



- 1Molecular Chemoprevention Group, Molecular Medicine Area, Regina Elena National Cancer Institute, Rome, Italy
- 2Translational Oncogenomic Unit, Molecular Medicine Area, Regina Elena National Cancer Institute, Rome, Italy
- 3Weis Center for Research, Geisinger Clinic, Danville, PA, USA
- 4Mount Sinai School of Medicine, New York, NY, USA
Growth arrest, inhibition of cell proliferation, apoptosis, senescence, and differentiation are the most characterized effects of a given tumor suppressor response. It is becoming increasingly clear that tumor suppression results from the integrated and synergistic activities of different pathways. This implies that tumor suppression includes linear, as well as lateral, crosstalk signaling. The latter may happen through the concomitant involvement of common nodal proteins. Here, we discuss the role of Promyelocytic leukemia protein (PML) in functional cross-talks with the HIPPO and the p53 family tumor suppressor pathways. PML, in addition to its own anti-tumor activity, contributes to the assembly of an integrated and superior network that may be necessary for the maximization of the tumor suppressor response to diverse oncogenic insults.
The HIPPO Pathway
First identified in Drosophila, the Hippo (Hpo) signaling pathway is a conserved string of molecular events, which regulates the proper size of organs through the balance of cell growth and cell death. Cell density and contact inhibition information are conveyed by membrane complexes through the Hpo pathway to specific transcriptional programs in the nucleus. In contact-inhibited cells, the Hpo pathway is activated, whereas in sparsely populated cells, it is inhibited. The pathway is characterized by a core kinase, Hpo (Mst1 and Mst2 in mammals), and its downstream effector, Warts (Wts) kinase (Lats1 and Lats2 in mammals), which regulate Yorkie (Yki), protein (YAP and TAZ in mammals), a transcriptional co-activator for transcription factors involved in the induction of cell proliferation, survival, and apoptosis (Figures 1A,B).
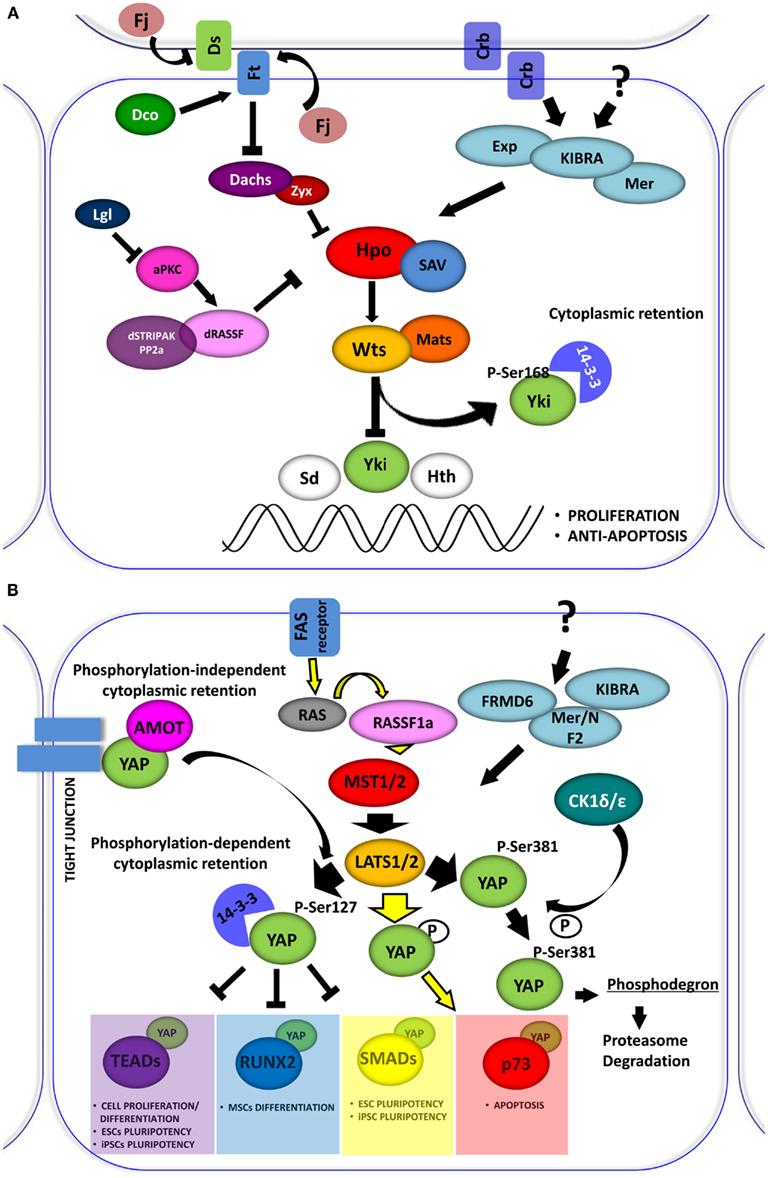
Figure 1. (A) The Drosophila Hippo pathway. Signaling diagram of Hippo (Hpo) kinases cascade and of its modulation by apical transmembrane protein complexes and proteins involved in cell polarity control (Arrowed or blunted ends indicate activation or inhibition, respectively). (B) Role of YAP in the Hpo-like pathway in human. In mammals the relationships between Drosophila Hpo and Wts, are conserved in Mst1/2 (Hpo homologs) and Lats1/2 (Wts homolog). Lats1/2 phosphorylates YAP on different conserved motifs. Phosphorylation-dependent 14-3-3 binding and cytoplasmic retention are conserved in YAP, which inhibits it to enhance the transcriptional activation of pro-proliferation genes. Depending on the cellular context Lats1/2 phosphorylates YAP, increasing its transcriptional support to p73 to induce apoptosis. YAP can also be phosphorylated by other kinase, such as CDK1δ/ε promoting its subsequent protein proteasome-degradation. The retention of YAP at cytoplasmic level can also be exerted by sequestering the protein at the junctional level through the interaction with the AMOT protein.
As the Hpo pathway plays an important role in organ size control. Loss of function of many individual members of the pathway leads to tumorigenesis. Accordingly, down-regulation of this pathway is frequently associated with human cancers. In addition, the Hpo pathway has been implicated in tissue homeostasis through the regulation of stem cells, cell differentiation, and tissue regeneration.
The Drosophila Hippo Pathway
The “blueprint” for the Hpo pathway emerged from genetic screens of Drosophila, Wts was the first identified Hpo pathway member (Justice et al., 1995), encoding a kinase of the Nuclear Dbf-2-related (NDR) family. Several years later, four tumor suppressors, named Hpo, Wts, Salvador (Sav), and Mats were implicated in the regulation of organ size in flies (Figure 1A). These proteins constitute the core kinase cassette of the pathway, whose products can affect proliferation without increasing susceptibility to apoptosis (Justice et al., 1995; Tapon et al., 2002; Wu et al., 2003; Lai et al., 2005). Wts activity, which is fundamental for the phosphorylation-dependent regulation of Yki (Huang et al., 2005; Oh and Irvine, 2008), is regulated through a series of phosphorylation events. Wts is directly phosphorylated by Hpo and this phosphorylation is facilitated by Sav protein (Tapon et al., 2002; Wu et al., 2003). Hpo is a member of the Sterile-20 family of Ser/Thr kinases. It interacts with and phosphorylates Sav, which in turn activates Hpo kinase function. Mob as tumor suppressor (MATS) is similar to Sav and acts as an adaptor protein. Mats also belongs to the NDR family and binds Wts, potentiating its intrinsic activity (Lai et al., 2005). Hpo phosphorylates Mats, increasing its binding affinity to Wts and potentiating Wts kinase activity (Praskova et al., 2008).
