hanish, J., Tempst, P., et al. (1995). A human telomeric protein. Science 270, 1663–1667.
Corcoran, L. M., Thompson, J. K., Walliker, D., and Kemp, D. J. (1988). Homologous recombination within subtelomeric repeat sequences generates chromosome size polymorphisms in P. falciparum. Cell 53, 807–813.
Cormack, B. P., Ghori, N., and Falkow, S. (1999). An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 285, 578–582.
Cornelissen, A. W., Bakkeren, G. A., Barry, J. D., Michels, P. A., and Borst, P. (1985). Characteristics of trypanosome variant antigen genes active in the tsetse fly. Nucleic Acids Res. 13, 4661–4676.
Cushion, M. T., and Stringer, J. R. (2010). Stealth and opportunism: alternative lifestyles of species in the fungal genus Pneumocystis. Annu. Rev. Microbiol. 64, 431–452.
De Bruin, D., Lanzer, M., and Ravetch, J. V. (1994). The polymorphic subtelomeric regions of Plasmodium falciparum chromosomes contain arrays of repetitive sequence elements. Proc. Natl. Acad. Sci. U.S.A. 91, 619–623.
de Lange, T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110.
de Lange, T., and Borst, P. (1982). Genomic environment of the expression-linked extra copies of genes for surface antigens of Trypanosoma brucei resembles the end of a chromosome. Nature 299, 451–453.
De Las Penas, A., Pan, S. J., Castano, I., Alder, J., Cregg, R., and Cormack, B. P. (2003). Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 17, 2245–2258.
Deitsch, K. W., Lukehart, S. A., and Stringer, J. R. (2009). Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 7, 493–503.
Denninger, V., Fullbrook, A., Bessat, M., Ersfeld, K., and Rudenko, G. (2010). The FACT subunit TbSpt16 is involved in cell cycle specific control of VSG expression sites in Trypanosoma brucei. Mol. Microbiol. 78, 459–474.
Dikmen, Z. G., Gellert, G. C., Jackson, S., Gryaznov, S., Tressler, R., Dogan, P., et al. (2005). In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res. 65, 7866–7873.
Domergue, R., Castano, I., De Las Penas, A., Zupancic, M., Lockatell, V., Hebel, J. R., et al. (2005). Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 308, 866–870.
Dreesen, O., and Cross, G. A. M. (2006). Consequences of telomere shortening at an active VSG expression site in telomerase-deficient Trypanosoma brucei. Eukaryot. Cell 5, 2114–2119.
Dreesen, O., Li, B., and Cross, G. A. M. (2007). Telomere structure and function in trypanosomes: a proposal. Nat. Rev. Microbiol. 5, 70–75.
Duraisingh, M. T., Voss, T. S., Marty, A. J., Duffy, M. F., Good, R. T., Thompson, J. K., et al. (2005). Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 121, 13–24.
Dzikowski, R., and Deitsch, K. W. (2009). Genetics of antigenic variation in Plasmodium falciparum. Curr. Genet. 55, 103–110.
Edman, J. C., Hatton, T. W., Nam, M., Turner, R., Mei, Q., Angus, C. W., et al. (1996). A single expression site with a conserved leader sequence regulates variation of expression of the Pneumocystis carinii family of major surface glycoprotein genes. DNA Cell Biol. 15, 989–999.
Eid, J. E., and Sollner-Webb, B. (1995). ST-1, a 39-kilodalton protein in Trypanosoma brucei, exhibits a dual affinity for the duplex form of the 29-base-pair subtelomeric repeat and its C-rich strand. Mol. Cell. Biol. 15, 389–397.
Eid, J. E., and Sollner-Webb, B. (1997). ST-2, a telomere and subtelomere duplex and G-strand binding protein activity in Trypanosoma brucei. J. Biol. Chem. 272, 14927–14936.
Figueiredo, L. M., and Cross, G. A. M. (2010). Nucleosomes are depleted at the VSG expression site transcribed by RNA polymerase I in African trypanosomes. Eukaryot. Cell 9, 148–154.
Freitas-Junior, L. H., Hernandez-Rivas, R., Ralph, S. A., Montiel-Condado, D., Ruvalcaba-Salazar, O. K., Rojas-Meza, A. P., et al. (2005). Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121, 25–36.
