


- 1Department of Stem Cell Biology and Regenerative Medicine, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, USA
- 2Department of Molecular Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland Clinic, Cleveland, OH, USA
- 3Department of Pathology, School of Medicine, Case Western Reserve University, Cleveland, OH, USA
- 4Department of Radiation Oncology, Cleveland Clinic, Cleveland, OH, USA
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults with a median survival of 12–15 months with treatment consisting of surgical resection followed by ionizing radiation (IR) and chemotherapy. Even aggressive treatment is often palliative due to near universal recurrence. Therapeutic resistance has been linked to a subpopulation of GBM cells with stem cell-like properties termed GBM initiating cells (GICs). Recent efforts have focused on elucidating resistance mechanisms activated in GICs in response to IR. Among these, GICs preferentially activate the DNA damage response (DDR) to result in a faster rate of double-strand break (DSB) repair induced by IR as compared to the bulk tumor cells. IR also activates NOTCH and the hepatic growth factor (HGF) receptor, c-MET, signaling cascades that play critical roles in promoting proliferation, invasion, and resistance to apoptosis. These pathways are preferentially activated in GICs and represent targets for pharmacologic intervention. While IR provides the benefit of improved survival, it paradoxically promotes selection of more malignant cellular phenotypes of GBM. As reviewed here, finding effective combinations of radiation and molecular inhibitors to target GICs and non-GICs is essential for the development of more effective therapies.
Introduction
Glioblastoma (GBM) is the most common and aggressive type of primary brain cancer in adults with approximately 18,000 patients diagnosed each year [(http://www.CBTRUS.org); Schwartzbaum et al., 2006]. GBM can arise as de novo (primary) cancer or may progress from lower grade gliomas (secondary). Despite aggressive multimodality treatment consisting of maximal safe resection, adjuvant chemoradiation with temozolomide, and maintenance temozolomide, median survival remains dismal at 12–15 months (Stupp et al., 2009). Patients typically respond initially to therapy, but ultimately relapse within the high-dose irradiation field (Hochberg and Pruitt, 1980; Lee et al., 1999), suggesting the presence of a subpopulation of resistant cells. While intertumoral heterogeneity between patients can, in part, explain differential patient responses (Maher et al., 2006; Phillips et al., 2006; Dang et al., 2009; Yan et al., 2009; Snuderl et al., 2011), intratumoral heterogeneity is now recognized as a critical factor in determining therapeutic response (Bao et al., 2006; Liu et al., 2006). GBM initiating cells (GICs) are a subgroup of cancer cells that exhibit the ability to self-renew and express putative stem cell markers such as CD133, SSEA-1 (CD15), L1CAM, and CD44high (Galli et al., 2004; Singh et al., 2004; Bao et al., 2008; Son et al., 2009; Anido et al., 2010). GICs are defined functionally by their ability to repopulate the tumor upon serial transplantation (Ignatova et al., 2002; Singh et al., 2003, 2004; Galli et al., 2004). When non-GICs are assayed in parallel, these cells fail to form tumors, even when their numbers are increased by orders of magnitude. Therefore, tumor recurrence is likely due to tumorigenic GICs equipped with resistance mechanisms to survive and proliferate following therapy (Figure 1A).
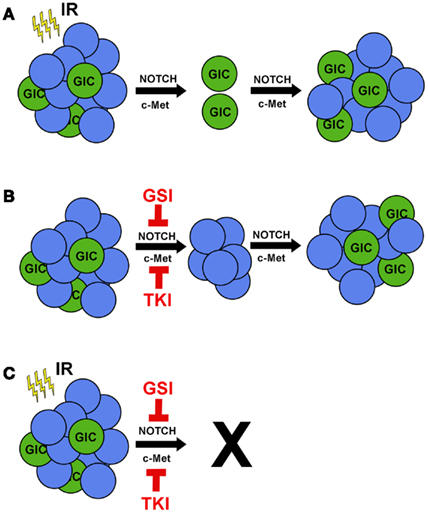
Figure 1. Ionizing radiation in combination with c-MET or NOTCH inhibitors prevents tumor recurrence. (A) Treating GBM with IR reduces tumor volume, but radioresistant GICs remain. IR promotes activation of the pro-survival pathways NOTCH and c-MET in GICs, leading to tumor recurrence. (B) Single treatment of GBM tumors with either gamma secretase inhibitors (GSIs) to target NOTCH or tyrosine kinase inhibitors (TKIs) to target c-MET would kill GICs specifically and have a minor effect on tumor volume. (C) Combinatorial treatment of GSIs or TKIs with IR would target both GICs and non-GICs and prevent tumor recurrence.
