Abstract
The PI3K-Akt pathway together with one of its downstream targets, the mechanistic target of rapamycin (mTOR; also known as the mammalian target of rapamycin) is a highly deregulated pathway in cancers. mTOR exists in two complexes, mTORC1 and mTORC2. Akt phosphorylated at T308 inhibits TSC1/2 complex to activate mTORC1; mTORC2 is recognized as the kinase phosphorylating Akt at S473. Inhibition of autophagy by mTORC1 was shown to rescue disheveled (Dvl) leading to activation of Wnt pathway. Cyclin D1 and the c-Myc are activated by the Wnt signaling. Cyclin D1 is a key player in initiation of cell cycle. c-Myc triggers metabolic reprograming in G1 phase of cell cycle, which also activates the transcription factors like FoxO and p53 that play key roles in promoting the progression of cell cycle. While the role of p53 in cancer cell metabolism in arresting glycolysis and inhibition of pentose phosphate pathway has come to be recognized, there are confusions in the literature on the role of FoxO and that of rictor. FoxO was shown to be the transcription factor of rictor, in addition to the cell cycle inhibitors like p21. Rictor has dual roles; inhibition of c-Myc and constitution of mTORC2, both of which are key factors in the exit of G1-S phase and entry into G2 phase of cell cycle. A model is presented in this article, which suggests that the PI3K-Akt-mTOR and Wnt pathways converge and regulate the progression of cell cycle through G0-G1-S-phases and reprogram the metabolism in cancer cells. This model is different from the conventional method of looking at individual pathways triggering the cell cycle.
Introduction
The protein kinase B (betweenproteinkinase A and C; Coffer and Woodgett, 1991), or Akt designated after the viral acute transforming retrovirus, Akt8 (Staal et al., 1977; Bellacosa et al., 1991; Downward, 1995) is recognized as the regulator of cell survival. Aberrant activation of the kinase is associated with many diseases, including cancer and diabetes (Pearce et al., 2010). Phosphorylation of Akt at threonine 308 (T308; in activation loop) and the serine 473 (S473; in hydrophobic motif) are considered important for its activity (Nicholson and Anderson, 2002). Akt is activated by insulin/insulin like growth factor (IGF) signaling (IIS). The autophosphorylation of the internal domains of IIS receptors leads to the recruitment of insulin receptor substrate (IRS) and activation of phosphatidylinositol 3-kinases (PI3K). PI3K phosphorylates phosphatidylinositol 4, 5 bisphosphate (PIP2) to PIP3 (Engelman et al., 2006; Manning and Cantley, 2007). PIP3 activates the phosphatidylinositol dependent protein kinase 1 (PDPK1) and recruits Akt to the plasma membrane. PDPK1 phosphorylates Akt T308 in the activation loop (Alessi et al., 1997). Several kinases, integrin-linked kinase (ILK), protein kinase Cα (PKCα), double-stranded DNA-dependent protein kinase (DNA-PK) ataxia telangiectasia mutated (ATM) gene product, and the mammalian target of rapamycin (mTOR) were proposed to phosphorylate Akt on Ser-473 (Dong and Liu, 2005). The mTORC2 (Sarbassov et al., 2005) is widely recognized as the key kinase that phosphorylates the Akt at S473. Ambiguity on the phosphorylation of this site however, remains; an atypical IκB kinase ε and TANK-binding kinase 1 (IKKε/TBK1) was reported to induce this phosphorylation in rictor (−/−) cells (Xie et al., 2011). There are reports that phosphorylation of Akt S473 could be cell specific (Riaz et al., 2012) or may not be required for full activation (Moore et al., 2011). T308 phosphorylation is considered a reliable biomarker of Akt activity especially for mTORC1 function (Jacinto et al., 2006; Breuleux et al., 2009; Vincent et al., 2011). Several tyrosine kinases are reported to phosphorylate Akt at different sites (Mahajan and Mahajan, 2012). Two phosphatases, the phosphatase and tensin homolog deleted from chromosome ten (PTEN) and the SH2 domain containing inositol-5-phosphatase 2 (SHIP2) regulate Akt function through dephosphorylation of 3-OH position of PIP3 (Leslie et al., 2003) and the 5-OH position (Aman et al., 1998) respectively.
The mechanistic target of rapamycin (mTOR: formerly known as mTOR; also known as FK506 binding protein 12-rapamycin associated protein 1 (FRAP1; Moore et al., 1996), in mammals exists in two multi protein complexes, mTORC1 and mTORC2, distinguished by their sensitivity to rapamycin. The catalytic cores of the two complexes have the kinase mTOR domain. While raptor (regulatory associated protein of mTOR) regulates the function of mTORC1, rictor (Rapamycin insensitive companion of mTOR) was shown to control the activity of mTORC2 (reviewed by Loewith et al., 2002; Laplante and Sabatini, 2009). DEPTOR is a negative regulator of the two complexes (Wang et al., 2012).
The complex mTORC1 responds to the nutrients and conditions that promote cellular growth. It is activated by AktT308 downstream of IIS (Wullschleger et al., 2006; Gamper and Powell, 2012). mTORC1 is activated both by the oncogenic PI3K-Akt as well as the Ras-Erk pathways, which inhibit the tuberous sclerosis complex (TSC1 and TSC2) (TSC complex) through the phosphorylation of the TSC2 (Manning and Cantley, 2007). The inhibition of TSC complex releases the inhibitory effect of TSC on the GTP-bound Rheb (Ras homolog enhanced in brain), which controls the activity of mTORC1. TSC is also inhibited by the Wnt pathway (Inoki et al., 2006). Activation of mTORC1 by amino acids is mediated by Rag GTPases. (Sancak et al., 2010), which is independent of IIS. AMP activated protein kinase (AMPK) inhibits mTORC1 by activating the TSC2 (Corradetti et al., 2004; Kwiatkowski and Manning, 2005; Inoki et al., 2006) and drugs that activate AMPK reverse the activation of mTORC1 (Guppy et al., 2011; He et al., 2011).
mTORC1 is a Feedback Regulator of IIS Pathway and it also Regulates mTORC2
One of the key downstream targets of mTORC1, the p70 ribosomal S6 Kinase (S6K) phosphorylates IRS and inhibits the IIS in a feedback regulatory step (Zhang et al., 2008; Veilleux et al., 2010; Kang et al., 2011). An inverse relation is reported both in relative abundance and activation of mTORC1 and mTORC2 in cells (Sarbassov et al., 2004). S6K also phosphorylates rictor and inhibits mTORC2 assembly (Dibble et al., 2009; Julien et al., 2010; Treins et al., 2010).