Located upstream of the Hpo pathway are Merlin (Mer) and Expanded (Ex; Boedigheimer et al., 1993; LaJeunesse et al., 1998; Hamaratoglu et al., 2006). These are considered tumor suppressors that cooperate with each other to control organ growth. Kibra, a newly discovered member of the Hpo pathway, is involved in formation of a Kibra-Mer-Ex, which constitutes an apical protein complex required for the association of the Hpo pathway with cellular membranes (Genevet et al., 2010; Yu et al., 2010). Crumbs (Crb) is a transmembrane protein involved in determining cell polarity. The loss of Crb expression was shown to further determine a phenotype characterized by overgrowth (Chen et al., 2010; Ling et al., 2010; Robinson et al., 2010). It is likely that Crb is one of the membrane receptors that regulate the Hpo pathway. Other proteins involved in cell polarity control have been linked to the Hpo pathway as well, including: Scribble (Scrib), Disk large (Dlg), Lethal giant larvae (Lgl), and a typical protein C (aPKC; Chen et al., 2010; Grzeschik et al., 2010). The atypical cadherin FAT (Ft) was the first transmembrane protein shown to affect Hpo signaling and was the first tumor suppressor gene isolated in Drosophila. Complete knock-out of the Ft protein induced cell death in Drosophila larvae with overgrown imaginal disks (Mahoney et al., 1991). Many lines of evidence suggest that the principal mechanism exerted by Ft is on the Wts function (Feng and Irvine, 2007, 2009). Many Ft partners were identified as potential modulators of the Hpo Pathway: (i) Dachsous (Ds), an atypical cadherin which binds to Ft (Matakatsu and Blair, 2004, 2006); (ii) Four-jointed (Fj), a kinase that typically localizes to the Golgi subcellular compartment (Ishikawa et al., 2008); (iii) a Casein I kinase, termed Dco (Disks overgrown), responsible for Ft phosphorylation in its cytoplasmatic segment (Cho et al., 2006; Feng and Irvine, 2009); and (iv) Dachs, which is an unconventional myosin that antagonizes Ft (Cho et al., 2006). All of these components are believed to be responsible for linking Hpo to extracellular stimuli (Harvey and Tapon, 2007).
Yorkie, a critical effector of the Hpo pathway, is not a direct transcriptional factor but is a potent transcriptional co-activator cooperating with different DNA binding proteins. Wts phosphorylates Yki at Ser 168, thus creating a binding site for 14-3-3 proteins, which in turn sequester Yki in the cytoplasm and prevent its nuclear import (Dong et al., 2007b; Oh and Irvine, 2008). Loss of Hpo signaling, as well as mutations in the 14-3-3 binding site for Yki, were shown to produce strong nuclear accumulation coupled with enhanced activity of Yki (Zhao et al., 2007). Interestingly, some binding partners of Yki are the same kinases that function upstream to it in the Hpo pathway (e.g., Ex, Wts, and Hpo; Badouel et al., 2009; Oh et al., 2009). Other partners are represented by transcription factors that govern different classes of genes. One class includes genes involved in cell survival and proliferation: scalloped (Sd), a member of TEAD/TEFs family (Wu et al., 2008; Zhang et al., 2008), and Homothorax (Hth), both of which promote cell proliferation (Peng et al., 2009). Sd, together with Yki, up-regulate transcription of Diap1 gene, an apoptosis inhibitor (Wu et al., 2008). Hth is involved in regulating the transcription of another Yki target, the growth promoting microRNA gene, bantam. Other Yki targets in this class are the cell-cycle regulators Cyc E, E2F1, and dMyc (Drosophila Myc; Goulev et al., 2008; Peng et al., 2009; Neto-Silva et al., 2010). The second class of genes are components of other signaling pathways including Notch, Wnt, EGFR, and Jak-Stat pathways. For example, Smad proteins are functional partners of Yki, potentiating the transcriptional response to BMP/TGF-β signaling (Alarcon et al., 2009). Finally, a third class of Yki targets consists of several proteins from its own Hpo cascade, including Ex, Mer, Kibra, Crb, and Fj (Cho et al., 2006; Hamaratoglu et al., 2006; Genevet et al., 2009, 2010).
The Mammalian Hippo Pathway
The Hpo pathway is evolutionarily conserved in mammals (Figure 1B). It was reported that loss of function in mutant flies can be rescued by expressing their respective human counterparts (Wu et al., 2003; Lai et al., 2005). One ortholog for the adaptor protein Sav, termed WW45 or SAV1, and two orthologs for Mats, termed MOBKL1A and MOBKL1B have been identified. These proteins form a conserved kinase cassette that phosphorylates and inactivates the mammalian Yki homologs, YAP and TAZ, preventing their nuclear translocation and consequently attenuating the transcription of genes involved in proliferation and differentiation (Huang et al., 2005; Zhao et al., 2007; Lei et al., 2008).
MST1/2 are serine-threonine kinases, which are able to initiate apoptosis when overexpressed (Lin et al., 2002; Ura et al., 2007). MSTs become activated by auto-phosphorylation on threonine residues within their activation loop domain. The inhibition of dimerization and auto-phosphorylation of MST2 exerted by the proto-oncogene serine/threonine protein kinase RAF1 was reported (O’Neill et al., 2004). In this latter context, expression of the scaffold protein RASSF1A is able to release MST2 from this inhibition, thus inducing apoptosis (Matallanas et al., 2007). Moreover, it was shown that PP2A phosphatase dephosphorylates MST1/2 kinases, thus inhibiting their function (Ribeiro et al., 2010). MST substrates include LATS and MOB1. LATS1/2 kinases control cellular homeostasis, negatively regulating cell division cycle 2 (CDC2) and favoring G2/M arrest (Yang et al., 2001; Xia et al., 2002; Yabuta et al., 2007). LATS2 was also reported to induce G1/S arrest (Li et al., 2003). The loss of LATS1/2 was shown to lead to a broad variety of tumors, such as soft tissue sarcoma and leukemia (St John et al., 1999). These proteins are strong tumor suppressors. LATS activity is supported by MOB1, which binds to and activates LATS kinases, favoring YAP and TAZ proto-oncogene phosphorylation and inhibiting their nuclear activity.
The mammalian genome contains orthologs for most of the reported upstream regulators of the Hpo pathway in flies. Notably, it encodes more than one paralog for most of the Drosophila Hpo components. Two homologs were identified for Kibra: KIBRA/WWC1 and WWC2, as well as for Ex: FRMD6 and FRMD1, while only one homologous was identified for Mer: NF2. Paralogs for MST and LATS kinases are well characterized and Yki orthologs, YAP and TAZ exhibit similar function in regulating signals related to cell growth or apotosis. The presence of conserved Drosophila orthologs in the mammalian Hpo pathway may very well serve as a mechanism of redundancy that protects the organism against cancer-causing mutations.