The factors that influence stem-like characteristics are more complex than previously recognized. Recently, studies have revealed the microenvironmental effects of hypoxia, low glucose, low pH, and perivascular niches in promoting GIC survival, maintenance, and cellular plasticity (Gatenby and Gillies, 2004; Calabrese et al., 2007; Heddleston et al., 2009; Soeda et al., 2009; Anido et al., 2010; Charles and Holland, 2010; Seidel et al., 2010; Zhu et al., 2011). For example, hypoxia has been shown to drive expression of stem cell genes and increase the tumorigenic capacity of GICs, particularly through hypoxia inducible factors (Heddleston et al., 2009; Soeda et al., 2009; Seidel et al., 2010). These effects were also seen in acidic conditions regardless of oxygen concentration (Hjelmeland et al., 2011). Under these conditions, non-GBM initiating cells (non-GICs) can assume stem-like features and initiate tumor formation in vivo (Heddleston et al., 2009; Hjelmeland et al., 2011), underscoring the plasticity of GBM cells (Figure 1B). Notably, many of these pro-GIC signaling components, such as c-MET and NOTCH, are activated by radiotherapy (Wang et al., 2010; Joo et al., 2012).
Exposure to ionizing radiation (IR) elicits a preferential activation of the DNA damage response (DDR) pathway, along with enhanced DNA repair kinetics in GICs compared to their non-GIC counterparts (Bao et al., 2006). These data suggest that GICs are better able to activate the DDR in response to genotoxic stress. Radiation causes extensive cellular damage, primarily through generation of reactive oxygen species leading to DNA double-strand breaks (DSBs). Activation of the DDR signaling cascade elicits a host of cellular responses including cell cycle regulation, DNA repair, autophagy, mitotic catastrophe, necrosis, senescence, and apoptosis. Moreover, irradiated (Bao et al., 2006) and temozolomide-treated (Firat et al., 2011) GICs have a lower percentage of apoptotic cells than their non-GIC counterparts, highlighting their intrinsic therapeutic resistance (Figure 1A). This expansion of GICs has been confirmed by histological analysis of recurrent GBM after initial treatment with chemoradiation at the time of salvage surgery (Tamura et al., 2010). Many, although not all, clinical trials have failed to show a benefit to radiation dose-escalation (Chan et al., 2002), radiosurgery boost (Souhami et al., 2004), or brachytherapy boost (Laperriere et al., 1998; Selker et al., 2002). Taken together, these studies suggest that GICs can overcome even high doses of radiation (Figure 1A). While traditional therapy may initially reduce the bulk of the tumor by targeting non-GICs, it ultimately selects for the outgrowth of a more aggressive tumor through expansion of GICs. This manifests as clinical and/or radiographic progression within several months.
Activation of the DNA Damage Response Pathway
Genotoxic stressors, including oncogenic stressors, induce DNA damage and activate the DDR pathway. The DDR pathway is a signaling cascade with multiple sensor, transducer, and effector proteins. Two such transducers are the serine/threonine protein kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related protein (ATR). ATM and ATR are members of the phosphatidylinositol 3-kinase (PI3K) family and are key regulators of DSB repair (Matsuoka et al., 2007). Upon DNA breakage, ATM senses the damage and the MRE11-RAD50-NBS1 (MRN) complex is recruited to the damaged site to accelerate phosphorylation of inactive ATM dimers. These dimers then dissociate and each phosphorylated ATM monomer further activates the protein by auto-phosphorylation in a feed-forward mechanism to activate effector proteins including CHK2 kinase (Matsuoka et al., 1998). CHK2 represents a molecular switch by directly activating various targets responsible for cell cycle progression, DNA repair, and, if the damage is extensive, apoptosis. Additionally, ATM-CHK2 activates transcription factors that alter the expression of numerous genes including the receptor tyrosine kinase c-MET (De Bacco et al., 2011). The implications of promoting c-MET expression will be explained below.
