
- Samantha Dickson Brain Cancer Unit, UCL Cancer Institute, University College London, London, UK
The promyelocytic leukemia (PML) protein has been implicated in regulation of multiple key cellular functions, from transcription to calcium homeostasis. PML pleiotropic role is in part related to its ability to localize to both the nucleus and cytoplasm. In the nucleus, PML is known to regulate gene transcription, a role linked to its ability to associate with transcription factors as well as chromatin-remodelers. A new twist came from the discovery that the PML-interacting protein death-associated protein 6 (DAXX) acts as chaperone for the histone H3.3 variant. H3.3 is found enriched at active genes, centromeric heterochromatin, and telomeres, and has been proposed to act as important carrier of epigenetic information. Our recent work has implicated DAXX in regulation of H3.3 loading and transcription in the central nervous system (CNS). Remarkably, driver mutations in H3.3 and/or its loading machinery have been identified in brain cancer, thus suggesting a role for altered H3.3 function/deposition in CNS tumorigenesis. Aberrant H3.3 deposition may also play a role in leukemia pathogenesis, given DAXX role in PML-RARα-driven transformation and the identification of a DAXX missense mutation in acute myeloid leukemia. This review aims to critically discuss the existing literature and propose new avenues for investigation.
The Promyelocytic Leukemia Protein
The Promyelocytic Leukemia (PML) gene was originally identified at the breakpoint of the t(15;17) translocation of Acute Promyelocytic Leukemia (APL), which generates the PML/retinoic acid receptor (RAR)α oncogene, an inhibitor of PML and RARα functions (Salomoni et al., 2008) [please refer to accompanying articles1 and reviews in the field, e.g. (Grimwade and Solomon, 1997; Brown et al., 2009; de The and Chen, 2010), for detailed information on APL pathogenesis]. PML can localize to the cytoplasm [for more extensive discussion on the role of cytoplasmic PML, see (Lin et al., 2004; Giorgi et al., 2010; Pinton et al., 2011) as well as this issue of Frontiers] and the nucleus, where it forms the PML nuclear body (PML-NB), of which it is the essential component (Salomoni and Khelifi, 2006; Salomoni et al., 2008). The PML-NB is a subnuclear structure associated with storage and post-translational modifications (PTMs) of several nuclear factors [(Salomoni et al., 2008) for more extensive discussion on the role and regulation of PML-NBs, see comprehensive reviews in the field (Zhong et al., 2000b; Bernardi and Pandolfi, 2007; Lallemand-Breitenbach and de The, 2012), including this issue of Frontiers]. The PML-NB is disrupted in APL cells by PML/RARα (Salomoni et al., 2008). Both PML and PML/RARα (via the PML moiety) can be targeted pharmacologically using arsenic trioxide (ATO), which, in part through direct binding, promotes their ubiquitin-dependent degradation (Jeanne et al., 2010; Zhang et al., 2010). ATO is used in APL therapy because of its ability to target the leukemic stem cell pool (de The and Chen, 2010).
Although the PML gene is rarely mutated in cancer, its protein expression is lost in a number of human tumors, suggesting that it acts as tumor suppressor. Indeed, PML limits tumorigenesis in APL, lung, and prostate cancer models (Salomoni and Pandolfi, 2002). However, recent studies have highlighted a potential role of PML in established tumors. In this respect, PML is required for maintenance of the leukemia-initiating stem cell pool in chronic myeloid leukemia (Ito et al., 2008). Notably, ATO phenocopies the effect of PML loss in leukemic stem cells and requires PML for this effect (Ito et al., 2008). Another growth suppressor, p21 controls leukemic stem cell maintenance via regulation of genomic stability (Viale et al., 2009). Furthermore, an additional study from the Pandolfi’s group showed that PML plays an important pro-survival role in cancer via regulation of tumor cell metabolism [(Carracedo et al., 2012); for more extensive discussion on the role of PML in tumorigenesis, see comprehensive reviews in the field (Salomoni and Pandolfi, 2002), including this issue of Frontiers].
What is PML nuclear function(s)? Several studies have implicated PML in regulation of transcription (Zhong et al., 2000b; Bernardi and Pandolfi, 2007; Salomoni et al., 2008). In this respect, PML-NBs localize in the proximity of active transcription sites in a cell cycle-dependent manner (Kiesslich et al., 2002). Notably, PML can directly regulate the function of several transcription factors (Bernardi and Pandolfi, 2007). For instance, PML interaction with the p53 tumor suppressor promotes p53-dependent transcription in a PML-NB-dependent as well as – independent manner (Bischof et al., 2002; Bernardi et al., 2004; Bernardi and Pandolfi, 2007; Salomoni et al., 2008). Furthermore, work from our group and others have shown that the tumor suppressor and transcriptional repressor retinoblastoma (pRb) also localizes to PML-NBs (Alcalay et al., 1998), resulting in alterations of its phosphorylation status (Ferbeyre et al., 2000; Regad et al., 2009). Interestingly, not only transcription factors are found in PML-NBs, as a number of chromatin regulators localize to these structures, such as the histone acetyltransferase CREB-binding protein (CBP)/p300, which can acetylate histones as well as transcription factors. In this respect, it has been proposed that in senescent cells PML promotes p53 acetylation via dynamic localization of CBP to PML-NBs (Pearson et al., 2000). It is presently unknown whether PML could affect CBP-mediated acetylation of histone tails, in addition to transcription factors. It is important to note that PML-NBs contain chromatin-associated factors with repressive activity, such as histone deacetylase 1 (HDAC1), the corepressors N-Cor, Sin3A (Khan et al., 2001), and the heterochromatin-associated protein 1 (HP1) (Seeler et al., 1998). Together, these findings suggest that PML could serve as scaffold for multiple chromatin-remodeling complexes, with potential implications for both transcriptional activation and repression. Interestingly, there is evidence that PML-NBs might be involved in regulation of chromatin architecture, as some genetic loci are non-randomly associated with the periphery of PML-NBs (Torok et al., 2009). Furthermore, PML has been implicated in special AT-rich sequence-binding protein 1 (SATB1)-mediated regulation of chromatin architecture and gene expression (Kumar et al., 2007). Although these studies suggest an involvement of PML in chromatin regulation via interaction with histone-modifying enzymes and other chromatin regulators, our understanding of PML and PML-NB role in this context remains limited.
New exciting studies now link PML to the histone loading machinery, with implications for chromatin remodeling and cancer pathogenesis. This will be the main focus of the present review article.
The PML-Interacting Protein DAXX is a Chaperone for the Histone Variant H3.3
The death-associated protein 6 (DAXX) interacts with PML and is found in PML-NBs as well as heterochromatin (Khelifi et al., 2005; Salomoni and Khelifi, 2006). DAXX recruitment to PML-NBs occurs via binding to SUMOylated PML with DAXX SUMO-interacting motif (SIM) (Zhong et al., 2000a; Lin et al., 2006). DAXX SIM is also required for its ability to localize to heterochromatin [(Kuo et al., 2005) and our unpublished data]. DAXX loss results in embryonic lethality (Michaelson et al., 1999; Garrick et al., 2006), indicating an essential role in embryogenesis. DAXX was originally identified as a CD95-interacting protein in the cytoplasm, affecting CD95-dependent activation of c-Jun-N-terminal kinase (JNK) (Yang et al., 1997). The link with JNK was reported by subsequent studies, which implicated an apoptosis signal-regulating kinase 1 (ASK1)-dependent mechanism for DAXX-mediated JNK activation (Ko et al., 2001; Perlman et al., 2001; Khelifi et al., 2005). However, it is presently unclear whether endogenous DAXX localizes to the cytoplasm in physiological conditions [for discussion see (Lindsay et al., 2009)].
What would be DAXX nuclear function? DAXX has been shown to regulate transcription, either indirectly or directly. On one hand, DAXX destabilizes p53 via inhibition of mouse double minute 2 homolog (MDM2) ubiquitylation, thus resulting in repression of p53 target gene expression (Tang et al., 2006). On the other hand, DAXX regulates transcription of mammalian and viral genes via its ability to interact with a number of transcription factors [for more details see (Preston and Nicholl, 2006; Saffert and Kalejta, 2006; Salomoni and Khelifi, 2006; Lindsay et al., 2008; Lukashchuk and Everett, 2010; Tsai et al., 2011; Rivera-Molina et al., 2012; Glass and Everett, 2013; Schreiner et al., 2013; Shalginskikh et al., 2013)] and epigenetic regulators. In particular, DAXX interacts with HDAC-II (Hollenbach et al., 2002), acetyltransferases (CBP) (Kuo et al., 2005), and DNA methyltransferase (Dnmt1) (Puto and Reed, 2008; Zhang et al., 2013), suggesting an important role in chromatin remodeling.
