 Juliana Vélez1 Numsen Hail Jr.2,3 Marina Konopleva2,3 Zhihong Zeng2,3 Kensuke Kojima2,3
Juliana Vélez1 Numsen Hail Jr.2,3 Marina Konopleva2,3 Zhihong Zeng2,3 Kensuke Kojima2,3 Ismael Samudio1*
Ismael Samudio1* Michael Andreeff2,3*
Michael Andreeff2,3*- 1Grupo de Terapia Celular y Molecular Laboratorio de Bioquimica, Pontificia Universidad Javeriana, Bogotá, Colombia
- 2Section of Molecular Hematology and Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
- 3Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Nearly 60 years ago Otto Warburg proposed, in a seminal publication, that an irreparable defect in the oxidative capacity of normal cells supported the switch to glycolysis for energy generation and the appearance of the malignant phenotype (Warburg, 1956). Curiously, this phenotype was also observed by Warburg in embryonic tissues, and recent research demonstrated that normal stem cells may indeed rely on aerobic glycolysis – fermenting pyruvate to lactate in the presence of ample oxygen – rather than on the complete oxidation of pyruvate in the Krebs cycle – to generate cellular energy (Folmes et al., 2012). However, it remains to be determined whether this phenotype is causative for neoplastic development, or rather the result of malignant transformation. In addition, in light of mounting evidence demonstrating that cancer cells can carry out electron transport and oxidative phosphorylation, although in some cases predominantly using electrons from non-glucose carbon sources (Bloch-Frankenthal et al., 1965), Warburg’s hypothesis needs to be revisited. Lastly, recent evidence suggests that the leukemia bone marrow microenvironment promotes the Warburg phenotype adding another layer of complexity to the study of metabolism in hematological malignancies. In this review we will discuss some of the evidence for alterations in the intermediary metabolism of leukemia cells and present evidence for a concept put forth decades ago by lipid biochemist Feodor Lynen, and acknowledged by Warburg himself, that cancer cell mitochondria uncouple ATP synthesis from electron transport and therefore depend on glycolysis to meet their energy demands (Lynen, 1951; Warburg, 1956).
Oxidizing Carbon Sources Other than Glucose
It has been shown that leukemia cells generate significant amounts of lactate even in the presence of adequate amounts of oxygen (Samudio et al., 2008, 2010) recapitulating Warburg’s observations in Ehrlich’s ascites tumor cells (Warburg et al., 1924). However, in contrast to what Warburg hypothesized, it is evident that leukemia cells have the ability to reduce molecular oxygen utilizing electrons from carbon sources other than pyruvate (Samudio et al., 2008, 2010). This finding is intriguing in light of the finding that in other tumor types, like glioblastoma, the anaplerotic (see Table 1) entry of glutamine-derived glutamate fuels the Krebs cycle and oxidative phosphorylation (OXPHOS) (DeBerardinis et al., 2007), suggesting that shunting pyruvate away from mitochondrial oxidation may be an acquired trait of different types of cancer. In the case of leukemia cells, recent evidence suggests that fatty acid-derived acetyl-CoA fuels Krebs cycle activity and the molecular reduction of oxygen (Samudio et al., 2010). Unpublished observations from our group suggest that glutaminolysis is essential to maintain oxygen consumption in these cells, supporting the notion that leukemia cell mitochondria are less prone to oxidize pyruvate (Figure 1). The question arises therefore, what could be the selective advantage of this metabolic shift?

Table 1. Glossary of terms.
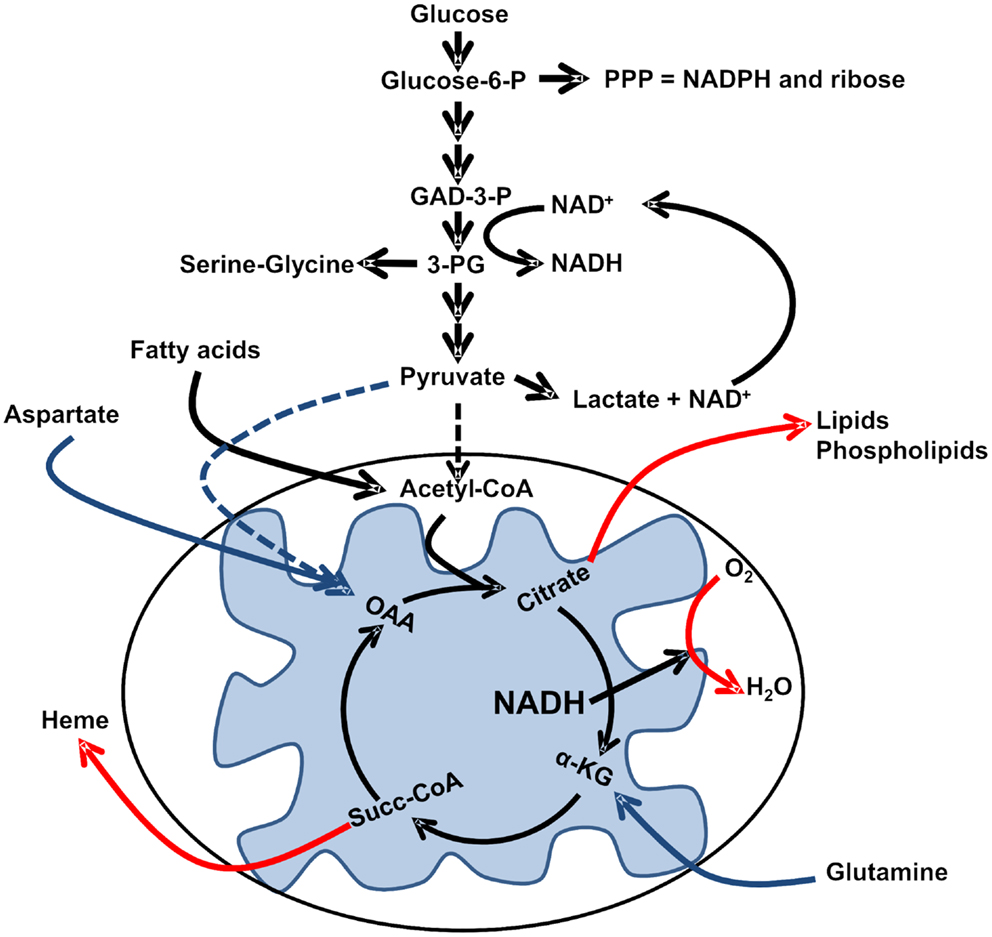
Figure 1. Reprogramed pathways of intermediary metabolism. Incomplete oxidation of glucose carbon skeletons provides intermediaries for biosynthesis, while fermentation of pyruvate to lactate regenerates NAD+ for the glycolytic conversion of glyceraldehydes-3-phosphate (GAD-3-P) to 1,3-bisphosphoglycerate and subsequent formation of 3-phosphoglycerate (3-PG). In order to maintain an adequate supply of biosynthetic intermediates in the absence of pyruvate-derived oxaloacetate (OAA), Krebs cycle activity relies on glutamine anaplerosis to regenerate alpha-ketoglutarate (α-KG; and potentially aspartate anaplerosis). Regeneration of cataplerotic citrate also relies on FAO-derived acetyl-CoA. Please refer to the text for additional details.
