

- Department of Pediatrics, Martin-Luther-University Halle-Wittenberg, Halle, Germany
Human endogenous retroviruses (ERV) are an integral part of our genome. Expression of ERV is usually switched off but reactivation of ERV has been observed in varying human diseases including cancer. Recently, reactivation of ERV associated promoters in Hodgkin’s lymphoma (HL) cells has been described. Despite relatively good prognosis, not all patients with HL can be cured with the established therapy and this therapy is associated with severe late side effects. Therefore, new targets are required for the development of future treatment strategies. Reactivated ERV might represent such target structures. Therefore, we asked which ERV loci are expressed in HL cells. Using DNA microarray analysis, we found no evidence for a general activation of ERV transcription in HL cells. In contrast, we observed down-regulation of ERV3, an ERV with potential tumor suppressor function, in HL cells in comparison to normal blood cells. Interestingly, ERV3 was also differentially expressed in published DNA microarray data from resting versus cycling B cells. Treatment of HL cells with the histone deacetylase inhibitor vorinostat strongly up-regulated ERV3 expression. In addition, we observed up-regulation in HL cells after treatment with hypoxia-mimetic cobalt(II) chloride. Like vorinostat, cobalt(II) chloride inhibited cell growth of HL cells. Our results suggest that cell cycle inhibition of HL cells is accompanied by up-regulation of ERV3.
Introduction
The exact etiology of Hodgkin’s lymphoma (HL) is unknown, but immunological and molecular properties suggest that the majority of HL are derived from B cells (1, 2). HL cells have a characteristic gene-expression profile that discriminates these cells from other normal and transformed cells (3, 4). Especially for pediatric HL patients the prognosis is relatively good, and with the combination of radio- and chemo-therapy the majority of patients with HL can be cured. However, the established therapy is associated with a plethora of late adverse side effects and some patients with chemo-resistant disease cannot be cured (5, 6). Therefore, it is important to search for new targets for treatment of patients with HL.
Recently, reactivation of endogenous retrovirus (ERV) activity has been observed in HL (7). This reactivation leads to expression of the receptor for macrophage colony-stimulating factor (colony-stimulating factor 1 receptor, CSF1R), a known oncogene, in HL cells. ERV are an integral part of the genome of virtually all eukaryotes, and ERV loci have been extensively analyzed in plants, insects, and vertebrates (8–13). In the human genome, ERV derived sequences constitute at least 8% of the complete DNA. Usually, ERV are silenced epigenetically and are not transcribed into RNA. Reactivation of ERV has been observed under pathological conditions, e.g., in cancer cells. Such reactivation can result even in the formation of virus particles (14, 15). A small number of ERV-encoded proteins can be found under normal physiological conditions. Such proteins can exert variable biological functions (16, 17). One notable example is ERVW-1 (also known as syncytin 1) which is required for proper formation of the syncytial layer of the placenta (16). In this ERV only the envelope protein is functional. Other open reading frames (ORF) have been inactivated by deleterious mutations. Such mutations destroy the ORF of the majority of ERV. Some ERV with intact ORF encode superantigens (18, 19). Such superantigens can activate a high percentage of all T cells. The activation of these T cells can lead to hyper-reactivity of the immune system but can also lead to the final deletion of the activated T cells.
In addition to the potential involvement in the pathogenesis of human diseases, ERV might also represent interesting target structures for the development of future treatment strategies. Immune responses against ERV-encoded antigens have been described in cancer patients (20, 21). In melanoma patients, antibodies against ERV are associated with shorter disease free survival (21). On the other hand, cytotoxic T cells with specificity for ERV-encoded antigens can kill melanoma cells (22), colorectal cancer cells (23), and renal cancer cells (24). In addition, antibodies against ERV products can inhibit growth of breast cancer cells in vitro and in an animal model (25). Successful immunization of rhesus macaques against simian ERV suggests that ERV derived antigens can be used as safe vaccines without development of auto-immunity (26). Interestingly, ERV-specific T cells have been detected in patients after allogeneic hematopoietic stem cell transplantation (alloHSCT) (24). These T cells can kill the tumor cells and might be responsible for graft-versus-tumor effects after alloHSCT (24). Graft-versus-tumor effects have also been described in HL patients after alloHSCT (27). It remains unclear whether ERV reactivation in HL cells (7) has an impact on such graft-versus-tumor effects. We asked whether ERV reactivation in HL is a phenomenon affecting multiple (or all) ERV loci or whether this reactivation is specific for single ERV loci. Therefore, we used DNA microarray data for the analysis of multiple ERV loci in HL cells. DNA microarrays can be used for the characterization of complete gene-expression profiles from normal and malignant cells in a single experiment (28). Modern DNA exon microarrays contain several probe sets with specificity for ERV and, therefore, can be used for analysis of expression of multiple ERV loci at once.
