
- Division of Molecular Oncology, The Netherlands Cancer Institute, Amsterdam, Netherlands
Telomeres are repetitive sequences at the natural ends of linear eukaryotic chromosomes that protect these from recognition as chromosome breaks. Their ability to do so critically depends on the binding of sufficient quantities of functional shelterin, a six-unit protein complex with specific and crucial roles in telomere maintenance and function. Insufficient telomere length, leading to insufficient concentration of shelterin at chromosome ends, or otherwise crippled shelterin function, causes telomere deprotection. While contributing to aging-related pathologies, loss of telomere protection can act as a barrier to tumorigenesis, as dysfunctional telomeres activate DNA-damage-like checkpoint responses that halt cell proliferation or trigger cell death. In addition, dysfunctional telomeres affect cancer development and progression by being a source of genomic instability. Reviewed here are the different approaches that are being undertaken to investigate the mammalian cellular response to telomere dysfunction and its consequences for cancer. Furthermore, it is discussed how current and future knowledge about the mechanisms underlying telomere damage responses might be applied for diagnostic purposes or therapeutic intervention.
Introduction
Linear eukaryotic chromosomes pose multiple challenges to cells that need to be properly taken care of to avoid loss of proliferative capacity and genome integrity. As conventional DNA-polymerases cannot replicate the very ends of chromosomes, each cell division chromosome ends lose a bit of DNA-sequence, potentially causing loss of genetic information. Furthermore, cells contain multiple DNA-damage recognition and repair activities that act on exposed DNA-ends to halt cell cycle progression and fix chromosome-internal DNA-breaks to prevent unequal separation of genetic information into daughter cells. If these activities would continuously act on natural chromosome ends, cells would not be able to divide or properly segregate chromosomes during mitosis. Telomeres are specialized nucleoprotein structures at chromosome ends that cope with these challenges (de Lange, 2009; O’Sullivan and Karlseder, 2010). By consisting of long stretches of TTAGGG DNA-repeats and lacking genes, telomeres represent buffers that prevent replication-associated sequence loss at chromosome termini from reaching nearby genes. Moreover, telomeres protect natural chromosome ends from being recognized and processed as damaged DNA. For this mammalian telomeres rely on binding shelterin, a unique set of proteins composed of TRF1, TRF2, RAP1, TIN2, TPP1, and POT1, and on specific structural features. Telomeres contain a 3′ G-rich single-strand overhang that binds POT1, folds back and invades the duplex telomere repeat array. This forms a loop (t-loop) that presumably hides the extreme end of the chromosome from multiple activities.