First, while complete oxidation of glucose in the Krebs cycle yields more ATP per mole of glucose than does glycolysis, it fails to safeguard carbon skeletons for anabolic reactions. For instance, pyruvate transamination produces alanine; alanine accumulation in turn allosterically inhibits pyruvate kinase, and results in accumulation of phosphoenolpyruvate which via equilibrium reactions is converted to 3-phosphoglycerate which serves as a precursor to serine and glycine (reviewed in DeBerardinis, 2011). Phosphoenolpyruvate is also an inhibitor of phosphofructokinase, which would shunt fructose-6-phosphate to glucose-6-phosphate that can now enter the pentose phosphate shunt to produce NADPH for biosynthesis and ribose-5-phosphate for nucleotide biosynthesis (Berg et al., 2002). Thus, non-oxidative metabolism of glucose carbon skeletons is essential for safeguarding biosynthetic capacity in rapidly proliferating cells.
Second, while the activity of the Krebs cycle has indeed a major influence in the regulation of OXPHOS-mediated ATP generation in transformed leukocytes, as these cells proliferate uncontrollably, OXPHOS is also likely to be affected by de novo pyrimidine synthesis. De novo pyrimidine synthesis is indispensable in rapidly proliferating cells in order to provide the heterocyclic aromatic precursors required for DNA, RNA, phospholipid, and glycoprotein formation (Evans and Guy, 2004; Hail et al., 2010b). This is especially true in vivo given the fact that the liver keeps the circulating levels of pyrimidines relatively low, thus limiting the role of pyrimidine salvage in the biochemical processes linked with cell proliferation (Traut, 1994).
Specifically, the oxidation of dihydroorotate via the activity of dihydroorotate dehydrogenase (DHODH, EC 1.3.99.11, the rate-limiting enzyme for the de novo pathway of pyrimidine synthesis) provides electrons for OXPHOS in a Krebs cycle- and glucose-independent manner thereby supporting mitochondrial bioenergetics and the proliferative capability of various cell types (Löffler, 1989), including hematopoietic cells (Xu et al., 1996; Rückemann et al., 1998; Sawamukai et al., 2007; Ringshausen et al., 2008). Coenzyme Q functions as the proximal electron acceptor for the oxidation of dihydroorotate to orotate by DHODH, and cytochrome c oxidase serves as the ultimate electron acceptor for this reaction. In this scenario, dihydroorotate functions as a reducing equivalent like NADH or succinate to modulate mitochondrial OXPHOS (Hail et al., 2010a) (Figure 2). In fact, the activity of DHODH is believed to be a major contributor to mitochondrial oxygen consumption in leukemia cells (Beuneu et al., 2000). Consequently, if DHODH and de novo pyrimidine biosynthesis are constitutively active in transformed hematopoietic cell this would not only affect their rate of proliferation (Shawver et al., 1997; Rückemann et al., 1998), but also their endogenous mitochondrial reactive oxygen species (ROS) production (Forman and Kennedy, 1975; Lakaschus et al., 1991; Lenaz, 2001). The very nature of this paired metabolic activity could serve as a feed-forward mechanism for leukemogenesis since ROS play an integral role in mutagenesis and oncogenic signaling (Hail and Lotan, 2009). Furthermore, DHODH activity in response to cell proliferation can function under a seemingly wide (i.e., ≥0.13%) range of oxygen tension, suggesting that aerobic conditions bordering on moderate hypoxia are theoretically sufficient to support OXPHOS and de novo pyrimidine synthesis (Löffler, 1989; Amellem et al., 1994).
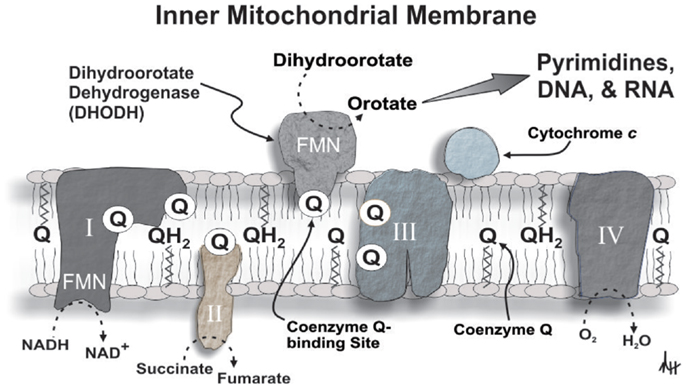
Figure 2. A diagrammatic depiction of DHODH in the inner mitochondrial membrane illustrating its role in mitochondrial bioenergetics and de novo pyrimidine synthesis. Please refer to the text for additional details (abbreviations: I, complex I; II, complex II; III, complex III; IV, complex IV; FMN, flavin mononucleotide).
