


- 1Department of Anatomy and Cell Biology, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 2Institute of Molecular Medicine, National Cheng Kung University, Tainan, Taiwan
- 3Department of Neurosurgery, Mackay Memorial Hospital, Taipei, Taiwan
- 4Graduate Institute of Injury Prevention and Control, Taipei Medical University, Taipei, Taiwan
- 5Advanced Optoelectronic Technology Center, National Cheng Kung University, Tainan, Taiwan
- 6Department of Neurochemistry, New York State Institute for Basic Research in Developmental Disabilities, New York, NY, USA
- 7Department of Neuroscience and Physiology, State University of New York Upstate Medical University, Syracuse, NY, USA
Unique astrocytic cell infiltrating growth and glial tumor growth in the confined skull make human glioblastoma (GBM) one of the most difficult cancers to treat in modern medicine. Prognosis for patients is very poor, as they die more or less within 12 months. Patients either die of the cancer itself, or secondary complications such as cerebral edema, herniations, or hemorrhages. GBMs rarely metastasize to other organs. However, GBM recurrence associated with resistance to therapeutic drugs is common. Patients die shortly after relapse. GBM is indeed an outstanding cancer model to search for potential mechanisms for drug resistance. Here, we reviewed the current cancer biology of gliomas and their pathophysiological events that contribute to the development of therapeutic resistance. We have addressed the potential roles of cancer stem cells, epigenetic modifications, and epithelial mesenchymal transition (EMT) in the development of resistance to inhibitor drugs in GBMs. The potential role of TIAF1 (TGF-β-induced antiapoptotic factor) overexpression and generation of intratumor amyloid fibrils for conferring drug resistance in GBMs is discussed.
Introduction
Glioblastoma (GBM) is one of the cancers most resistant to treatments and is associated with extremely poor prognosis. The current standard of care includes surgery with or without combined radiation/chemotherapy. Alternative treatment protocols and targeted therapies have been applied to GBM patients, but have unfortunately yielded to limited efficacies (Omuro et al., 2007; Lukas et al., 2009). Most cancer therapies have focused on attacking the key biological constituents that relate to the tumor growth and survival. It includes complex mechanisms of signal transduction and gene expression that contribute to the formation of microenvironment for supporting tumor growth (Omuro et al., 2007). Targeted therapies could be complicated by interconnecting signal networks with multiple interferences and convergences (Lukas et al., 2009). New methods and techniques for the targeted treatment of gliomas such as interruption of signaling pathways, nanoparticles targeting, and boron neutron capture therapy have been underway (Takagaki et al., 2001; Desai et al., 2006; Barth et al., 2012; Nduom et al., 2012). However, the clinical outcomes have made little progress in malignant gliomas to date (Omuro et al., 2007). It is thus necessary to reevaluate current strategies to find alternative approaches to eradicate malignant gliomas, or revisit the fundamental biology to explore the potential cancer resistance mechanisms in GBMs.
GBM Classification and Common Markers
Histologically, GBMs can be derived from low-grade gliomas in younger patients, according to the classification of World Health Organization (WHO). Or, they can directly develop de novo in elderly patients (Ohgaki and Kleihues, 2007). Among the complex genetic pathways in the development of gliomas, oligodendrocyte transcription factor 2 (olig2) and vascular endothelial growth factor (VEGF) are expressed in all high-grade gliomas (Ohgaki and Kleihues, 2007). Therefore, classifying GBMs based on the activation of signal pathways or mutations of genes in the glioma-relevant pathways may help establish targeted therapies. For example, alterations in the activation of epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDFGR), or loss of the RAS regulator NF1, are frequently observed in primary GBMs (Brennan et al., 2009). These findings implicate that these molecules as potential therapeutic targets. Although these classifications do not include all clinical GBMs, clinical trials targeting EGFR or PDFGR have been underway. EGFR, EGFR variant III (EGFRvIII), phosphatase, and tensin homolog deleted on chromosome 10 (PTEN), and O6-methylguanine–DNA methyltransferase (MGMT) have been regarded as common markers for GBMs (Camara-Quintana et al., 2012).