While acting as buffers to prevent loss of genetic information, telomeres themselves shorten with every round of replication (Harley et al., 1990). Progressive shortening eventually causes telomeres to lose their protective activity, even before all telomere repeats are lost (Figure 1). Dividing cells can only avoid telomere deprotection when able to induce sufficient activity of the telomerase enzyme to add telomere repeats (Bodnar et al., 1998). Alternatively cells can engage the ALT mechanism of “alternative lengthening of telomeres” to lengthen telomeres through recombination (Cesare and Reddel, 2010). Normal human somatic cells do not have sufficient telomerase or ALT activity, which confronts them with short dysfunctional telomeres after a certain number of cell divisions.
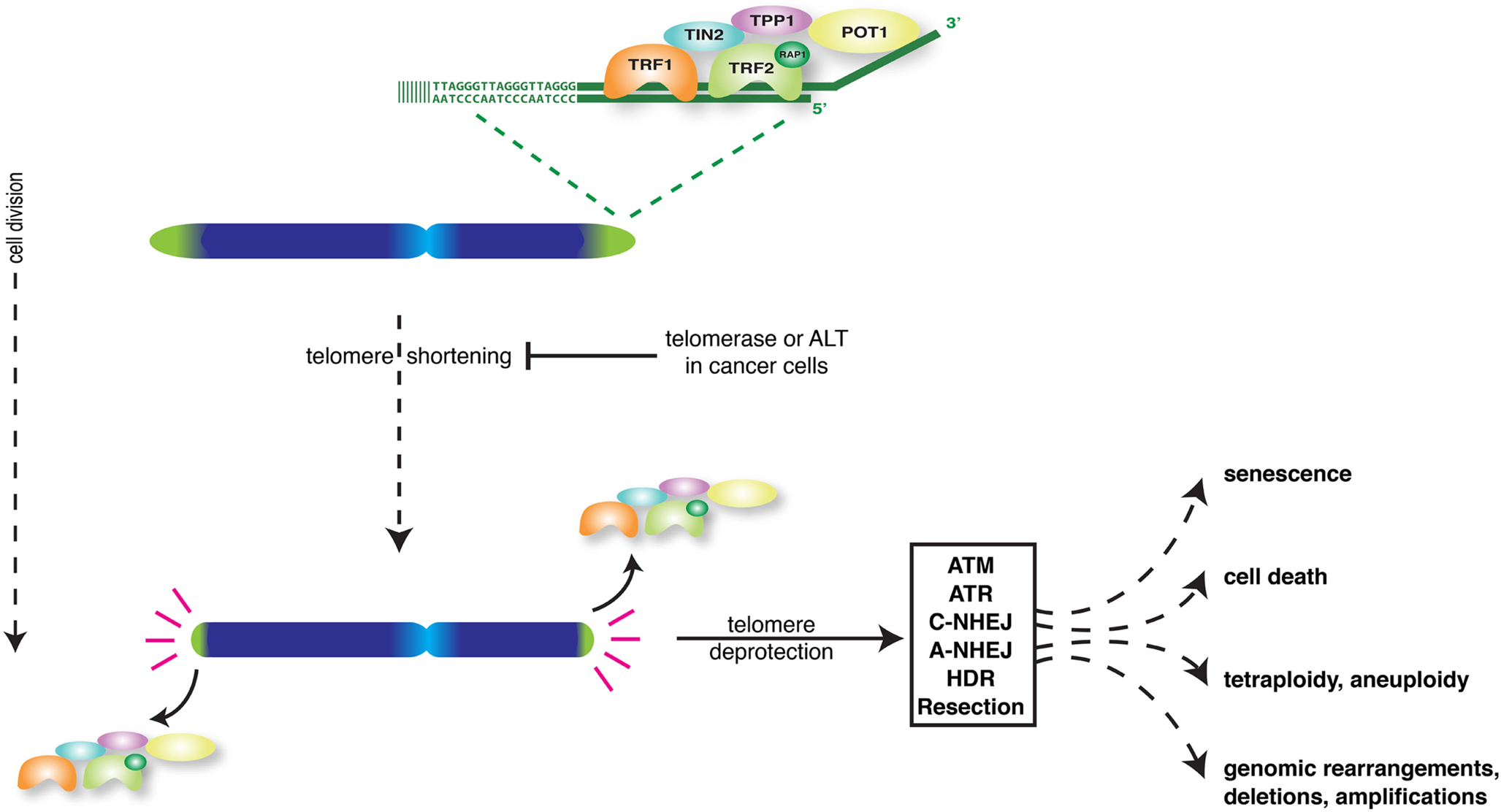
Figure 1. Overview of the major consequences of loss of telomere protection in mammalian cells.
Loss of telomere protection activates a DNA-damage-like signaling response that through induction of tumor suppressors p53 and p16 forces cells into senescence or apoptosis (d’Adda di Fagagna et al., 2003; Takai et al., 2003; Jacobs and de Lange, 2004, 2005; de Lange, 2010; O’Sullivan and Karlseder, 2010). This serves as an important tumor suppressor mechanism as it prevents the outgrowth of potentially cancerous cells. However telomere dysfunction can also be a source of genomic instability and put cells at risk of developing into cancer (Artandi and DePinho, 2010; Martinez and Blasco, 2010; Davoli and de Lange, 2011; Shay and Wright, 2011). This because DNA-repair activities at deprotected chromosome ends generate chromosome end-to-end fusions. In cells that escape senescence or apoptosis and divide, such fusions initiate breakage-fusion-bridge cycles that generate complex unbalanced chromosome rearrangements (Murnane, 2012). In addition, telomere dysfunction can lead to tetraploidization and chromosome mis-segregation (Davoli et al., 2010; Davoli and de Lange, 2012). Thus, depending on the effectiveness of the DNA-damage checkpoint in arresting or eliminating cells, telomere dysfunction either inhibits or promotes the development of cancer.
Understanding the Consequences of Telomere Deprotection
Multiple approaches have contributed to our current understanding of the molecular events and consequences associated with loss of telomere protection (Figure 2). These can roughly be grouped into three categories: (1) models of telomere uncapping in mammalian cells or mice; (2) analyses on patient-derived material; (3) unbiased and genome-wide approaches.