Third, although OXPHOS is metabolically more efficient than glycolysis in terms of ATP generation, glycolysis occurs in the cytosol which typically represents >70% of the cell volume (Luby-Phelps, 2000) and thus has the potential of matching net ATP contributions from OXPHOS which occurs in the smaller volume of the mitochondrial matrix and inner membrane. In addition, OXPHOS depends on adequate amounts of oxygen and may not be sustainable under high rates of electron transport in rapidly dividing cells. Furthermore, oxygen levels are markedly reduced in the leukemic bone marrow niche (Benito et al., 2011). In discussing this latter point, it is important to consider that although glycolysis depends on the availability of NAD+ for the reaction catalyzed by glyceraldehyde-3-phosphate dehydrogenase, limiting the availability of oxygen – the canonical final acceptor of electrons from NADH – may not be critical as this oxidized cofactor can be readily formed by the fermentation of pyruvate to lactate. As such, depending on glycolysis instead of OXPHOS becomes a distinct advantage for proliferating leukemia cells in the hypoxic bone marrow niche.
Fourth, reduced dependence on OXPHOS for ATP generation favors the use of Krebs cycle intermediates for biosynthetic reactions. For instance, citrate cataplerosis from the Krebs cycle provides cytosolic acetyl-CoA for de novo fatty acid synthesis (FAS), and succinyl-CoA cataplerosis from the cycle provides carbon skeletons for the synthesis of heme groups (Berg et al., 2002) (Figure 1). These cataplerotic reactions of the Krebs cycle not only provide intermediates for biosynthesis, but when followed by anaplerotic reactions to regenerate oxaloacetate, also reduce the amount of NADH generated in the cycle. One should consider that citrate cataplerosis followed by α-ketoglutarate anaplerosis via glutaminolysis produces one less NADH than if citrate continued in the cycle; similarly, succinyl-CoA cataplerosis followed by oxaloacetate anaplerosis via transamination of aspartate or carboxylation of pyruvate produces one less NADH and one less FADH2 than if succinyl-CoA continued in the cycle. Taken together, it is intriguing to postulate that a “leaky” or truncated Krebs cycle can bypass in large part the requirement for sustained oxygen levels while at the same time providing critical intermediates for biosynthetic reactions.
Lastly, seminal work by Bonnet et al. (2007) demonstrated that in solid tumor cell lines the mitochondrial oxidation of pyruvate – induced pharmacologically by dichloroacetate (DCA) – promotes ROS generation and cell death, suggesting that oxidizing other carbon sources different from glucose may ipso facto be favorable in terms of cell survival. While there is no experimental evidence of a similar toxic effect of pyruvate oxidation in leukemia cells, it is important to consider the possibility that leukemia cells may also forgo pyruvate oxidation to maintain cell viability.
Regulatory Adaptations
Leukemia cells must have acquired mechanisms to bypass the inhibitory effect of ATP accumulation on glycolysis, particularly at the point of phosphofructokinase 1 (PFK-1), the canonical regulator of glycolysis that carries out the conversion of fructose-6-phosphate into fructose-1,6-bisphosphate. Recent evidence suggests that in lymphoma (Nb2-11) and leukemia (TF-1) cell lines, PFK-1 has been post-translationally modified via a proteinase K-type of mechanism resulting in a shorter polypeptide which maintains catalytic activity, but is missing the allosteric sites required for inhibition by citrate, ATP, and phosphoenolpyruvate (Smerc et al., 2011). Intriguingly, the truncated enzyme is super-activated by fructose-2,6-bisphosphate (Smerc et al., 2011). Although similar modifications have not been reported in primary leukemia samples, it is intriguing to speculate that bypassing inhibitory effects of intracellular ATP accumulation could generate a critical growth advantage of leukemia vs. normal progenitors.
In addition to resisting the inhibitory effects of ATP accumulation on glycolysis, leukemia cells also use mitochondria-derived ATP to accelerate the rate of glucose activation and hence glycolysis. Over two decades ago, Arany et al. (1988) reported that the glycolytic enzyme hexokinase promoted oxygen consumption in leukemia cells when it associated with the outer mitochondrial membrane, suggesting perhaps a novel link between glycolysis and electron transport. More recent evidence (reviewed in Mathupala et al., 2010) indeed supports the notion that a particular isoform of hexokinase (HK), HKII associates with mitochondria to attain direct access to OXPHOS generated ATP for the phosphorylation of glucose. Additionally, the kinase AKT – a known activator of glycolysis in leukemia cells – phosphorylates HKII and is thus promoting its localization to the outer mitochondrial membrane (Majewski et al., 2004a,b). Phosphorylated HKII associates with VDAC, the outer mitochondrial membrane anion channel that allows entry of most metabolites, but only permits egress of pro-apoptotic proteins when the intrinsic pathway of programed cell death is activated (reviewed in Shoshan-Barmatz et al., 2010; Shoshan-Barmatz and Ben-Hail, 2012). Since mitochondria-bound HKII inhibits apoptosis in leukemia and lymphoma cell lines (Chen et al., 2009), it is intriguing to speculate that HKII prevents cytochrome c release by blocking the channel space in VDAC (Abu-Hamad et al., 2008), although conflicting reports suggest that the antiapoptotic effect of HKII may be instead dependent on the presence and/or activity of Cyclophilin D and ANT (Chiara et al., 2008). Regardless of the precise mechanisms, it is clear that HKII critically links mitochondrial metabolism, glycolysis, and survival in leukemia cells.