ATR functions in response to endogenous DNA damage; however, it may also be activated in response to DSBs induced by IR, albeit to a lesser extent than ATM. The signaling cascade activated by ATR works through a second checkpoint kinase, CHK1 (Guo et al., 2000). CHK1 and CHK2 demonstrate both overlapping and non-redundant roles, such as those affecting cell cycle progression, DNA repair, and apoptosis (Zhou and Elledge, 2000). The contributions of the ATM-CHK2 and ATR-CHK1 signaling pathways to GIC radiation resistance remain unclear. The ATM-CHK2 pathway is preferentially activated in GICs and targeting CHK1/2 results in improved response to DNA damaging agents (Bao et al., 2006). In addition, ATM overexpression in GBM patient specimens correlates with better overall survival. Taken together, these results indicate a potential role for CHK1/2 kinase inhibitors in the treatment of GBM. Indeed CHK1 inhibitors are currently being investigated in phase I trials for advanced cancers (LY2606368, Eli Lilly and Company, 2000–2013; LY2603618, Eli Lilly and Company, 2000–2013). Further studies are needed to elucidate the mechanisms by which checkpoint kinases can be therapeutic targets or have cellular-protective roles.
c-MET
MET undergoes focal amplification in ∼5% of GBM patients (Maher et al., 2006; Brennan et al., 2009; Dunn et al., 2012). Overexpression of c-MET occurs in ∼29% of GBM and directly correlates with poor patient prognosis (Maher et al., 2006; Cancer Genome Atlas Research, 2008; Brennan et al., 2009; Kong et al., 2009; Verhaak et al., 2010; Snuderl et al., 2011; Dunn et al., 2012; Joo et al., 2012). c-MET becomes activated upon interaction with its ligand, hepatocyte growth factor/scatter factor (HGF/SF), which is secreted in an autocrine fashion by GICs (Joo et al., 2012). This autocrine/paracrine loop helps maintain the GIC phenotype and underscores the significance of this signaling pathway in GBM. Enrichment of c-METhigh-expressing cells from primary GBM display stem-like characteristics including in vivo tumor initiation (Li et al., 2011; De Bacco et al., 2012; Joo et al., 2012). Activation of c-MET stimulates proliferation, migration, and invasion (Kong et al., 2009; Joo et al., 2012; Kim et al., 2013). c-MET also stimulates angiogenesis through the induction of vascular endothelial growth factor (VEGF) expression (Abounader et al., 1999), and resistance to bevacizumab, an anti-VEGF monoclonal antibody, occurs by c-MET activation of pro-survival and invasion mechanisms (Lu et al., 2012).
IR increases c-MET expression, activation, and ligand secretion in GBM (De Bacco et al., 2011) and GICs (Joo et al., 2012). These effects were abrogated by treatment with an ATM inhibitor (De Bacco et al., 2011). Collectively, this suggests that blocking IR-induced c-MET up-regulation may provide therapeutic benefit (Figure 1B). This hypothesis was tested both in vitro and in pre-clinical models by targeting c-MET receptor with genetic approaches in combination with IR. The combinatorial approach decreased cell proliferation and tumor volumes compared to IR or c-MET inhibition alone, highlighting the synergistic benefit of combined treatment (Abounader et al., 1999; Jin et al., 2011). Targeting HGF specifically with three neutralizing antibodies also decreased tumor volume (Cao et al., 2001). Furthermore, dual inhibition of c-MET receptor and HGF-ligand expression together with IR not only reduced proliferation and tumor volume, but also increased apoptosis, DNA fragmentation, and survival (Lal et al., 2005; Li et al., 2009). These findings provide a foundation for investigating c-MET inhibitors, such as cabozantinib (XL-184; Exelixis), in combination with conventional GBM therapy.
Many new drugs targeting HGF/c-MET signaling are progressing into clinical trials. Some of these studies have been completed in other solid tumors, including skin, lung, and thyroid cancers, which are often driven by similar molecular mechanisms found in GBM. Multiple c-MET pathway inhibitors are in the developmental pipeline (Liu et al., 2010). Those that have been evaluated in GBM are listed in Table 1. Most notably, cabozantinib, a pan-tyrosine kinase inhibitor with high affinity for c-MET and VEGFR2, is being tested in a phase II clinical trial for recurrent GBM with encouraging tumor responses and acceptable toxicity (Zhang et al., 2010). Other tyrosine kinase inhibitors that secondarily target c-MET are in various stages of clinical evaluation (Table 1). The HGF/c-MET pathway may also be targeted by ligand sequestration. Rilotumumab (AMG-102; Amgen), a monoclonal antibody against HGF-ligand, has shown promise in a phase II trial in patients with solid tumors (Amgen, 2012).