Inhibitors of Receptor Tyrosine Kinases and Resistance in GBMs
Recent studies have shown that EGFR inhibitors fail to yield significant clinical outcomes in GBM patients. Simultaneous activation of multiple receptor tyrosine kinases (RTKs), which generates redundant activation of phosphoinositide-3′-kinase (PI3K) signaling, may explain for the drug failure (Fenton et al., 2012). Tumor suppressor PTEN, a phosphatidylinositol-3,4,5-trisphosphate 3 (PIP3) phosphatase, can be phosphorylated at a conserved tyrosine 240 (Y240). The phosphorylated PTEN (p-PTEN) is associated with shortened survival and resistance to therapy with EGFR inhibitors in GBM patients (Fenton et al., 2012). Both fibroblast growth factor receptors (FGFRs) and SRC family kinases (SFKs) phosphorylate PTEN, and p-PTEN fails to antagonize the PI3K signaling (Fenton et al., 2012), suggesting that loss of control of PI3K signaling is associated with resistance to EGFR inhibitors in GBM.
Amplification and/or mutation of a specific RTK gene in GBMs could confer resistance to RTK inhibitors. For example, genes encoding EGFR, platelet-derived growth factor receptor α (PDGFRα), hepatocyte growth factor receptor (MET), and/or others are frequently altered (Cancer Genome Atlas Research, 2008; Huse and Holland, 2010). It is not surprising to predict the failure in therapy using small-molecule inhibitors in targeting the mutated and/or amplified RTKs due in part to constitutive and concurrent activations of signal pathways in GBMs (Stommel et al., 2007; De Witt Hamer, 2010; Hasselbalch et al., 2010; Paulsson et al., 2011). Worse, growth factors could further enhance the drug resistance in subpopulations of GBM cells harboring amplifications of EGFR and PDGFR-α genes (Szerlip et al., 2012; Wilson et al., 2012). These observations highlight the role of RTK ligands and extensive redundancy of RTK-transduced signaling in innate and acquired resistance of GBMs to drugs targeting oncogenic kinases (Wilson et al., 2012).
Cancer Stem Cells Confer Intrinsic Drug Resistance
Cancer stem cells (CSCs) are crucial in the initiation, progression, and angiogenesis for GBMs and essentially all cancers (Wen and Kesari, 2008; Dietrich et al., 2010). GBM CSCs express CD133 and nestin, which are also expressed by normal stem cells or progenitor cells. Whether drugs can specifically select against CSCs in the brain without affecting normal stem cells is not quite understood (Yilmaz et al., 2006; Calabrese et al., 2007). How CSCs develop into highly vascular GBMs is largely unknown. Expression of Olig2 and VEGF in all high-grade gliomas and glioma stem cells may render them highly vascular (Plate et al., 1992; Ohgaki and Kleihues, 2007; Takano, 2012). Anti-angiogenesis strategies to block CSC expansion have been utilized. However, the benefit of anti-angiogenesis therapy has been questionable in both preclinical and clinical trials.
The failure of anti-angiogenesis therapy may be due to evasive (adaptive) and/or intrinsic (pre-existing) resistance in GBM cells (Bergers and Hanahan, 2008). Simultaneous inhibition of cancer survival targets, along with potential escape pathways, may have a great potential in eliminating drug-resistant cancer cells. One such example is targeting both FGFRs and VEGF. Combined VEGF and FGFR–Fc fusion protein (FGF-trap) treatment attenuated revascularization and slowed tumor growth, which indicates that FGF signaling is involved in regulating angiogenesis (Casanovas et al., 2005). It has been reported that AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates cerebral edema in GBM patients. This study implies analogous evasive resistance may be mediated by FGF-dependent revascularization (Batchelor et al., 2007). Given that FGFRs also participate in EGFR and PDGFR-mediated cancer resistance, designing therapeutic regimes that simultaneously target activation-reactivation, and amplification of FGFRs and RTKs may be beneficial in resolving therapeutic resistance in GBM patients.