Mitochondrial Uncoupling: Lessons from the Microenvironment
In 1956, Otto Warburg acknowledged an alternative hypothesis put forth by Feodor Lynen suggesting that the increased dependence of cancer cells on glycolysis stems not from their inability to reduce oxygen, but rather from their inability to synthesize ATP in response to the mitochondrial proton gradient [ΔΨM (Warburg, 1956)]. Lynen’s hypothesis was based on his own observations (Lynen, 1951) and previous work by Ronzoni and Ehrenfest (1936) using the prototypical protonophore 2,4-dinitrophenol (2,4-DNP), which causes a “short circuit” in the electrochemical gradient that abolishes the mitochondrial synthesis of ATP, and decreases the entry of pyruvate into the Krebs cycle. Since Ronzoni and Ehrenfest (1936) observed that 2,4-DNP induced an increase in oxygen consumption, and at the same time an increase in pyruvate conversion to lactate, it must have been tempting to hypothesize (although they did not) that electrons for the molecular reduction of oxygen must be coming from carbon sources different from glucose. Recent work has supported this hypothesis by demonstrating that the abrogation of ATP synthesis in response to ΔΨM (“mitochondrial uncoupling”) results in a preferential oxidation of non-glucose carbon sources to maintain mitochondrial function and an increase in lactate generation under aerobic conditions (Samudio et al., 2008, 2010; Sheets et al., 2008).
In leukemia cells mitochondrial uncoupling has been reported to recapitulate the Warburg effect and support a shift toward fatty acid oxidation (FAO) (Samudio et al., 2008, 2010). On one hand, it is important to point out that FAO induces an uncoupling and/or thermogenic phenotype in various cell types (reviewed in Gambert and Ricquier, 2007); on the other hand it is also evident that increased FAO during mitochondrial uncoupling may require anaplerotic reactions – in particular pyruvate carboxylation, glutaminolysis, and aspartate transamination – to replenish oxaloacetate for efficient Krebs cycle use of fatty acid-derived acetyl-CoA (Goodwin and Taegtmeyer, 2000). Indeed glutaminolysis has been reported in lymphoma cell lines (Le et al., 2012), and unpublished observations from our group suggest that glutamine supports oxygen consumption in leukemia cells. Taken together, the above data support the concept – and indirectly, Lynen’s hypothesis – that the Warburg effect could be the result of glutaminolysis-dependent FAO.
Mitochondrial Uncoupling and Cell Survival: The Role of Bcl-2
In leukemia cells mitochondrial uncoupling promotes resistance to intrinsic apoptosis via, in part, antagonism of Bax/Bak oligomerization (Samudio et al., 2010). Uncoupling and chemoresistance are exacerbated in leukemia cells cultured on mesenchymal stromal feeder layers (Samudio et al., 2008), and previous reports indicate that stromal coculture upregulates Bcl-2 and Bcl-xL in leukemia cells (Konopleva et al., 2002) suggesting that the leukemia microenvironment promotes chemoresistance via, in part, by potentiation of the antiapoptotic function and/or expression of Bcl-2 family members. In addition, since mitochondrial uncoupling in leukemia cells is associated with a shift toward FAO and away from glucose oxidation (Samudio et al., 2010), it is tempting to speculate that the leukemia microenvironment also signals leukemia cells to spare glucose carbon skeletons for the generation of biomass (Samudio et al., 2009). Notably, pharmacologic inhibition of FAO was cytotoxic and chemosensitizing to leukemia cells cultured on bone marrow stromal feeder layers, promoting Bax/Bak oligomerization (Samudio et al., 2010). Most importantly, pharmacologic inhibition of FAO demonstrated a therapeutic benefit in combination with chemotherapy in mouse models of human leukemia (Samudio et al., 2010), suggesting that the shift toward FAO is a bona fide target for the treatment of hematological malignancies. Nevertheless, it is unclear whether FAO or electron transport per se antagonizes apoptosis in leukemia cells. In addition, the mechanisms by which FAO and/or electron transport regulate Bax/Bak oligomerization remain to be elucidated.
Recent work has suggested that Bcl-2 overexpression defines a subset of OXPHOS-dependent, ROS-low, quiescent leukemia stem cells (LSC), in which pharmacologic or genetic inhibition of Bcl-2 rapidly diminishes oxygen consumption capacity and increases ROS generation prior to the onset of apoptosis (Lagadinou et al., 2013). Although the precise mechanisms by which pharmacologic inhibition of Bcl-2 antagonize electron transport in LSC remain unclear, it is known that Bcl-2 displays an antioxidant function in mitochondria by facilitating the import of reduced glutathione into the mitochondrial matrix (Wilkins et al., 2012), and by directly reducing ROS generation (Low et al., 2010). In addition, Bcl-2 protects leukemic cells against mitochondrial uncoupling-induced apoptosis (Armstrong et al., 2001), and multiple other cell types against palmitate-induced apoptosis (de Pablo et al., 1999; Kui et al., 2009), suggesting that Bcl-2 may protect cells under metabolic overload of the electron transport chain. In light of the above, it is tempting to speculate that dependence on OXPHOS or increased rates of electron transport – such as observed in mitochondrial uncoupling – necessitates the accompaniment of Bcl-2 to safeguard mitochondrial integrity. It is of utmost importance to elucidate the precise molecular events that connect Bcl-2, apoptosis, and electron transport in leukemia cells in order to develop effective therapies to eradicate the elusive LSC.