Recent exciting studies have implicated DAXX in direct chromatin regulation via its ability to act as chaperone for a histone 3 (H3) variant called H3.3. Best understood for PTMs of histones, chromatin modification also occurs via incorporation of histone variants. Unlike canonical H3, H3.3 can be loaded on DNA in a replication-independent manner. H3.3 is believed to be an important carrier of epigenetic information (Szenker et al., 2011). H3.3 is encoded by two genes, H3F3A and H3F3B. H3F3A inactivation via gene trap leads to perinatal lethality (Couldrey et al., 1999), whereas H3F3B knockout embryos display partial embryonic lethality and infertility in surviving homozygous animals (Bush et al., 2013). DAXX acts as a H3.3 chaperone as part of a nuclear complex containing the α-thalassemia and mental retardation X-linked (ATRX) DNA helicase (Drané et al., 2010; Lewis et al., 2010; Dawson and Kouzarides, 2012). ATRX, like DAXX, can associate with PML-NBs (Bérubé et al., 2007) and has been proposed to contribute to DAXX/H3.3 targeting to chromatin, potentially via its ability to bind histone repressive marks in heterochromatin and G-rich DNA repeats (Elsaesser et al., 2010; Law et al., 2010; Iwase et al., 2011). DAXX and ATRX mediate H3.3 loading onto telomeres and pericentric heterochromatin, with implications for transcription of telomeric and centromeric repeats (Drané et al., 2010; Goldberg et al., 2010; Lewis et al., 2010). Furthermore, H3.3 loading at telomeres has been suggested to play an important role in maintaining chromatin structure (Wong et al., 2009, 2010). Loading of H3.3 may affect transcription also at euchromatin, as it is enriched at transcriptionally active genes and has been proposed to regulate epigenetic memory of transcriptional competence (Henikoff, 2008; Ng and Gurdon, 2008; Jullien et al., 2012). Loading of H3.3 at transcription start site (TSS) and body of active gene is dependent on the chaperone HIRA (Goldberg et al., 2010). However, H3.3 is also enriched at regulatory regions not immediately adjacent to TSS (Mito et al., 2007; Jin et al., 2009; Goldberg et al., 2010). Deposition at those sites is in part HIRA-independent (Goldberg et al., 2010), but the histone chaperone involved was not known. In this respect, our recent work implicated DAXX in the regulation of H3.3 deposition at promoters and enhancers of immediate early genes (IEGs) in neurons (Michod et al., 2012), thus demonstrating that DAXX is one of the previously unidentified H3.3 chaperones at regulatory regions (Michod et al., 2012). Work from Genevieve Almouzni, John Gurdon, and Peter Adams groups (Ray-Gallet et al., 2011; Jullien et al., 2012; Pchelintsev et al., 2013) showed that HIRA could also mediate H3.3 loading at regulatory regions. Notably, DAXX-dependent H3.3 deposition correlates with its ability to modulate transcription, thus suggesting a link between H3.3 loading and transcription (Michod et al., 2012). Among the IEGs analyzed, only a subset of them displayed dependence on DAXX for H3.3 loading and transcriptional activation, thus suggesting that other H3.3 chaperones are involved in IEG regulation in neurons, such HIRA or DEK (Sawatsubashi et al., 2010; Jullien et al., 2012). Finally, both DAXX-dependent loading and transcription are controlled by a calcium-dependent phosphorylation switch affecting serine 669 (S669) (Michod et al., 2012), which is a target of homeodomain-interacting protein kinases (HIPKs) (Hofmann et al., 2003) (Figure 1). In particular, upon neuronal activation DAXX S669 is dephosphorylated by the calcium-dependent phosphatase calcineurin (CaN), leading to increased loading activity and transcription (Michod et al., 2012). Although H3.3 is preferentially found associated with hypophosphorylated DAXX, S669 dephosphorylation does not affect DAXX affinity for H3.3, suggesting that when in complex with H3.3 DAXX is either more effectively dephosphorylated or its HIPK-dependent phosphorylation is inhibited. Since CaN is believed to be mainly cytosolic, it is most likely that DAXX dephosphorylation occurs outside the nucleus, whereas one could speculate that its HIPK-dependent phosphorylation could be nuclear. It is important to note that DAXX S669 phosphorylation status does not affect its chromatin association. One could speculate that HIPKs could associate with DAXX on chromatin and inhibit its chaperone activity. Interestingly, the HIRA chaperone complex contains the CaN-binding protein CABIN1, a CaN regulator (Rai et al., 2011), suggesting that calcium-dependent signaling could regulate multiple H3.3 chaperone complexes.
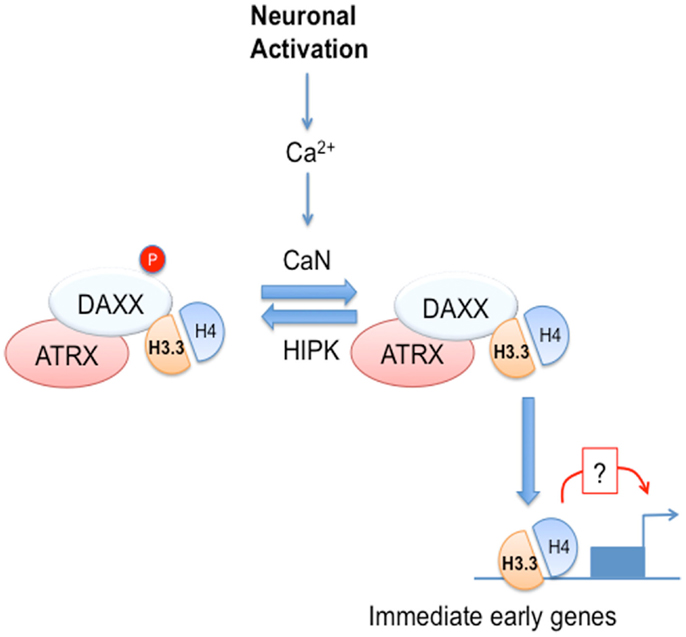
Figure 1. DAXX chaperone activity is regulated by calcium-dependent signaling in neural cells. Neuronal activation leads to calcium (Ca2+) entry and activation of the Ca2+-dependent phosphatase, calcineurin (CaN). In turn, CaN dephosphorylates DAXX at serine 669, leading to increased H3.3 loading at selected immediate early genes (IEGs). DAXX loss not only affects H3.3 loading, but also leads to impaired induction of IEGs, thus suggesting that H3.3 loading may modulate IEG transcriptional induction.
Together, these studies suggest that DAXX-mediated loading of H3.3 at regulatory regions may affect transcription. One could argue that DAXX ability to regulate transcription could be H3.3-independent, for instance via its interaction with HDAC-II (Hollenbach et al., 2002), CBP (Kuo et al., 2005), or Dnmt1 (Puto and Reed, 2008; Zhang et al., 2013). However, DAXX loss fails to promote any significant changes in histone acetylation or DNA methylation at the BDNF Exon IV promoter (Michod et al., 2012). Another possibility is that DAXX regulates key transcription factors involved in activity-dependent IEG induction, in particular CREB and MEF2 (Hong et al., 2005; Flavell et al., 2008). For instance, DAXX has been recently reported to repress CREB transcriptional activity via direct interaction via its C-terminus (Huang et al., 2012) and HIPK2 is known to phosphorylate CREB (Sakamoto et al., 2010). However, based on these findings DAXX loss would result in increased CREB-mediated transcription, opposite to what we have observed in neurons (Michod et al., 2012). To incontrovertibly assess the role of DAXX-mediated H3.3 loading in transcription, one should test the ability of recently described DAXX mutants impaired in histone binding (Eustermann et al., 2011; Elsasser et al., 2012) to rescue transcriptional defects observed in DAXX-deficient cells (Michod et al., 2012). It is important to note that histone chaperones are often components of chromatin remodeling complexes, such as the nucleosome remodeling and deacetylation (NuRD) and Polycomb complexes (Lai and Wade, 2011; Margueron and Reinberg, 2011). Thus, DAXX could load H3.3 while being part of a larger chromatin-remodeling complex containing histone- and/or DNA-modifying enzymes, which could cooperate with histone loading in promoting chromatin modification and transcriptional changes.