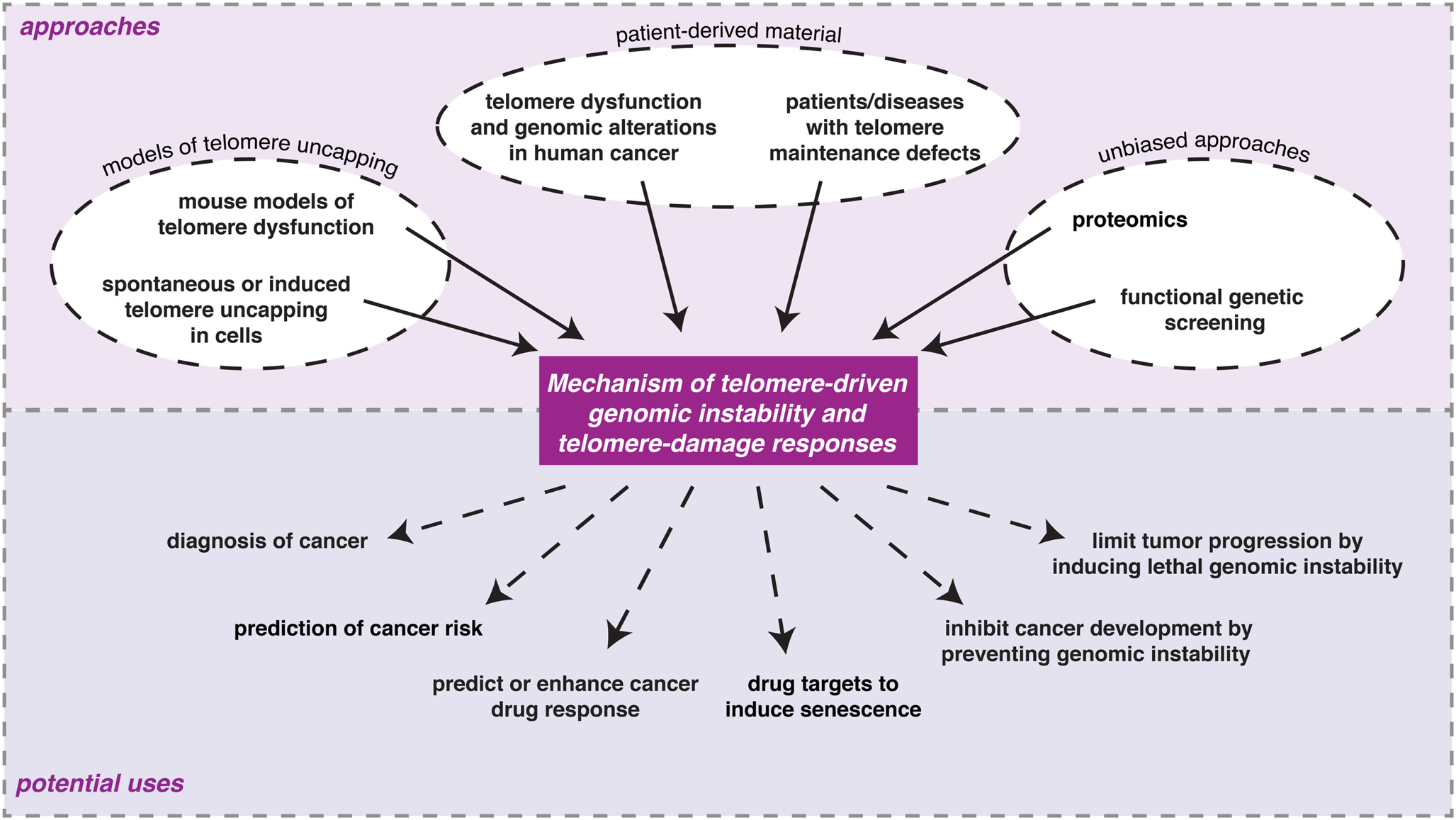
Figure 2. Examples of the different experimental approaches being undertaken to understand the consequences of loss of telomere protection in mammalian cells and the potential ways in which information from such studies might eventually be used in the clinic.
Models of Telomere Uncapping in Mammalian Cells or Mice
Telomere uncapping occurs when telomeres become critically short upon continued proliferation in the absence of telomerase. Telomere dysfunction is therefore ideally studied in a replicative senescence model where primary human cells are propagated in vitro until they reach their maximum lifespan, the so-called Hayflick limit, and stop proliferating. Analysis of the molecular changes induced at replicative senescence has yielded critical insights, such as that critically short telomeres are recognized by DNA-damage response (DDR) proteins (d’Adda di Fagagna et al., 2003). Despite the advantage of following the natural course of telomere uncapping, this approach also has limitations. One problem is that prolonged culturing of cells in vitro is associated with culture stress inducing complex telomere-independent cellular responses that partially overlap with telomere-mediated responses. Equally important, telomere uncapping due to shortening is not a synchronous process, complicating studies on telomere-dependent effects in a population of cells. Only a subset of cells experiences critically short telomeres at a given time and one has to wait until the majority of cells has senesced. This precludes detection of immediate effects of telomere uncapping. Studies on natural telomere deprotection in the context of an entire organism face additional challenges. These are caused by the variability in telomere lengths between individuals and between individual cells, but also by the slow speed of telomere shortening. A particular problem arises with the use of inbred laboratory mouse strains. While being a tremendously useful model system to study many different biological pathways, laboratory mice are not a good model to study the consequences of natural telomere shortening because, unlike humans, commonly used laboratory mouse strains have extremely long telomeres and high telomerase activity in all cells. During their normal lifespan such mice do not experience significant telomere uncapping (Blasco et al., 1997). Importantly, this also implies that most studies in laboratory mice, including those modeling cancer, do not incorporate the contribution of a telomere dysfunction component that would apply to humans.