When tumors outgrow their blood supply, CSCs upregulate pro-inflammatory proteins to help tumor survival under hypoxic conditions (Tafani et al., 2011). In essence, CSCs play multi-faceted roles in allowing a tumor to escape complete eradication. Specific molecular markers that are capable of distinguishing CSCs from normal stem cells or progenitor cells in the brain is not completely resolved (Yilmaz et al., 2006; Calabrese et al., 2007). Therefore, before CSCs can be clearly identified, therapeutic approaches designated to target them may actually cause more harm than good to GBM patients.
Epigenetic Modifications and GBM Resistance to Temozolomide
Temozolomide (TMZ) with or without radiation is the current standard treatment for GBMs. Methylation of MGMT (O6-methylguanine–DNA methyltransferase) promoter prevents gene translation, and thereby prevents DNA repair in cancer cells. The methylation status of the MGMT promoter is currently considered one of the strongest predictors of outcome and benefit to TMZ treatment (Stupp et al., 2009). Mutant forms of metabolic enzyme isocitrate dehydrogenase (IDH1) are found in a great proportion of secondary gliomas, but are absent in the primary glioblastomas. The mutations are rarely found in primary high-grade glioblastoma multiforme. Clinical data have demonstrated that IDH1 may be a reliable prognostic marker for GBM. Intriguingly, the presence of IDH1 mutation(s) in patients with newly diagnosed GBMs showed prolonged, progression-free survival (Weller et al., 2009; Wick et al., 2009). GBMs with the CpG island methylator phenotype (CIMP) have been shown to possess extensive epigenetic aberrations to define a distinct subgroup of gliomas (Noushmehr et al., 2010). Recent data showed that IDH mutation and the CIMP phenotype are two very common features in cancer (Turcan et al., 2012). IDH mutation is the cause of CIMP and leads to the CIMP phenotype by stably reshaping the epigenome. This genome-wide remodeling involves modulating patterns of methylation, changing transcriptional programs, and alteration of cell differentiation (Turcan et al., 2012). These data highlight the interplay between genomic and epigenomic changes in human cancers and may prove to be helpful in the development of novel therapies for cancers. Shown in the Figure 1 is the summary of MGMT repair and epigenetic modification in TMZ treatment.
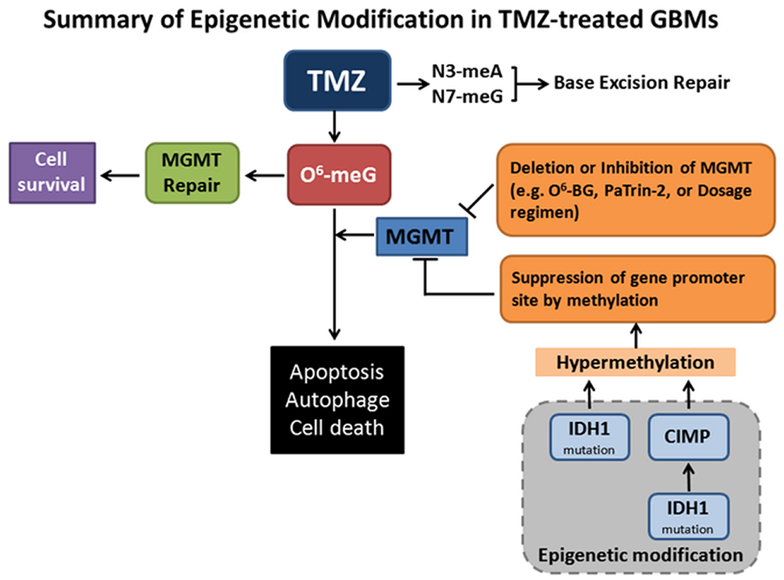
Figure 1. Summary of epigenetic modifications in Temozolomide (TMZ)-treated GBMs. TMZ is an oral alkylating agent used for the treatment of GBMs. TMZ causes cytotoxic DNA lesions such as O6-methylguanine (O6-meG), and N3-methyladenine and N7-methylguanine (N3-meA, N7-meG). The latter two lesions can be repaired by base excision repair (BER) pathway. O6-meG DNA methyltransferase (MGMT) removes the O6-alkylguanine and restores the guanine to normal, which promotes tumor cell survival. When MGMT transfers and accepts an alkyl-group from inhibitors O6-benzylguanine (O6-BG) and O6-(4-bromothenyl) guanine (PaTrin-2), MGMT is inactivated and subjected to ubiquitin-mediated degradation, thereby promoting cell death. MGMT functions may also be impaired by gene deletion, altered therapeutic regimen, and suppression of gene methylation promoter site. Notably, isocitrate dehydrogenase 1 (IDH1) mutation is the cause of CpG island methylator phenotype (CIMP) via stably reshaping the epigenome. IDH1 mutation by itself is also sufficient to hypermethylate MGMT. MGMT hypermethylation causes DNA strand breaks, apoptosis, autophagy, and tumor cell death. This is the mechanistic rationale for the usage of the methylating therapeutic drugs.