What is the evidence for a role of H3.3 loading in transcriptional regulation? Our work and other studies discussed above suggest a potential role for H3.3 in transcription and/or regulation of the transcriptional state (Ng and Gurdon, 2008; Jullien et al., 2012; Michod et al., 2012). Furthermore, H3.3 downregulation in B cells results in transcriptional repression at the Igh locus (Aida et al., 2013). In contrast, loss of H3f3b does not dramatically alter the transcriptome of mouse embryo fibroblasts (Bush et al., 2013) and HIRA deficiency in ES cells has limited impact on transcription (Goldberg et al., 2010). It is plausible that the impact of H3.3 loading on transcription could depend on the cell type, developmental stage, and environmental cues (e.g., neuronal activation, B cell differentiation stimuli, etc). In this respect, the concept of H3.3 deposition contributing to transcriptional memory at selected loci during development is particularly intriguing. Overall, there is accumulating evidence that H3.3 might regulate transcription. A key question is how. Most active genes are associated with variant nucleosomes containing H3.3 and the histone 2A variant H2A.z, which promotes nucleosome instability (Jin et al., 2009). These properties of H3.3 may explain its enrichment at bivalent genes in flies (Henikoff, 2008) and, along with H2A.z, in mammalian cells (Creyghton et al., 2008; Goldberg et al., 2010). Many genes involved in brain development and postnatal neurogenesis are characterized by bivalent chromatin (Valk-Lingbeek et al., 2004; Marino, 2005; Lim et al., 2009; Sawarkar and Paro, 2010; Schuettengruber et al., 2011; Dawson and Kouzarides, 2012). Bivalency is defined by the presence of both active and repressive histone marks, which keep genes in a poised state. These are the repressive mark trimethylated H3 lysine 27 (H3K27me3) and the active mark H3K4me3, which are generated via the action of Polycomb Repressive Complex 2 (PRC2) and Trithorax complexes, respectively. H3K27me3 has a dual role in amplification of PRC2-mediated K27 methylation and recruitment of the Polycomb repressive complex 1 (PRC1), which mediates ubiquitylation of H2A (another repressive mark) (Wang et al., 2004). H3K4me3 is found associated with several chromatin-remodelers, as well as H3K27 demethylases (Dawson and Kouzarides, 2012). Polycomb group (PcG) and Trithorax group (TrxG) proteins are key regulators of stem cell fate in both the embryonic and postnatal brain (Simon and Kingston, 2009; Margueron and Reinberg, 2011). In particular, the PRC1 component Bmi1 and the TrxG complex component Mll1 are important regulators of neural stem cell self-renewal and neurogenesis (Valk-Lingbeek et al., 2004; Marino, 2005; Lim et al., 2009). Notably, H3.3 is enriched in the H3K4me3 mark (Henikoff, 2008). Furthermore, H2A.z has been proposed to regulate targeting of both PcG and TrxG complexes to chromatin (Hu et al., 2012). Overall, these studies suggest a potential functional involvement of H3.3 and H2A.z loading in regulation of bivalency. In particular, it is conceivable that H3.3 deposition could affect bivalent domains at IEGs in neurons, as potential mechanism for DAXX-mediated transcriptional changes (Michod et al., 2012). In general, it is of key importance to generate new genetic systems to better define the molecular function of H3.3 and its impact on fundamental biological processes. In this respect, Jeffrey Mann’s group has been involved in generation of new models based on conditional allelic replacement, which bear great promise for advancing our understanding of H3.3 function in vivo (Tang et al., 2013).
H3.3 Loading and Disease Pathogenesis
An even greater interest in H3.3 and its chaperones has arisen from the discovery that H3.3 itself, DAXX and ATRX are mutated in human cancer. In this respect, driver heterozygous mutations in the H3F3A gene are found in pediatric glioblastoma multiforme (GBM) (Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012) (H3F3B is expressed at much lower levels in neural cells; our unpublished observation). H3.3 is mutated at K27 (K27M) and G34 (G34R or V), with the former found in brainstem tumors of young children (Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012) and the latter in the cerebral hemispheres of older children and adolescents (Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012). ATRX is mutated in pediatric (Schwartzentruber et al., 2012) and adult GBM (Heaphy et al., 2011), and DAXX in pediatric GBM, albeit very infrequently (Schwartzentruber et al., 2012). ATRX is also found mutated in neuroblastoma, while both DAXX and ATRX are mutated in neuroendocrine tumors of the pancreas (Elsasser et al., 2011; Heaphy et al., 2011; Jiao et al., 2011; Molenaar et al., 2012). Most DAXX and ATRX mutations are mutually exclusive and result in loss of expression (Elsasser et al., 2011; Heaphy et al., 2011; Jiao et al., 2011; Schwartzentruber et al., 2012; Wu et al., 2012), apart from a missense mutation found in acute myeloid leukemia (AML) (Ding et al., 2012). It is important to note that pediatric GBM also display mutations of ATRX in the absence of H3.3 mutations, as observed in adult GBM, neuroblastoma and pancreatic tumors, suggesting that alterations in loading of WT H3.3 may per se lead to cancer. H3.3/ATRX- and ATRX-only-mutated GBM tumors often carry p53 mutations, suggesting that loss of p53 tumor suppressive function cooperates with H3.3 and/or ATRX mutations for tumorigenesis. Finally, in pancreatic tumors carrying DAXX mutations, it is conceivable that H3.3 loading could be mediated by other chaperones, thus leading to alterations in its genome-wide distribution, with potential consequences for tumorigenesis. At present, it is unclear what are the expression levels of the H3.3 chaperones HIRA and DEK in pancreatic tumors and other cancers displaying alterations in H3.3 and DAXX/ATRX loading machinery.
The key question is how alterations of H3.3 function can drive/contribute to neoplastic transformation. Analysis of gene expression changes in GBM neoplasms carrying H3.3 mutations showed that H3.3 K27M and G34R/V tumors display distinct transcriptional changes. In this respect, H3.3K27M GBM tumors display deregulation of some PcG targets (Schwartzentruber et al., 2012). Furthermore, mutations of the TrxG component multiple endocrine neoplasia type 1 (MEN1) are found in neuroendocrine pancreatic tumors and is mutually exclusive with DAXX and ATRX mutations, suggesting similar functional roles (Jiao et al., 2011). Together, these data indicate that alteration of bivalent gene expression may represent one of the mechanisms underlying the transforming role of the H3.3 K27M mutation. It is important to note that deregulation of the machinery controlling the H3K27me3 epigenetic mark has been implicated in pathogenesis of another pediatric brain tumor, medulloblastoma, and adult GBM. In most cases, this leads to increased H3K27me3 (Bruggeman et al., 2007; van Haaften et al., 2009; Jones et al., 2012; Lu et al., 2012; Robinson et al., 2012), via deregulated expression/mutations of H3K27me3 methylases/demethylases, inactivation of chromatin-remodeling factors or metabolic enzymes (Bruggeman et al., 2007; van Haaften et al., 2009; Jones et al., 2012; Lu et al., 2012; Robinson et al., 2012). Increased H3K27me3 in medulloblastoma and adult GBM is expected to lead to increased repression. However, it was unclear what would be the consequences of pediatric GBM mutation of H3.3 at K27 on H3K27me3 and transcription. A very recent study from David Allis group has provided important clues (Lewis et al., 2013). In particular, this work shows that the presence of H3.3K27M negatively affects PRC2-mediated amplification of K27 trimethylation in cis and trans. This occurs via inhibition of the enzymatic activity of the PRC2 methyltransferase EZH2. Interestingly, introduction of K-to-M mutations at other known methylated H3 residues (H3K9 and H3K36) has similar negative effects on enzymatic activity of the dedicated methyltransferases. Together, these data suggest that H3.3K27M acts at least in part as a gain-of-function mutant. Notably, the gain-of-function effect of the K-to-M mutation is not restricted to H3.3, as also canonical H3 is found mutated in GBM and H3K27M displays similar EZH2 inhibitory activity. What would be the effect of H3.3K27M loading on chromatin? Some clues came from another recent study, which showed that H3.3K27M is associated with loss of H3K27me3 at many loci (Chan et al., 2013), as expected based on David Allis work. However, several genomic regions gained this mark along with H3K4me3, leading to repression of genes involved in cancer development, such as the tumour suppressor p16INK4a (Chan et al., 2013). Thus, the consequences of this mutation on chromatin structure/modifications are more complex than previously thought.
How would G34R/V mutations function? Notably, H3.3 G34 mutations almost invariably coexist with ATRX mutations (Henikoff, 2008; Schwartzentruber et al., 2012) and are associated with the alternative lengthening of telomeres (ALT) mechanism (Heaphy et al., 2011; Bower et al., 2012; Liu et al., 2012; Lovejoy et al., 2012), a recombinogenic mechanism for telomere elongation. ALT cells contain a modified PML-NB called ALT-associated PML-NB (APB), which we and others have implicated in telomeric damage response and potentially telomere recombination (Stagno D’Alcontres et al., 2007; Lovejoy et al., 2012). Thus, it is conceivable that H3.3 G34 mutations could lead to ATRX loss and alteration of telomere maintenance mechanisms, thus in turn contributing to transformation and GBM development. Recent work from Chris Jones laboratory shows that H3.3G34 mutations alter transcription and enrichment of the H3K36me3 active mark at a number of developmentally regulated genes linked to forebrain development and stem cell self-renewal (Bjerke et al., 2013). Remarkably, these mutations lead to increased expression of the MYCN proto-oncogene (Bjerke et al., 2013), suggesting a potential link between histone variant loading and MYCN-mediated transformation.