S6K is also shown to inhibit glycogen synthase kinase3β (GSK3β) (Zhang et al., 2006). Recognized as one of the key targets of Akt, GSK3β was also shown to phosphorylate rictor (Chen et al., 2011). GSK3β has multiple roles ranging from glucose homeostasis (Kim and Kimmel, 2000) to inflammation (Wang et al., 2011), and it plays a key role in Wnt signaling (Wu and Pan, 2010). GSK3β phosphorylates the voltage-dependent anion channel (VDAC) and regulates the mitochondrial metabolite exchange and apoptosis (Shoshan-Barmatz et al., 2010); its depletion was shown to increase the beta cell proliferation (Stein et al., 2011). GSK3β cooperates with AMPK in activation of TSC complex that leads to inactivation of mTORC1 (Kwiatkowski and Manning, 2005).
Regulation of protein synthesis is recognized as one of the conserved role of mTORC1; it phosphorylates and inhibits, the eukaryotic initiation factor 4E (eIF4E)-binding proteins (4E-BP1/2), which are the inhibitors of translation (Castellvi et al., 2006; Ma and Blenis, 2009). The two functions of mTORC1, phosphorylation of S6kinase and inhibition of 4E-BP, have come to be accepted as routine markers for its activity and activation of protein synthesis in cells (Miron et al., 2003).
RAS-Erk Map Kinase Signaling also Activates mTORC1
Over expression of epidermal growth factor receptors belonging to the proto-oncogene erbB (Thompson and Gill, 1985) and abberrant activation of RAS-Erk MAP kinase signaling was recognized as the cause of several cancers and antibodies targeting the receptors were developed during early 1980s (Sato et al., 1983; Schlessinger, 2000; Mendelsohn and Baselga, 2003; Lemmon and Schlessinger, 2010). The MAP kinase Erk was shown to phosphorylate and inactivate TSC2 (Ma et al., 2005) leading to activation of mTORC1. Several drugs that target the nutrient and growth factor (PI3K-Akt and Ras-Erk MAP kinase) pathways claim that the targeted drugs arrest the progression of cell cycle. The convergence of the two pathways at mTORC1 led to a surge in the activity in targeting of mTORC1 for the control of carcinogenesis. Rapamycin, which was initially recognized as an immunosuppressant for its ability to reduce organ rejection (Abraham and Wiederrecht, 1996) was subsequently found useful in treatment of cancers (Mita et al., 2003). But, the realization that rapamycin is inadequate in completely inhibiting mTORC1 functions (Shor et al., 2009) led to a search for the ATP competitive inhibitors (Bhagwat and Crew, 2010; Schenone et al., 2011). These inhibitors are claimed to arrest the cells in quiescent or gap1 (G0/G1) phase of the cell cycle (Evangelisti et al., 2011). But the exact link between the growth factor, mTOR pathways and cell cycle remains unexplained.
Wnt Pathway is the Key Pathway in Activation of Cell Cycle
Wnt pathway is the key pathway in activation of cell cycle. Wnt signaling in general activates the Cyclin D, the c-Myc; matrix metalloproteinases, COX-2, peroxisome proliferator-activated receptors (PPARs), and the growth factors, and their receptors, and down regulates E-Cadherin, the cell cycle inhibitor P16ink4A (ARF) and p53 (http://www.stanford.edu/group/nusselab/cgi-bin/wnt/human_genetic_diseases; 2010). Wnt pathway thus, regulates the cancer cells entry into the cell cycle through the production of cyclin D. Cyclin D complexes with cyclin-dependent kinase 4/6 (Cdk4/6), inactivates the tumor suppressor protein retinoblastoma (Rb), and promotes the entry of the cell from G0 to G1 phase of cell cycle. E2F uncoupled from the phosphorylated Rb transcribes the cyclin E, which binds to Cdk2 and promotes the progression of the cell cycle. The up regulation of cyclin E/CDK2 is reported to correlate with the G1/S transition (Stott et al., 1998; Arima et al., 2004; Soto Martinez et al., 2005; Sun et al., 2007). Aberrant activation of Wnt pathway was shown to lead cells to malignant transformation (Polakis, 2012).
Activation of Wnt pathway is usually based on destabilizing the commonly known “destruction complex” comprising of the APC, the Axin, and the casein kinase I (CKI) and GSK3β. The disheveled (Dvl; Dsh gene homolog in mammals) protein is recognized as the key component in the signaling (Nusse, 2005; Gao and Chen, 2010).
Autophagy Promotes Degradation of Dvl and Negatively Regulates the Wnt Pathway
The process of autophagy involves the fusion of phagophores with lysosomes (Yang and Klionsky, 2010); it plays a key role in human diseases like immune disorders (Deretic, 2011), neurodegenerative disorders (Weihl, 2011), and also in cancers (Brech et al., 2009; Stipanuk, 2009; White and Lowe, 2009; Chen and Klionsky, 2011; Nyfeler et al., 2011). mTORC1 was shown to inhibit autophagy by phosphorylation of ULK1 at Ser 757 (Kim et al., 2011).
Gao et al. (2010) demonstrated that autophagy is a negative regulator of the Wnt pathway by promoting the degradation of Dvl, a component of Wnt pathway. All the three isoforms of Dvl (Dvl1, Dvl2, and Dvl3) were shown to be degraded. Inhibition of autophagy by mTORC1 therefore, releases the Dvl. Autophagy mediated down regulation of Wnt signaling was confirmed by rapamycin treatment, which resulted in down regulation of the Wnt target genes axin2, c-Myc, and cyclin D1. Dvl appears to be the link for the cooperative interaction between the MAP kinase and PI3K-Akt-mTOR pathways converging at autophagy to activate cell proliferation. In addition, c-Myc, a downstream target of the Wnt signaling, was shown to be involved in carcinogenesis along with erbB2 as early as in 1980s (Dotto et al., 1986; Land et al., 1986). Pacheco-Pinedo et al. (2011) recently demonstrated that cooperation between K-Ras mutant and the Wnt/β-catenin signaling is the cause of aggressive lung tumor phenotype. Heallen et al. (2011) demonstrated that Hippo pathway inhibits the Wnt signaling to prevent cardiomyocyte proliferation; Dvl was shown to be the link between the two pathways. Apart from reusing Dvl from autophagic degradation, mTORC1 inactivation of GSK3β by S6K (Zhang et al., 2006) blocks the β-catenin degradation. Dvl was also shown to translocate into nucleus and in conjugation with the transcription factor of AP1 complex, c-jun is reported to stabilize the β-catenin-TCF/LEF signaling (Gan et al., 2008).