Epigenetic inactivation of tumor suppressor genes has been shown mainly in human malignant cancers. DKK1 gene expression, for example, is decreased in GBM tumor samples, as compared with non-tumor brain tissues. Restoration of DKK1 expression by a demethylating agent 5-azacytidine in T98 GBM cells enhances their susceptibility to camptothecin- and etoposide-induced apoptosis (Foltz et al., 2010), suggesting that treatment of GBM cells with inhibitors of promoter methylation for tumor suppressors facilitates drug efficacy. Also, regulatory enzymes for epigenetic alterations may be used for cancer therapy. When GBM cells were treated with histone deacetylase (HDAC) inhibitors, accumulation of methylated histone occurred. This histone methylation can be removed by lysine specific demethylase 1 (LSD1). Intriguingly, simultaneous inhibition of HDACs and LSD1 leads to synergistic apoptotic cell death in GBM cells (Singh et al., 2011).
Epithelial Mesenchymal Transition and Drug Resistance in GBMs
Epithelial mesenchymal transition (EMT) is a critical biologic process that allows cancer cells to become metastatic. That is, there is an increased cell migratory capacity, invasiveness, and resistance to apoptosis (Kalluri and Neilson, 2003). EMT may occur in tumor cells that have previously undergone genetic and epigenetic changes, specifically in genes that favor clonal outgrowth, development of localized tumors, and enhancement of carcinoma invasion and metastasis (Jin et al., 2011). Growth factors are known to induce EMT include members of the EGF family, FGF, insulin-like growth factor, and MET (De Wever et al., 2008). Transforming growth factor beta (TGF-β) is released by glioma cells in large quantities in vitro. It has been implicated in the malignant progression of glial tumors and the immune dysfunction in patients with GBM (Xu et al., 2009). TGF-β promotes tumor-associated angiogenesis, tumor invasion, and suppression of T cell-mediated immune responses (Xu et al., 2009). Proto-oncogene MET (c-met) amplification in human gliomas causes increased activity of its downstream targets (e.g., Wnt/β-catenin signaling) (Moon et al., 2000; Kim et al., 2013), and prolongs the cancer survival (Halatsch et al., 2006). MET inhibitors appear to lead to aberrant MET amplification in gliomas (Moon et al., 2000; Chi et al., 2012). Dysregulation and prominent pathophysiological roles of EGFR have been identified in GBMs. EGFR is overexpressed in about 40% of GBM cases, and half of these co-express a mutated, activated subtype, EGFRvIII (Halatsch et al., 2006). Targeted therapy against EFGR and EGFRvIII has no survival benefit when compared to standard therapy. Stemness and invasiveness of migrating glioma cells regulated by Frizzled 4 (FZD4), which promotes expression of the EMT transition regulator SNAI1, are considered as an important mechanism contributing to the failure of this approach (Jin et al., 2011; Pala et al., 2012). In its complexity, EMT encompasses pathways that promote the continuous acquisition of malignant biological features by glioma cells. A better understanding of the molecular mechanism of EMT may help identify and rationally design combined therapeutic regimes as well as selected patient subgroups that may benefit from EGFR inhibition in GBMs (Tektonidis et al., 2011; Pala et al., 2012).