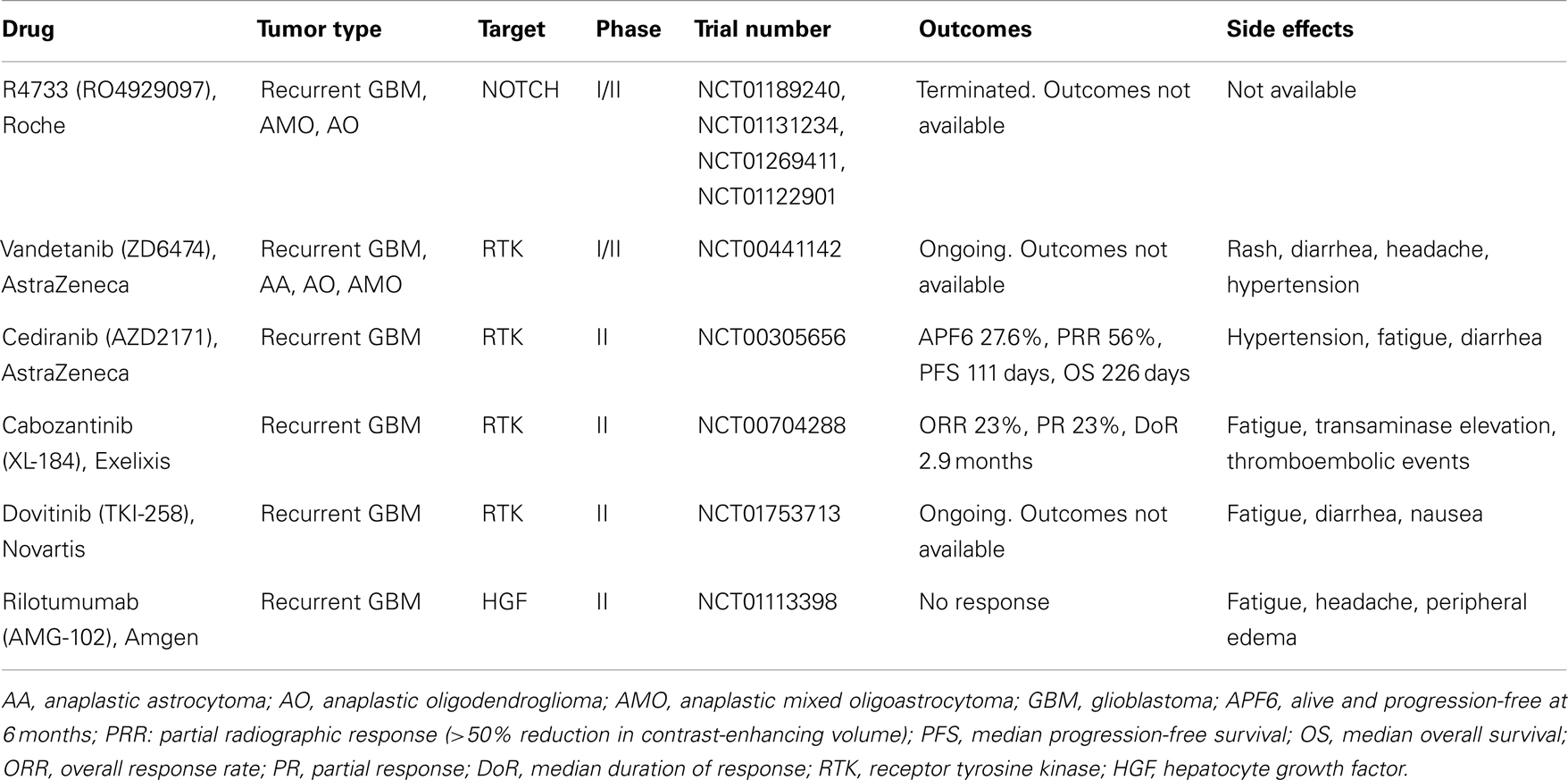
Table 1. Clinical trials of GBM targeting c-MET or NOTCH.