A solution to problems associated with studying natural telomere shortening came from strategies in which telomere dysfunction is experimentally induced. Following the identification of the telomerase reverse transcriptase and RNA components and the different shelterin factors, significant knowledge about their function has come from experimental manipulation of telomerase or shelterin in tissue culture cells and mice. Apart from proving that telomeres control replicative lifespan and affect the development of cancer and aging-related pathologies, such studies also revealed many underlying molecular details (Artandi and DePinho, 2010; Martinez and Blasco, 2010; Sahin and Depinho, 2010; Shay and Wright, 2011; Tumpel and Rudolph, 2012). We now know that telomere function depends on both unique and redundant roles of shelterin components in protecting chromosome termini against six major threats: ATM-kinase activation, ATR-kinase activation, DNA-Ligase 4- and Ku70/80-dependent classical non-homologous end-joining (c-NHEJ), DNA-Ligase 3- and PARP-dependent alternative NHEJ (a-NHEJ), homologous recombination (HR), and end-resection (Figure 1) (Sfeir and de Lange, 2012). Activation of ATM and ATR results in checkpoint activation and proliferation arrest or apoptosis. c-NHEJ generates chromosome-end-to-end fusions without apparent major end-processing, leaving significant amounts of telomere repeats at the fusion sites. On the other hand, a-NHEJ, the primary pathway causing chromosomal translocations, uses microhomology and results in chromosome-end-to-end fusion with significant telomeric and subtelomeric deletion. Resection and HR both threaten telomere integrity, the latter for instance via unequal exchanges between sister telomeres that change telomere length.
Of the six threats, ATM and c-NHEJ are specifically blocked by TRF2, ATR is inhibited by POT1, while HR is inhibited by either POT1 or RAP1, and on top by Ku70/80. On the other hand, repression of a-NHEJ and end-resection depend on redundant functions of shelterin and in addition are repressed by Ku70/80 and 53BP1, respectively. While the TTAGGG-repeats provided by telomerase are needed to concentrate enough shelterin at chromosome ends, telomerase activity itself is regulated by shelterin, as well as by other factors and processes, together contributing to complex control of telomere maintenance (Cifuentes-Rojas and Shippen, 2012).
Studies in mice with manipulated shelterin or telomerase have shown the consequences of telomere deprotection on an organismal level. These studies clearly illustrate the opposite effects dysfunctional telomeres can have on the development of cancer, depending on the DNA-damage checkpoint status and stage of tumor development. Indeed, short telomeres in late-generation telomerase-deficient mice protect against cancer, while predisposing to aging (Greenberg et al., 1999; Rudolph et al., 1999). That is, when DNA-damage checkpoint activity by p53 is intact. In a p53-deficient background, where cells escape senescence or apoptosis, short telomeres accelerate the onset of epithelial tumorigenesis through increased genomic instability (Artandi et al., 2000). While telomere-driven genomic instability can promote cancer initiation, it can limit further cancer progression. Re-establishment of telomere protection by activation of telomerase or ALT facilitates further malignant progression by genome stabilization and improved cell viability (Begus-Nahrmann et al., 2012; Ding et al., 2012; Hu et al., 2012).
Analyses on Patient-Derived Material
Analyses of human cancers revealed that telomere shortening is widespread during human tumorigenesis. In addition, it was found that during their development all human tumors activate mechanisms to counteract further telomere shortening. Most tumors activate telomerase while 10–15% of tumors activate ALT (Bryan et al., 1997; Shay and Bacchetti, 1997). There is mounting evidence that concomitant with shortening, telomeres become dysfunctional during human cancer development (Artandi and DePinho, 2010). For example, anaphase bridges, a hallmark of dysfunctional telomeres leading to genomic instability, become more abundant at (pre)-invasive stages of colon and breast cancer. Moreover, in breast cancer the presence of short telomeres coincides with a sudden rise in genomic instability at the transition from ductal hyperplasia to carcinoma in situ and before the activation of telomerase (Chin et al., 2004). This, together with the recent PCR-based detection of telomere fusions in early breast cancer, provides strong evidence for the occurrence of telomere crisis during breast cancer development (Tanaka et al., 2012). In addition, syndromes associated with telomere replication defects, such as dyskeratosis congenita and Werner syndrome, are characterized by short telomeres, genomic instability, and an elevated incidence of spontaneous cancer (Armanios and Blackburn, 2012). Similarly, diseases accompanied by increased cell turnover, such as ulcerative colitis or liver cirrhosis, are associated with accelerated telomere shortening, telomere dysfunction, and cancer-predisposition. However, the currently most convincing data supporting a role for telomere dysfunction in cancer development stems from the molecular analysis of chronic lymphocytic leukemia (CLL) (Lin et al., 2010). A major advantage is that this cancer allows analysis of samples from patients at different stages of the disease. Single-molecule telomere length and telomere fusion analysis showed that short telomeres and fusions increase with more advanced disease, but are already present in a subset of patients prior to disease progression. Specifically patients with dysfunctional telomeres, not those with longer telomeres, were found to have large-scale genomic rearrangements concentrated in telomeric regions. Together the data strongly support the model that telomere attrition and fusion contribute to the progression of CLL. Moreover, the recent discovery of frequent POT1 mutations and associated telomeric and chromosomal abnormalities in CLL further reinforces the involvement of telomere dysfunction in the progression of this cancer (Ramsay et al., 2013).