The Hypoxic Bone Marrow Niche and Metabolism: The Reverse Krebs Cycle
It is intriguing that in the bone marrow, although it constitutes a highly vascularized sinusoidal cavity, the oxygen concentration appears to be similar to that found in venous blood (i.e., 6%) (Fiegl et al., 2009) or even lower in the stem cell niche (i.e., 1%) (Benito et al., 2011), suggesting the notion that the bone marrow is ipso facto hypoxic, at least physiologically. However, if leukemia cell mitochondria are indeed uncoupled in vivo, the mechanism by which oxygen tension is reduced in the bone marrow may be directly related to the increased oxygen reducing capacity of leukemia cells. Still, this presents an interesting paradox: the reduced oxygen tension would eventually lead to an inhibition of Krebs cycle activity – via accumulation of NADH – and the reduced generation of ATP and biomass. Or will it? Intriguing recent research has suggested that under hypoxic conditions the Krebs cycle can actually go in reverse, starting from glutamine-derived α-ketoglutarate to form citrate via reductive carboxylation, to support the de novo synthesis of fatty acids (Wise et al., 2011; Metallo et al., 2012). Although no evidence of this effect has been reported in leukemia cells under hypoxia (Figure 2 speculates on the possibility of this event), it is tempting to consider that reductive carboxylation of α-ketoglutarate may rescue biomass generation from the Krebs cycle in leukemia cells under reduced oxygen tension, whether this resulted from cell autonomous consumption of available oxygen, or from yet unidentified anatomical and/or microenvironmental effects. Likewise, it is intriguing to consider that if reductive carboxylation of α-ketoglutarate is accompanied by increased conversion of NADH to NADPH (via NADH kinase), it may also represent a rescue mechanism against the inhibitory effects of NADH accumulation on the Krebs cycle under conditions of reduced oxygen partial pressure. Nevertheless, the reverse Krebs cycle faces some theoretical hurdles that would have to be addressed. First, it is unclear if sufficient amounts of α-ketoglutarate, , and NADPH (reductive carboxylation of α-ketoglutarate requires NADPH via mitochondrial IDH2) are available in the mitochondrial matrix to promote the thermodynamically spontaneous formation of isocitrate. Second, it is kinetically improbable that isocitrate dehydrogenase, an enzyme that does not contain biotin, can catalyze carboxylation of α-ketoglutarate to form isocitrate. Perhaps a human ortholog of 2-oxoglutarate carboxylase (found in a variety of microorganisms; Reaction: ATP + 2-oxoglutarate + = ADP + phosphate + oxalosuccinate; EC 6.4.1.7) that can more readily catalyze carboxylation of α-ketoglutarate (2-oxoglutarate) into oxalosuccinate, which is then converted to isocitrate by IDH2, could account for the observed reversal of the Krebs cycle. However, whether this enzyme activity is expressed in leukemia cells, and whether leukemia cells can in fact generate isocitrate from – ketoglutarate, remains to be determined.
Concluding Remarks: Warburg and Weinhouse Reconcile
A preponderance of evidence supports Warburg’s observation that cancer cells rely on glycolysis to meet their energy demands, although common, generalized defects in mitochondrial oxidative capacity have yet to be identified for the vast majority of cancers. In fact, more recent evidence has confirmed that cancer cells maintain the ability to oxidize pyruvate, albeit under conditions that were pharmacologically induced by DCA. Nevertheless, the concomitant ROS generation caused by this process is conceivably cytotoxic and, as such, could impinge on cell viability (Bonnet et al., 2007). At first, it may seem intriguing that Warburg’s hypothesis – that an irreparable damage to the oxygen reduction capacity of tumor cells promotes the origin of cancer – survived for nearly 50 years, especially as experimental evidence generated by contemporary biochemist Sydney Weinhouse demonstrated that tumor cells consumed oxygen at a rate similar, or higher than, their normal counterparts (reviewed in Weinhouse, 1972). Additionally, to biochemistry students familiar with the amphibolic importance of the Krebs cycle, and its apparent dependence on oxygen to accept electrons from NADH for it normal “clockwise” functioning, it must seem like an obvious conclusion that proliferating cells – tumor or normal – require an intact oxygen reduction capacity to generate important metabolic intermediates such as citrate, α-ketoglutarate, succinyl-CoA, and oxaloacetate. However, one must remember that the Krebs cycle was unknown to Warburg for nearly 10 years after his initial observations of respiratory impairment of cancer cells, when his former research assistant, Sir Hans Adolf Krebs, published two seminal papers that described the citric acid cycle (Krebs and Johnson, 1937; Krebs et al., 1938). Moreover, it then took several decades beyond that to fully understand the amphibolic nature of the Krebs cycle. In the end, Warburg’s imposing personality and his outstanding achievements ensured that his hypothesis survived – nearly unchallenged if not for Weinhouse – for almost 50 years.
Can the observations of Weinhouse and Warburg be reconciled? The evidence presented here suggests that, at least in leukemia cells, they can. On one hand, our recent data support Feodor Lynen’s hypothesis that increased lactate generation can be promoted under normal oxygen tension through mitochondrial uncoupling – induced by coculture with bone marrow mesenchymal stromal cells – in the absence of permanent, transmissible defects in oxygen consumption capacity. On the other hand, our observation that leukemia cells sustain molecular oxygen reduction rates utilizing electrons derived from FAO is in agreement with the work of Weinhouse utilizing transplantable hepatomas (Bloch-Frankenthal et al., 1965). Indeed, it would appear that Warburg and Weinhouse were looking at two sides of the same coin: aerobic glycolysis and FAO, which are elegantly connected by Feodor Lynen’s mitochondrial uncoupling theory.
While is important to point out that this review has focused predominantly in hematological malignancies, our data parallels recent observations in solid tumors where glutamine-dependent, glucose-independent Krebs cycle activity has been reported in glioblastoma and melanoma cells (Scott et al., 2011). In addition, our observation that mitochondrial uncoupling in leukemia cells is associated with increased resistance to apoptosis is consistent with the work of Derdak et al. (2008) who demonstrated a similar paradigm in colon cancer cells. As such, we hypothesize that our model may indeed be broadly applicable to other tumor types since there is presently insufficient evidence to suggest otherwise.