Gardner, M. J., Hall, N., Fung, E., White, O., Berriman, M., and Hyman, R. W. (2002). Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511.
Gellert, G. C., Dikmen, Z. G., Wright, W. E., Gryaznov, S., and Shay, J. W. (2006). Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res. Treat. 96, 73–81.
Glover, L., Alsford, S., Beattie, C., and Horn, D. (2007). Deletion of a trypanosome telomere leads to loss of silencing and progressive loss of terminal DNA in the absence of cell cycle arrest. Nucleic Acids Res. 35, 872–880.
Glover, L., and Horn, D. (2006). Repression of polymerase I-mediated gene expression at Trypanosoma brucei telomeres. EMBO Rep. 7, 93–99.
Gottschling, D. E., Aparicio, O. M., Billington, B. L., and Zakian, V. A. (1990). Position effect at S. cerevisiae telomeres: reversible repression of pol II transcription. Cell 63, 751–762.
Greider, C. W., and Blackburn, E. H. (1987). The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51, 887–898.
Gunzl, A., Bruderer, T., Laufer, G., Schimanski, B., Tu, L. C., Chung, H. M., et al. (2003). RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot. Cell 2, 542–551.
Hartley, C. L., and McCulloch, R. (2008). Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination. Mol. Microbiol. 68, 1237–1251.
Herbert, B. S., Gellert, G. C., Hochreiter, A., Pongracz, K., Wright, W. E., Zielinska, D., et al. (2005). Lipid modification of GRN163, an N3′→P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 24, 5262–5268.
Hertz-Fowler, C., Figueiredo, L. M., Quail, M. A., Becker, M., Jackson, A., Bason, N., et al. (2008). Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 3, e3527. doi: 10.1371/journal.pone.0003527
Holloman, W. K. (2011). Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 18, 748–754.
Hong, S. T., Steele, P. E., Cushion, M. T., Walzer, P. D., Stringer, S. L., and Stringer, J. R. (1990). Pneumocystis carinii karyotypes. J. Clin. Microbiol. 28, 1785–1795.
Hoppe, G. J., Tanny, J. C., Rudner, A. D., Gerber, S. A., Danaie, S., Gygi, S. P., et al. (2002). Steps in assembly of silent chromatin in yeast: Sir3-independent binding of a Sir2/Sir4 complex to silencers and role for Sir2-dependent deacetylation. Mol. Cell. Biol. 22, 4167–4180.
Horn, D., and McCulloch, R. (2010). Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Curr. Opin. Microbiol. 13, 700–705.
Horn, D., and Cross, G. A. M. (1997a). Position-dependent and promoter-specific regulation of gene expression in Trypanosoma brucei. EMBO J. 16, 7422–7431.
Horn, D., and Cross, G. A. M. (1997b). Analysis of Trypanosoma brucei vsg expression site switching in vitro. Mol. Biochem. Parasitol. 84, 189–201.
Horn, D., Spence, C., and Ingram, A. K. (2000). Telomere maintenance and length regulation in Trypanosoma brucei. EMBO J. 19, 2332–2339.
Houghtaling, B. R., Cuttonaro, L., Chang, W., and Smith, S. (2004). A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 14, 1621–1631.
Hovel-Miner, G. A., Boothroyd, C. E., Mugnier, M., Dreesen, O., Cross, G. A., and Papavasiliou, F. N. (2012). Telomere length affects the frequency and mechanism of antigenic variation in Trypanosoma brucei. PLoS Pathog. 8, e1002900. doi: 10.1371/journal.ppat.1002900
Hughes, K., Wand, M., Foulston, L., Young, R., Harley, K., Terry, S., et al. (2007). A novel ISWI is involved in VSG expression site downregulation in African trypanosomes. EMBO J. 26, 2400–2410.
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD- dependent histone deacetylase. Nature 403, 795–800.
Iraqui, I., Garcia-Sanchez, S., Aubert, S., Dromer, F., Ghigo, J. M., d’Enfert, C., et al. (2005). The Yak1p kinase controls expression of adhesins and biofilm formation in Candida glabrata in a Sir4p-dependent pathway. Mol. Microbiol. 55, 1259–1271.