YAP: A Critical Component of the Hippo Pathway
Yes-associated protein (YAP) was identified as Yes (and Src) kinase-interacting protein (Sudol, 1994; Sudol et al., 1995). The complex between YAP and Yes was recently validated functionally in colon cancer cells by the Bill Hahn laboratory (Rosenbluh et al., 2012). Originally, YAP was shown as a transcriptional co-activator that binds the PPxY motif in Runx1 (or AML1 for acute myeloid leukemia 1; Chen and Sudol, 1995; Yagi et al., 1999). Soon after, it was reported that YAP, like its paralog TAZ, binds 14-3-3 in a phospho-dependent manner (Kanai et al., 2000) and YAP plays a role as a co-activator for the TEAD/TEF family of transcription factors (Vassilev et al., 2001; Basu et al., 2003). The conserved Hpo pathway kinase cassette triggers the phosphorylation and inactivation of YAP, preventing its nuclear import and consequently attenuating the transcription of genes regulating proliferation (Huang et al., 2005; Zhao et al., 2007; Lei et al., 2008).
At least eight different splicing isoforms of YAP exist (Komuro et al., 2003; Gaffney et al., 2012). YAP orthologs in lower organisms, such as Drosophila, have tandem WW domains, indicating that the double WW domain organization is conserved from the fly to humans. The WW is a small modular protein domain that has two conserved Tryptophan (W) residues (Bork and Sudol, 1994; Sudol et al., 1995). The WW domain shares a similar function with the SH3 domain, able to bind short peptides that are Proline-rich. Initially, the WW domain was shown to bind to Proline-rich peptides terminated with Tyrosine (Y), so called PY motifs (Chen and Sudol, 1995; Einbond and Sudol, 1996; Sudol and Hunter, 2000). YAP has been found to interact with various proteins of diverse functions and the majority of these interactions occur through the YAPs WW domains (reviewed in Bertini et al., 2009; Sudol, 2010, 2012; Sudol and Harvey, 2010). Apart from WW domains, the modular structure of YAP contains a TEAD factor-binding domain (TB), which is located at the amino-terminal portion of the protein (Vassilev et al., 2001), a carboxy-terminal SH3 binding region, transcriptional activation domain and PDZ-binding motif, which is critical for nuclear translocation and indispensable for binding the PDZ motif of ZO-1 and ZO-2 (Oka and Sudol, 2009; Oka et al., 2010). None of the signaling pathways known is as rich in WW domain-containing proteins as the Hpo pathway (Sudol and Harvey, 2010). The intact WW domains of YAP are required for their co-transcriptional activator function and promoting cell proliferation (Zhao et al., 2009).
The activation of the Hpo pathway triggers the inhibition of YAP activity (Figure 1B). YAP is phosphorylated by LATS on different serine residues, among them, serine 127 appears to be the most critical for its function (Basu et al., 2003). This phosphorylation generates a 14-3-3 binding site on YAP, which induces its cytoplasmic retention, thus inhibiting its co-transcriptional activity (Basu et al., 2003; Zhao et al., 2007; Hao et al., 2008). YAP is also phosphorylated by LATS on Ser 381 (Zhao et al., 2010). The Ser 381 modification primes YAP to subsequent CK1δ/ε phosphorylation and degradation via the SCF (beta-TRCP) E3 ubiquitin ligase complex.
Hippo pathway activation is also triggered by cell–cell contacts. High-cell density induces YAP phosphorylation and cytoplasmic retention (Zhao et al., 2007) and the disruption of cell–cell junctions in epithelial tissue, induces nuclear translocation of YAP and TAZ (Varelas et al., 2010).
There are phosphorylation-independent mechanisms through which YAP could be sequestered in the cytoplasm. It was observed that YAP was able to interact with the angiomotin (AMOT) family of proteins. This interaction occurs via the WW domains of YAP and sequesters the protein in the cytoplasm and junctional complexes (Chan et al., 2011; Wang et al., 2011; Zhao et al., 2011). The existence of a tripartite complex between AmotL1, YAP, and ZO-2, which regulates YAP function, apparently in opposite directions, was highlighted recently (Oka et al., 2012). The cytoplasmic retention of YAP, exerted by AmotL1, disabled YAP in its nuclear function, whereas ZO-2 enhanced the nuclear translocation of YAP (Oka et al., 2012).
Yes-associated protein serves as a co-regulator for various transcriptional factors, including members of the TEAD family factors, Smad proteins, RUNX, Erb4, and p73 (reviewed in (Sudol, 2010; Mauviel et al., 2012). Among these targets, only the interaction with the TEFs/TAEDs appears to be conserved from flies to humans (Vassilev et al., 2001; Wu et al., 2001; Mahoney et al., 2005; Sawada et al., 2008). Two major target genes for YAP-TEADs complexes were described in mammals: CTGF and Cyr61 (Lai et al., 2011). CTFG is a direct target of YAP-TEADs complex and was shown to be critical for YAP-induced proliferation and anchorage-independent growth. Another YAP target is Myc. Similar to the Dropsophila Yki-Sd effect on dMyc expression (Neto-Silva et al., 2010), YAP was shown to up-regulate the expression of Myc gene in transgenic mouse liver (Dong et al., 2007a). Therefore, it is not surprising that the targeted disruption of the transcriptional complex of YAP-TEADs by genetic approaches or pharmacological compounds is the main focus of drug discovery efforts aimed at cancers with amplified YAP oncogene (Liu-Chittenden et al., 2012; Sudol et al., 2012).
As the transcriptional co-activator of SMAD proteins, YAP seems to also be involved in embryonic stem cell (ESC) pluripotency. The maintenance of this ability by ESC is governed by coordinating multiple pathways, including TGFβ and BMP pathways. Both YAP and TAZ were shown to be critical mediators of both these pathways. YAP interacts physically with SMADs, driven by TGFβ and BMP toward their transcriptional function (Varelas et al., 2008, 2010; Alarcon et al., 2009; Sudol, 2012). The ESC pluripotency is described as the ability to give rise to all types of cells and tissues maintained in adults. Thus, differentiated cells can also be reprogrammed in ESC-like cells, termed induced pluripotent stem cells (iPSC), by activating a given set of transcription factors. iPSC cells can then self-renew and differentiate again in every type of cell and tissue. This ability is due to the activity of distinct transcription factors such as KLF4, cMYC, OCT4, and SOX2 (Takahashi and Yamanaka, 2006). SMADs proteins were shown to co-occupy the same genomic site of OCT4 and SOX2 in human ESC (Mullen et al., 2011). Moreover, YAP was shown to favor, possibly on a transcriptional level, reprogramming mouse embryonic fibroblasts in an iPSC-like state (Lian et al., 2010). The majority of genomic sites for ESCs transcription factors also contain a consensus for TEF/TEADs transcriptional binding factors, suggesting that YAP could be implicated in this mechanism also through its interaction with TEADs. Interestingly, it was observed that simultaneous knocking down of TEAD1, TEAD3 and TEAD4 inhibits the ESC pluripotency in mice (Lian et al., 2010).