Potential Role of TIAF1 in GBM Progression
We have recently demonstrated that accumulation of extracellular proteins, which are generated by both cancer cells and neural cells, is critical for the self-protection, progression, and expansion of brain metastatic cancer cells (Lee et al., 2010; Chang et al., 2012). TGF-β-induced antiapoptotic factor (TIAF1) is an intracellular protein, whose self-aggregation intracellularly may induce generation of toxic amyloid beta (Aβ) and formation of amyloid fibrils (Lee et al., 2010; Chang et al., 2012). For example, when TIAF1 undergoes self-aggregation, the aggregating protein may stimulate caspase activation, subsequent phosphorylation of membrane amyloid precursor protein (APP), breakdown of APP, generation of toxic Aβ (e.g., Aβ42), and formation of amyloid fibrils (Lee et al., 2010; Chang et al., 2012). Aβ does not appear to cause cancer death. However, Aβ42 causes damage to neuronal death or induces neurodegeneration. TIAF1 is known to participate in the signaling of TGF-β/Smad proteins (Chang et al., 1998, 2012; Khera and Chang, 2003; Lee et al., 2010).
TIAF1 is shown to be significantly upregulated in the malignant glioma cells in patients (Chang et al., 2012). A postulated model for GBM malignancy is that upregulation of TIAF1 occurs in the proliferating GBM stem cells, probably in response to extracellular stress (Figure 2). TIAF1 is then becoming self-aggregated intracellularly for leading to APP degradation and Aβ generation. Aβ is then released to the extracellular matrix, or polymerizes further to become amyloid fibrils, which are accumulated intracellularly. For example, when metastatic U87-MG glioma cells were inoculated in two subcutaneous sides in both flanks of nude mice, the cells were metastatic to the lung. Both aggregated TIAF1 and amyloid fibrils are overly expressed in the growing solid tumor (Chang et al., 2012) (Figure 2). The intracellular TIAF1 and amyloid fibrils may provide resistance to drug penetration into cancer cells, thereby promoting cancer cell survival.

Figure 2. A postulated model for GBM malignancy. (A) U87-MG glioma cells were inoculated in both flanks of nude mice. Two months later, U87-MG cells were shown to metastasize to the lung. The solid tumor lesion has overexpressed TIAF1 and amyloid fibrils, compared to untreated normal lung. Specific antibodies against TIAF1 and amyloid fibril were used (Lee et al., 2010; Chang et al., 2012). (B) TIAF1 expression is frequently upregulated in the proliferating GBM cells, probably due to the stimulation of micro-environmental factors in the brain. The intracellular TIAF1 undergoes self-aggregation, may induce caspase activation, and leads to phosphorylation and degradation of membrane amyloid precursor protein (APP), generation of amyloid beta (Aβ), and formation of amyloid fibrils (Lee et al., 2010; Chang et al., 2012). Secreted Aβ42 is toxic to neurons. Also, Aβ42 undergoes self-polymerization and binds secreted TIAF1, and the complexes are probably detrimental to neurons. Aβ is indeed localized intracellularly. Cancer cells are resistant to the toxic effect of polymerized TIAF1 and Aβ. The presence of intracellular aggregated TIAF1 and amyloid fibrils in the tumor is likely to prevent the penetration and efficacy of therapeutic drugs.
Conclusion
Human GBM is one of most challenging tumors to treat. Failure of therapeutic approaches to target specific signaling transduction or glioma-relevant mutation pathway genes in GBMs is mainly due to reactivation of certain common signaling pathways that are involved in cell growth and proliferation. Unpredictable clonal growth and specific gene amplification occur in CSCs. CSCs may also participate in treatment-induced evasive and/or intrinsic resistance and therefore therapeutic outcomes.