Are H3.3 mutations transforming per se? It is important to note that H3.3K27M is unable to promote glioma even in a p53 null background (Lewis et al., 2013), suggesting that either other genetic events are needed or, more likely, the cell targeted in this model is not the correct one. In this respect, it is possible that the type of progenitor and/or the developmental stage are crucial for transformation by H3.3 mutant proteins. Definite answers to these outstanding questions will be achieved only upon development of more sophisticated genetic models, which are currently in the pipeline in many laboratories in the field.
Alterations of the H3.3 chaperone complex might extend to non-neoplastic conditions, such as the ATR-X syndrome, which is driven by ATRX mutations (Gibbons and Higgs, 2000). Furthermore, ATRX interacts with MeCP2 and cohesin, mutated in the Rett and Cornelia de Lange (CdLS) syndromes, respectively (Kernohan et al., 2010). It is presently unknown whether alterations of H3.3 loading may participate in the pathogenesis of these conditions.
Role of PML and PML-RARα in Regulation of H3.3 Loading?
As mentioned earlier, DAXX localizes to PML-NB via a SUMO-dependent mechanism involving its SIM (Figure 2). Thus, it is conceivable that PML could regulate DAXX function by altering its subcellular localization. In this respect, PML was reported to negatively regulate DAXX repressive function through recruitment to PML-NBs (Li et al., 2000). However, PML role in regulation of H3.3 deposition is still unclear. Clues have come from a recent study reporting H3.3/H4 dimers localization to PML-NBs (Figure 2) (Delbarre et al., 2012). The authors of this study report that exogenously expressed H3.3 along with H4 and DAXX localizes to PML-NBs in G1-enriched mesenchymal stem cells, thus potentially regulating the nucleoplasmic pool of H3.3. Although exogenous H3.1 and H3.2 failed to localize to PML-NBs, it is still possible that H3.3 could be targeted to PML-NBs only when expressed at supraphysiological levels. One obvious question is whether PML regulates incorporation of endogenous H3.3 into chromatin. In this respect, it could hypothesized that PML-mediated localization of H3.3 and its chaperones to PML-NBs inhibits H3.3 loading into chromatin (Delbarre et al., 2012). Another recent report implicated PML and PML-NBs in regulation of ATRX and H3.3 association at telomeres during S-phase in ES cells (Chang et al., 2013). As a result, PML downregulation caused telomere dysfunction and altered telomeric enrichment of selected epigenetic marks. Although it cannot be excluded that PML-NBs are sites for localization of extrachromosomal telomeric DNA, this work suggests that PML is either directly or indirectly involved in regulation of telomere replication/maintenance potentially via an H3.3/ATRX-dependent mechanism. A question arising from this study is whether PML role in regulating H3.3 association with telomeric DNA is maintained also in ALT cells, which are often ATRX-negative, in particular in G34R/V, ATRX-deficient GBM cells. Finally, PML could play a more indirect role in regulation of H3.3 loading via modulation of DAXX PTMs, in particular its phosphorylation at S669 (Michod et al., 2012). In this respect, while the S669 phosphatase CaN is mainly cytosolic, the S669 kinase HIPK2 has been shown to localize to PML-NBs (Krieghoff-Henning and Hofmann, 2008), suggesting that localization of HIPK2 and DAXX to PML-NBs could affect its phosphorylation and as a result its chaperone activity.
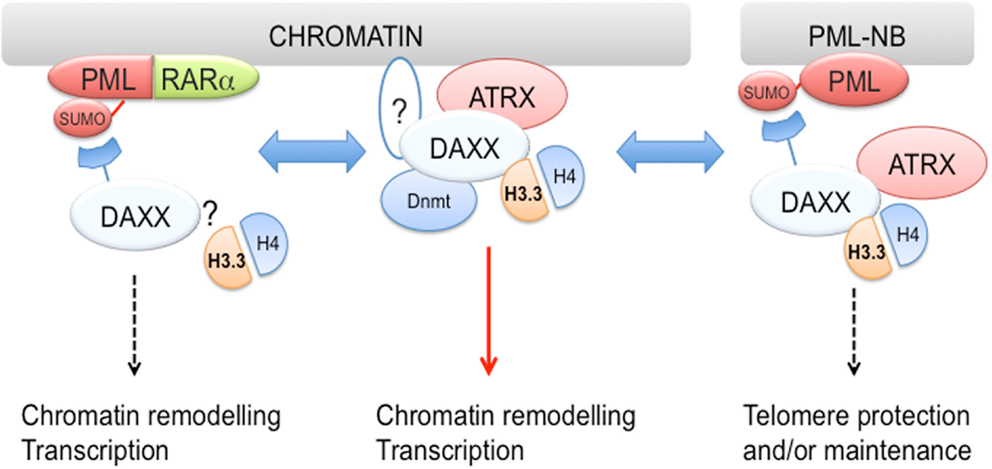
Figure 2. DAXX associates with both PML and PML-RARα?. DAXX and H3.3/H4 dimers are found at PML-NBs, suggesting that PML may regulate H3.3 loading. Furthermore, DAXX association with PML-RARα is required for transformation in vitro. Although it is presently unknown whether H3.3 also associates with PML-RARα, it is conceivable that PML-RARα via interaction with DAXX could modulate H3.3 loading. While DAXX is recruited to PML-NBs via a SUMO-interacting motif (SIM)-dependent mechanism, it is still unclear whether a similar mechanism is implicated in its targeting to chromatin (see question mark in middle panel). DAXX also interacts with DNA methyltransferase 1 (Dnmt1) and targets its activity to chromatin, suggesting that DAXX coordinates multiple epigenetic modifications.
Promyelocytic leukemia-mediated regulation of DAXX function could be particular relevant in the central nervous system (CNS), given the roles played by the two proteins in this context. In this respect, our previous work has shown that PML is expressed in neural progenitor/stem cells (NPCs) in the developing neocortex as well as in postnatal neurogenic niches [(Regad et al., 2009; Salomoni and Betts-Henderson, 2011) and our unpublished data]. As a result, PML loss leads to alterations of corticogenesis and smaller brains (Regad et al., 2009), as well as aberrant postnatal neurogenesis (our unpublished data). While PML and DAXX are expressed in the germinal area of developing neocortex and in NPCs within adult neurogenic niches, PML expression is downregulated in postmitotic neuroblasts and neurons (Regad et al., 2009; Michod et al., 2012) (and our unpublished data). It is therefore possible that PML could regulate DAXX chaperone function in NPCs, thus potentially affecting epigenetic changes driven by H3.3 loading. In turn, this could have implications for cell fate regulation and neurogenesis. In contrast, PML-mediated control of DAXX function would be absent in differentiated neurons.
What about the oncogenic form of PML, PML-RARα? SUMOylation within the PML moiety of PML-RARα is required for transformation (Zhu et al., 2005) and is responsible for recruitment of DAXX via its C-terminal SIM (Figure 2; see also accompanying review articles by Hugues de The in this issue of Frontiers). Mutation of the critical SUMOylation site within PML-RARα (K160R) releases DAXX and results in defective differentiation block (Zhu et al., 2005). In contrast, fusion of K160R PML-RARα with DAXX restores its transforming capacity (Zhu et al., 2005). A subsequent study from de The’s laboratory reported that a DAXX-RARα chimera carrying a multimerization domain can repress RA-dependent transcription, inhibit differentiation, and promote transformation. In contrast, a multimerization-prone RARα mutant, despite inhibiting RA-dependent transcription and differentiation, was unable to transform hematopoietic progenitors (Zhou et al., 2006), suggesting that molecular determinants of the differentiation block and transformation may not be identical. However, a separate study by Eric So’s group showed that fusion of the FKBP oligodimerisation sequence with RARα can promote transformation (Kwok et al., 2006). The presence of a SUMOylation site (Rodriguez et al., 2001) in FKBP (which would in principle still recruit DAXX) and non-identical experimental settings may explain the different results. Overall, these findings suggest that SUMO-dependent PML-RARα association with DAXX contributes to block of differentiation and transformation.
PML-RARα has been reported to associate with epigenetic regulators and mediate epigenetic changes: (i) PML-RARα multimerization properties lead to increased density of corepressors and chromatin remodeling factors at retinoic acid (RA) target genes (Lin and Evans, 2000; Minucci et al., 2000; de The and Chen, 2010); (ii) PML-RARα interacts with DNA methyltransferases, thus leading to DNA methylation of a number of RA target genes (Di Croce et al., 2002). Interaction with the H3.3 chaperone DAXX could provide PML-RARα with additional weaponry to promote epigenetic changes. One could argue that H3.3 is mainly associated with active genes, not with repression. In this respect, it is important to note that there is little in vivo evidence that repression of RA target genes is sufficient to initiate APL (de The and Chen, 2010), and it is now recognized that PML-RARα also possess gain-of-function properties through its ability to bind target sequences that are not recognized by the normal RARα-RXRα heterodimers (de The and Chen, 2010). Among these sites, there are many genes controlling stem cell self-renewal and myeloid differentiation (Purton et al., 2006; Viale et al., 2009; de The and Chen, 2010). Finally, mouse APL leukemias express high levels of the IEG c-Fos (Yuan et al., 2007), which we have shown to be regulated by DAXX in neural cells (Michod et al., 2012). Overall, the existing literature suggests that PML-RARα promotes transformation through a combination of dominant-negative and gain-of-function activities. Interaction with DAXX could contribute to the latter.