Despite all analyses on human tumors the relative contribution of telomere deprotection to cancer development remains unclear. While next-generation sequencing approaches rapidly generate large amounts of data on (epi)genetic and transcriptional aberrations in human cancers, such approaches cannot easily reveal how many tumors developed under facilitation by telomere-driven genomic instability. For one, telomere repeats are filtered out during the analysis of such sequencing data, as they cannot be mapped to specific chromosomes. Also, current PCR-based methods do not detect all telomere fusions. Furthermore, telomere-driven genomic instability follows a hit-and-run mode. One single fusion of an uncapped telomere to another uncapped telomere or a DNA-double-strand break is sufficient to initiate breakage-fusion-bridge cycles where subsequent breaks and fusions do not necessarily involve telomeres but involve the entire genome. Thus both quantitative assessment of telomere fusion events and evaluation of the full extent of genomic alterations triggered by telomere uncapping, as opposed to other causes, remain difficult.
Unbiased and Genome-Wide Approaches
Biochemical fishing expeditions using mass-spectrometry or yeast-two-hybrid screens have tremendously contributed to our current knowledge about telomere biology by identifying multiple factors that interact with mammalian telomeres. These consist of shelterin components as well as many telomere-associated proteins that are not considered as part of shelterin because they have main functions outside of telomeres (Palm and de Lange, 2008; Dejardin and Kingston, 2009; Nittis et al., 2010). Among telomere-associated factors are nucleases, helicases, DNA-replication proteins, DDR factors, and the CST (CTC1, STN1, and TEN1) complex, which plays critical roles in controlling telomeric G-overhang length, telomere replication, and telomerase activity (Palm and de Lange, 2008; Chen et al., 2012; Gu et al., 2012; Sfeir, 2012; Wu et al., 2012). The finding that DDR factors are already present at telomeres that are sufficiently long to prevent activation of DNA-damage signaling responses was surprising. Subsequent work revealed that some of these factors in fact play protective roles. For instance Ku assists in protecting telomeres against HR and a-NHEJ, whereas ATM and ATR have been proposed to facilitate completion of telomere replication and formation of a proper telomere structure (Verdun et al., 2005; Celli et al., 2006; Verdun and Karlseder, 2006; Sfeir and de Lange, 2012).
Not all telomere-associated factors interact with telomeres constantly. Some interactions are restricted to certain cell cycle phases or specific conditions. For instance, when telomeres become critically short, some protein interactions disappear whereas new proteins now intensively associate with telomeres, such as 53BP1 and other DDR components. Likewise, differences exist between proteins present at telomeres maintained by telomerase versus those present at telomeres maintained by ALT. If proteomics-based approaches would be applied to compare different telomere states, such as capped versus uncapped, this could significantly increase our understanding of the mechanisms underlying control of telomere damage responses. A major challenge in such studies is that many signaling regulators are low in abundance or might only transiently or weakly associate with telomeres. Conventional immunoprecipitation and mass-spectrometry might therefore have to be complemented with more sensitive techniques, such as bimolecular fluorescent complementation that enables analysis of protein interactions in living cells and has recently been applied to identify proteins interacting with shelterin (Lee et al., 2011).
A powerful approach that proved very successful to identify key components of multiple cellular pathways, including the response to DNA-double-strand breaks, is gain-of-function or loss-of-function genetic screening in mammalian cells (Jacobs et al., 2000; Brummelkamp et al., 2004; Kolas et al., 2007; Paulsen et al., 2009; Ashworth and Bernards, 2010; Hurov et al., 2010; Cotta-Ramusino et al., 2011; Gudjonsson et al., 2012; Nguyen et al., 2012). Functional genetic screening has also proven powerful to identify novel telomere regulators in lower organisms, leading for instance to implication of the yeast KEOPs complex in telomere regulation (Downey et al., 2006). Since recently, functional genetic screening is also being applied in the field of mammalian telomere biology. For instance, RNA-interference screens have been initiated to identify factors involved in regulation of telomerase activity (Coussens et al., 2010; Cerone et al., 2011), ALT (Osterwald et al., 2012), telomere protection (Lackner et al., 2011), or telomere-driven genomic instability (Jacobs et al., unpublished). As an example, high-throughput RNA-interference screening identified multiple kinases, including ERK8, with no previous association to telomere biology as novel regulators of telomerase activity and attractive new drug targets for cancer therapy (Cerone et al., 2011). Given the previous successes in multiple experimental settings, recently initiated or future unbiased functional genetic screening approaches in mammalian systems hold significant potential for identifying new factors with important roles in the control of cancer or aging by telomeres.