NOTCH
NOTCH receptor is over-expressed in multiple types of cancer initiating cells including GICs (Rizzo et al., 2008; Wang et al., 2012). Upon DELTA/JAGGED ligand binding, the NOTCH receptor is proteolytically cleaved by γ-secretase to promote the release and subsequent nuclear translocation of the NOTCH intracellular domain (NICD) (Guruharsha et al., 2012). This event promotes activation of the PI3K/AKT pathway and expression of NOTCH-regulated genes (Stockhausen et al., 2010; Wang et al., 2010, 2012). These target genes, including c-myc, hes1, and hey1, are responsible for promoting self-renewal and GIC maintenance (Hitoshi et al., 2002; Jeon et al., 2008; Wang et al., 2010; Zhu et al., 2011; Guruharsha et al., 2012).
IR induction of NOTCH activation results in an expansion of GICs (Wang et al., 2010). Combining TGF-β inhibition and IR failed to induce the DDR and NOTCH activation, underlining the interplay between the DDR and NOTCH signaling pathways (Hardee et al., 2012). In vitro studies of glioma cells with γ-secretase inhibitors (GSIs) decreased cell proliferation, viability, and percentage of CD133-positive cells, while inducing cell death exclusively in GICs (Fan et al., 2010; Hovinga et al., 2010). Exogenous expression of NICD2 in GICs was able to rescue the phenotype even in the presence of GSIs (Wang et al., 2010). Furthermore, in vivo studies of GBM xenografts treated with GSIs impaired tumor growth and increased survival (Fan et al., 2010), and these effects synergized with radiation (Hovinga et al., 2010; Lin et al., 2010; Wang et al., 2010). Collectively, these data indicate that GSIs effectively target GICs and may be synergistic with IR (Figure 1C).
Currently, there are several phase I or phase I/II clinical trials examining GSIs for the treatment of patients with GBM (Table 1). RO4929097 is a GSI that has shown early promise in a phase I trial with chemoradiation for newly diagnosed glioma [Princess Margaret Hospital, National Cancer Institute (NCI), 2000b; National Cancer Institute (NCI), 2000]. Single agent or neoadjuvant use of RO4929097 has moved into a phase II trial for recurrent or progressive GBM [Sydney Kimmel Comprehensive Cancer Center, National Cancer Institute (NCI), 2000]. RO4929097 is also being used in combination with the tyrosine kinase inhibitor cediranib (AZD2171/AstraZeneca) in multiple solid tumors, including high grade gliomas [Princess Margaret Hospital, National Cancer Institute (NCI), 2000a] as well as with bevacizumab in patients with recurrent or progressive high grade gliomas NCT01189240 [National Cancer Institute (NCI), 2000]. We eagerly await the results of these studies.
Conclusion
Glioblastoma initiating cells have evolved the ability to activate c-MET and NOTCH pathways after IR, highlighting the cunning ways by which GICs overcome standard cytotoxic treatment. Pre-clinical data on targeting of these pathways have shown potential and have led to multiple clinical trials. Ultimately, too many single agents have failed due to the presence of multiple resistance mechanisms that render single agent therapies ineffective. Combined modality therapy with radiation, chemotherapy, and inhibitors of growth factor signaling will likely be necessary to improve therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Jeremy Rich and Dr. Monica Venere for critical reading of this manuscript. We regret not being able to cite additional relevant references due to space limitations.
References
Abounader, R., Ranganathan, S., Lal, B., Fielding, K., Book, A., Dietz, H., et al. (1999). Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J. Natl. Cancer Inst. 91, 1548–1556.
Amgen. (2012). Analysis of Rilotumumab (AMG 102) Data Identifies a Potential Predictive Biomarker for Patients with Gastric or Gastroesophageal Cancer [Press Release]. Available at: http://www.amgen.com/media/media_pr_detail.jsp?releaseID=1696824
Anido, J., Saez-Borderias, A., Gonzalez-Junca, A., Rodon, L., Folch, G., Carmona, M. A., et al. (2010). TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18, 655–668.
Bao, S., Wu, Q., Li, Z., Sathornsumetee, S., Wang, H., McLendon, R. E., et al. (2008). Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 68, 6043–6048.