Considering H3.3 enrichment at bivalent genes and the alteration of PcG/TrxG activities in hematopoietic tumors (Mills, 2010; Muntean and Hess, 2012), it could be hypothesized that DAXX-mediated H3.3 loading could affect bivalent gene expression in APL cells. In this respect, there is a functional crosstalk between RA-dependent transcription and the PcG machinery, as many homeobox genes contain RA responsive elements (RARE) (Mainguy et al., 2003; Ringrose and Paro, 2004) and RA targets are also PcG targets (e.g., CYP26a1 and RARβ). Notably, PML-RARα-regulated loci display increased H3K4me3 (Hoemme et al., 2008), thus suggesting that PML-RARα may regulate this epigenetic mark. As H3.3 is enriched in H3K4me3, it is conceivable that PML-RARα could direct DAXX-mediated H3.3 deposition at a number of its gene targets. As H3K4me3 is lost from a number of bivalent loci during differentiation of human hematopoietic stem cells (Cui et al., 2009), it is possible that PML-RARα could re-establish/maintain H3K4me3 at these loci through modification of TrxG complex activity and/or loading of H3.3. PML-RARα is also found in complex with PRC2 components (Villa et al., 2007; Martens et al., 2010), suggesting it could affect H3K27 trimethylation. However, subsequent genome-wide analysis showed that RA treatment fails to significantly affect H3K27me3 (Martens et al., 2010). More work is needed to define PML-RARα role in regulation of bivalent chromatin and the contribution of H3.3 loading to its epigenetic activity. It is important to note that a DAXX missense mutation of unknown functional consequences has been identified in AML (Ding et al., 2012), suggesting that alterations of H3.3 loading may occur in non-APL hematopoietic neoplasms.
Finally, both PML and PML-RARα have been linked to another H3.3 chaperone, HIRA (Ye et al., 2007; Delbarre et al., 2012). In this respect, localization of HIRA to PML-NBs is required for formation of senescence-associated heterochromatic foci (SAHF). PML-RARα, which disrupts PML-NBs, inhibits SAHF generation. Although it is presently unknown whether H3.3 is loaded as SAHF, these data suggest that PML and its oncogenic version may regulate H3.3 loading by acting on multiple chaperones.
Conclusion
The discovery of DAXX chaperone function provides the fascinating possibility that PML and its oncogenic form PML-RARα could promote epigenetic changes in part via regulation of H3.3 loading. In this respect, PML-RARα could utilize DAXX chaperone activity to modify the epigenetic and transcriptional status of its target genes, as part of its gain-of-function activities in transformation of hematopoietic progenitors. In contrast, PML could play a more indirect role in regulation of loading of wild-type H3.3 as well as its GBM-associated mutants by controlling the availability of soluble H3.3/H4 dimers and/or DAXX PTMs, with potential implications for cell fate regulation and transformation (Figure 3). In this respect, pharmacological degradation of PML via ATO treatment could represent a strategy to affect H3.3 loading in cancer cells. More broadly, an increased understanding of H3.3 loading, its function and regulatory pathways has the potential to lead to a paradigm shift in the field of cancer epigenetics.
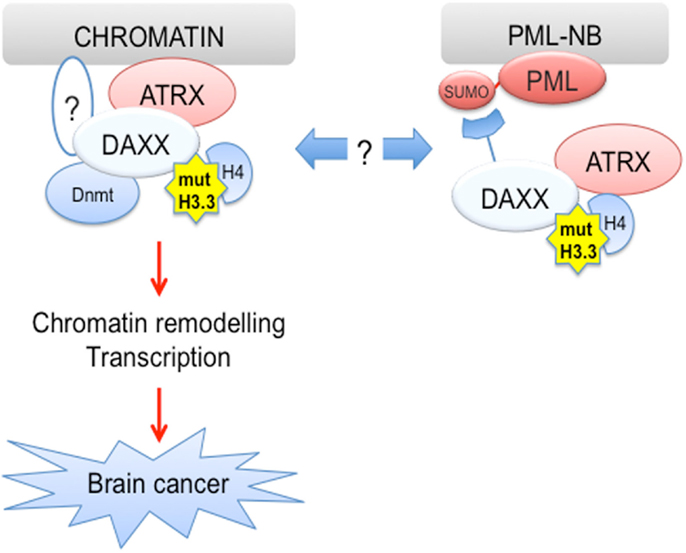
Figure 3. H3.3 is mutated in human cancer. Driver H3.3 mutations are found in pediatric glioblastoma (pGBM), suggesting that alterations of H3.3 function may lead to brain cancer. It is possible that PML via its ability to recruit DAXX to PML-NBs could regulate loading of mutant H3.3 proteins, thus potentially affecting brain tumorigenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank David Michod (Institute of Child Health, UCL), the members of my laboratory. I also would like to thank my numerous collaborators, in particular Pierluigi Nicotera (DZNE, Bonn, Germany), for support and stimulating scientific discussion. The funding bodies supporting H3.3- and PML-related work in my laboratory are: the Medical Research Council (MRC), The Brain Tumor Charity (formerly known as Samantha Dickson Brain Tumor Trust), the Association for International Cancer Research (AICR) and Cancer Research UK (CRUK). Finally, a special thank to the Brian Cross family for their generous support of my laboratory through The Brain Tumor Charity.
Footnote
References
Aida, M., Hamad, N., Stanlie, A., Begum, N. A., and Honjo, T. (2013). Accumulation of the FACT complex, as well as histone H3.3, serves as a target marker for somatic hypermutation. Proc. Natl. Acad. Sci. U.S.A. 110, 7784–7789. doi:10.1073/pnas.1305859110
Alcalay, M., Tomassoni, L., Colombo, E., Stoldt, S., Grignani, F., Fagioli, M., et al. (1998). The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol. Cell. Biol. 18, 1084–1093.
Bernardi, R., and Pandolfi, P. P. (2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8, 1006–1016. doi:10.1038/nrm2277
Bernardi, R., Scaglioni, P. P., Bergmann, S., Horn, H. F., Vousden, K. H., and Pandolfi, P. P. (2004). PML regulates p53 stability by sequestering MDM2 to the nucleolus. Nat. Cell Biol. 6, 665–672. doi:10.1038/ncb1147
Bérubé, N. G., Healy, J., Medina, C. F., Wu, S., Hodgson, T., Jagla, M., et al. (2007). Patient mutations alter ATRX targeting to PML nuclear bodies. Eur. J. Hum. Genet. 16, 192–201. doi:10.1038/sj.ejhg.5201943
Bischof, O., Kirsh, O., Pearson, M., Itahana, K., Pelicci, P. G., and Dejean, A. (2002). Deconstructing PML-induced premature senescence. EMBO J. 21, 3358–3369. doi:10.1093/emboj/cdf341
Bjerke, L., Mackay, A., Nandhabalan, M., Burford, A., Jury, A., Popov, S., et al. (2013). Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. [Epub ahead of print]. doi:10.1158/2159-8290.CD-12-0426
Bower, K., Napier, C. E., Cole, S. L., Dagg, R. A., Lau, L. M., Duncan, E. L., et al. (2012). Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLoS ONE 7:e50062. doi:10.1371/journal.pone.0050062
Brown, N. J., Ramalho, M., Pedersen, E. W., Moravcsik, E., Solomon, E., and Grimwade, D. (2009). PML nuclear bodies in the pathogenesis of acute promyelocytic leukemia: active players or innocent bystanders? Front. Biosci. 14, 1684–1707. doi:10.2741/3333
Bruggeman, S. W., Hulsman, D., Tanger, E., Buckle, T., Blom, M., Zevenhoven, J., et al. (2007). Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 12, 328–341. doi:10.1016/j.ccr.2007.08.032
Bush, K. M., Yuen, B. T., Barrilleaux, B. L., Riggs, J. W., O’Geen, H., Cotterman, R. F., et al. (2013). Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin 6, 7. doi:10.1186/1756-8935-6-7
Carracedo, A., Weiss, D., Leliaert, A. K., Bhasin, M., de Boer, V. C., Laurent, G., et al. (2012). A metabolic prosurvival role for PML in breast cancer. J. Clin. Invest. 122, 3088–3100. doi:10.1172/JCI62129
Chan, K. M., Fang, D., Gan, H., Hashizume, R., Yu, C., Schroeder, M., et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990. doi:10.1101/gad.217778.113
Chang, F. T., McGhie, J. D., Chan, F. L., Tang, M. C., Anderson, M. A., Mann, J. R., et al. (2013). PML bodies provide an important platform for the maintenance of telomeric chromatin integrity in embryonic stem cells. Nucleic Acids Res. 41, 4447–4458. doi:10.1093/nar/gkt114
Couldrey, C., Carlton, M. B., Nolan, P. M., Colledge, W. H., and Evans, M. J. (1999). A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum. Mol. Genet. 8, 2489–2495.