Janzen, C. J., Hake, S. B., Lowell, J. E., and Cross, G. A. M. (2006). Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol. Cell 23, 497–507.
Kapteyn, J. C., Van Den Ende, H., and Klis, F. M. (1999). The contribution of cell wall proteins to the organization of the yeast cell wall. Biochim. Biophys. Acta 1426, 373–383.
Kaur, R., Domergue, R., Zupancic, M. L., and Cormack, B. P. (2005). A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 8, 378–384.
Keely, S. P., Renauld, H., Wakefield, A. E., Cushion, M. T., Smulian, A. G., Fosker, N., et al. (2005). Gene arrays at Pneumocystis carinii telomeres. Genetics 170, 1589–1600.
Keely, S. P., and Stringer, J. R. (2009). Complexity of the MSG gene family of Pneumocystis carinii. BMC Genomics 10, 367. doi: 10.1186/1471-2164-10-367
Kim, H. S., and Cross, G. A. M. (2010). TOPO3α influences antigenic variation by monitoring expression-site-associated VSG switching in Trypanosoma brucei. PLoS Pathog. 6, e1000992. doi: 10.1371/journal.ppat.1000992
Kim, H. S., and Cross, G. A. M. (2011). Identification of Trypanosoma brucei RMI1/BLAP75 homologue and its roles in antigenic variation. PLoS ONE 6, e25313. doi: 10.1371/journal.pone.0025313
Kim, S. H., Kaminker, P., and Campisi, J. (1999). TIN2, a new regulator of telomere length in human cells. Nat. Genet. 23, 405–412.
Koering, C. E., Pollice, A., Zibella, M. P., Bauwens, S., Puisieux, A., Brunori, M., et al. (2002). Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep. 3, 1055–1061.
Kraemer, S. M., and Smith, J. D. (2003). Evidence for the importance of genetic structuring to the structural and functional specialization of the Plasmodium falciparum var gene family. Mol. Microbiol. 50, 1527–1538.
Labandeira-Rey, M., and Skare, J. T. (2001). Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 69, 446–455.
Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L., et al. (2000). The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811.
Lavstsen, T., Salanti, A., Jensen, A. T., Arnot, D. E., and Theander, T. G. (2003). Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar. J. 2, 27.
Lenardo, M. J., Rice-Ficht, A. C., Kelly, G., Esser, K. M., and Donelson, J. E. (1984). Characterization of the genes specifying two metacyclic variable antigen types in Trypanosoma brucei rhodesiense. Proc. Natl. Acad. Sci. U.S.A. 81, 6642–6646.
Lewis, K. A., and Wuttke, D. S. (2012). Telomerase and telomere-associated proteins: structural insights into mechanism and evolution. Structure 20, 28–39.
Li, B., Espinal, A., and Cross, G. A. M. (2005). Trypanosome telomeres are protected by a homologue of mammalian TRF2. Mol. Cell. Biol. 25, 5011–5021.
Li, B., Oestreich, S., and de Lange, T. (2000). Identification of human Rap1: implications for telomere evolution. Cell 101, 471–483.
Li, B., and Lustig, A. J. (1996). A novel mechanism for telomere size control in Saccharomyces cerevisiae. Genes Dev. 10, 1310–1326.
Liu, D., Safari, A., O’Connor, M. S., Chan, D. W., Laegeler, A., Qin, J., et al. (2004). PTOP interacts with POT1 and regulates its localization to telomeres. Nat. Cell Biol. 6, 673–680.
Longtine, M. S., Wilson, N. M., Petracek, M. E., and Berman, J. (1989). A yeast telomere binding activity binds to two related telomere sequence motifs and is indistinguishable from RAP1. Curr. Genet. 16, 225–239.
Luo, K., Vega-Palas, M. A., and Grunstein, M. (2002). Rap1-Sir4 binding independent of other Sir, yKu, or histone interactions initiates the assembly of telomeric heterochromatin in yeast. Genes Dev. 16, 1528–1539.
Mancio-Silva, L., Rojas-Meza, A. P., Vargas, M., Scherf, A., and Hernandez-Rivas, R. (2008). Differential association of Orc1 and Sir2 proteins to telomeric domains in Plasmodium falciparum. J. Cell Sci. 121, 2046–2053.