Many lines of evidence indicate that the Hpo pathway may play an important role also in mesenchymal stem cells (MSC), which are able to differentiate into osteoblasts, adipocytes, chondrocytes, and myoblasts. Consistent with this observation is the discovery of another important YAP transcriptional partner, RUNX2, which is essential for skeletal mineralization because it stimulates osteoblast differentiation of MSC, promotes chondrocyte hypertrophy, and contributes to endothelial cell migration and vascular invasion in bone development. Like other RUNT-domain proteins, RUNX2 is a context-dependent transcriptional activator and repressor of genes that regulate cellular proliferation and differentiation. YAP was shown to interact with full-length RUNX2 in osseous cells via co-immunoprecipitation of endogenous proteins and co-immunofluorescence (Zaidi et al., 2004). RUNX2 recruits YAP to subnuclear foci and to the osteocalcin gene promoter, but does not affect its nucleo-cytoplasmic shuffling. The signature Y residue in the PPPYP motif of RUNX2 is essential for interacting with YAP (Chen and Sudol, 1995; Yagi et al., 1999). YAP-mediated repression of RUNX2 activity on the osteocalcin promoter is cell-type independent. YAP-mediated repression of RUNX2 instead seems to be dependent on the promoter context: YAP blocked RUNX2-dependent activation of TGFβR1 promoter and enhanced RUNX2-dependent repression of its own promoter; however it did not affect RUNX2 transcriptional effects on the p6OSE2 or p21 promoters. These data indicate that RUNX2 can recruit YAP to promoter regions, but the effects of YAP on the expression of RUNX2 target genes is dependent on the cohort of other DNA binding proteins and co-factors brought to the gene by specific DNA sequences and protein–protein interactions. Transcriptional repression of RUNX2 by YAP depends on Src-induced activation and tyrosine phosphorylation of YAP (Zaidi et al., 2004). Dominant-negative Src and YAP proteins, as well as Src kinase inhibitors, increased RUNX2 transcriptional activation of the osteocalcin promoter in ROS 17/2.8 cells (Zaidi et al., 2004). The idea that YAP could support RUNX2 function as co-activator has been confirmed by a recent study in which authors observed that a nuclear localized mutant form of YAP can induce osteogenesis. It was observed that YAP is part of a signaling complexes that forms its response to mechanical signals exerted by extracellular matrix (ECM) rigidity and cell shape. This regulation requires Rho GTPase activity and tension of the actomyosin cytoskeleton, but is independent of the canonical Hpo pathway. YAP is required for differentiation of MSC induced by ECM stiffness and for survival of endothelial cells regulated by cell geometry (Dupont et al., 2011).
PML and p53: A Close Link in Tumor Suppression
In humans, a complete or partial loss of PML expression has been observed in multiple types of cancers, including breast, colon, and prostate (Gurrieri et al., 2004a,b). PML KO mice develop normally, but are resistant to lethal doses of γ-IR. In addition, they are prone to tumorigenesis in response to carcinogens, or an additional oncogenic event, such as the loss of PTEN (Wang et al., 1998; Trotman et al., 2006). PML is a key factor in the formation of PML nuclear bodies (PML-NBs), which are distinct nuclear multi-protein complexes that have been associated with critical cellular processes, including tumor suppression, gene regulation, post-translational modifications, and protein catabolism. The stabilization of PML protein levels leads to recruitment of p53 to the PML-NBs, an event that facilitates p53 acetylation and transcriptional activation (Pearson et al., 2000; Bernardi and Pandolfi, 2007). It was demonstrated that in conditions of oncogenic stress, PML is a p53 transcriptional target gene (de Stanchina et al., 2004). PML contains three putative p53 responsive elements (REs) in its promoter region (de Stanchina et al., 2004). p53 is able to induce PML protein as well as PML mRNA, and to increase the number and size of PML-NBs. PML also contributes to cellular p53-dependent processes like senescence, cell-cycle arrest, and p53-mediated apoptosis, and additionally PML arranges p53 tumor suppressor functions (de Stanchina et al., 2004). Recently, it has been reported that PML expression is also regulated at the translational level during oncogenic K-RAS-induced OIS in a p53-independent manner (Scaglioni et al., 2012). The authors show that mTOR-dependent translational control mechanisms are important in modulating PML protein levels during oncogenic K-RAS-induced OIS and that the PML 5′ untranslated mRNA region plays a key role in mediating PML protein upregulation and OIS induction (Scaglioni et al., 2012). Haupt and colleagues found that PML interacts and co-localizes with mutant p53. PML activates mutant p53 transcriptional activity and is important for its gain of function in cultured human cancer cells. These findings support the notion that as is the case for wtp53, PML is a key regulator of mutant p53 (Haupt et al., 2009).
p73/YAP and PML/YAP: Two Cooperative Pro-Apoptotic Protein Complexes
p53 protein is known as the “guardian of the genome” and is the focus of studies in understanding tumorigenesis. Another member of the p53 family, p73, was recently discovered for its pivotal role in DNA damage signaling. The main activities of the p53 family occur through the transcriptional activation or repression of target genes that encode key proteins involved in cell growth inhibition, apoptosis, senescence, and differentiation (Vousden and Lu, 2002). However, the members of the family differ in their upstream regulation by different kinases. The YAP is a critical mediator of p73 function. It binds p73 to regulate its transcriptional activity (Strano et al., 2001) and subsequent cell death induction (Basu et al., 2003). This binding is negatively regulated by Akt-mediated YAP phosphorylation (Basu et al., 2003; Figure 2) and enhanced by DNA damage (Strano et al., 2005). Furthermore, YAP stabilizes p73 protein in a post-translational way by competing with the ITCH E3-ligase for binding to p73 (Levy et al., 2007) and inducing its transcriptional activity via the p300 acteyltansferase (Strano et al., 2005). Rossi et al. (2005) have shown that Itch, a human ubiquitin protein ligase that belongs to the Nedd4-like E3 family containing a WW domain, binds, and ubiquitinates p73 and determines its rapid proteosome-dependent degradation.
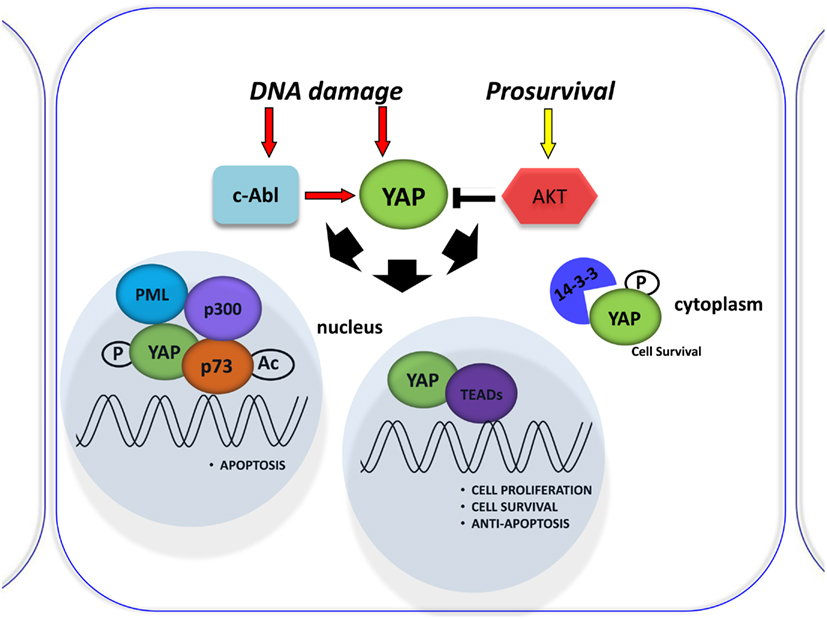
Figure 2. Involvement of YAP in the DNA damage response. Schematic representation of the DNA damage response induced by Cisplatin (CDDP) treatment. It was shown to signal c-Abl-mediated YAP phosphorylation, resulting in YAP nuclear localization and increased p73 binding and activation of pro-apoptotic genes. By contrast, active Akt counters this effect by phosphorylating YAP and sequestering it at the cytoplasmic level.