Creyghton, M. P., Markoulaki, S., Levine, S. S., Hanna, J., Lodato, M. A., Sha, K., et al. (2008). H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 135, 649–661. doi:10.1016/j.cell.2008.09.056
Cui, K., Zang, C., Roh, T. Y., Schones, D. E., Childs, R. W., Peng, W., et al. (2009). Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4, 80–93. doi:10.1016/j.stem.2008.11.011
Dawson, M. A., and Kouzarides, T. (2012). Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. doi:10.1016/j.cell.2012.06.013
de The, H., and Chen, Z. (2010). Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783. doi:10.1038/nrc2943
Delbarre, E., Ivanauskiene, K., Kuntziger, T., and Collas, P. (2012). DAXX-dependent supply of soluble (H3.3-H4) dimers into PML bodies pending deposition into chromatin. Genome Res. 23, 440–451.
Di Croce, L., Raker, V. A., Corsaro, M., Fazi, F., Fanelli, M., Faretta, M., et al. (2002). Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295, 1079–1082. doi:10.1126/science.1065173
Ding, L., Ley, T. J., Larson, D. E., Miller, C. A., Koboldt, D. C., Welch, J. S., et al. (2012). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510. doi:10.1038/nature10738
Drané, P., Ouararhni, K., Depaux, A., Shuaib, M., and Hamiche, A. (2010). The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 24, 1253–1265. doi:10.1101/gad.566910
Elsaesser, S. J., Goldberg, A. D., and Allis, C. D. (2010). New functions for an old variant: no substitute for histone H3.3. Curr. Opin. Genet. Dev. 20, 110–117. doi:10.1016/j.gde.2010.01.003
Elsasser, S. J., Allis, C. D., and Lewis, P. W. (2011). Cancer. New epigenetic drivers of cancers. Science 331, 1145–1146. doi:10.1126/science.1203280
Elsasser, S. J., Huang, H., Lewis, P. W., Chin, J. W., Allis, C. D., and Patel, D. J. (2012). DAXX envelops an H3.3-H4 dimer for H3.3-specific recognition. Nature 491, 560–565. doi:10.1038/nature11608
Eustermann, S., Yang, J. C., Law, M. J., Amos, R., Chapman, L. M., Jelinska, C., et al. (2011). Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 18, 777–782. doi:10.1038/nsmb.2070
Ferbeyre, G., De Stanchina, E., Querido, E., Baptiste, N., Prives, C., and Lowe, S. W. (2000). PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 14, 2015–2027.
Flavell, S. W., Kim, T. K., Gray, J. M., Harmin, D. A., Hemberg, M., Hong, E. J., et al. (2008). Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022–1038. doi:10.1016/j.neuron.2008.11.029
Garrick, D., Sharpe, J. A., Arkell, R., Dobbie, L., Smith, A. J., Wood, W. G., et al. (2006). Loss of ATRX affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet. 2:e58. doi:10.1371/journal.pgen.0020058
Gibbons, R. J., and Higgs, D. R. (2000). Molecular-clinical spectrum of the ATR-X syndrome. Am. J. Med. Genet. 97, 204–212. doi:10.1002/1096-8628(200023)97:3<204::AID-AJMG1038>3.0.CO;2-X
Giorgi, C., Ito, K., Lin, H. K., Santangelo, C., Wieckowski, M. R., Lebiedzinska, M., et al. (2010). PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251. doi:10.1126/science.1189157
Glass, M., and Everett, R. D. (2013). Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 87, 2174–2185. doi:10.1128/JVI.02950-12
Goldberg, A. D., Banaszynski, L. A., Noh, K. M., Lewis, P. W., Elsaesser, S. J., Stadler, S., et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691. doi:10.1016/j.cell.2010.01.003
Grimwade, D., and Solomon, E. (1997). Characterisation of the PML/RAR alpha rearrangement associated with t(15;17) acute promyelocytic leukaemia. Curr. Top. Microbiol. Immunol. 220, 81–112. doi:10.1007/978-3-642-60479-9_6
Heaphy, C. M., de Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., Shi, C., et al. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. doi:10.1126/science.1207313
Henikoff, S. (2008). Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 9, 15–26. doi:10.1038/nrg2206
Hoemme, C., Peerzada, A., Behre, G., Wang, Y., McClelland, M., Nieselt, K., et al. (2008). Chromatin modifications induced by PML-RARalpha repress critical targets in leukemogenesis as analyzed by ChIP-Chip. Blood 111, 2887–2895. doi:10.1182/blood-2007-03-079921
Hofmann, T. G., Stollberg, N., Schmitz, M. L., and Will, H. (2003). HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 63, 8271–8277.
Hollenbach, A. D., McPherson, C. J., Mientjes, E. J., Iyengar, R., and Grosveld, G. (2002). DAXX and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell. Sci. 115, 3319–3330.
Hong, E. J., West, A. E., and Greenberg, M. E. (2005). Transcriptional control of cognitive development. Curr. Opin. Neurobiol. 15, 21–28. doi:10.1016/j.conb.2005.01.002
Hu, G., Cui, K., Northrup, D., Liu, C., Wang, C., Tang, Q., et al. (2012). H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell 12, 180–192. doi:10.1016/j.stem.2012.11.003
Huang, Y. S., Chang, C. C., Huang, T. C., Hsieh, Y. L., and Shih, H. M. (2012). DAXX interacts with and modulates the activity of CREB. Cell Cycle 11, 99–108. doi:10.4161/cc.11.1.18430
Ito, K., Bernardi, R., Morotti, A., Matsuoka, S., Saglio, G., Ikeda, Y., et al. (2008). PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453, 1072–1078. doi:10.1038/nature07016
Iwase, S., Xiang, B., Ghosh, S., Ren, T., Lewis, P. W., Cochrane, J. C., et al. (2011). ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 18, 769–776. doi:10.1038/nsmb.2062
Jeanne, M., Lallemand-Breitenbach, V., Ferhi, O., Koken, M., Le Bras, M., Duffort, S., et al. (2010). PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell 18, 88–98. doi:10.1016/j.ccr.2010.06.003
Jiao, Y., Shi, C., Edil, B. H., de Wilde, R. F., Klimstra, D. S., Maitra, A., et al. (2011). DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–1203. doi:10.1126/science.1200609
Jin, C., Zang, C., Wei, G., Cui, K., Peng, W., Zhao, K., et al. (2009). H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 41, 941–945. doi:10.1038/ng.409
Jones, D. T., Jager, N., Kool, M., Zichner, T., Hutter, B., Sultan, M., et al. (2012). Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105. doi:10.1038/nature11284
Jullien, J., Astrand, C., Szenker, E., Garrett, N., Almouzni, G., and Gurdon, J. B. (2012). HIRA dependent H3.3 deposition is required for transcriptional reprogramming following nuclear transfer to Xenopus oocytes. Epigenetics Chromatin 5, 17. doi:10.1186/1756-8935-5-17
Kernohan, K. D., Jiang, Y., Tremblay, D. C., Bonvissuto, A. C., Eubanks, J. H., Mann, M. R., et al. (2010). ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 18, 191–202. doi:10.1016/j.devcel.2009.12.017
Khan, M. M., Nomura, T., Kim, H., Kaul, S. C., Wadhwa, R., Shinagawa, T., et al. (2001). Role of PML and PML-RARalpha in Mad-mediated transcriptional repression. Mol. Cell 7, 1233–1243. doi:10.1016/S1097-2765(01)00257-X
Khelifi, A. F., D’Alcontres, M. S., and Salomoni, P. (2005). DAXX is required for stress-induced cell death and JNK activation. Cell Death Differ. 12, 724–733. doi:10.1038/sj.cdd.4401559
Kiesslich, A., Von Mikecz, A., and Hemmerich, P. (2002). Cell cycle-dependent association of PML bodies with sites of active transcription in nuclei of mammalian cells. J. Struct. Biol. 140, 167–179. doi:10.1016/S1047-8477(02)00571-3
Ko, Y. G., Kang, Y. S., Park, H., Seol, W., Kim, J., Kim, T., et al. (2001). Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (DAXX) in the cytoplasm. J. Biol. Chem. 276, 39103–39106. doi:10.1074/jbc.M105928200
Krieghoff-Henning, E., and Hofmann, T. G. (2008). Role of nuclear bodies in apoptosis signalling. Biochim. Biophys. Acta 1783, 2185–2194. doi:10.1016/j.bbamcr.2008.07.002
Kumar, P. P., Bischof, O., Purbey, P. K., Notani, D., Urlaub, H., Dejean, A., et al. (2007). Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat. Cell Biol. 9, 45–56. doi:10.1038/ncb1516