Oncogenes and Tumor Suppressors are Both Involved in Metabolic Reprograming
Progression of cell cycle also requires the activation of metabolic pathways in G1 phase. Glycolysis, Krebs cycle, and the pentose phosphate pathways are the key pathways involved in metabolic reprograming of cells. Warburg, in the early twentieth century, was the first to suggest that cancer cells utilize the aerobic glycolysis for promotion of tumorigenesis (Warburg, 1956). A re-examination of Warburg hypothesis in the last part of twentieth century led to a search for the role of oncogenes and tumor suppressors in metabolism (Figure 1A). Akt was named as the “Warburg enzyme” (Robey and Hay, 2009), p53 was recognized to suppress both glycolysis through TIGAR (TP53-induced glycolysis and apoptosis regulator; Bensaad et al., 2006) and Pentose phosphate pathway by inhibiting the G6PD (Jiang et al., 2011). It was shown to activate glutamine metabolism and control the ROS production (Gottlieb, 2011; Maddocks and Vousden, 2011). HIF and c-Myc were shown to up regulate the enzymes of glycolysis (Kim et al., 2007). The signature of cancer cells is recognized by loss of function of the tumor suppressors p53, PTEN and by activation of Akt, Myc, HIF-1α, and NFkB (Markert et al., 2012). The oncogene c-Myc is recognized to play an important role in activation of genes of enzymes of glycolysis and Krebs cycle as well as those involved in chromatin structure, and its transcriptional networks that are involved predominantly in cell cycle regulation and cellular metabolism and protein synthesis specific to the G0-G1-S transition in cancer cells, lymphocytes, and in embryonic stem cell (Kim et al., 2010; Swami, 2010; Lin et al., 2012).
Figure 1
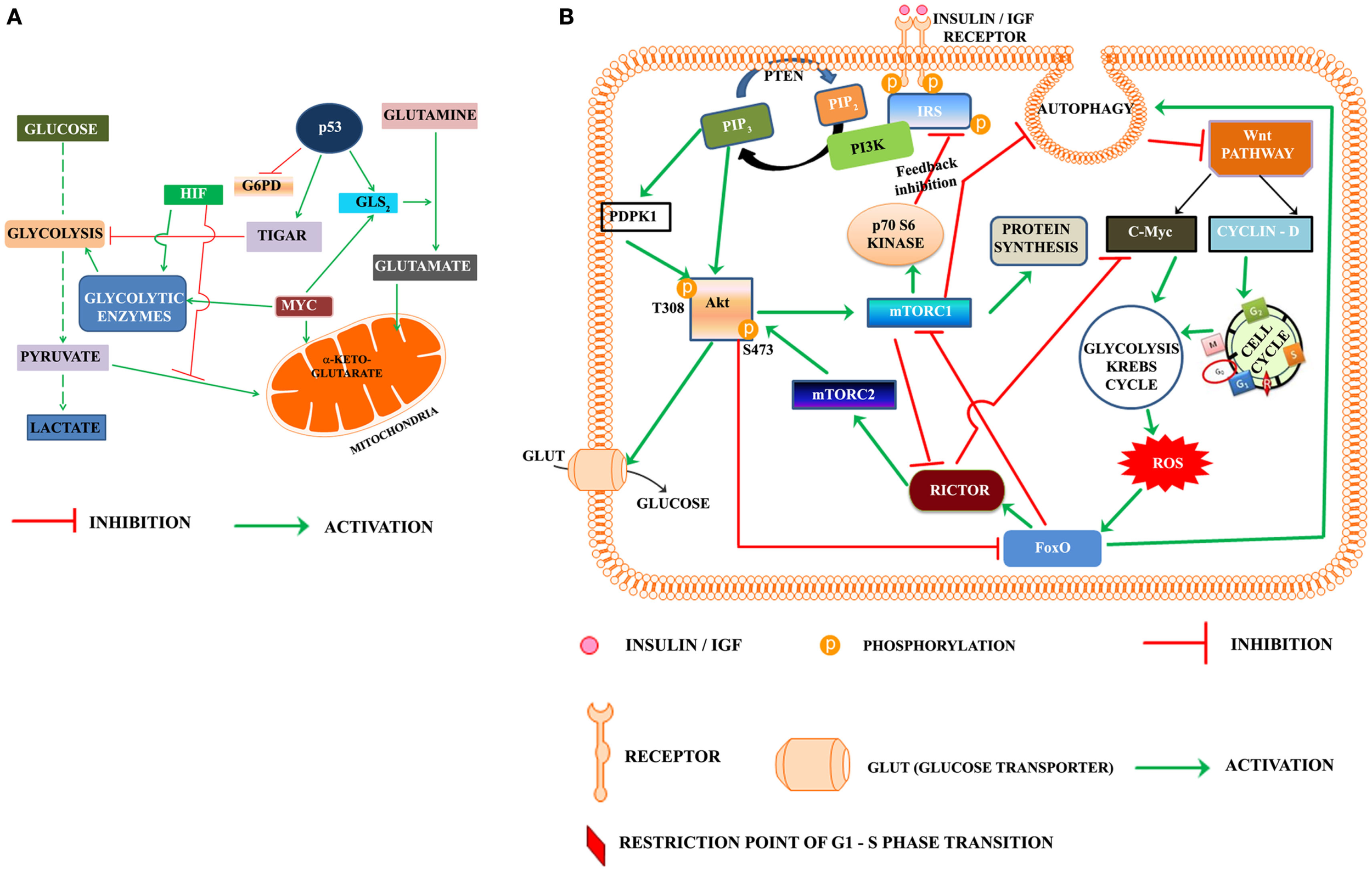
(A) Oncogenes and tumor suppressor modulate metabolic reprograming in cancer cells. HIF, hypoxia inducible factor; TIGAR, TP53-induced glycolysis and apoptosis regulator; MYC, Proto oncogene c-Myc; G6PD, glucose-6-phosphate dehydrogenase; GLS2, glutaminase 2. (B) Proposed model highlighting the role of PI3K-Akt-mTOR and Wnt pathways in regulation of the cell cycle progression in cancer cell: according to the presented model, activation of insulin/IGF receptor by nutrients/growth factors activates PI3K-Akt pathway. Akt phosphorylated on T308 activates mTORC1. One of the downstream target of mTORC1, the p70S6Kinase, phosphorylates the serine residues on IRS and inhibits the insulin/IGF signaling in a regulatory feedback control. The activated mTORC1 activates the protein synthesis, but inhibits autophagy. Inhibition of autophagy rescues Dvl and this lead to activation of Wnt pathway. The activated Wnt pathway up regulates cyclin D and C-Myc, which trigger the activation of cell cycle and metabolic reprograming. The metabolic activation in cancer cells leads to the production of reaction oxygen species (ROS), which activate the FoxO. FoxO is the transcription factor of rictor, one of the key components of mTORC2 recognized as the kinase phosphorylating Akt on S473. Rictor also inhibits c-Myc, which according to the present model is required for exit of G1/S restriction point. Akt, protein kinase B (T308, S473 are the phosphorylated sites Threonine 308 and Serine 473 of Akt), c-Myc is the oncoprotein activated by Wnt signaling, FoxO, fork head transcription factors of O group; GLUT, glucose transporter; IGF, insulin growth factor; IRS, insulin receptor substrate; mTORC1, 2, mammalian target of rapamycin (mTOR) Complex 1 and 2, PIP2, phosphoinositide 4,5 bisphosphate; PIP3, phosphoinositide 3,4,5 trisphosphate; rictor, a component of mTORC2; P70S6K, the p70 ribosomal S6K; PTEN, phosphatase and tensin homolog deleted from chromosome ten; ROS, reaction oxygen species; PDPK1 (also abbreviated as PDK1), phosphoinositide dependent protein kinase.
The Role of FoxO Transcription Factors