The promyelocytic leukemia protein (PML) has been shown to bind p73 in the nuclear bodies (NBs) to promote p300-mediated acetylation of p73 and to regulate its transcriptional activity (Bernassola et al., 2004). PML is the key component of PML-NBs that regulate a large number of cellular processes by facilitating post-translational modification of target proteins, promoting protein–protein contacts, or by sequestering proteins. It functions as a tumor suppressor, required for normal, caspase-dependent apoptosis in response to DNA damage, FAS, TNF, or interferons. It plays a role in transcription regulation, DNA damage response, DNA repair, and chromatin organization (Salomoni and Pandolfi, 2002; Bernardi and Pandolfi, 2007; Alsheich-Bartok et al., 2008). Therefore, Blandino’s group described the PML tumor suppressor protein as a mediator of the YAP-p73 complex (Strano et al., 2005). Furthermore, they demonstrated that PML was a direct transcriptional target of p73/YAP, showing that PML transcriptional activation by p73/YAP is under the negative control of the proto-oncogenic Akt/PKB kinase (Figure 2). Importantly, PML and YAP physically interact through their PVPVY and WW domains. This direct interaction provides part of the mechanism by which PML can enhance the activity of the YAP-p73 complex (Lapi et al., 2008). Binding assays and site-specific mutagenesis have shown that the PY “XPPXY” motif consensus binds with relatively high affinity to the WW domain of YAP (Chen and Sudol, 1995; Sudol et al., 1995). The PVPVY motif of PML is not a canonical consensus sequence for YAP WW domain binding but the VPxY peptide, along with LPxY, SPxYs peptides were shown to be exceptions that were able to bind to YAP WW domain (Chen and Sudol, 1995; Chen et al., 1997). This evidence may be important in suggesting the possibility of interactions of YAP with a plethora of proteins, which contain non-canonical WW domain binding sequences and participating in various cellular and metabolic pathways. Together with the specificity of the cellular contexts in which YAP binds to its canonical or non-canonical partners, these observations may also explain the dual role of YAP as both an oncogene and tumor suppressor.
It has also been demonstrated that PML contains a SUMO binding motif that is independent of its sumoylation sites necessary for PML-NBs formation (Shen et al., 2006). According to a large number of proteins associated with the PML, NBs are sumoylated, and/or contain SUMO binding motifs. This suggests the possibility that these proteins are recruited to the PML networks through non-covalent interactions mediated by covalently bound SUMO and SUMO binding motifs present in PML. Furthermore, YAP was identified as a target for PML-mediated sumoylation that blocks YAP polyubiquitylation and its subsequent degradation. This could also be important in mediating the recruitment of YAP/p73 complex to the PML-NBs (Lapi et al., 2008). In fact, the stabilized YAP in the protein complex with p73, is then recruited to PML-NBs to achieve full transcriptional activation of p73. The spatial and temporal kinetics of the recruitment and competition for binding must be elucidated to have a more complete picture of this dynamic complex. All these views contribute to the idea that YAP/PML complex could act as a bridge at the crossroads of many signaling pathways, where the actual biological read-out might lay in the cellular context of binding partners, including cell-specific transcription factors or effectors (apoptosis, DNA damage response, senescence, cancerogenesis). In any case, it is important to stress the importance of post-translational modifications in the regulation of YAP activity. Phosphorylation is the most common described modification of YAP, but as demonstrated by Lapi and colleagues, different afferent signals may modulate the choice of the partners during physiological cell life and the onset of cancer.
The Transcriptional Axis YAP/PML/p73 in DNA Damage-Induced Apoptosis
The regulation of p73 activity by YAP has mainly been investigated in the field of DNA damage signaling. As an activating cofactor for a pro-apoptotic transcription factor, one line of thinking has assumed that YAP plays a tumor suppressor role in cancer and cell culture models (Oka et al., 2008; Yuan et al., 2008). However, YAP has also been clearly identified as an oncogene and cell size regulator in both Drosophila m. and mammalian cells (Dong et al., 2007b; Zhao et al., 2007; Cordenonsi et al., 2011; Halder et al., 2012).
Lapi et al. (2008) demonstrated that active Akt counters cisplatin-induced increases in PML transcription via the YAP-p73 complex. Cisplatin (CDDP) was shown to signal c-Abl-mediated YAP phosphorylation, resulting in YAP nuclear localization and increased p73 binding and activation of pro-apoptotic genes (Levy et al., 2008; Figure 2). Those investigating the role of YAP phosphorylation events downstream of cisplatin have so far focused on its subcellular localization for its effect on p73 binding, but perhaps different phosphorylations can affect YAP stability (Levy et al., 2007). However, the molecular mechanism was not described. The upregulation of YAP protein, upon CDDP treatment, is not due to transcriptional regulation, but is a post-translational event that correlates with an increase in YAP sumoylation mediated by PML, since it is completely abrogated in PML/MEFs. Elevation of YAP levels in response to DNA damage demonstrates another level of regulation of this pathway, suggesting that p73 activation must be tightly controlled to ensure quick and efficient activation of p73 target genes in response to stress conditions (Lapi et al., 2008).
The last findings demonstrate the existence of a positive regulatory loop between the p73/YAP protein complex and PML during apoptosis triggered by CDDP in HCT116 cells (Lapi et al., 2008).
In fact PML, YAP, and p73 can be recruited on Bax and p53AIP1 apoptotic gene promoters, which contain p73 binding sites within their promoter regions, in response to CDDP and PML binds to its own promoter and first intron where YAP and p73 were also recruited, and promotes its own transcriptional activation (Figure 3).
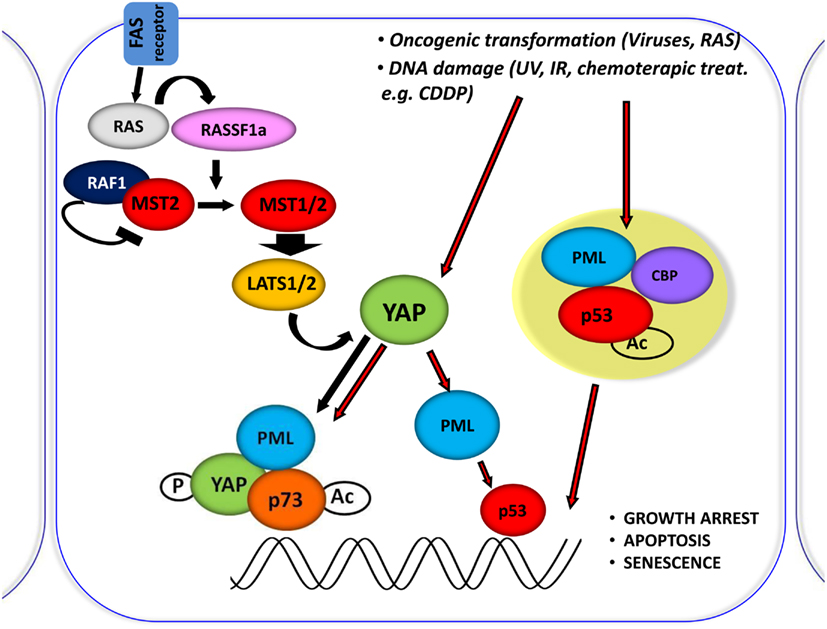
Figure 3. Central role of YAP in proliferation inhibiting pathways. Schematic representation of the central role of YAP in inducing the inhibition of proliferation, by integrating and mediating different upstream stimuli. It was shown that RASSF1A disrupts the inhibitory complex between RAF1 and MST2 and favors the physical association between MST2 and LATS1 concomitantly; therefore, leading to YAP1 phosphorylation and nuclear re-localization where it binds to p73 and potentiates its apoptotic activity. Oncogenic transformation as well as DNA damage (achieved by CDDP treatment) leads to YAP accumulation and to its functional activation.