Bao, S., Wu, Q., McLendon, R. E., Hao, Y., Shi, Q., Hjelmeland, A. B., et al. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760.
Brennan, C., Momota, H., Hambardzumyan, D., Ozawa, T., Tandon, A., Pedraza, A., et al. (2009). Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 4:e7752. doi:10.1371/journal.pone.0007752
Calabrese, C., Poppleton, H., Kocak, M., Hogg, T. L., Fuller, C., Hamner, B., et al. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82.
Cancer Genome Atlas Research, N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068.
Cao, B., Su, Y., Oskarsson, M., Zhao, P., Kort, E. J., Fisher, R. J., et al. (2001). Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc. Natl. Acad. Sci. U.S.A. 98, 7443–7448.
Chan, J. L., Lee, S. W., Fraass, B. A., Normolle, D. P., Greenberg, H. S., Junck, L. R., et al. (2002). Survival and failure patterns of high-grade gliomas after three-dimensional conformal radiotherapy. J Clin. Oncol. 20, 1635–1642.
Charles, N., and Holland, E. C. (2010). The perivascular niche microenvironment in brain tumor progression. Cell Cycle 9, 3012–3021.
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744.
De Bacco, F., Casanova, E., Medico, E., Pellegatta, S., Orzan, F., Albano, R., et al. (2012). The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 72, 4537–4550.
De Bacco, F., Luraghi, P., Medico, E., Reato, G., Girolami, F., Perera, T., et al. (2011). Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 103, 645–661.
Dunn, G. P., Rinne, M. L., Wykosky, J., Genovese, G., Quayle, S. N., Dunn, I. F., et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 26, 756–784.
Fan, X., Khaki, L., Zhu, T. S., Soules, M. E., Talsma, C. E., Gul, N., et al. (2010). NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28, 5–16.
Firat, E., Gaedicke, S., Tsurumi, C., Esser, N., Weyerbrock, A., and Niedermann, G. (2011). Delayed cell death associated with mitotic catastrophe in gamma-irradiated stem-like glioma cells. Radiat. Oncol. 6, 71.
Galli, R., Binda, E., Orfanelli, U., Cipelletti, B., Gritti, A., De Vitis, S., et al. (2004). Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 64, 7011–7021.
Gatenby, R. A., and Gillies, R. J. (2004). Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899.
Guo, Z., Kumagai, A., Wang, S. X., and Dunphy, W. G. (2000). Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 14, 2745–2756.
Guruharsha, K. G., Kankel, M. W., and Artavanis-Tsakonas, S. (2012). The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 13, 654–666.
Hardee, M. E., Marciscano, A. E., Medina-Ramirez, C. M., Zagzag, D., Narayana, A., Lonning, S. M., et al. (2012). Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 72, 4119–4129.
Heddleston, J. M., Li, Z., McLendon, R. E., Hjelmeland, A. B., and Rich, J. N. (2009). The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8, 3274–3284.
Hitoshi, S., Alexson, T., Tropepe, V., Donoviel, D., Elia, A. J., Nye, J. S., et al. (2002). Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 16, 846–858.
Hjelmeland, A. B., Wu, Q., Heddleston, J. M., Choudhary, G. S., MacSwords, J., Lathia, J. D., et al. (2011). Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 18, 829–840.
Hochberg, F. H., and Pruitt, A. (1980). Assumptions in the radiotherapy of glioblastoma. Neurology 30, 907–911.
Hovinga, K. E., Shimizu, F., Wang, R., Panagiotakos, G., Van Der Heijden, M., Moayedpardazi, H., et al. (2010). Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 28, 1019–1029.
Ignatova, T. N., Kukekov, V. G., Laywell, E. D., Suslov, O. N., Vrionis, F. D., and Steindler, D. A. (2002). Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39, 193–206.
Jeon, H. M., Jin, X., Lee, J. S., Oh, S. Y., Sohn, Y. W., Park, H. J., et al. (2008). Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev. 22, 2028–2033.
Jin, J., Bae, K. H., Yang, H., Lee, S. J., Kim, H., Kim, Y., et al. (2011). In vivo specific delivery of c-Met siRNA to glioblastoma using cationic solid lipid nanoparticles. Bioconjug. Chem. 22, 2568–2572.