One of the consequences of metabolic reprograming in cancer cells is activation of FoxO transcription factors (Ronnebaum and Patterson, 2012). The FoxO transcription factors are reported to sequester β-catenin away from the TCF/LEF transcription factors (Hoogeboom et al., 2008; Hoogeboom and Burgering, 2009). Although the exact mechanism is still enigmatic, deregulation of adherens junction (AJ) was reported to result in translocation of the FoxO transcription factors into nucleus (Fournier et al., 2009). One of the critical steps in the progression of cell cycle is the crossover of restriction point in G1, which is regulated by the E2F-pRb. The role of FoxO in up regulating cell cycle inhibitors p15 (INK4b) and p19 (INK4d) is viewed as an arrest of cell cycle (Katayama et al., 2008). Rictor transcribed by FoxO is a key component of the mTORC2 (Chen et al., 2010). Guo et al. (2012) reported that rictor promotes ubiquitination and degradation of c-Myc and cyclin E and suggested that it leads to the arrest of cell cycle in G1 phase. Rictor is also reported to be involved in regulation of Rho GTPases (Jacinto et al., 2004) and RhoA activation is crucial for G1-S progression of cell cycle (Zhang et al., 2009). The up regulation of cell cycle inhibitors (p21WAF1/CIP1 and p27KIP1) and transcription of rictor by FoxOs and inhibition of c-Myc and cyclin E reported by Guo et al. (2012) should therefore, be viewed as a regulation of the exit of G1/S phase of cell cycle rather than inhibition of cell cycle in G1 phase. FoxO is also reported to transcribe the antioxidant genes like sestrins (Chen et al., 2010; Lee et al., 2010) which inhibit mTORC1 and inhibit the mitochondrial metabolism (Ferber et al., 2012). Young et al. (2009) demonstrated a feedback link between mTORC1 and C2 signaling and the timing of inhibition of mTORC1 correlated with activation of autophagy and cyclin A. These results suggest the role of FoxO in progression of cell cycle and assembly of mTORC2. mTORC2 was shown to be required for proliferation and survival of TSC2-Null cells (Goncharova et al., 2011). The hypothesis that FoxOs are involved in the progression of cell cycle is further strengthened by the fact that FoxOs also regulate the expression of mitotic genes such as cyclin B, polo-like kinase (Plk) (Alvarez et al., 2001). In addition, recent reports indicate the role of FoxO1 in dedifferentiation of pancreatic β-cells (Talchai et al., 2012) and in osteoblast proliferation (Kode et al., 2012).
The Proposed Model
Based on the foregone discussion, we propose that activation of PI3K-Akt-mTORC1 leads to inhibition of autophagy and rescues Dvl, which activates the Wnt pathway (Figure 1B). The transcriptional activation of Cyclin D by Wnt pathway triggers the entry of cells from G0 to G1 phase. c-Myc promotes reprograming of cancer cell metabolism in the G1 phase, which apart from generating ROS, activates the transcription factors like p53 and FoxO and autophagy. The transcription of rictor by FoxO leads to the inhibition of c-Myc and promotes exit of the restriction point of G1-S phase of cell cycle. Rictor also constitutes mTORC2 in G2 phase. Phosphorylation of Akt at S473 by mTORC2 leads to feedback inhibition of FoxO.
Some Unanswered Questions
Cancer cells consume lots of glucose, but it is reported that glucose transporters are activated only following phosphorylation of Akt at S473 (Kumar et al., 2010) and it coincides with the inactivation of FoxO proteins. Under hypoxic conditions, loss of p53 promotes the expression of mono carboxylate transporters (MCT1) and lactate export which is reported to promote cell proliferation by fueling mitochondrial respiration (Boidot et al., 2012). Do cancer cells exiting the divisional phase depend on excess glucose and glycolytic flux fuels respiration, while actively proliferating cells depend on lactate as a fuel resulting in a Warburg effect?