Ongoing “normal” epigenetic processes are associated with differentiation (Gan et al., 2007). Emerging data suggest that GBM patients with epigenetic MGMT methylation respond to TZM better and have prolonged survival than the patients without it. However, it is reported that patients without MGMT methylation may also be beneficial to TZM treatment (von Deimling et al., 2011), which suggests multiple mechanisms are involved in GBM pathogenesis. Direct genetic disruptions within the core epigenetic machinery may occur, such as the recently identified mutations within IDH1/2 and variant histone genes H3.3/H3F3A (Carén et al., 2012). IDH1 mutation alone is enough for the development of the glioma hypermethylate phenotype (Turcan et al., 2012). Therefore, IDH1 may be used as a prognostic marker in low-grade and high-grade gliomas and aid in the differentiation and diagnosis of various tumors with histologic ambiguity. Insights into MGMT, IDH1 mutation may help decode GBM resistance in both primary and secondary GBMs. EMT helps the continuous acquisition of malignant phenotype in GBMs, and may also contribute to the failure of targeted therapy.
In summary, development of GBM resistance involves multiple mechanisms with interplayed redundancies that determine the survival or death of cancer. Designing therapeutic regimes that target the aforementioned potential resistance mechanisms may likely be the primary focus of GBM treatment in the future.
Authors Contribution
Wan-Pei Su, Chen-Yu Lu, and Yu-An Chen: worked on graphic arts, literature search, and proofread manuscript; Ming-Fu Chiang: proofread manuscript; Chun-I Sze and Nan-Shan Chang: wrote the manuscript, conceived ideas, and discussed thoroughly.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported to Nan-Shan Chang by the Department of Defense USA (W81XWH-08-1-0682), the Research the National Science Council, Taiwan, ROC (NSC96-2320-B-006-014, 98-2628-B-006-045-MY3, 98-2628-B-006-041-MY3, and 99-2320-B-006-012-MY3), the National Health Research Institute, Taiwan, ROC (NHRI-EX99-9704BI), and the Department of Health, Taiwan, ROC (DOH101-TD-PB-111-TM010).
References
Barth, R. F., Vicente, M. G., Harling, O. K., Kiger, W. S. III, Riley, K. J., Binns, P. J., et al. (2012). Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat. Oncol. 7, 146.
Batchelor, T. T., Sorensen, A. G., Di Tomaso, E., Zhang, W. T., Duda, D. G., Cohen, K. S., et al. (2007). AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11, 83–95.
Bergers, G., and Hanahan, D. (2008). Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603.
Brennan, C., Momota, H., Hambardzumyan, D., Ozawa, T., Tandon, A., Pedraza, A., et al. (2009). Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 4:e7752. doi:10.1371/journal.pone.0007752
Calabrese, C., Poppleton, H., Kocak, M., Hogg, T. L., Fuller, C., Hamner, B., et al. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82.
Camara-Quintana, J. Q., Nitta, R. T., and Li, G. (2012). Pathology: commonly monitored glioblastoma markers: EFGR, EGFRvIII, PTEN, and MGMT. Neurosurg. Clin. N. Am. 23, 237–246, viii.
Cancer Genome Atlas Research, N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068.
Carén, H., Pollard, S. M., and Beck, S. (2012). The good, the bad and the ugly: epigenetic mechanisms in glioblastoma. Mol. Aspects Med.. doi:10.1016/j.mam.2012.06.007. [Epub ahead of print].
Casanovas, O., Hicklin, D. J., Bergers, G., and Hanahan, D. (2005). Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8, 299–309.
Chang, J. Y., Chiang, M. F., Lin, S. R., Lee, M. H., He, H., Chou, P. Y., et al. (2012). TIAF1 self-aggregation in peritumor capsule formation, spontaneous activation of SMAD-responsive promoter in p53-deficient environment, and cell death. Cell Death Dis. 3, e302.
Chang, N. S., Mattison, J., Cao, H., Pratt, N., Zhao, Y., and Lee, C. (1998). Cloning and characterization of a novel transforming growth factor-beta1-induced TIAF1 protein that inhibits tumor necrosis factor cytotoxicity. Biochem. Biophys. Res. Commun. 253, 743–749.
Chi, A. S., Batchelor, T. T., Kwak, E. L., Clark, J. W., Wang, D. L., Wilner, K. D., et al. (2012). Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 30, e30–e33.