Based on analysis of the literature and our own data, we propose a paradigm (Figure 3) for mitochondrial uncoupling in hematological malignancies built on four salient foundations: (1) mitochondrial uncoupling promotes an adequate energy balance through aerobic glycolysis, and sustains biomass generation through cataplerotic reactions from the Krebs cycle; (2) mitochondrial uncoupling can orchestrate functional changes in mitochondria that accompany resistance to intrinsic apoptotic stimuli, mediated, in part, by modulation of the Bcl-2 rheostat; (3) mitochondrial uncoupling is regulated by, and regulates the bone marrow microenvironment, perhaps contributing to cellular hypoxia; and (4) mitochondrial uncoupling depends on anaplerotic reactions to regenerate oxaloacetate in the Krebs cycle to support fatty acid-dependent oxygen consumption. Whether or not this paradigm can be targeted for therapeutic benefit in patients with leukemia will largely depend on a thorough understanding of the limiting reactions that support the altered metabolic behavior of leukemia cells.
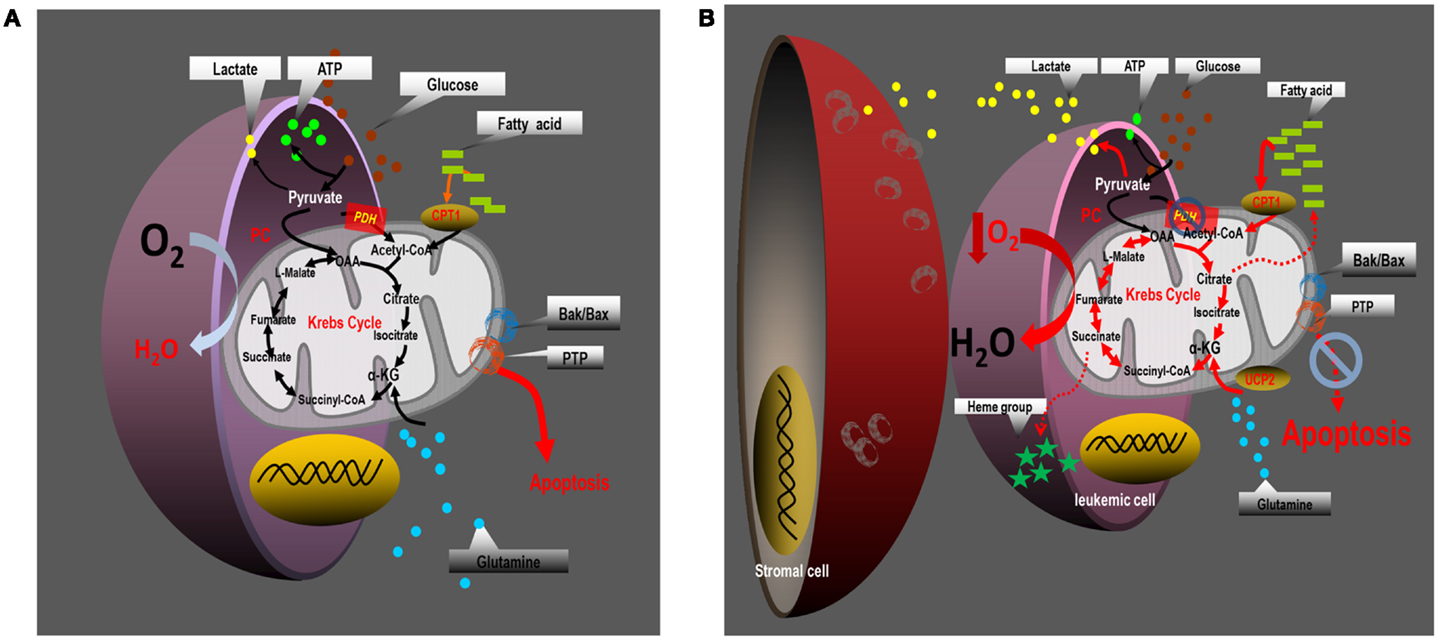
Figure 3. Proposed paradigm for the reprograming of intermediary metabolism in leukemia cells. (A) Leukemia cells cultured alone engage in aerobic glycolysis, and maintain an intact Krebs cycle which is capable of oxidizing pyruvate-derived acetyl-CoA, fatty acid-derived acetyl-CoA, and possibly glutamine carbon skeletons. Their energy demands are met through glycolysis and perhaps to some extent through mitochondrial OXPHOS, although mitochondria may display a proclivity for uncoupling electron transport from ATP synthesis. In monoculture, leukemia cells are most sensitive to apoptosis induction. (B) Coculture of leukemia cells with bone marrow-derived stromal cells promotes mitochondrial uncoupling. Under coculture conditions leukemia cells augment aerobic glycolysis and increase Krebs cycle activity in a manner dependent on anaplerotic reactions – in particular glutaminolysis, and to a lesser extent pyruvate carboxylation. The increased Krebs cycle activity does not oxidize pyruvate-derived acetyl-CoA, but instead metabolizes large quantities of fatty acid-derived acetyl-CoA to sustain increased rates of oxygen consumption, that may contribute to the generation of local hypoxic conditions. Mitochondrial uncoupling in leukemia cells is associated with increased expression of UCP-2, and increased resistance to apoptotic stimuli that induce Bax/Bak-dependent MOMP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funds from the Oficina para el Fomento de la Investigación, Pontificia Universidad Javeriana (OFI; Project ID 4049) (to Ismael Samudio); a grant from the Departamento Administrativo de Ciencia y Tecnologia COLCIENCIAS (Project ID 120351929072) (to Ismael Samudio); a doctoral scholarship from COLCIENCIAS (to Juliana Vélez); by the National Institutes of Health grants CA155056, CA100632, and LLSA 2189-12 (to Marina Konopleva), CA55164, CA016672, CA100632, CA136411, and CA049639, and the Paul and Mary Haas Chair in Genetics (to Michael Andreeff).
References
Abu-Hamad, S., Zaid, H., Israelson, A., Nahon, E., and Shoshan-Barmatz, V. (2008). Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 283, 13482–13490.