Statements
Acknowledgments
Dr. Lakshmipathi Vadlakonda is thankful to the Director of CR RAO AIMSCS, Prof. Allam Apparao, and the DST, New Delhi for providing a placement as the adjunct faculty. Dr. Mukesh Pasupuleti thanks the management of SRM University for providing funds and facilities for carrying out the research work at SRM Research Institute. The financial support by the Department of Biotechnology and Indian Council of Medical Research to Dr. Pallu Reddanna is gratefully acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Akt, protein kinase B (T308, S473 – phosphorylated sites Threonine 308 and Serine 473); APC, adenomatous polyposis coli; AXIN, axis inhibition protein; CDK2, cyclin-dependent kinase 2; CK, casein kinase; CKI, cyclin-dependent kinase inhibitors; c-Myc, the oncoprotein activated by Wnt signaling; COX-2, cyclooxygenase-2; Dvl, Dsh homolog in mammals; 4E-BP, eukaryotic translation initiation factor (eIF4E) binding protein1; FoxO, fork head transcription factors of O group; G0, G1, and S are phases of cell cycle; GLUT, glucose transporter; GSK3β, glycogen synthase kinase3β; HIF, hypoxia inducible factor; IGF, insulin like growth factor; IIS, insulin/insulin like growth factor signaling; IRS, insulin receptor substrate; LEF, lymphoid enhancer-binding factor; LRP, LDL receptor protein; mTORC1, 2, mechanistic target of rapamycin complex 1 and 2 (mTOR, formerly known as mammalian target of rapamycin); p16INK 4a ARF, cyclin-dependent kinase inhibitor 2A family of cell cycle inhibitors; p16INK 4aARF, cyclin-dependent kinase inhibitor 2A family of cell cycle inhibitors; PDPK1, phosphoinositide dependent kinase 1 (the abbreviation PDPK1 is preferred over the original PDK1 in the article to avoid confusion with the pyruvate dehydrogenase kinase, which is also abbreviated as PDK1 in the literature); PDK2, phosphoinositide dependent kinase2 (a putative kinase thought to phosphorylate Akt on S473); PI3K, phosphatidylinositol 3-kinases; PIP2, phosphatidylinositol 4,5 bisphosphate; PIP3, phosphatidylinositol 3,4,5 trisphosphate; PPP, pentose phosphate pathway; PTEN, phosphatase and tensin homolog deleted from chromosome ten; rictor, a component of mTORC2; ROS, reaction oxygen species; S6K, the p70 ribosomal S6K; sestrins, stress response proteins; snail/slug, transcriptional inhibitors of E-Cadherin; TCF, T-cell factor; TIGAR, TP53-induced glycolysis and apoptosis regulator.
References
1
Abraham R. T. Wiederrecht G. J. (1996). Immunopharmacology of rapamycin. Annu. Rev. Immunol.14, 483–510.10.1146/annurev.immunol.14.1.483
2
Alessi D. R. James S. R. Downes C. P. Holmes A. B. Gaffney P. R. Reese C. B. et al (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol.7, 261–269.10.1016/S0960-9822(06)00336-8
3
Alvarez B. Martinez A. C. Burgering B. M. Carrera A. C. (2001). Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature413, 744–747.10.1038/35099574
4
Aman M. J. Lamkin T. D. Okada H. Kurosaki T. Ravichandran K. S. (1998). The inositol phosphatase SHIP inhibits Akt/PKB activation in B cells. J. Biol. Chem.273, 33922–33928.10.1074/jbc.273.51.33922
5
Arima Y. Hirota T. Bronner C. Mousli M. Fujiwara T. Niwa S. et al (2004). Down-regulation of nuclear protein ICBP90 by p53/p21Cip1/WAF1-dependent DNA-damage checkpoint signals contributes to cell cycle arrest at G1/S transition. Genes Cells9, 131–142.10.1111/j.1356-9597.2004.00710.x
6
Bellacosa A. Testa J. R. Staal S. P. Tsichlis P. N. (1991). A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science254, 274–277.10.1126/science.1833819
7
Bensaad K. Tsuruta A. Selak M. A. Vidal M. N. Nakano K. Bartrons R. et al (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell126, 107–120.10.1016/j.cell.2006.05.036
8
Bhagwat S. V. Crew A. P. (2010). Novel inhibitors of mTORC1 and mTORC2. Curr. Opin. Investig. Drugs11, 638–645.
9
Boidot R. Vegran F. Meulle A. Le Breton A. Dessy C. Sonveaux P. et al (2012). Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res.72, 939–948.10.1158/0008-5472.CAN-11-2474
10
Brech A. Ahlquist T. Lothe R. A. Stenmark H. (2009). Autophagy in tumour suppression and promotion. Mol. Oncol.3, 366–375.10.1016/j.molonc.2009.05.007
11
Breuleux M. Klopfenstein M. Stephan C. Doughty C. A. Barys L. Maira S. M. et al (2009). Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther.8, 742–753.10.1158/1535-7163.MCT-08-0668
12
Castellvi J. Garcia A. Rojo F. Ruiz-Marcellan C. Gil A. Baselga J. et al (2006). Phosphorylated 4E binding protein 1: a hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer107, 1801–1811.10.1002/cncr.22195
13
Chen C. C. Jeon S. M. Bhaskar P. T. Nogueira V. Sundararajan D. Tonic I. et al (2010). FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev. Cell18, 592–604.10.1016/j.devcel.2010.03.008
14
Chen C. H. Shaikenov T. Peterson T. R. Aimbetov R. Bissenbaev A. K. Lee S. W. et al (2011). ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci. Signal.4, ra10.10.1126/scisignal.2001731
15
Chen Y. Klionsky D. J. (2011). The regulation of autophagy – unanswered questions. J. Cell. Sci.124, 161–170.10.1242/jcs.078170
16
Coffer P. J. Woodgett J. R. (1991). Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur. J. Biochem.201, 475–481.10.1111/j.1432-1033.1991.tb16305.x
17
Corradetti M. N. Inoki K. Bardeesy N. DePinho R. A. Guan K. L. (2004). Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev.18, 1533–1538.10.1101/gad.1199104
18
Deretic V. (2011). Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev.240, 92–104.10.1111/j.1600-065X.2010.00995.x
19
Dibble C. C. Asara J. M. Manning B. D. (2009). Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol.29, 5657–5670.10.1128/MCB.00735-09
20
Dong L. Q. Liu F. (2005). PDK2: the missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am. J. Physiol. Endocrinol. Metab.289, E187–E196.10.1152/ajpendo.00011.2005
21
Dotto G. P. Gilman M. Z. Maruyama M. Weinberg R. A. (1986). c-Myc and c-fos expression in differentiating mouse primary keratinocytes. EMBO J.5, 2853–2857.