It has been previously demonstrated that YAP requires PML and NBs localization to coactivate p73 (Strano et al., 2005). We describe that p73 and YAP are required not only for the transcriptional activation of PML during the apoptotic response but also for the subsequent accumulation of PML protein and formation of NBs. As a result, PML can contribute to the p73-dependent apoptotic response by promoting both p300-mediated acetylation of p73 (Bernassola et al., 2004) and stabilizing YAP by inhibiting its ubiquitin-mediated degradation.
Whether YAP-p73 regulation of PML gene transcription or PML regulation of the YAP-p73 complex is signaled by other stresses besides cisplatin, needs to be addressed in the future.
Based on what we have previously described, YAP is not a bona-fide tumor suppressor, but a transcriptional co-activator that impinges directly or indirectly on different tumor suppressor pathways, here represented by p53 family, PML, and HIPPO (Figure 3). As for transcription factors with tumor suppressor function, YAP could also reinforce either by binding directly or indirectly the activity of transcription factors whose gene targets encode for oncogenic proteins. Further studies of when and how YAP is stabilized and to which transcription factor complexes it is recruited will help us understand the paradoxical activities of YAP in DNA damage and tissue growth, as well as its contradictory roles in tumorigenesis in different tissues.
Concluding Remarks
Our work has revealed a direct crosstalk between PML and YAP, one of the two main effectors of the Hpo tumor suppressor pathway. This finding has several important ramifications for our understanding of the molecular mechanism of cancer. A good example comes from a recent study of glioblastoma multiforme (GBM), one of the most aggressive human tumors (Okazaki et al., 2012). The authors showed that the level of PML protein in human glioma tissue decreased as the degree of malignancy increased. A complete loss of PML was observed in 11.1% of GBM biopsies. The report described solid evidence that after the treatment of TMZ (temozolomide)-resistant glioma cells with both interferon-β (IFN-β) and TMZ, the expression of the endogenous PML gene and its associated protein increased. This effect was primarily attributable to IFN-β, which activates and translocates p73/YAP complex into the nucleus for interaction with PML to induce glioma cell apoptosis, explaining in part the observed in vivo anti-tumor effects. The knowledge of precise mechanisms underlying the activation of the endogenous PML/p73/YAP axis is essential to induce the apoptosis of specific cancer cells, in addition to the DNA damage inflicted by the combination therapy.
We hope that the unexpected finding of “PML surfing into the YAP-Hpo tumor suppressor pathway” will allow us to better understand the entire Hpo signaling network, and to design systems biology approaches to fight cancers caused by the genetic lesions that map within, and affect the network.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues Tania Merlino and Virginia Mazack for valuable comments on the first version of the manuscript. This work was supported by AIRC grant (11/30/R/42; to Giovanni Blandino) and PA Breast Cancer Coalition Grants (#60707 an #920093) plus the Geisinger Clinic (to Marius Sudol).
References
Alarcon, C., Zaromytidou, A. I., Xi, Q., Gao, S., Yu, J., Fujisawa, S., et al. (2009). Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 139, 757–769.
Alsheich-Bartok, O., Haupt, S., Alkalay-Snir, I., Saito, S., Appella, E., and Haupt, Y. (2008). PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene 27, 3653–3661.
Badouel, C., Gardano, L., Amin, N., Garg, A., Rosenfeld, R., Le Bihan, T., et al. (2009). The FERM-domain protein expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev. Cell 16, 411–420.
Basu, S., Totty, N. F., Irwin, M. S., Sudol, M., and Downward, J. (2003). Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 11, 11–23.
Bernardi, R., and Pandolfi, P. P. (2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8, 1006–1016.
Bernassola, F., Salomoni, P., Oberst, A., Di Como, C. J., Pagano, M., Melino, G., et al. (2004). Ubiquitin-dependent degradation of p73 is inhibited by PML. J. Exp. Med. 199, 1545–1557.
Bertini, E., Oka, T., Sudol, M., Strano, S., and Blandino, G. (2009). YAP: at the crossroad between transformation and tumor suppression. Cell Cycle 8, 49–57.
Boedigheimer, M., Bryant, P., and Laughon, A. (1993). Expanded, a negative regulator of cell proliferation in Drosophila, shows homology to the NF2 tumor suppressor. Mech. Dev. 44, 83–84.
Bork, P., and Sudol, M. (1994). The WW domain: a signalling site in dystrophin? Trends Biochem. Sci. 19, 531–533.
Chan, S. W., Lim, C. J., Chong, Y. F., Pobbati, A. V., Huang, C., and Hong, W. (2011). Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J. Biol. Chem. 286, 7018–7026.
Chen, C. L., Gajewski, K. M., Hamaratoglu, F., Bossuyt, W., Sansores-Garcia, L., Tao, C., et al. (2010). The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 107, 15810–15815.
Chen, H. I., Einbond, A., Kwak, S. J., Linn, H., Koepf, E., Peterson, S., et al. (1997). Characterization of the WW domain of human yes-associated protein and its polyproline-containing ligands. J. Biol. Chem. 272, 17070–17077.
Chen, H. I., and Sudol, M. (1995). The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. U.S.A. 92, 7819–7823.
Cho, E., Feng, Y., Rauskolb, C., Maitra, S., Fehon, R., and Irvine, K. D. (2006). Delineation of a Fat tumor suppressor pathway. Nat. Genet. 38, 1142–1150.
Cordenonsi, M., Zanconato, F., Azzolin, L., Forcato, M., Rosato, A., Frasson, C., et al. (2011). The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772.
de Stanchina, E., Querido, E., Narita, M., Davuluri, R. V., Pandolfi, P. P., Ferbeyre, G., et al. (2004). PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 13, 523–535.
Dong, J., Feldmann, G., Huang, J., Wu, S., Zhang, N., Comerford, S. A., et al. (2007a). Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133.
Dong, S., Kang, S., Gu, T. L., Kardar, S., Fu, H., Lonial, S., et al. (2007b). 14-3-3 Integrates prosurvival signals mediated by the AKT and MAPK pathways in ZNF198-FGFR1-transformed hematopoietic cells. Blood 110, 360–369.
Dupont, S., Morsut, L., Aragona, M., Enzo, E., Giulitti, S., Cordenonsi, M., et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183.
Einbond, A., and Sudol, M. (1996). Towards prediction of cognate complexes between the WW domain and proline-rich ligands. FEBS Lett. 384, 1–8.
Feng, Y., and Irvine, K. D. (2007). Fat and expanded act in parallel to regulate growth through Warts. Proc. Natl. Acad. Sci. U.S.A. 104, 20362–20367.
Feng, Y., and Irvine, K. D. (2009). Processing and phosphorylation of the Fat receptor. Proc. Natl. Acad. Sci. U.S.A. 106, 11989–11994.
Gaffney, C. J., Oka, T., Mazack, V., Hilman, D., Gat, U., Muramatsu, T., et al. (2012). Identification, basic characterization and evolutionary analysis of differentially spliced mRNA isoforms of human YAP1 gene. Gene 509, 215–222.
Genevet, A., Polesello, C., Blight, K., Robertson, F., Collinson, L. M., Pichaud, F., et al. (2009). The Hippo pathway regulates apical-domain size independently of its growth-control function. J. Cell. Sci. 122, 2360–2370.
Genevet, A., Wehr, M. C., Brain, R., Thompson, B. J., and Tapon, N. (2010). Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev. Cell 18, 300–308.