Opportunities
Telomerase-Based Approaches
The strong dependence on telomere maintenance for sustained tumor growth and the presence of telomerase in 90% of cancers, but not most somatic cells, have inspired development of telomerase-based strategies to inhibit cancer cell growth and triggered evaluation of telomerase as a biomarker or prognostic marker for cancer (Ruden and Puri, 2012; Mocellin et al., 2013). Multiple telomerase-based strategies are currently evaluated in clinical trials, but it is still unclear how beneficial such strategies are to cancer patients. This is in part due to incomplete understanding of the effects of inhibiting telomerase on both cancer cells and normal cells. Telomerase is important for the renewal capacity of normal stem and progenitor cells, which could cause unwanted side-effects. Telomerase has also been implicated in telomere-independent pathways, making the outcome of its inhibition less predictable until these pathways and roles of telomerase therein are fully understood (Martinez and Blasco, 2011; Ghosh et al., 2012). In addition, as telomere uncapping can cause genomic instability, there is a risk that telomerase inhibition might in fact accelerate progression of some tumor cells (Pereira and Ferreira, 2013). Moreover, dysfunctional telomeres cause impaired mitochondrial function and promote oxidative stress, which might lead to accumulation of additional mutations that promote tumor progression (Sahin et al., 2011). Furthermore, the generation of senescent cells by telomere uncapping to limit outgrowth of the targeted tumor, comes at the cost of generating a permissive environment for tumor progression. Namely, senescent cells are known to secrete multiple factors that can promote tumor growth and invasion of surrounding (pre-)malignant cells (Rodier and Campisi, 2011). The risks associated with telomerase inhibition and telomere uncapping might be less of a concern in patients with advanced cancer, but they could limit the use of telomerase-based strategies in young patients or patients with early disease.
Alternative Strategies
Understanding the molecular mechanisms underlying telomere protection, telomere-driven senescence and genomic instability will not only increase predictability and understanding of the efficacy of telomerase-based strategies, but will also facilitate development of novel therapeutic strategies for cancer and (premature) aging-related disease (Figure 2). For cancer these could aim to provoke senescence, as with telomerase inhibition. However, particularly interesting would be to think of alternative strategies that avoid unwanted side-effects or risks of existing approaches, such as associated with induction of senescence. A potential, but challenging strategy could be to inhibit telomere-driven genomic instability before obvious cancer presentation to prevent or slow down cancer development. This might for instance be applied to people predisposed to cancer because of a condition associated with accelerated telomere shortening. Inhibition of telomere-driven genomic instability could be achieved by inhibiting telomere fusion by interfering with DNA-repair activities at uncapped telomeres. Such a strategy should not impair regular error-free DNA-repair at other sites, to avoid increasing cancer risk. An intriguing opposite strategy might in fact be to increase genomic instability such that cancer cells die of crisis, by “tipping the balance.” High levels of genomic instability, that may drive initiation of early lesions, need to be reduced to allow efficient tumor progression. Nevertheless, many cancers are hallmarked by an instable genome, hyper-sensitizing them to genome-destabilizing strategies. Interestingly, increased and ongoing chromosomal instability is also common for tumors utilizing ALT, along with telomeres that are recognized as DNA-damage, but appear not to fuse (Scheel et al., 2001; Cesare et al., 2009; Lovejoy et al., 2012). Besides interfering with the ALT mechanism, alleviating repression of telomere fusion might be an effective way to kill ALT tumor cells by inducing rampant genomic instability. This is an especially intriguing possibility given that ALT tumors do not rely on telomerase for telomere maintenance and would thus be resistant to telomerase-based therapeutics.
The development and adequate application of such new therapeutic strategies, require thorough investigation and understanding of the mechanisms underlying control of DDR and repair activities at telomeres and DNA-lesions. Apart from revealing new drug targets, studies addressing this might also contribute to prediction of therapy responses to DNA-damage inducing anti-cancer drugs or to development of strategies to enhance cancer drug sensitivity. With respect to the latter, it is interesting to note that dysfunctional telomeres have been shown to increase the sensitivity of cells to ionizing radiation and DNA-damaging chemotherapeutics (Wong et al., 2000; Lee et al., 2001; Soler et al., 2009). In addition, insights into the precise events associated with telomere uncapping could contribute to the establishment of diagnostic tools, such as for early detection of cancers and identification of cancer-predisposed individuals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank the members of my group for their comments and apologize to colleagues whose work was not cited due to space constraints. Work in my group is supported by the Dutch Cancer Society (NKI2012-5305), the Netherlands Organization for Scientific Research (ALW822.02.004), the European Research Council (ERCStG 311565), and the EMBO Young Investigator Program.
References
Artandi, S. E., Chang, S., Lee, S. L., Alson, S., Gottlieb, G. J., Chin, L., et al. (2000). Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406, 641–645.
Artandi, S. E., and DePinho, R. A. (2010). Telomeres and telomerase in cancer. Carcinogenesis 31, 9–18.
Ashworth, A., and Bernards, R. (2010). Using functional genetics to understand breast cancer biology. Cold Spring Harb. Perspect. Biol. 2, a003327.
Begus-Nahrmann, Y., Hartmann, D., Kraus, J., Eshraghi, P., Scheffold, A., Grieb, M., et al. (2012). Transient telomere dysfunction induces chromosomal instability and promotes carcinogenesis. J. Clin. Invest. 122, 2283–2288.
Blasco, M. A., Lee, H. W., Hande, M. P., Samper, E., Lansdorp, P. M., Depinho, R. A., et al. (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34.
Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G. B., et al. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352.
Brummelkamp, T. R., Berns, K., Hijmans, E. M., Mullenders, J., Fabius, A., Heimerikx, M., et al. (2004). Functional identification of cancer-relevant genes through large-scale RNA interference screens in mammalian cells. Cold Spring Harb. Symp. Quant. Biol. 69, 439–445.
Bryan, T. M., Englezou, A., Dalla-Pozza, L., Dunham, M. A., and Reddel, R. R. (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 3, 1271–1274.
Celli, G. B., Denchi, E. L., and De Lange, T. (2006). Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 8, 885–890.
Cerone, M. A., Burgess, D. J., Naceur-Lombardelli, C., Lord, C. J., and Ashworth, A. (2011). High-throughput RNAi screening reveals novel regulators of telomerase. Cancer Res. 71, 3328–3340.
Cesare, A. J., Kaul, Z., Cohen, S. B., Napier, C. E., Pickett, H. A., Neumann, A. A., et al. (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 16, 1244–1251.
Cesare, A. J., and Reddel, R. R. (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 11, 319–330.
Chen, L. Y., Redon, S., and Lingner, J. (2012). The human CST complex is a terminator of telomerase activity. Nature 488, 540–544.
Chin, K., De Solorzano, C. O., Knowles, D., Jones, A., Chou, W., Rodriguez, E. G., et al. (2004). In situ analyses of genome instability in breast cancer. Nat. Genet. 36, 984–988.
Cotta-Ramusino, C., McDonald, E. R. III, Hurov, K., Sowa, M. E., Harper, J. W., and Elledge, S. J. (2011). A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317.
Coussens, M., Davy, P., Brown, L., Foster, C., Andrews, W. H., Nagata, M., et al. (2010). RNAi screen for telomerase reverse transcriptase transcriptional regulators identifies HIF1alpha as critical for telomerase function in murine embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 13842–13847.
Davoli, T., and de Lange, T. (2011). The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 27, 585–610.
Davoli, T., and de Lange, T. (2012). Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 21, 765–776.
Davoli, T., Denchi, E. L., and De Lange, T. (2010). Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 141, 81–93.
de Lange, T. (2010). How shelterin solves the telomere end-protection problem. Cold Spring Harb. Symp. Quant. Biol. 75, 167–177.
Dejardin, J., and Kingston, R. E. (2009). Purification of proteins associated with specific genomic loci. Cell 136, 175–186.
d’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198.
Ding, Z., Wu, C. J., Jaskelioff, M., Ivanova, E., Kost-Alimova, M., Protopopov, A., et al. (2012). Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 148, 896–907.
Downey, M., Houlsworth, R., Maringele, L., Rollie, A., Brehme, M., Galicia, S., et al. (2006). A genome-wide screen identifies the evolutionarily conserved KEOPS complex as a telomere regulator. Cell 124, 1155–1168.
Ghosh, A., Saginc, G., Leow, S. C., Khattar, E., Shin, E. M., Yan, T. D., et al. (2012). Telomerase directly regulates NF-kappaB-dependent transcription. Nat. Cell Biol. 14, 1270–1281.
Greenberg, R. A., Chin, L., Femino, A., Lee, K. H., Gottlieb, G. J., Singer, R. H., et al. (1999). Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 97, 515–525.
Gu, P., Min, J. N., Wang, Y., Huang, C., Peng, T., Chai, W., et al. (2012). CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 31, 2309–2321.
Gudjonsson, T., Altmeyer, M., Savic, V., Toledo, L., Dinant, C., Grofte, M., et al. (2012). TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709.
Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460.
Hu, J., Hwang, S. S., Liesa, M., Gan, B., Sahin, E., Jaskelioff, M., et al. (2012). Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148, 651–663.
Hurov, K. E., Cotta-Ramusino, C., and Elledge, S. J. (2010). A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 24, 1939–1950.
Jacobs, J. J., and de Lange, T. (2004). Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr. Biol. 14, 2302–2308.
Jacobs, J. J., and de Lange, T. (2005). p16INK4a as a second effector of the telomere damage pathway. Cell Cycle 4, 1364–1368.
Jacobs, J. J., Keblusek, P., Robanus-Maandag, E., Kristel, P., Lingbeek, M., Nederlof, P. M., et al. (2000). Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat. Genet. 26, 291–299.
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640.
Lackner, D. H., Durocher, D., and Karlseder, J. (2011). A siRNA-based screen for genes involved in chromosome end protection. PLoS ONE 6:e21407. doi:10.1371/journal.pone.0021407
Lee, K. H., Rudolph, K. L., Ju, Y. J., Greenberg, R. A., Cannizzaro, L., Chin, L., et al. (2001). Telomere dysfunction alters the chemotherapeutic profile of transformed cells. Proc. Natl. Acad. Sci. U.S.A. 98, 3381–3386.
Lee, O. H., Kim, H., He, Q., Baek, H. J., Yang, D., Chen, L. Y., et al. (2011). Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells. Mol. Cell Proteomics 10, M110001628.