22
Downward J. (1995). Signal transduction. A target for PI(3) kinase. Nature376, 553–554.10.1038/376553a0
23
Engelman J. A. Luo J. Cantley L. C. (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet.7, 606–619.10.1038/ni1334
24
Evangelisti C. Ricci F. Tazzari P. Tabellini G. Battistelli M. Falcieri E. et al (2011). Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia25, 781–791.10.1038/leu.2011.20
25
Ferber E. C. Peck B. Delpuech O. Bell G. P. East P. Schulze A. (2012). FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ.19, 968–979.10.1038/cdd.2011.179
26
Fournier M. V. Fata J. E. Martin K. J. Yaswen P. Bissell M. J. (2009). Interaction of E-cadherin and PTEN regulates morphogenesis and growth arrest in human mammary epithelial cells. Cancer Res.69, 4545–4552.10.1158/0008-5472.CAN-08-1694
27
Gamper C. J. Powell J. D. (2012). All PI3Kinase signaling is not mTOR: dissecting mTOR-dependent and independent signaling pathways in T cells. Front. Immunol.3:312.10.3389/fimmu.2012.00312
28
Gan X. Q. Wang J. Y. Xi Y. Wu Z. L. Li Y. P. Li L. (2008). Nuclear Dvl, c-Jun, beta-catenin, and TCF form a complex leading to stabilization of beta-catenin-TCF interaction. J. Cell Biol.180, 1087–1100.10.1083/jcb.200710050
29
Gao C. Cao W. Bao L. Zuo W. Xie G. Cai T. et al (2010). Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol.12, 781–790.10.1038/ncb2082
30
Gao C. Chen Y. G. (2010). Dishevelled: the hub of Wnt signaling. Cell. Signal.22, 717–727.10.1016/j.cellsig.2009.11.010
31
Goncharova E. A. Goncharov D. A. Li H. Pimtong W. Lu S. Khavin I. et al (2011). mTORC2 is required for proliferation and survival of TSC2-null cells. Mol. Cell. Biol.31, 2484–2498.10.1128/MCB.01061-10
32
Gottlieb E. (2011). p53 guards the metabolic pathway less travelled. Nat. Cell Biol.13, 195–197.10.1038/ncb2177
33
Guo Z. Zhou Y. Evers B. M. Wang Q. (2012). Rictor regulates FBXW7-dependent c-Myc and cyclin E degradation in colorectal cancer cells. Biochem. Biophys. Res. Commun.418, 426–432.10.1016/j.bbrc.2012.01.054
34
Guppy A. Jamal-Hanjani M. Pickering L. (2011). Anticancer effects of metformin and its potential use as a therapeutic agent for breast cancer. Future Oncol.7, 727–736.10.2217/fon.11.49
35
He X. Wang Y. Zhu J. Orloff M. Eng C. (2011). Resveratrol enhances the anti-tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin-induced AKT signaling. Cancer Lett.301, 168–176.10.1016/j.canlet.2010.11.012
36
Heallen T. Zhang M. Wang J. Bonilla-Claudio M. Klysik E. Johnson R. L. et al (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science332, 458–461.10.1126/science.1199010
37
Hoogeboom D. Burgering B. M. (2009). Should I stay or should I go: beta-catenin decides under stress. Biochim. Biophys. Acta1796, 63–74.
38
Hoogeboom D. Essers M. A. Polderman P. E. Voets E. Smits L. M. Burgering B. M. (2008). Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J. Biol. Chem.283, 9224–9230.10.1074/jbc.M706638200
39
Inoki K. Ouyang H. Zhu T. Lindvall C. Wang Y. Zhang X. et al (2006). TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell126, 955–968.10.1016/j.cell.2006.06.055
40
Jacinto E. Facchinetti V. Liu D. Soto N. Wei S. Jung S. Y. et al (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell127, 125–137.10.1016/j.cell.2006.08.033
41
Jacinto E. Loewith R. Schmidt A. Lin S. Ruegg M. A. Hall A. et al (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol.6, 1122–1128.10.1038/ncb1183
42
Jiang P. Du W. Wang X. Mancuso A. Gao X. Wu M. et al (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol.13, 310–316.10.1038/ncb2172
43
Julien L. A. Carriere A. Moreau J. Roux P. P. (2010). mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol.30, 908–921.10.1128/MCB.00601-09
44
Kang S. Chemaly E. R. Hajjar R. J. Lebeche D. (2011). Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J. Biol. Chem.286, 18465–18473.10.1074/jbc.M110.210120
45
Katayama K. Nakamura A. Sugimoto Y. Tsuruo T. Fujita N. (2008). FOXO transcription factor-dependent p15(INK4b) and p19(INK4d) expression. Oncogene27, 1677–1686.10.1038/sj.onc.1210813
46
Kim J. Kundu M. Viollet B. Guan K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol.13, 132–141.10.1038/ncb2183
47
Kim J. Woo A. J. Chu J. Snow J. W. Fujiwara Y. Kim C. G. et al (2010). A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell143, 313–324.10.1016/j.cell.2010.11.018
48
Kim J. W. Gao P. Liu Y. C. Semenza G. L. Dang C. V. (2007). Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell. Biol.27, 7381–7393.10.1128/MCB.01315-06
49
Kim L. Kimmel A. R. (2000). GSK3, a master switch regulating cell-fate specification and tumorigenesis. Curr. Opin. Genet. Dev.10, 508–514.10.1016/S0959-437X(00)00120-9
50
Kode A. Mosialou I. Silva B. C. Rached M. T. Zhou B. Wang J. et al (2012). FOXO1 orchestrates the bone-suppressing function of gut-derived serotonin. J. Clin. Invest.122, 3490–3503.10.1172/JCI64906
51
Kumar A. Lawrence J. C. Jr. Jung D. Y. Ko H. J. Keller S. R. Kim J. K. et al (2010). Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes59, 1397–1406.10.2337/db09-1061
52
Kwiatkowski D. J. Manning B. D. (2005). Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet.14, 251–258.10.1093/hmg/ddi260
53
Land H. Chen A. C. Morgenstern J. P. Parada L. F. Weinberg R. A. (1986). Behavior of myc and ras oncogenes in transformation of rat embryo fibroblasts. Mol. Cell. Biol.6, 1917–1925.