Goulev, Y., Fauny, J. D., Gonzalez-Marti, B., Flagiello, D., Silber, J., and Zider, A. (2008). SCALLOPED interacts with YORKIE, the nuclear effector of the Hippo tumor-suppressor pathway in Drosophila. Curr. Biol. 18, 435–441.
Grzeschik, N. A., Parsons, L. M., Allott, M. L., Harvey, K. F., and Richardson, H. E. (2010). Lgl, aPKC, and crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr. Biol. 20, 573–581.
Gurrieri, C., Capodieci, P., Bernardi, R., Scaglioni, P. P., Nafa, K., Rush, L. J., et al. (2004a). Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 96, 269–279.
Gurrieri, C., Nafa, K., Merghoub, T., Bernardi, R., Capodieci, P., Biondi, A., et al. (2004b). Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood 103, 2358–2362.
Halder, G., Dupont, S., and Piccolo, S. (2012). Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13, 591–600.
Hamaratoglu, F., Willecke, M., Kango-Singh, M., Nolo, R., Hyun, E., Tao, C., et al. (2006). The tumour-suppressor genes NF2/Merlin and expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 8, 27–36.
Hao, Y., Chun, A., Cheung, K., Rashidi, B., and Yang, X. (2008). Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509.
Harvey, K., and Tapon, N. (2007). The Salvador-Warts-Hippo pathway – an emerging tumour-suppressor network. Nat. Rev. Cancer 7, 182–191.
Haupt, S., Di Agostino, S., Mizrahi, I., Alsheich-Bartok, O., Voorhoeve, M., Damalas, A., et al. (2009). Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 69, 4818–4826.
Huang, J., Wu, S., Barrera, J., Matthews, K., and Pan, D. (2005). The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122, 421–434.
Ishikawa, H. O., Takeuchi, H., Haltiwanger, R. S., and Irvine, K. D. (2008). Four-jointed is a Golgi kinase that phosphorylates a subset of cadherin domains. Science 321, 401–404.
Justice, R. W., Zilian, O., Woods, D. F., Noll, M., and Bryant, P. J. (1995). The Drosophila tumor suppressor gene Warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546.
Kanai, F., Marignani, P. A., Sarbassova, D., Yagi, R., Hall, R. A., Donowitz, M., et al. (2000). TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791.
Komuro, A., Nagai, M., Navin, N. E., and Sudol, M. (2003). WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J. Biol. Chem. 278, 33334–33341.
Lai, D., Ho, K. C., Hao, Y., and Yang, X. (2011). Taxol resistance in breast cancer cells is mediated by the Hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 71, 2728–2738.
Lai, Z. C., Wei, X., Shimizu, T., Ramos, E., Rohrbaugh, M., Nikolaidis, N., et al. (2005). Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120, 675–685.
LaJeunesse, D. R., McCartney, B. M., and Fehon, R. G. (1998). Structural analysis of Drosophila Merlin reveals functional domains important for growth control and subcellular localization. J. Cell Biol. 141, 1589–1599.
Lapi, E., Di Agostino, S., Donzelli, S., Gal, H., Domany, E., Rechavi, G., et al. (2008). PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 32, 803–814.
Lei, Q. Y., Zhang, H., Zhao, B., Zha, Z. Y., Bai, F., Pei, X. H., et al. (2008). TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the Hippo pathway. Mol. Cell. Biol. 28, 2426–2436.
Levy, D., Adamovich, Y., Reuven, N., and Shaul, Y. (2007). The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 14, 743–751.
Levy, D., Reuven, N., and Shaul, Y. (2008). A regulatory circuit controlling Itch-mediated p73 degradation by Runx. J. Biol. Chem. 283, 27462–27468.
Li, Y., Pei, J., Xia, H., Ke, H., Wang, H., and Tao, W. (2003). Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 22, 4398–4405.
Lian, I., Kim, J., Okazawa, H., Zhao, J., Zhao, B., Yu, J., et al. (2010). The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 24, 1106–1118.
Lin, Y., Khokhlatchev, A., Figeys, D., and Avruch, J. (2002). Death-associated protein 4 binds MST1 and augments MST1-induced apoptosis. J. Biol. Chem. 277, 47991–48001.
Ling, C., Zheng, Y., Yin, F., Yu, J., Huang, J., Hong, Y., et al. (2010). The apical transmembrane protein crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc. Natl. Acad. Sci. U.S.A. 107, 10532–10537.
Liu-Chittenden, Y., Huang, B., Shim, J. S., Chen, Q., Lee, S. J., Anders, R. A., et al. (2012). Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305.
Mahoney, P. A., Weber, U., Onofrechuk, P., Biessmann, H., Bryant, P. J., and Goodman, C. S. (1991). The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell 67, 853–868.
Mahoney, W. M. Jr., Hong, J. H., Yaffe, M. B., and Farrance, I. K. (2005). The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J. 388, 217–225.
Matakatsu, H., and Blair, S. S. (2004). Interactions between Fat and Dachsous and the regulation of planar cell polarity in the Drosophila wing. Development 131, 3785–3794.
Matakatsu, H., and Blair, S. S. (2006). Separating the adhesive and signaling functions of the fat and dachsous protocadherins. Development 133, 2315–2324.
Matallanas, D., Romano, D., Yee, K., Meissl, K., Kucerova, L., Piazzolla, D., et al. (2007). RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975.
Mauviel, A., Nallet-Staub, F., and Varelas, X. (2012). Integrating developmental signals: a Hippo in the (path)way. Oncogene 31, 1743–1756.
Mullen, A. C., Orlando, D. A., Newman, J. J., Loven, J., Kumar, R. M., Bilodeau, S., et al. (2011). Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576.
Neto-Silva, R. M., De Beco, S., and Johnston, L. A. (2010). Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell 19, 507–520.
Oh, H., and Irvine, K. D. (2008). In vivo regulation of Yorkie phosphorylation and localization. Development 135, 1081–1088.
Oh, H., Reddy, B. V., and Irvine, K. D. (2009). Phosphorylation-independent repression of Yorkie in fat-Hippo signaling. Dev. Biol. 335, 188–197.
Oka, T., Mazack, V., and Sudol, M. (2008). Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 283, 27534–27546.
Oka, T., Remue, E., Meerschaert, K., Vanloo, B., Boucherie, C., Gfeller, D., et al. (2010). Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem. J. 432, 461–472.
Oka, T., Schmitt, A. P., and Sudol, M. (2012). Opposing roles of angiomotin-like-1 and zona occludens-2 on pro-apoptotic function of YAP. Oncogene 31, 128–134.
Oka, T., and Sudol, M. (2009). Nuclear localization and pro-apoptotic signaling of YAP2 require intact PDZ-binding motif. Genes Cells 14, 607–615.
Okazaki, T., Kageji, T., Kuwayama, K., Kitazato, K. T., Mure, H., Hara, K., et al. (2012). Up-regulation of endogenous PML induced by a combination of interferon-beta and temozolomide enhances p73/YAP-mediated apoptosis in glioblastoma. Cancer Lett. 323, 199–207.
O’Neill, E., Rushworth, L., Baccarini, M., and Kolch, W. (2004). Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 306, 2267–2270.
Pearson, M., Carbone, R., Sebastiani, C., Cioce, M., Fagioli, M., Saito, S., et al. (2000). PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210.
Peng, H. W., Slattery, M., and Mann, R. S. (2009). Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev. 23, 2307–2319.
Praskova, M., Xia, F., and Avruch, J. (2008). MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 18, 311–321.
Ribeiro, P. S., Josue, F., Wepf, A., Wehr, M. C., Rinner, O., Kelly, G., et al. (2010). Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol. Cell 39, 521–534.