Amellem, O., Löffler, M., and Pettersen, E. O. (1994). Regulation of cell proliferation under extreme and moderate hypoxia: the role of pyrimidine (deoxy)nucleotides. Br. J. Cancer 70, 857–866.
Arany, I., Ember, I., and Rady, P. (1988). Subcellular distribution of hexokinase in leukemic and stimulated lymphoid cells of mice. Haematologia (Budap.) 21, 109–114.
Armstrong, J. S., Steinauer, K. K., French, J., Killoran, P. L., Walleczek, J., Kochanski, J., et al. (2001). Bcl-2 inhibits apoptosis induced by mitochondrial uncoupling but does not prevent mitochondrial transmembrane depolarization. Exp. Cell Res. 262, 170–179.
Benito, J., Shi, Y., Szymanska, B., Carol, H., Boehm, I., Lu, H., et al. (2011). Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS ONE 6:e23108. doi:10.1371/journal.pone.0023108
Beuneu, C., Auger, R., Loffler, M., Guissani, A., Lemaire, G., and Lepoivre, M. (2000). Indirect inhibition of mitochondrial dihydroorotate dehydrogenase activity by nitric oxide. Free Radic. Biol. Med. 28, 1206–1213.
Bloch-Frankenthal, L., Langan, J., Morris, H. P., and Weinhouse, S. (1965). Fatty acid oxidation and ketogenesis in transplantable liver tumors. Cancer Res. 25, 732–736.
Bonnet, S., Archer, S. L., Allalunis-Turner, J., Haromy, A., Beaulieu, C., Thompson, R., et al. (2007). A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37–51.
Chen, Z., Zhang, H., Lu, W., and Huang, P. (2009). Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. Biophys. Acta 1787, 553–560.
Chiara, F., Castellaro, D., Marin, O., Petronilli, V., Brusilow, W. S., Juhaszova, M., et al. (2008). Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE 3:e1852. doi:10.1371/journal.pone.0001852
de Pablo, M. A., Susin, S. A., Jacotot, E., Larochette, N., Costantini, P., Ravagnan, L., et al. (1999). Palmitate induces apoptosis via a direct effect on mitochondria. Apoptosis 4, 81–87.
DeBerardinis, R. J. (2011). Serine metabolism: some tumors take the road less traveled. Cell Metab. 14, 285–286.
DeBerardinis, R. J., Mancuso, A., Daikhin, E., Nissim, I., Yudkoff, M., Wehrli, S., et al. (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350.
Derdak, Z., Mark, N. M., Beldi, G., Robson, S. C., Wands, J. R., and Baffy, G. (2008). The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 68, 2813–2819.
Evans, D. R., and Guy, H. I. (2004). Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem. 279, 33035–33038.
Fiegl, M., Samudio, I., Clise-Dwyer, K., Burks, J. K., Mnjoyan, Z., and Andreeff, M. (2009). CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 113, 1504–1512.
Folmes, C. D., Dzeja, P. P., Nelson, T. J., and Terzic, A. (2012). Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11, 596–606.
Forman, J. H., and Kennedy, J. (1975). Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. J. Biol. Chem. 250, 4322–4326.
Gambert, S., and Ricquier, D. (2007). Mitochondrial thermogenesis and obesity. Curr. Opin. Clin. Nutr. Metab. Care 10, 664–670.
Goodwin, G. W., and Taegtmeyer, H. (2000). Improved energy homeostasis of the heart in the metabolic state of exercise. Am. J. Physiol. Heart Circ. Physiol. 279, H1490–1501.
Hail, N. Jr., Chen, P., Rower, J., and Bushman, L. R. (2010a). Teriflunomide encourages cytostatic and apoptotic effects in premalignant and malignant cutaneous keratinocytes. Apoptosis 15, 1234–1246.
Hail, N. Jr., Chen, P., and Wempe, M. F. (2010b). The hydroxyl functional group of N-(4-hydroxyphenyl)retinamide mediates cellular uptake and cytotoxicity in premalignant and malignant human epithelial cells. Free Radic. Biol. Med. 49, 2001–2009.
Hail, N. Jr., and Lotan, R. (2009). Cancer chemoprevention and mitochondria: targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Mol. Nutr. Food Res. 53, 49–67.
Konopleva, M., Konoplev, S., Hu, W., Zaritskey, A. Y., Afanasiev, B. V., and Andreeff, M. (2002). Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 16, 1713–1724.
Krebs, H. A., and Johnson, W. A. (1937). Acetopyruvic acid (alphagamma-diketovaleric acid) as an intermediate metabolite in animal tissues. Biochem. J. 31, 772–779.
Krebs, H. A., Salvin, E., and Johnson, W. A. (1938). The formation of citric and alpha-ketoglutaric acids in the mammalian body. Biochem. J. 32, 113–117.
Kui, L., Weiwei, Z., Ling, L., Daikun, H., Guoming, Z., Linuo, Z., et al. (2009). Ghrelin inhibits apoptosis induced by high glucose and sodium palmitate in adult rat cardiomyocytes through the PI3K-Akt signaling pathway. Regul. Pept. 155, 62–69.
Lagadinou, E. D., Sach, A., Callahan, K., Rossi, R. M., Neering, S. J., Minhajuddin, M., et al. (2013). BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341.
Lakaschus, G., Krüger, H., Heese, D., and Löffler, M. (1991). Evidence from in vitro studies that dihydroorotate dehydrogenase may be a source of toxic oxygen species. Adv. Exp. Med. Biol. 309A, 361–364.
Le, A., Lane, A. N., Hamaker, M., Bose, S., Gouw, A., Barbi, J., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121.
Lenaz, G. (2001). The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52, 159–164.
Löffler, M. (1989). The biosynthetic pathway of pyrimidine (deoxy)nucleotides: a sensor of oxygen tension necessary for maintaining cell proliferation? Exp. Cell Res. 182, 637–680.