Marcello, L., and Barry, J. D. (2007). Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 17, 1344–1352.
Martin, S. G., Laroche, T., Suka, N., Grunstein, M., and Gasser, S. M. (1999). Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97, 621–633.
Martino, F., Kueng, S., Robinson, P., Tsai-Pflugfelder, M., van Leeuwen, F., Ziegler, M., et al. (2009). Reconstitution of yeast silent chromatin: multiple contact sites and O-AADPR binding load SIR complexes onto nucleosomes in vitro. Mol. Cell. 33, 323–334.
Matthews, K. R. (2005). The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 118, 283–290.
McCulloch, R., and Barry, J. D. (1999). A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 13, 2875–2888.
McEachern, M. J., and Haber, J. E. (2006). “Telomerase-independent telomere maintenance in yeast,” in Telomeres, eds T. de lange, V. Lundblad, and E. H. Blackburn (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Mehlert, A., Zitzmann, N., Richardson, J. M., Treumann, A., and Ferguson, M. A. J. (1998). The glycosylation of the variant surface glycoproteins and procyclic acidic repetitive proteins of Trypanosoma brucei. Mol. Biochem. Parasitol. 91, 145–152.
Melville, S. E., Leech, V., Navarro, M., and Cross, G. A. M. (2000). The molecular karyotype of the megabase chromosomes of Trypanosoma brucei stock 427. Mol. Biochem. Parasitol. 111, 261–273.
Merrick, C. J., and Duraisingh, M. T. (2007). Plasmodium falciparum Sir2: an unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot. Cell 6, 2081–2091.
Miyake, Y., Nakamura, M., Nabetani, A., Shimamura, S., Tamura, M., Yoneha-ra, S., et al. (2009). RPA-like mamma-lian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell. 36, 193–206.
Miyoshi, T., Kanoh, J., Saito, M., and Ishikawa, F. (2008). Fission yeast Pot1-Tpp1 protects telomeres and regulates telomere length. Science 320, 1341–1344.
Moazed, D., Kistler, A., Axelrod, A., Rine, J., and Johnson, A. D. (1997). Silent information regulator protein complexes in Saccharomyces cerevisiae – a sir2/sir4 complex and evidence for a regulatory domain in sir4 that inhibits its interaction with sir3. Proc. Natl. Acad. Sci. U.S.A. 94, 2186–2191.
Moretti, P., Freeman, K., Coodly, L., and Shore, D. (1994). Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 8, 2257–2269.
Moretti, P., and Shore, D. (2001). Multiple interactions in sir protein recruitment by Rap1p at silencers and telomeres in yeast. Mol. Cell. Biol. 21, 8082–8094.
Myler, P. J., Aline, R. F. Jr., Scholler, J. K., and Stuart, K. D. (1988). Changes in telomere length associated with antigenic variation in Trypanosoma brucei. Mol. Biochem. Parasitol. 29, 243–250.
Nabetani, A., and Ishikawa, F. (2011). Alternative lengthening of telomeres pathway: recombination-mediated telomere maintenance mechanism in human cells. J. Biochem. 149, 5–14.
Navarro, M., and Gull, K. (2001). A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414, 759–763.
Norris, S. J. (2006). Antigenic variation with a twist – the Borrelia story. Mol. Microbiol. 60, 1319–1322.
Park, M. J., Jang, Y. K., Choi, E. S., Kim, H. S., and Park, S. D. (2002). Fission yeast Rap1 homolog is a telomere-specific silencing factor and interacts with Taz1p. Mol. Cells 13, 327–333.
Pays, E., Laurent, M., Delinte, K., Van Meirvenne, N., and Steinert, M. (1983a). Differential size variations between transcriptionally active and inactive telomeres of Trypanosoma brucei. Nucleic Acids Res. 11, 8137–8147.
Pays, E., van Assel, S., Laurent, M., Dero, B., Michiels, F., Kronenberger, P., et al. (1983b). At least two transposed sequences are associated in the expression site of a surface antigen gene in different trypanosome clones. Cell 34, 359–369.