Lin, T. T., Letsolo, B. T., Jones, R. E., Rowson, J., Pratt, G., Hewamana, S., et al. (2010). Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: evidence for a telomere crisis. Blood 116, 1899–1907.
Lovejoy, C. A., Li, W., Reisenweber, S., Thongthip, S., Bruno, J., De Lange, T., et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 8:e1002772. doi:10.1371/journal.pgen.1002772
Martinez, P., and Blasco, M. A. (2010). Role of shelterin in cancer and aging. Aging Cell 9, 653–666.
Martinez, P., and Blasco, M. A. (2011). Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 11, 161–176.
Mocellin, S., Pooley, K. A., and Nitti, D. (2013). Telomerase and the search for the end of cancer. Trends. Mol. Med. 19, 125–133.
Nguyen, C. L., Possemato, R., Bauerlein, E. L., Xie, A., Scully, R., and Hahn, W. C. (2012). Nek4 regulates entry into replicative senescence and the response to DNA damage in human fibroblasts. Mol. Cell. Biol. 32, 3963–3977.
Nittis, T., Guittat, L., Leduc, R. D., Dao, B., Duxin, J. P., Rohrs, H., et al. (2010). Revealing novel telomere proteins using in vivo cross-linking, tandem affinity purification, and label-free quantitative LC-FTICR-MS. Mol. Cell Proteomics 9, 1144–1156.
Osterwald, S., Worz, S., Reymann, J., Sieckmann, F., Rohr, K., Erfle, H., et al. (2012). A three-dimensional colocalization RNA interference screening platform to elucidate the alternative lengthening of telomeres pathway. Biotechnol. J. 7, 103–116.
O’Sullivan, R. J., and Karlseder, J. (2010). Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 11, 171–181.
Palm, W., and de Lange, T. (2008). How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334.
Paulsen, R. D., Soni, D. V., Wollman, R., Hahn, A. T., Yee, M. C., Guan, A., et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 35, 228–239.
Pereira, B., and Ferreira, M. G. (2013). Sowing the seeds of cancer: telomeres and age-associated tumorigenesis. Curr. Opin. Oncol. 25, 93–98.
Ramsay, A. J., Quesada, V., Foronda, M., Conde, L., Martinez-Trillos, A., Villamor, N., et al. (2013). POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. doi:10.1038/ng.2584.
Ruden, M., and Puri, N. (2012). Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. doi:10.1016/j.ctrv.2012.06.007
Rudolph, K. L., Chang, S., Lee, H. W., Blasco, M., Gottlieb, G. J., Greider, C., et al. (1999). Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96, 701–712.
Sahin, E., Colla, S., Liesa, M., Moslehi, J., Muller, F. L., Guo, M., et al. (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365.
Sahin, E., and Depinho, R. A. (2010). Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464, 520–528.
Scheel, C., Schaefer, K. L., Jauch, A., Keller, M., Wai, D., Brinkschmidt, C., et al. (2001). Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 20, 3835–3844.
Sfeir, A., and de Lange, T. (2012). Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597.
Shay, J. W., and Bacchetti, S. (1997). A survey of telomerase activity in human cancer. Eur. J. Cancer 33, 787–791.
Shay, J. W., and Wright, W. E. (2011). Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 21, 349–353.
Soler, D., Pampalona, J., Tusell, L., and Genesca, A. (2009). Radiation sensitivity increases with proliferation-associated telomere dysfunction in nontransformed human epithelial cells. Aging Cell 8, 414–425.
Takai, H., Smogorzewska, A., and De Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556.
Tanaka, H., Abe, S., Huda, N., Tu, L., Beam, M. J., Grimes, B., et al. (2012). Telomere fusions in early human breast carcinoma. Proc. Natl. Acad. Sci. U.S.A. 109, 14098–14103.
Tumpel, S., and Rudolph, K. L. (2012). The role of telomere shortening in somatic stem cells and tissue aging: lessons from telomerase model systems. Ann. N. Y. Acad. Sci. 1266, 28–39.
Verdun, R. E., Crabbe, L., Haggblom, C., and Karlseder, J. (2005). Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol. Cell 20, 551–561.
Verdun, R. E., and Karlseder, J. (2006). The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127, 709–720.
Wong, K. K., Chang, S., Weiler, S. R., Ganesan, S., Chaudhuri, J., Zhu, C., et al. (2000). Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat. Genet. 26, 85–88.
Keywords: telomeres, DNA-damage, genome instability, cancer, therapy
Citation: Jacobs JJL (2013) Loss of telomere protection: consequences and opportunities. Front. Oncol. 3:88. doi: 10.3389/fonc.2013.00088
Received: 15 January 2013; Accepted: 02 April 2013;
Published online: 15 April 2013.
Reviewed by:
Min H. Kang, Texas Tech University Health Sciences Center, USAHiroyuki Seimiya, Japanese Foundation for Cancer Research, Japan
Copyright: © 2013 Jacobs. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Jacqueline J. L. Jacobs, Division of Molecular Oncology, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, Netherlands. e-mail:ai5qYWNvYnNAbmtpLm5s