Robinson, B. S., Huang, J., Hong, Y., and Moberg, K. H. (2010). Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein expanded. Curr. Biol. 20, 582–590.
Rosenbluh, J., Nijhawan, D., Cox, A. G., Li, X., Neal, J. T., Schafer, E. J., et al. (2012). beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473.
Rossi, M., De Laurenzi, V., Munarriz, E., Green, D. R., Liu, Y. C., Vousden, K. H., et al. (2005). The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 24, 836–848.
Sawada, A., Kiyonari, H., Ukita, K., Nishioka, N., Imuta, Y., and Sasaki, H. (2008). Redundant roles of Tead1 and Tead2 in notochord development and the regulation of cell proliferation and survival. Mol. Cell. Biol. 28, 3177–3189.
Scaglioni, P. P., Rabellino, A., Yung, T. M., Bernardi, R., Choi, S., Konstantinidou, G., et al. (2012). Translation-dependent mechanisms lead to PML upregulation and mediate oncogenic K-RAS-induced cellular senescence. EMBO Mol. Med. 4, 594–602.
Shen, T. H., Lin, H. K., Scaglioni, P. P., Yung, T. M., and Pandolfi, P. P. (2006). The mechanisms of PML-nuclear body formation. Mol. Cell 24, 331–339.
St John, M. A., Tao, W., Fei, X., Fukumoto, R., Carcangiu, M. L., Brownstein, D. G., et al. (1999). Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 21, 182–186.
Strano, S., Monti, O., Pediconi, N., Baccarini, A., Fontemaggi, G., Lapi, E., et al. (2005). The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 18, 447–459.
Strano, S., Munarriz, E., Rossi, M., Castagnoli, L., Shaul, Y., Sacchi, A., et al. (2001). Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 276, 15164–15173.
Sudol, M. (1994). Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 9, 2145–2152.
Sudol, M. (2010). Newcomers to the WW domain-mediated network of the Hippo tumor suppressor pathway. Genes Cancer 1, 1115–1118.
Sudol, M., Bork, P., Einbond, A., Kastury, K., Druck, T., Negrini, M., et al. (1995). Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 270, 14733–14741.
Sudol, M., and Harvey, K. F. (2010). Modularity in the Hippo signaling pathway. Trends Biochem. Sci. 35, 627–633.
Sudol, M., Shields, D. C., and Farooq, A. (2012). Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin. Cell Dev. Biol. 23, 827–833.
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676.
Tapon, N., Harvey, K. F., Bell, D. W., Wahrer, D. C., Schiripo, T. A., Haber, D. A., et al. (2002). Salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110, 467–478.
Trotman, L. C., Alimonti, A., Scaglioni, P. P., Koutcher, J. A., Cordon-Cardo, C., and Pandolfi, P. P. (2006). Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441, 523–527.
Ura, S., Nishina, H., Gotoh, Y., and Katada, T. (2007). Activation of the c-Jun N-terminal kinase pathway by MST1 is essential and sufficient for the induction of chromatin condensation during apoptosis. Mol. Cell. Biol. 27, 5514–5522.
Varelas, X., Sakuma, R., Samavarchi-Tehrani, P., Peerani, R., Rao, B. M., Dembowy, J., et al. (2008). TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 10, 837–848.
Varelas, X., Samavarchi-Tehrani, P., Narimatsu, M., Weiss, A., Cockburn, K., Larsen, B. G., et al. (2010). The crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev. Cell 19, 831–844.
Vassilev, A., Kaneko, K. J., Shu, H., Zhao, Y., and Depamphilis, M. L. (2001). TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 15, 1229–1241.
Vousden, K. H., and Lu, X. (2002). Live or let die: the cell’s response to p53. Nat. Rev. Cancer 2, 594–604.
Wang, W., Huang, J., and Chen, J. (2011). Angiomotin-like proteins associate with and negatively regulate YAP1. J. Biol. Chem. 286, 4364–4370.
Wang, Z. G., Delva, L., Gaboli, M., Rivi, R., Giorgio, M., Cordon-Cardo, C., et al. (1998). Role of PML in cell growth and the retinoic acid pathway. Science 279, 1547–1551.
Wu, J., Duggan, A., and Chalfie, M. (2001). Inhibition of touch cell fate by egl-44 and egl-46 in C. elegans. Genes Dev. 15, 789–802.
Wu, S., Huang, J., Dong, J., and Pan, D. (2003). Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with Salvador and Warts. Cell 114, 445–456.
Wu, S., Liu, Y., Zheng, Y., Dong, J., and Pan, D. (2008). The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell 14, 388–398.
Xia, H., Qi, H., Li, Y., Pei, J., Barton, J., Blackstad, M., et al. (2002). LATS1 tumor suppressor regulates G2/M transition and apoptosis. Oncogene 21, 1233–1241.
Yabuta, N., Okada, N., Ito, A., Hosomi, T., Nishihara, S., Sasayama, Y., et al. (2007). Lats2 is an essential mitotic regulator required for the coordination of cell division. J. Biol. Chem. 282, 19259–19271.
Yagi, R., Chen, L. F., Shigesada, K., Murakami, Y., and Ito, Y. (1999). A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 18, 2551–2562.
Yang, X., Li, D. M., Chen, W., and Xu, T. (2001). Human homologue of Drosophila lats, LATS1, negatively regulate growth by inducing G(2)/M arrest or apoptosis. Oncogene 20, 6516–6523.
Yu, J., Zheng, Y., Dong, J., Klusza, S., Deng, W. M., and Pan, D. (2010). Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 18, 288–299.
Yuan, M., Tomlinson, V., Lara, R., Holliday, D., Chelala, C., Harada, T., et al. (2008). Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 15, 1752–1759.
Zaidi, S. K., Sullivan, A. J., Medina, R., Ito, Y., Van Wijnen, A. J., Stein, J. L., et al. (2004). Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23, 790–799.
Zhang, L., Ren, F., Zhang, Q., Chen, Y., Wang, B., and Jiang, J. (2008). The TEAD/TEF family of transcription factor scalloped mediates Hippo signaling in organ size control. Dev. Cell 14, 377–387.
Zhao, B., Kim, J., Ye, X., Lai, Z. C., and Guan, K. L. (2009). Both TEAD-binding and WW domains are required for the growth stimulation and oncogenic transformation activity of yes-associated protein. Cancer Res. 69, 1089–1098.
Zhao, B., Li, L., Lu, Q., Wang, L. H., Liu, C. Y., Lei, Q., et al. (2011). Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 25, 51–63.
Zhao, B., Li, L., Tumaneng, K., Wang, C. Y., and Guan, K. L. (2010). A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24, 72–85.
Keywords: tumor suppression, pathway, interaction, signaling, transcription
Citation: Strano S, Fausti F, Di Agostino S, Sudol M and Blandino G (2013) PML surfs into HIPPO tumor suppressor pathway. Front. Oncol. 3:36. doi: 10.3389/fonc.2013.00036
Received: 17 December 2012; Accepted: 09 February 2013;
Published online: 01 March 2013.
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Rosa Bernardi, Fondazione Centro San Raffaele del Monte Tabor, ItalyArkaitz Carracedo, CIC bioGUNE, Spain
Copyright: © 2013 Strano, Fausti, Di Agostino, Sudol and Blandino. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Giovanni Blandino, Translational Oncogenomic Unit, Molecular Medicine Area, Regina Elena National Cancer Institute, Via Elio Chianesi, 53, 00144 Rome, Italy. e-mail:YmxhbmRpbm9AaWZvLml0