Kuo, H. Y., Chang, C. C., Jeng, J. C., Hu, H. M., Lin, D. Y., Maul, G. G., et al. (2005). SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of DAXX. Proc. Natl. Acad. Sci. U.S.A. 102, 16973–16978. doi:10.1073/pnas.0504460102
Kwok, C., Zeisig, B. B., Dong, S., and So, C. W. (2006). Forced homo-oligomerization of RARalpha leads to transformation of primary hematopoietic cells. Cancer Cell 9, 95–108. doi:10.1016/j.ccr.2006.01.005
Lai, A. Y., and Wade, P. A. (2011). Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer 11, 588–596. doi:10.1038/nrc3091
Lallemand-Breitenbach, V., and de The, H. (2012). PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2, a000661. doi:10.1101/cshperspect.a000661
Law, M. J., Lower, K. M., Voon, H. P., Hughes, J. R., Garrick, D., Viprakasit, V., et al. (2010). ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143, 367–378. doi:10.1016/j.cell.2010.09.023
Lewis, P. W., Elsaesser, S. J., Noh, K. M., Stadler, S. C., and Allis, C. D. (2010). DAXX is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U.S.A. 107, 14075–14080. doi:10.1073/pnas.1008850107
Lewis, P. W., Muller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., et al. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. doi:10.1126/science.1232245
Li, H., Leo, C., Zhu, J., Wu, X., O’Neil, J., Park, E. J., et al. (2000). Sequestration and inhibition of DAXX-mediated transcriptional repression by PML. Mol. Cell. Biol. 20, 1784–1796. doi:10.1128/MCB.20.5.1784-1796.2000
Lim, D. A., Huang, Y. C., Swigut, T., Mirick, A. L., Garcia-Verdugo, J. M., Wysocka, J., et al. (2009). Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458, 529–533. doi:10.1038/nature07726
Lin, D. Y., Huang, Y. S., Jeng, J. C., Kuo, H. Y., Chang, C. C., Chao, T. T., et al. (2006). Role of SUMO-interacting motif in DAXX SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 24, 341–354. doi:10.1016/j.molcel.2006.10.019
Lin, H. K., Bergmann, S., and Pandolfi, P. P. (2004). Cytoplasmic PML function in TGF-beta signalling. Nature 431, 205–211. doi:10.1038/nature02783
Lin, R. J., and Evans, R. M. (2000). Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol. Cell 5, 821–830. doi:10.1016/S1097-2765(00)80322-6
Lindsay, C. R., Giovinazzi, S., and Ishov, A. M. (2009). DAXX is a predominately nuclear protein that does not translocate to the cytoplasm in response to cell stress. Cell Cycle 8, 1544–1551. doi:10.4161/cc.8.10.8379
Lindsay, C. R., Morozov, V. M., and Ishov, A. M. (2008). PML NBs (ND10) and DAXX: from nuclear structure to protein function. Front. Biosci. 13, 7132–7142. doi:10.2741/3216
Liu, X. Y., Gerges, N., Korshunov, A., Sabha, N., Khuong-Quang, D. A., Fontebasso, A. M., et al. (2012). Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 124, 615–625. doi:10.1007/s00401-012-1031-3
Lovejoy, C. A., Li, W., Reisenweber, S., Thongthip, S., Bruno, J., de Lange, T., et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 8:e1002772. doi:10.1371/journal.pgen.1002772
Lu, C., Ward, P. S., Kapoor, G. S., Rohle, D., Turcan, S., Abdel-Wahab, O., et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. doi:10.1038/nature10860
Lukashchuk, V., and Everett, R. D. (2010). Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDAXX. J. Virol. 84, 4026–4040. doi:10.1128/JVI.02597-09
Mainguy, G., In Der Rieden, P. M., Berezikov, E., Woltering, J. M., Plasterk, R. H., Durston, A. J., et al. (2003). A position-dependent organisation of retinoid response elements is conserved in the vertebrate Hox clusters. Trends Genet. 19, 476–479. doi:10.1016/S0168-9525(03)00202-6
Margueron, R., and Reinberg, D. (2011). The polycomb complex PRC2 and its mark in life. Nature 469, 343–349. doi:10.1038/nature09784
Marino, S. (2005). Medulloblastoma: developmental mechanisms out of control. Trends Mol. Med. 11, 17–22. doi:10.1016/j.molmed.2004.11.008
Martens, J. H., Brinkman, A. B., Simmer, F., Francoijs, K. J., Nebbioso, A., Ferrara, F., et al. (2010). PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 17, 173–185. doi:10.1016/j.ccr.2009.12.042
Michaelson, J. S., Bader, D., Kuo, F., Kozak, C., and Leder, P. (1999). Loss of DAXX, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 13, 1918–1923. doi:10.1101/gad.13.15.1918
Michod, D., Bartesaghi, S., Khelifi, A., Bellodi, C., Berliocchi, L., Nicotera, P., et al. (2012). Calcium-dependent dephosphorylation of the histone chaperone DAXX regulates H3.3 loading and transcription upon neuronal activation. Neuron 74, 122–135. doi:10.1016/j.neuron.2012.02.021
Mills, A. A. (2010). Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 10, 669–682. doi:10.1038/nrc2931
Minucci, S., Maccarana, M., Cioce, M., De Lucas, P., Gelmetti, V., Segalla, S., et al. (2000). Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol. Cell 5, 811–820. doi:10.1016/S1097-2765(00)80321-4
Mito, Y., Henikoff, J. G., and Henikoff, S. (2007). Histone replacement marks the boundaries of cis-regulatory domains. Science 315, 1408–1411. doi:10.1126/science.1134004
Molenaar, J. J., Koster, J., Zwijnenburg, D. A., van Sluis, P., Valentijn, L. J., van der Ploeg, I., et al. (2012). Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593. doi:10.1038/nature10910
Muntean, A. G., and Hess, J. L. (2012). The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 7, 283–301. doi:10.1146/annurev-pathol-011811-132434
Ng, R. K., and Gurdon, J. B. (2008). Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat. Cell Biol. 10, 102–109. doi:10.1038/ncb1674
Pchelintsev, N. A., McBryan, T., Rai, T. S., van Tuyn, J., Ray-Gallet, D., Almouzni, G., et al. (2013). Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 3, 1012–1019. doi:10.1016/j.celrep.2013.03.026
Pearson, M., Carbone, R., Sebastiani, C., Cioce, M., Fagioli, M., Saito, S., et al. (2000). PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210. doi:10.1038/35018127
Perlman, R., Schiemann, W. P., Brooks, M. W., Lodish, H. F., and Weinberg, R. A. (2001). TGF-beta-induced apoptosis is mediated by the adapter protein DAXX that facilitates JNK activation. Nat. Cell Biol. 3, 708–714. doi:10.1038/35087019
Pinton, P., Giorgi, C., and Pandolfi, P. P. (2011). The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ. 18, 1450–1456. doi:10.1038/cdd.2011.31
Preston, C. M., and Nicholl, M. J. (2006). Role of the cellular protein hDAXX in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 87, 1113–1121. doi:10.1099/vir.0.81566-0
Purton, L. E., Dworkin, S., Olsen, G. H., Walkley, C. R., Fabb, S. A., Collins, S. J., et al. (2006). RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 203, 1283–1293. doi:10.1084/jem.20052105
Puto, L. A., and Reed, J. C. (2008). DAXX represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 22, 998–1010. doi:10.1101/gad.1632208
Rai, T. S., Puri, A., McBryan, T., Hoffman, J., Tang, Y., Pchelintsev, N. A., et al. (2011). Human CABIN1 is a functional member of the human HIRA/UBN1/ASF1a histone H3.3 chaperone complex. Mol. Cell. Biol. 31, 4107–4118. doi:10.1128/MCB.05546-11
Ray-Gallet, D., Woolfe, A., Vassias, I., Pellentz, C., Lacoste, N., Puri, A., et al. (2011). Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 44, 928–941. doi:10.1016/j.molcel.2011.12.006
Regad, T., Bellodi, C., Nicotera, P., and Salomoni, P. (2009). The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat. Neurosci. 12, 132–140. doi:10.1038/nn.2251
Ringrose, L., and Paro, R. (2004). Epigenetic regulation of cellular memory by the polycomb and trithorax group proteins. Annu. Rev. Genet. 38, 413–443. doi:10.1146/annurev.genet.38.072902.091907
Rivera-Molina, Y. A., Rojas, B. R., and Tang, Q. (2012). Nuclear domain 10-associated proteins recognize and segregate intranuclear DNA/protein complexes to negate gene expression. Virol. J. 9, 222. doi:10.1186/1743-422X-9-222