54
Laplante M. Sabatini D. M. (2009). mTOR signaling at a glance. J. Cell. Sci.122, 3589–3594.10.1242/jcs.051011
55
Lee J. H. Budanov A. V. Park E. J. Birse R. Kim T. E. Perkins G. A. et al (2010). Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science327, 1223–1228.10.1126/science.1182228
56
Lemmon M. A. Schlessinger J. (2010). Cell signaling by receptor tyrosine kinases. Cell141, 1117–1134.10.1016/j.cell.2010.06.011
57
Leslie N. R. Bennett D. Lindsay Y. E. Stewart H. Gray A. Downes C. P. (2003). Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J.22, 5501–5510.10.1093/emboj/cdg513
58
Lin C. Y. Loven J. Rahl P. B. Paranal R. M. Burge C. B. Bradner J. E. et al (2012). Transcriptional amplification in tumor cells with elevated C-Myc. Cell151, 56–67.10.1016/j.cell.2012.10.026
59
Loewith R. Jacinto E. Wullschleger S. Lorberg A. Crespo J. L. Bonenfant D. et al (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell10, 457–468.10.1016/S1097-2765(02)00636-6
60
Ma L. Chen Z. Erdjument-Bromage H. Tempst P. Pandolfi P. P. (2005). Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell121, 179–193.10.1016/j.cell.2005.02.031
61
Ma X. M. Blenis J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol.10, 307–318.10.1038/nrm2672
62
Maddocks O. D. Vousden K. H. (2011). Metabolic regulation by p53. J. Mol. Med.89, 237–245.10.1007/s00109-011-0735-5
63
Mahajan K. Mahajan N. P. (2012). PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J. Cell. Physiol.227, 3178–3184.10.1002/jcp.24065
64
Manning B. D. Cantley L. C. (2007). AKT/PKB signaling: navigating downstream. Cell129, 1261–1274.10.1016/j.cell.2007.06.009
65
Markert E. K. Levine A. J. Vazquez A. (2012). Proliferation and tissue remodeling in cancer: the hallmarks revisited. Cell Death Dis.3, e397.10.1038/cddis.2012.140
66
Mendelsohn J. Baselga J. (2003). Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol.21, 2787–2799.10.1200/JCO.2003.01.504
67
Miron M. Lasko P. Sonenberg N. (2003). Signaling from Akt to FRAP/TOR targets both 4E-BP and S6K in Drosophila melanogaster. Mol. Cell. Biol.23, 9117–9126.10.1128/MCB.23.24.9117-9126.2003
68
Mita M. M. Mita A. Rowinsky E. K. (2003). The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol. Ther.2, S169–S177.10.4161/cbt.2.2.334
69
Moore P. A. Rosen C. A. Carter K. C. (1996). Assignment of the human FKBP12-rapamycin-associated protein (FRAP) gene to chromosome 1p36 by fluorescence in situ hybridization. Genomics33, 331–332.10.1006/geno.1996.0206
70
Moore S. F. Hunter R. W. Hers I. (2011). mTORC2-mediated Akt Ser473 phosphorylation is not required for Akt1 activity in human platelets. J. Biol. Chem.36, 374–387.
71
Nicholson K. M. Anderson N. G. (2002). The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal.14, 381–395.10.1016/S0898-6568(01)00271-6
72
Nusse R. (2005). Wnt signaling in disease and in development. Cell Res.15, 28–32.10.1038/sj.cr.7290260
73
Nyfeler B. Bergman P. Triantafellow E. Wilson C. J. Zhu Y. Radetich B. et al (2011). Relieving autophagy and 4EBP1 from rapamycin resistance. Mol. Cell. Biol.14, 2867–2876.10.1128/MCB.05430-11
74
Pacheco-Pinedo E. C. Durham A. C. Stewart K. M. Goss A. M. Lu M. M. Demayo F. J. et al (2011). Wnt/beta-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J. Clin. Invest.121, 1935–1945.10.1172/JCI44871
75
Pearce L. R. Komander D. Alessi D. R. (2010). The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol.11, 9–22.10.1038/nrg2695
76
Polakis P. (2012). Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol.4, a008052.10.1101/cshperspect.a008052
77
Riaz A. Zeller K. S. Johansson S. (2012). Receptor-specific mechanisms regulate phosphorylation of AKT at Ser473: role of RICTOR in beta1 integrin-mediated cell survival. PLoS ONE7:e32081.10.1371/journal.pone.0032081
78
Robey R. B. Hay N. (2009). Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol.19, 25–31.10.1016/j.semcancer.2008.11.010
79
Ronnebaum S. M. Patterson C. (2012). The FoxO family in cardiac function and dysfunction. Annu. Rev. Physiol.72, 81–94.10.1146/annurev-physiol-021909-135931
80
Sancak Y. Bar-Peled L. Zoncu R. Markhard A. L. Nada S. Sabatini D. M. (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell141, 290–303.10.1016/j.cell.2010.02.024
81
Sarbassov D. D. Ali S. M. Kim D. H. Guertin D. A. Latek R. R. Erdjument-Bromage H. et al (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol.14, 1296–1302.10.1016/j.cub.2004.06.054
82
Sarbassov D. D. Guertin D. A. Ali S. M. Sabatini D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science307, 1098–1101.10.1126/science.1106148
83
Sato J. D. Kawamoto T. Le A. D. Mendelsohn J. Polikoff J. Sato G. H. (1983). Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol. Biol. Med.1, 511–529.