Pedram, M., Sprung, C. N., Gao, Q., Lo, A. W., Reynolds, G. E., and Murnane, J. P. (2006). Telomere position effect and silencing of transgenes near telomeres in the mouse. Mol. Cell. Biol. 26, 1865–1878.
Perez-Toledo, K., Rojas-Meza, A. P., Mancio-Silva, L., Hernandez-Cuevas, N. A., Delgadillo, D. M., Vargas, M., et al. (2009). Plasmodium falciparum heterochromatin protein 1 binds to tri-methylated histone 3 lysine 9 and is linked to mutually exclusive expression of var genes. Nucleic Acids Res. 37, 2596–2606.
Pham, V. P., Qi, C. C., and Gottesdiener, K. M. (1996). A detailed mutational analysis of the VSG gene expression site promoter. Mol. Biochem. Parasitol. 75, 241–254.
Pologe, L. G., and Ravetch, J. V. (1988). Large deletions result from breakage and healing of P. falciparum chromosomes. Cell 55, 869–874.
Proudfoot, C., and McCulloch, R. (2005). Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucleic Acids Res. 33, 6906–6919.
Purser, J. E., and Norris, S. J. (2000). Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U.S.A. 97, 13865–13870.
Riha, K., Heacock, M. L., and Shippen, D. E. (2006). The role of the nonhomologous end-joining DNA double-strand break repair pathway in telomere biology. Annu. Rev. Genet. 40, 237–277.
Roberts, D. J., Craig, A. G., Berendt, A. R., Pinches, R., Nash, G., Marsh, K., et al. (1992). Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature 357, 689–692.
Robinson, N. P., Burman, N., Melville, S. E., and Barry, J. D. (1999). Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol. Cell. Biol. 19, 5839–5846.
Rudenko, G., McCulloch, R., Dirksmulder, A., and Borst, P. (1996). Telomere exchange can be an important mechanism of variant surface glycoprotein gene switching in Trypanosoma brucei. Mol. Biochem. Parasitol. 80, 65–75.
Rudenko, G., and Van der Ploeg, L. H. (1989). Transcription of telomere repeats in protozoa. EMBO J. 8, 2633–2638.
Schwan, T. G., Karstens, R. H., Schrumpf, M. E., and Simpson, W. J. (1991). Changes in antigenic reactivity of Borrelia burgdorferi, the Lyme disease spirochete, during persistent infection in mice. Can. J. Microbiol. 37, 450–454.
Shi, H. F., Djikeng, A., Mark, T., Wirtz, E., Tschudi, C., and Ullu, E. (2000). Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA 6, 1069–1076.
Smith, J. D., Chitnis, C. E., Craig, A. G., Roberts, D. J., Hudson-Taylor, D. E., Peterson, D. S., et al. (1995). Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell 82, 101–110.
Stanne, T. M., Kushwaha, M., Wand, M., Taylor, J. E., and Rudenko, G. (2011). TbISWI regulates multiple polymerase I (Pol I)-transcribed loci and is present at Pol II transcription boundaries in Trypanosoma brucei. Eukaryot. Cell 10, 964–976.
Stanne, T. M., and Rudenko, G. (2010). Active VSG expression sites in Trypanosoma brucei are depleted of nucleosomes. Eukaryot. Cell 9, 136–147.
Steere, A. C., Coburn, J., and Glickstein, L. (2004). The emergence of Lyme disease. J. Clin. Invest. 113, 1093–1101.
Stewart, J. A., Chaiken, M. F., Wang, F., and Price, C. M. (2011). Maintaining the end: roles of telomere proteins in end-protection, telomere replication and length regulation. Mutat. Res. 730, 12–19.
Strahl-Bolsinger, S., Hecht, A., Luo, K., and Grunstein, M. (1997). SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 11, 83–93.
Stringer, J. R. (2005). “Surface antigens,” in Pneumocystis Pneumonia, eds P. D. Walzer, and M. T. Cushion (New York, NY: Marcel Dekker).
Su, X., Heatwole, V. M., Wertheimer, S. P., Guinet, F., Herrfeldt, J. A., Peterson, D. S., et al. (1995). The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 82, 89–100.