Joo, K. M., Jin, J., Kim, E., Ho Kim, K., Kim, Y., Gu Kang, B., et al. (2012). MET signaling regulates glioblastoma stem cells. Cancer Res. 72, 3828–3838.
Kim, K. H., Seol, H. J., Kim, E. H., Rheey, J., Jin, H. J., Lee, Y., et al. (2013). Wnt/beta-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro-oncology 15, 161–171.
Kong, D. S., Song, S. Y., Kim, D. H., Joo, K. M., Yoo, J. S., Koh, J. S., et al. (2009). Prognostic significance of c-Met expression in glioblastomas. Cancer 115, 140–148.
Lal, B., Xia, S., Abounader, R., and Laterra, J. (2005). Targeting the c-Met pathway potentiates glioblastoma responses to gamma-radiation. Clin. Cancer Res. 11, 4479–4486.
Laperriere, N. J., Leung, P. M., McKenzie, S., Milosevic, M., Wong, S., Glen, J., et al. (1998). Randomized study of brachytherapy in the initial management of patients with malignant astrocytoma. Int. J. Radiat. Oncol. Biol. Phys. 41, 1005–1011.
Lee, S. W., Fraass, B. A., Marsh, L. H., Herbort, K., Gebarski, S. S., Martel, M. K., et al. (1999). Patterns of failure following high-dose 3-D conformal radiotherapy for high-grade astrocytomas: a quantitative dosimetric study. Int. J. Radiat. Oncol. Biol. Phys. 43, 79–88.
Li, Y., Guessous, F., Zhang, Y., Dipierro, C., Kefas, B., Johnson, E., et al. (2009). MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 69, 7569–7576.
Li, Y., Li, A., Glas, M., Lal, B., Ying, M., Sang, Y., et al. (2011). c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. U.S.A. 108, 9951–9956.
Lin, J., Zhang, X. M., Yang, J. C., Ye, Y. B., and Luo, S. Q. (2010). gamma-secretase inhibitor-I enhances radiosensitivity of glioblastoma cell lines by depleting CD133+ tumor cells. Arch. Med. Res. 41, 519–529.
Liu, G., Yuan, X., Zeng, Z., Tunici, P., Ng, H., Abdulkadir, I. R., et al. (2006). Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 5, 67.
Liu, X., Newton, R. C., and Scherle, P. A. (2010). Developing c-MET pathway inhibitors for cancer therapy: progress and challenges. Trends Mol. Med. 16, 37–45.
Lu, K. V., Chang, J. P., Parachoniak, C. A., Pandika, M. M., Aghi, M. K., Meyronet, D., et al. (2012). VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22, 21–35.
Maher, E. A., Brennan, C., Wen, P. Y., Durso, L., Ligon, K. L., Richardson, A., et al. (2006). Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res. 66, 11502–11513.
Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R. III, Hurov, K. E., Luo, J., et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166.
Matsuoka, S., Huang, M., and Elledge, S. J. (1998). Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282, 1893–1897.
National Cancer Institute (NCI). (2000). “RO4929097 and bevacizumab in treating patients with progressive or recurrent malignant glioma,” in ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine. Available at: http://clinicaltrials.gov/ct2/show/NCT01189240 [cited Dec 12, 2012].
Phillips, H. S., Kharbanda, S., Chen, R., Forrest, W. F., Soriano, R. H., Wu, T. D., et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173.
Princess Margaret Hospital, National Cancer Institute (NCI). (2000a). “Gamma-secretase inhibitor RO4929097 and cediranib maleate in treating patients with advanced solid tumors,” in ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine. Available at: http://clinicaltrials.gov/ct2/show/NCT01131234 [cited Dec 12, 2012].
Princess Margaret Hospital, National Cancer Institute (NCI). (2000b). “RO4929097 in treating patients with recurrent invasive gliomas,” in ClinicalTrials.gov[Internet]. Bethesda, MD: National Library of Medicine. Available at: http://clinicaltrials.gov/ct2/show/NCT01269411 [cited Dec 12, 2012].
Rizzo, P., Osipo, C., Foreman, K., Golde, T., Osborne, B., and Miele, L. (2008). Rational targeting of Notch signaling in cancer. Oncogene 27, 5124–5131.