84
Schenone S. Brullo C. Musumeci F. Radi M. Botta M. (2011). ATP-competitive inhibitors of mTOR: an update. Curr. Med. Chem.18, 2995–3014.10.2174/092986711796150432
85
Schlessinger J. (2000). Cell signaling by receptor tyrosine kinases. Cell103, 211–225.10.1016/S0092-8674(00)00114-8
86
Shor B. Gibbons J. J. Abraham R. T. Yu K. (2009). Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle8, 3831–3837.10.4161/cc.8.23.10070
87
Shoshan-Barmatz V. De Pinto V. Zweckstetter M. Raviv Z. Keinan N. Arbel N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med.31, 227–285.10.1016/j.mam.2010.03.002
88
Soto Martinez J. L. Cabrera Morales C. M. Serrano Ortega S. Lopez-Nevot M. A. (2005). Mutation and homozygous deletion analyses of genes that control the G1/S transition of the cell cycle in skin melanoma: p53, p21, p16 and p15. Clin. Transl. Oncol.7, 156–164.10.1007/BF02708753
89
Staal S. P. Hartley J. W. Rowe W. P. (1977). Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc. Natl. Acad. Sci. U.S.A.74, 3065–3067.10.1073/pnas.74.7.3065
90
Stein J. Milewski W. M. Hara M. Steiner D. F. Dey A. (2011). GSK-3 inactivation or depletion promotes beta-cell replication via down regulation of the CDK inhibitor, p27 (Kip1). Islets3, 21–34.10.4161/isl.3.1.14435
91
Stipanuk M. H. (2009). Macroautophagy and its role in nutrient homeostasis. Nutr. Rev.67, 677–689.10.1111/j.1753-4887.2009.00252.x
92
Stott F. J. Bates S. James M. C. McConnell B. B. Starborg M. Brookes S. et al (1998). The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J.17, 5001–5014.10.1093/emboj/17.17.5001
93
Sun A. Bagella L. Tutton S. Romano G. Giordano A. (2007). From G0 to S phase: a view of the roles played by the retinoblastoma (Rb) family members in the Rb-E2F pathway. J. Cell. Biochem.102, 1400–1404.10.1002/jcb.21609
94
Swami M. (2010). MYC matters. Nat. Rev. Cancer10, 812.10.1038/nrc2879
95
Talchai C. Xuan S. Lin H. V. Sussel L. Accili D. (2012). Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell150, 1223–1234.10.1016/j.cell.2012.07.029
96
Thompson D. M. Gill G. N. (1985). The EGF receptor: structure, regulation and potential role in malignancy. Cancer Surv.4, 767–788.
97
Treins C. Warne P. H. Magnuson M. A. Pende M. Downward J. (2010). Rictor is a novel target of p70 S6 kinase-1. Oncogene29, 1003–1016.10.1038/onc.2009.401
98
Veilleux A. Houde V. P. Bellmann K. Marette A. (2010). Chronic inhibition of the mTORC1/S6K1 pathway increases insulin-induced PI3K activity but inhibits Akt2 and glucose transport stimulation in 3T3-L1 adipocytes. Mol. Endocrinol.24, 766–778.10.1210/me.2009-0328
99
Vincent E. E. Elder D. J. Thomas E. C. Phillips L. Morgan C. Pawade J. et al (2011). Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer104, 1755–1761.10.1038/bjc.2011.132
100
Wang H. Brown J. Gu Z. Garcia C. A. Liang R. Alard P. et al (2011). Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-beta-signaling pathways regulates the innate inflammatory response. J. Immunol.186, 5217–5226.10.4049/jimmunol.1001252
101
Wang Z. Zhong J. Inuzuka H. Gao D. Shaik S. Sarkar F. H. et al (2012). An evolving role for DEPTOR in tumor development and progression. Neoplasia14, 368–375.
102
Warburg O. (1956). On the origin of cancer cells. Science123, 309–314.10.1126/science.123.3191.309
103
Weihl C. C. (2011). Valosin containing protein associated fronto-temporal lobar degeneration: clinical presentation, pathologic features and pathogenesis. Curr. Alzheimer Res.8, 252–260.10.2174/156720511795563773
104
White E. Lowe S. W. (2009). Eating to exit: autophagy-enabled senescence revealed. Genes Dev.23, 784–787.10.1101/gad.1795309
105
Wu D. Pan W. (2010). GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci.35, 161–168.10.1016/j.tibs.2009.10.002
106
Wullschleger S. Loewith R. Hall M. N. (2006). TOR signaling in growth and metabolism. Cell124, 471–484.10.1016/j.cell.2006.01.016
107
Xie X. Zhang D. Zhao B. Lu M. K. You M. Condorelli G. et al (2011). IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc. Natl. Acad. Sci. U.S.A.108, 6474–6479.10.1073/pnas.1011953108
108
Yang Z. Klionsky D. J. (2010). Eaten alive: a history of macroautophagy. Nat. Cell Biol.12, 814–822.10.1038/ncb2099
109
Young A. R. Narita M. Ferreira M. Kirschner K. Sadaie M. Darot J. F. et al (2009). Autophagy mediates the mitotic senescence transition. Genes Dev.23, 798–803.10.1101/gad.519709
110
Zhang H. H. Lipovsky A. I. Dibble C. C. Sahin M. Manning B. D. (2006). S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell24, 185–197.10.1016/j.molcel.2006.10.025
111
Zhang J. Gao Z. Yin J. Quon M. J. Ye J. (2008). S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem.283, 35375–35382.10.1074/jbc.M802578200
112
Zhang S. Tang Q. Xu F. Xue Y. Zhen Z. Deng Y. et al (2009). RhoA regulates G1-S progression of gastric cancer cells by modulation of multiple INK4 family tumor suppressors. Mol. Cancer Res.7, 570–580.10.1158/1541-7786.MCR-08-0533
Summary
Keywords
mTORC1, autophagy, Wnt, G1-S, cell cycle
Citation
Vadlakonda L, Pasupuleti M and Pallu R (2013) Role of PI3K-AKT-mTOR and Wnt Signaling Pathways in Transition of G1-S Phase of Cell Cycle in Cancer Cells. Front. Oncol. 3:85. doi: 10.3389/fonc.2013.00085
Received
07 February 2013
Accepted
31 March 2013
Published
12 April 2013
Volume
3 - 2013
Edited by
Alexandre Arcaro, University of Bern, Switzerland
Reviewed by
Deborah Stroka, University of Bern, Switzerland; Alexandre Arcaro, University of Bern, Switzerland
Copyright
© 2013 Vadlakonda, Pasupuleti and Pallu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Lakshmipathi Vadlakonda, CR Rao Advanced Institute of Mathematics Statistics and Computer Science, University of Hyderabad, Hyderabad 500046, India. e-mail: lvadlakonda@crraoaimscs.res.in
†Present address: Lakshmipathi Vadlakonda, CR Rao Advanced Institute of Mathematics Statistics and Computer Science, University of Hyderabad, Hyderabad, India.
This article was submitted to Frontiers in Molecular and Cellular Oncology, a specialty of Frontiers in Oncology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.