Sun, D., Thompson, B., Cathers, B. E., Salazar, M., Kerwin, S. M., Trent, J. O., et al. (1997). Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 40, 2113–2116.
Sunkin, S. M., Linke, M. J., McCormack, F. X., Walzer, P. D., and Stringer, J. R. (1998). Identification of a putative precursor to the major surface glycoprotein of Pneumocystis carinii. Infect. Immun. 66, 741–746.
Sunkin, S. M., and Stringer, J. R. (1997). Residence at the expression site is necessary and sufficient for the transcription of surface antigen genes of Pneumocystis carinii. Mol. Microbiol. 25, 147–160.
Tanny, J. C., Dowd, G. J., Huang, J., Hilz, H., and Moazed, D. (1999). An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99, 735–745.
Tonkin, C. J., Carret, C. K., Duraisingh, M. T., Voss, T. S., Ralph, S. A., Hommel, M., et al. (2009). Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 7, e84. doi: 10.1371/journal.pbio.1000084
van der Ploeg, L. H. T., Liu, A. Y. C., and Borst, P. (1984). Structure of the growing telomeres of trypanosomes. Cell 36, 459–468.
Vanhamme, L., Poelvoorde, P., Pays, A., Tebabi, P., Van Xong, H., and Pays, E. (2000). Differential RNA elongation controls the variant surface glycoprotein gene expression sites of Trypanosoma brucei. Mol. Microbiol. 36, 328–340.
Voss, T. S., Thompson, J. K., Waterkeyn, J., Felger, I., Weiss, N., Cowman, A. F., et al. (2000). Genomic distribution and functional characterisation of two distinct and conserved Plasmodium falciparum var gene 5′ flanking sequences. Mol. Biochem. Parasitol. 107, 103–115.
Wada, M., Sunkin, S. M., Stringer, J. R., and Nakamura, Y. (1995). Antigenic variation by positional control of major surface glycoprotein gene expression in Pneumocystis carinii. J. Infect. Dis. 171, 1563–1568.
Wada, M., and Nakamura, Y. (1996). Unique telomeric expression site of major-surface-glycoprotein genes of Pneumocystis carinii. DNA Res. 3, 55–64.
Wan, M., Qin, J., Songyang, Z., and Liu, D. (2009). OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J. Biol. Chem. 284, 26725–26731.
Wang, Q. P., Kawahara, T., and Horn, D. (2010). Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol. Microbiol. 77, 1237–1245.
Yang, X., Figueiredo, L. M., Espinal, A., Okubo, E., and Li, B. (2009). RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell 137, 99–109.
Ye, J. Z., Hockemeyer, D., Krutchinsky, A. N., Loayza, D., Hooper, S. M., Chait, B. T., et al. (2004). POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 18, 1649–1654.
Zhang, J. R., Hardham, J. M., Barbour, A. G., and Norris, S. J. (1997). Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89, 275–285.
Zhang, J. R., and Norris, S. J. (1998a). Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect. Immun. 66, 3698–3704.
Zhang, J. R., and Norris, S. J. (1998b). Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect. Immun. 66, 3689–3697.
Zomerdijk, J. C. B. M., Kieft, R., Duyndam, M., Shiels, P. G., and Borst, P. (1991). Antigenic variation in Trypanosoma brucei: a telomeric expression site for variant-specific surface glycoprotein genes with novel features. Nucleic Acids Res. 19, 1359–1368.
Keywords: telomere, virulence, microbial pathogen, antigenic variation, infectious diseases
Citation: Li B (2012) Telomere components as potential therapeutic targets for treating microbial pathogen infections. Front. Oncol. 2:156. doi: 10.3389/fonc.2012.00156
Received: 03 August 2012; Accepted: 16 October 2012;
Published online: 01 November 2012.
Edited by:
Claus M. Azzalin, Institute of Biochemistry, ETH Zurich, SwitzerlandSusan M. Bailey, Colorado State University, USA
Copyright: © 2012 Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Bibo Li, Center for Gene Regulation in Health and Disease, Department of Biological, Geological, and Environmental Sciences, Cleveland State University, 2121 Euclid Avenue, Cleveland, OH 44115, USA. e-mail:Yi5saTM3QGNzdW9oaW8uZWR1