De Wever, O., Pauwels, P., De Craene, B., Sabbah, M., Emami, S., Redeuilh, G., et al. (2008). Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 130, 481–494.
De Witt Hamer, P. C. (2010). Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncology 12, 304–316.
Desai, N., Trieu, V., Yao, Z., Louie, L., Ci, S., Yang, A., et al. (2006). Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 12, 1317–1324.
Dietrich, J., Diamond, E. L., and Kesari, S. (2010). Glioma stem cell signaling: therapeutic opportunities and challenges. Expert Rev. Anticancer Ther. 10, 709–722.
Fenton, T. R., Nathanson, D., Ponte De Albuquerque, C., Kuga, D., Iwanami, A., Dang, J., et al. (2012). Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc. Natl. Acad. Sci. U.S.A. 109, 14164–14169.
Foltz, G., Yoon, J. G., Lee, H., Ma, L., Tian, Q., and Hood, L. (2010). Epigenetic regulation of Wnt pathway antagonists in human glioblastoma multiforme. Genes Cancer 1, 81–90.
Gan, Q., Yoshida, T., McDonald, O. G., and Owens, G. K. (2007). Concise review: epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells 25, 2–9.
Halatsch, M. E., Schmidt, U., Behnke-Mursch, J., Unterberg, A., and Wirtz, C. R. (2006). Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat. Rev. 32, 74–89.
Hasselbalch, B., Eriksen, J. G., Broholm, H., Christensen, I. J., Grunnet, K., Horsman, M. R., et al. (2010). Prospective evaluation of angiogenic, hypoxic and EGFR-related biomarkers in recurrent glioblastoma multiforme treated with cetuximab, bevacizumab and irinotecan. APMIS 118, 585–594.
Huse, J. T., and Holland, E. C. (2010). Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 10, 319–331.
Jin, X., Jeon, H. Y., Joo, K. M., Kim, J. K., Jin, J., Kim, S. H., et al. (2011). Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 71, 3066–3075.
Kalluri, R., and Neilson, E. G. (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112, 1776–1784.
Khera, S., and Chang, N. S. (2003). TIAF1 participates in the transforming growth factor beta1 – mediated growth regulation. Ann. N. Y. Acad. Sci. 995, 11–21.
Kim, K. H., Seol, H. J., Kim, E. H., Rheey, J., Jin, H. J., Lee, Y., et al. (2013). Wnt/β-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro-oncology 15, 161–171.
Lee, M. H., Lin, S. R., Chang, J. Y., Schultz, L., Heath, J., Hsu, L. J., et al. (2010). TGF-β induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid β plaques in Alzheimer’s disease. Cell Death Dis. 1, e110.
Lukas, R. V., Boire, A., and Nicholas, M. K. (2009). Targeted therapy in the treatment of malignant gliomas. Onco Targets Ther. 2, 115–133.
Moon, Y. W., Weil, R. J., Pack, S. D., Park, W. S., Pak, E., Pham, T., et al. (2000). Missense mutation of the MET gene detected in human glioma. Mod. Pathol. 13, 973–977.
Nduom, E. K., Bouras, A., Kaluzova, M., and Hadjipanayis, C. G. (2012). Nanotechnology applications for glioblastoma. Neurosurg. Clin. N. Am. 23, 439–449.
Noushmehr, H., Weisenberger, D. J., Diefes, K., Phillips, H. S., Pujara, K., Berman, B. P., et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522.
Ohgaki, H., and Kleihues, P. (2007). Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 170, 1445–1453.
Omuro, A. M., Faivre, S., and Raymond, E. (2007). Lessons learned in the development of targeted therapy for malignant gliomas. Mol. Cancer Ther. 6, 1909–1919.
Pala, A., Karpel-Massler, G., Kast, R. E., Wirtz, C. R., and Halatsch, M. C. (2012). Epidermal to mesenchymal transition and failure of EGFR-targeted therapy in glioblastoma. Cancers 4, 523–530.