Low, I. C., Chen, Z. X., and Pervaiz, S. (2010). Bcl-2 modulates resveratrol-induced ROS production by regulating mitochondrial respiration in tumor cells. Antioxid. Redox Signal. 13, 807–819.
Luby-Phelps, K. (2000). Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 192, 189–221.
Lynen, F. (1951). Die Rolle der Phosphorsaeure bei Dehydrierungsvorgaengen und ihre biologische Bedeutung. Naturwissenschaften 30, 398–406.
Majewski, N., Nogueira, V., Bhaskar, P., Coy, P. E., Skeen, J. E., Gottlob, K., et al. (2004a). Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 16, 819–830.
Majewski, N., Nogueira, V., Robey, R. B., and Hay, N. (2004b). Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 24, 730–740.
Mathupala, S. P., Ko, Y. H., and Pedersen, P. L. (2010). The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 1797, 1225–1230.
Metallo, C. M., Gameiro, P. A., Bell, E. L., Mattaini, K. R., Yang, J., Hiller, K., et al. (2012). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384.
Ringshausen, I., Oelsner, M., Bogner, C., Peschel, C., and Decker, T. (2008). The immunomodulatory drug Leflunomide inhibits cell cycle progression of B-CLL cells. Leukemia 22, 635–638.
Ronzoni, E., and Ehrenfest, E. (1936). The effect of dinitrophenol on the metabolism of frog muscle. J. Biol. Chem. 15, 749.
Rückemann, K., Fairbanks, L. D., Carrey, E. A., Hawrylowicz, C. M., Richards, D. F., Kirschbaum, B., et al. (1998). Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J. Biol. Chem. 273, 21682–21691.
Samudio, I., Fiegl, M., and Andreeff, M. (2009). Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 69, 2163–2166.
Samudio, I., Fiegl, M., McQueen, T., Clise-Dwyer, K., and Andreeff, M. (2008). The Warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 68, 5198–5205.
Samudio, I., Harmancey, R., Fiegl, M., Kantarjian, H., Konopleva, M., Korchin, B., et al. (2010). Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Invest. 120, 142–156.
Sawamukai, N., Saito, K., Yamaoka, K., Nakayamada, S., Ra, C., and Tanaka, Y. (2007). Leflunomide inhibits PDK1/Akt pathway and induces apoptosis of human mast cells. J. Immunol. 179, 6479–6484.
Scott, D. A., Richardson, A. D., Filipp, F. V., Knutzen, C. A., Chiang, G. G., Ronai, Z. A., et al. (2011). Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J. Biol. Chem. 286, 42626–42634.
Shawver, L. K., Schwartz, D. P., Mann, E., Chen, H., Tsai, J., Chu, L., et al. (1997). Inhibition of platelet-derived growth factor-mediated signal transduction and tumor growth by N-[4-(trifluoromethyl)-phenyl]5-methylisoxazole-4-carboxamide. Clin. Cancer Res. 3, 1167–1177.
Sheets, A. R., Fulop, P., Derdak, Z., Kassai, A., Sabo, E., Mark, N. M., et al. (2008). Uncoupling protein-2 modulates the lipid metabolic response to fasting in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1017–G1024.
Shoshan-Barmatz, V., and Ben-Hail, D. (2012). VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12, 24–34.
Shoshan-Barmatz, V., De Pinto, V., Zweckstetter, M., Raviv, Z., Keinan, N., and Arbel, N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285.
Smerc, A., Sodja, E., and Legisa, M. (2011). Posttranslational modification of 6-phosphofructo-1-kinase as an important feature of cancer metabolism. PLoS ONE 6:e19645. doi:10.1371/journal.pone.0019645
Traut, T. W. (1994). Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22.
Warburg, O., Posener, K., and Negelein, E. (1924). Über den Stoffwechsel der Tumoren. Biochem. Z. 152, 319–344.
Weinhouse, S. (1972). Glycolysis, respiration, and anomalous gene expression in experimental hepatomas: G.H.A. Clowes memorial lecture. Cancer Res. 32, 2007–2016.
Wilkins, H. M., Marquardt, K., Lash, L. H., and Linseman, D. A. (2012). Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Radic. Biol. Med. 52, 410–419.
Wise, D. R., Ward, P. S., Shay, J. E., Cross, J. R., Gruber, J. J., Sachdeva, U. M., et al. (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U.S.A. 108, 19611–19616.
Keywords: Krebs cycle, mitochondrial uncoupling, electron transport, OXPHOS, apoptosis, anaplerosis, cataplerosis, microenvironment
Citation: Vélez J, Hail Jr. N, Konopleva M, Zeng Z, Kojima K, Samudio I and Andreeff M (2013) Mitochondrial uncoupling and the reprograming of intermediary metabolism in leukemia cells. Front. Oncol. 3:67. doi: 10.3389/fonc.2013.00067
Received: 21 February 2013; Paper pending published: 07 March 2013;
Accepted: 14 March 2013; Published online: 02 April 2013.
Edited by:
Jozsef Dudas, Medical University Innsbru, AustriaReviewed by:
Daniel Christian Hoessli, International Center for Chemical and Biological Sciences, SwitzerlandJozsef Dudas, Medical University Innsbruck, Austria
Copyright: © 2013 Vélez, Hail Jr., Konopleva, Zeng, Kojima, Samudio and Andreeff. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Ismael Samudio, Laboratorio de Bioquimica #301, Edificio Jesús Emilio Ramírez, Pontificia Universidad Javeriana, Cra 7 No. 43–82, Bogotá, Colombia. e-mail:c2FtdWRpb2lAamF2ZXJpYW5hLmVkdS5jbw==;
Michael Andreeff, Departments of Leukemia and Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd., Unit 448, Houston, TX 77030, USA. e-mail:bWFuZHJlZWZAbWRhbmRlcnNvbi5vcmc=