Robinson, G., Parker, M., Kranenburg, T. A., Lu, C., Chen, X., Ding, L., et al. (2012). Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48. doi:10.1038/nature11213
Rodriguez, M. S., Dargemont, C., and Hay, R. T. (2001). SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659. doi:10.1074/jbc.M009476200
Saffert, R. T., and Kalejta, R. F. (2006). Inactivating a cellular intrinsic immune defense mediated by DAXX is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80, 3863–3871. doi:10.1128/JVI.80.8.3863-3871.2006
Sakamoto, K., Huang, B. W., Iwasaki, K., Hailemariam, K., Ninomiya-Tsuji, J., and Tsuji, Y. (2010). Regulation of genotoxic stress response by homeodomain-interacting protein kinase 2 through phosphorylation of cyclic AMP response element-binding protein at serine 271. Mol. Biol. Cell 21, 2966–2974. doi:10.1091/mbc.E10-01-0015
Salomoni, P., and Betts-Henderson, J. (2011). The role of PML in the nervous system. Mol. Neurobiol. 43, 114–123. doi:10.1007/s12035-010-8156-y
Salomoni, P., Ferguson, B. J., Wyllie, A. H., and Rich, T. (2008). New insights into the role of PML in tumour suppression. Cell Res. 18, 622–640. doi:10.1038/cr.2008.58
Salomoni, P., and Khelifi, A. F. (2006). DAXX: death or survival protein? Trends Cell Biol. 16, 97–104. doi:10.1016/j.tcb.2005.12.002
Salomoni, P., and Pandolfi, P. P. (2002). The role of PML in tumor suppression. Cell 108, 165–170. doi:10.1016/S0092-8674(02)00626-8
Sawarkar, R., and Paro, R. (2010). Interpretation of developmental signaling at chromatin: the polycomb perspective. Dev. Cell 19, 651–661. doi:10.1016/j.devcel.2010.10.012
Sawatsubashi, S., Murata, T., Lim, J., Fujiki, R., Ito, S., Suzuki, E., et al. (2010). A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 24, 159–170. doi:10.1101/gad.1857410
Schreiner, S., Bürck, C., Glass, M., Groitl, P., Wimmer, P., Kinkley, S., et al. (2013). Control of human adenovirus type 5 gene expression by cellular DAXX/ATRX chromatin-associated complexes. Nucleic Acids Res. 41, 3532–3550. doi:10.1093/nar/gkt064
Schuettengruber, B., Martinez, A. M., Iovino, N., and Cavalli, G. (2011). Trithorax group proteins: switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 12, 799–814. doi:10.1038/nrm3230
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. doi:10.1038/nature10833
Seeler, J. S., Marchio, A., Sitterlin, D., Transy, C., and Dejean, A. (1998). Interaction of SP100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc. Natl. Acad. Sci. U.S.A. 95, 7316–7321. doi:10.1073/pnas.95.13.7316
Shalginskikh, N., Poleshko, A., Skalka, A. M., and Katz, R. A. (2013). Retroviral DNA methylation and epigenetic repression are mediated by the antiviral host protein DAXX. J. Virol. 87, 2137–2150. doi:10.1128/JVI.02026-12
Simon, J. A., and Kingston, R. E. (2009). Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 10, 697–708. doi:10.1038/nrm2763
Stagno D’Alcontres, M., Mendez-Bermudez, A., Foxon, J. L., Royle, N. J., and Salomoni, P. (2007). Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J. Cell Biol. 179, 855–867. doi:10.1083/jcb.200703020
Sturm, D., Witt, H., Hovestadt, V., Khuong-Quang, D. A., Jones, D. T., Konermann, C., et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437. doi:10.1016/j.ccr.2012.08.024
Szenker, E., Ray-Gallet, D., and Almouzni, G. (2011). The double face of the histone variant H3.3. Cell Res. 21, 421–434. doi:10.1038/cr.2011.14
Tang, J., Qu, L. K., Zhang, J., Wang, W., Michaelson, J. S., Degenhardt, Y. Y., et al. (2006). Critical role for DAXX in regulating MDM2. Nat. Cell Biol. 8, 855–862. doi:10.1038/ncb1442
Tang, M. C., Jacobs, S. A., Wong, L. H., and Mann, J. R. (2013). Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis 51, 142–146. doi:10.1002/dvg.22366
Torok, D., Ching, R. W., and Bazett-Jones, D. P. (2009). PML nuclear bodies as sites of epigenetic regulation. Front. Biosci. 14, 1325–1336. doi:10.2741/3311
Tsai, K., Thikmyanova, N., Wojcechowskyj, J. A., Delecluse, H. J., and Lieberman, P. M. (2011). EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 7:e1002376. doi:10.1371/journal.ppat.1002376
Valk-Lingbeek, M. E., Bruggeman, S. W., and Van Lohuizen, M. (2004). Stem cells and cancer; the polycomb connection. Cell 118, 409–418. doi:10.1016/j.cell.2004.08.005
van Haaften, G., Dalgliesh, G. L., Davies, H., Chen, L., Bignell, G., Greenman, C., et al. (2009). Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 41, 521–523. doi:10.1038/ng.349
Viale, A., De Franco, F., Orleth, A., Cambiaghi, V., Giuliani, V., Bossi, D., et al. (2009). Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 457, 51–56. doi:10.1038/nature07618
Villa, R., Pasini, D., Gutierrez, A., Morey, L., Occhionorelli, M., Viré, E., et al. (2007). Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 11, 513–525. doi:10.1016/j.ccr.2007.04.009
Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R. S., et al. (2004). Role of histone H2A ubiquitination in polycomb silencing. Nature 431, 873–878. doi:10.1038/nature02985
Wong, L. H., McGhie, J. D., Sim, M., Anderson, M. A., Ahn, S., Hannan, R. D., et al. (2010). ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 20, 351–360. doi:10.1101/gr.101477.109
Wong, L. H., Ren, H., Williams, E., McGhie, J., Ahn, S., Sim, M., et al. (2009). Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 19, 404–414. doi:10.1101/gr.084947.108
Wu, G., Broniscer, A., McEachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253. doi:10.1038/ng.1102
Yang, X., Khosravi-Far, R., Chang, H. Y., and Baltimore, D. (1997). DAXX, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89, 1067–1076. doi:10.1016/S0092-8674(00)80294-9
Ye, X., Zerlanko, B., Zhang, R., Somaiah, N., Lipinski, M., Salomoni, P., et al. (2007). Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 27, 2452–2465. doi:10.1128/MCB.01592-06
Yuan, W., Payton, J. E., Holt, M. S., Link, D. C., Watson, M. A., DiPersio, J. F., et al. (2007). Commonly dysregulated genes in murine APL cells. Blood 109, 961–970. doi:10.1182/blood-2006-07-036640
Zhang, H., He, J., Li, J., Tian, D., Gu, L., and Zhou, M. (2013). Methylation of RASSF1A gene promoter is regulated by p53 and DAXX. FASEB J. 27, 232–242. doi:10.1096/fj.12-215491
Zhang, X. W., Yan, X. J., Zhou, Z. R., Yang, F. F., Wu, Z. Y., Sun, H. B., et al. (2010). Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 328, 240–243. doi:10.1126/science.1183424
Zhong, S., Müller, S., Ronchetti, S., Freemont, P. S., Dejean, A., and Pandolfi, P. P. (2000a). Role of SUMO-1-modified PML in nuclear body formation. Blood 95, 2748–2752.
Zhong, S., Salomoni, P., and Pandolfi, P. P. (2000b). The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2, E85–E90. doi:10.1038/35010583
Zhou, J., Pérès, L., Honoré, N., Nasr, R., Zhu, J., and de The, H. (2006). Dimerization-induced corepressor binding and relaxed DNA-binding specificity are critical for PML/RARA-induced immortalization. Proc. Natl. Acad. Sci. U.S.A. 103, 9238–9243. doi:10.1073/pnas.0603324103
Keywords: PML, DAXX, cancer, epigenetics, histone variant
Citation: Salomoni P (2013) The PML-interacting protein DAXX: histone loading gets into the picture. Front. Oncol. 3:152. doi: 10.3389/fonc.2013.00152
Received: 11 April 2013; Accepted: 24 May 2013;
Published online: 07 June 2013.
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Keisuke Ito, Albert Einstein College of Medicine, USAGenevieve Almouzni, Centre National de la Recherche Scientifique, France
Copyright: © 2013 Salomoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Paolo Salomoni, Samantha Dickson Brain Cancer Unit, UCL Cancer Institute, University College London, 72 Huntley Street, London WC1E 6BT, UK e-mail:cC5zYWxvbW9uaUBjYW5jZXIudWNsLmFjLnVr