Paulsson, J., Lindh, M. B., Jarvius, M., Puputti, M., Nister, M., Nupponen, N. N., et al. (2011). Prognostic but not predictive role of platelet-derived growth factor receptors in patients with recurrent glioblastoma. Int. J. Cancer 128, 1981–1988.
Plate, K. H., Breier, G., Weich, H. A., and Risau, W. (1992). Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 359, 845–848.
Singh, M. M., Manton, C. A., Bhat, K. P., Tsai, W. W., Aldape, K., Barton, M. C., et al. (2011). Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro-oncology 13, 894–903.
Stommel, J. M., Kimmelman, A. C., Ying, H., Nabioullin, R., Ponugoti, A. H., Wiedemeyer, R., et al. (2007). Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318, 287–290.
Stupp, R., Hegi, M. E., Mason, W. P., van den Bent, M. J., Taphoorn, M. J., Janzer, R. C., et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466.
Szerlip, N. J., Pedraza, A., Chakravarty, D., Azim, M., McGuire, J., Fang, Y., et al. (2012). Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. U.S.A. 109, 3041–3046.
Tafani, M., Di Vito, M., Frati, A., Pellegrini, L., De Santis, E., Sette, G., et al. (2011). Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflammation 8, 32.
Takagaki, M., Powell, W., Sood, A., Spielvogel, B. F., Hosmane, N. S., Kirihata, M., et al. (2001). Boronated dipeptide borotrimethylglycylphenylalanine as a potential boron carrier in boron neutron capture therapy for malignant brain tumors. Radiat. Res. 156, 118–122.
Takano, S. (2012). Glioblastoma angiogenesis: VEGF resistance solutions and new strategies based on molecular mechanisms of tumor vessel formation. Brain Tumor Pathol. 29, 73–86.
Tektonidis, M., Hatzikirou, H., Chauviere, A., Simon, M., Schaller, K., and Deutsch, A. (2011). Identification of intrinsic in vitro cellular mechanisms for glioma invasion. J. Theor. Biol. 287, 131–147.
Turcan, S., Rohle, D., Goenka, A., Walsh, L. A., Fang, F., Yilmaz, E., et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483.
von Deimling, A., Korshunov, A., and Hartmann, C. (2011). The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol. 21, 74–87.
Weller, M., Felsberg, J., Hartmann, C., Berger, H., Steinbach, J. P., Schramm, J., et al. (2009). Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J. Clin. Oncol. 27, 5743–5750.
Wick, W., Hartmann, C., Engel, C., Stoffels, M., Felsberg, J., Stockhammer, F., et al. (2009). NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J. Clin. Oncol. 27, 5874–5880.
Wilson, T. R., Fridlyand, J., Yan, Y., Penuel, E., Burton, L., Chan, E., et al. (2012). Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509.
Xu, J., Lamouille, S., and Derynck, R. (2009). TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172.
Keywords: glioblastoma multiforme, temozolomide, resistance mechanisms, cancer stem cell, TIAF1 expression, extracellular matrix
Citation: Sze C-I, Su W-P, Chiang M-F, Lu C-Y, Chen Y-A and Chang N-S (2013) Assessing current therapeutic approaches to decode potential resistance mechanisms in glioblastomas. Front. Oncol. 3:59. doi: 10.3389/fonc.2013.00059
Received: 18 October 2012; Accepted: 04 March 2013;
Published online: 19 March 2013.
Edited by:
Gaspar Kitange, Mayo Clinic, USAReviewed by:
Michael W. Epperly, University of Pittsburgh Cancer Institute, USAHsin-Ell Wang, National Yang-Ming University, Taiwan
Copyright: © 2013 Sze, Su, Chiang, Lu, Chen and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Chun-I Sze, Department of Anatomy and Cell Biology, College of Medicine, National Cheng Kung University, 1 University Road, Tainan 70101, Taiwan. e-mail:c3plY0BtYWlsLm5ja3UuZWR1LnR3;
Nan-Shan Chang, Institute of Molecular Medicine, National Cheng Kung University, 1 University Road, Tainan 70101, Taiwan. e-mail:Y2hhbmduc0BtYWlsLm5ja3UuZWR1LnR3