Schwartzbaum, J. A., Fisher, J. L., Aldape, K. D., and Wrensch, M. (2006). Epidemiology and molecular pathology of glioma. Nat. Clin. Pract. Neurol. 2, 494–503. [quiz 491 p. following 516].
Seidel, S., Garvalov, B. K., Wirta, V., Von Stechow, L., Schanzer, A., Meletis, K., et al. (2010). A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133, 983–995.
Selker, R. G., Shapiro, W. R., Burger, P., Blackwood, M. S., Arena, V. C., Gilder, J. C., et al. (2002). The Brain Tumor Cooperative Group NIH Trial 87-01: a randomized comparison of surgery, external radiotherapy, and carmustine versus surgery, interstitial radiotherapy boost, external radiation therapy, and carmustine. Neurosurgery 51, 343–355; discussion 355–347.
Singh, S. K., Clarke, I. D., Terasaki, M., Bonn, V. E., Hawkins, C., Squire, J., et al. (2003). Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828.
Singh, S. K., Hawkins, C., Clarke, I. D., Squire, J. A., Bayani, J., Hide, T., et al. (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401.
Snuderl, M., Fazlollahi, L., Le, L. P., Nitta, M., Zhelyazkova, B. H., Davidson, C. J., et al. (2011). Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20, 810–817.
Soeda, A., Park, M., Lee, D., Mintz, A., Androutsellis-Theotokis, A., McKay, R. D., et al. (2009). Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 28, 3949–3959.
Son, M. J., Woolard, K., Nam, D. H., Lee, J., and Fine, H. A. (2009). SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 4, 440–452.
Souhami, L., Seiferheld, W., Brachman, D., Podgorsak, E. B., Werner-Wasik, M., Lustig, R., et al. (2004). Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93-05 protocol. Int. J. Radiat. Oncol. Biol. Phys. 60, 853–860.
Stockhausen, M. T., Kristoffersen, K., and Poulsen, H. S. (2010). The functional role of Notch signaling in human gliomas. Neuro-oncology 12, 199–211.
Stupp, R., Hegi, M. E., Mason, W. P., Van Den Bent, M. J., Taphoorn, M. J., Janzer, R. C., et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466.
Sydney Kimmel Comprehensive Cancer Center, National Cancer Institute (NCI). (2000). “RO4929097 in treating patients with recurrent or progressive glioblastoma,” in ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine. Available at: http://clinicaltrials.gov/ct2/show/NCT01122901 [cited Dec 12, 2012].
Tamura, K., Aoyagi, M., Wakimoto, H., Ando, N., Nariai, T., Yamamoto, M., et al. (2010). Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J. Neurosurg. 113, 310–318.
Verhaak, R. G., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M. D., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110.
Wang, J., Sullenger, B. A., and Rich, J. N. (2012). Notch signaling in cancer stem cells. Adv. Exp. Med. Biol. 727, 174–185.
Wang, J., Wakeman, T. P., Lathia, J. D., Hjelmeland, A. B., Wang, X. F., White, R. R., et al. (2010). Notch promotes radioresistance of glioma stem cells. Stem Cells 28, 17–28.
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773.
Zhang, Y., Guessous, F., Kofman, A., Schiff, D., and Abounader, R. (2010). XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs 13, 112–121.
Zhou, B. B., and Elledge, S. J. (2000). The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439.
Keywords: glioma initiating cells, radiation, c-MET, NOTCH, DNA damage response
Citation: Rivera M, Sukhdeo K and Yu J (2013) Ionizing radiation in glioblastoma initiating cells. Front. Oncol. 3:74. doi: 10.3389/fonc.2013.00074
Received: 15 December 2012; Paper pending published: 27 January 2013;
Accepted: 23 March 2013; Published online: 08 April 2013.
Edited by:
Daphne Haas-Kogan, University of California San Francisco, USAReviewed by:
Ji-Hong Hong, Chang Gung Memorial Hospital, TaiwanHsin-Ell Wang, National Yang-Ming University, Taiwan
Copyright: © 2013 Rivera, Sukhdeo and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Jennifer Yu, Department of Radiation Oncology and Department of Stem Cell Biology and Regenerative Medicine, Cleveland Clinic, 9500 Euclid Avenue, T28 Cleveland, OH 44195, USA. e-mail:eXVqMkBjY2Yub3Jn