Materials and Methods
Cell Lines and Cell Culture
Hodgkin’s lymphoma-cell lines HDLM-2, KM-H2, L-1236, L-428, and L-540 (29–33) were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), Braunschweig, Germany. P439-6 cells were kindly provided by G. W. Bornkamm and G. Laux, Munich, Germany. P493-6 cells carry an EBV nuclear antigen 2 (EBNA2)-estrogen receptor fusion gene and MYC under control of a promoter which can be regulated by tetracycline (34–36). All cells were cultured in RPMI-1640 (Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (PAA, Pasching, Germany) at 37°C in a humidified atmosphere at 5% CO2. Treatment of HL cells with 1 μM vorinostat was carried out as described (37) at a cell density of 1 × 106 cells/mL for 24 h. Dimethyl sulfoxide (DMSO) was used as control. For simulation of hypoxia, HL cells were treated for 2 days at a cell density of 1 × 106 cells/mL with 200 μM cobalt(II) chloride. P439-6 cells were cultured for 4 days in medium with or without 2 μM estradiol and/or 1 μg/mL tetracycline.
Gene-Expression Analysis
RNA from cell lines were isolated using TriFast reagent (peqlab, Erlangen, Germany) following manufacturer’s protocol. Two micrograms of the RNA were transcribed into cDNA and used as template for polymerase chain reaction (PCR). The following primer combinations were used for real-time quantitative reverse transcription-PCR (qRT-PCR): actin beta (ACTB): 5′-GGC ATC GTG ATG GAC TCC G-3′, 5′-GCT GGA AGG TGG ACA GCG A-3′; ERV3: 5′-GGG AGT ATG CGG AAA GTT CA-3′, 5′-CTC CAA GGG ATG AGA ACC AA-3′. Quantitative RT-PCR was performed using the Go Taq qPCR Master Mix (Promega, Mannheim, Germany). The reaction was performed with 10 μL Go Taq qPCR Master Mix, 6 μL water, 1 μL primer combination, and 2 μL cDNA using the following conditions: 94°C, 30 s; 60°C, 30 s; 72°C, 45 s (40 cycles). Determination of gene expression was performed using the method (38). Global gene expression in HL cell lines was analyzed by using Affymetrix Human Exon 1.0ST arrays (Affymetrix, Santa Clara, USA). In addition to microarray data from HL cell lines L-540, HDLM-2 and L-428 (39), microarray data from normal peripheral blood cells (40), P493-6 cells (41), and normal B cells (42, 43) were used for comparative analysis. These cel files were down-loaded from the gene-expression omnibus (GEO) data base. All cel files were processed together using the robust microarray analysis (RMA) algorithm with Expression Console 1.1 (Affymetrix). Cel files from DNA microarrays from HL cell lines have been submitted to the GEO data base (accession number GSE47686). Signal intensities from ERV-specific probe sets were visualized with the Genesis software (44).
Results
Analysis of ERV Expression in DNA Microarray Data
We analyzed expression of human ERV in DNA microarray data from HL cell lines HDLM-2, L-428, and L-540 in comparison to normal blood cells, normal B cells, and the conditionally immortalized B cell line P493-6. A total of 169 probe sets with specificity for ERV sequences were analyzed (Figure 1). Signal intensities (RMA normalized, linear values) above 100 were considered to be expressed in the corresponding samples. According to this threshold, HDLM-2 cells expressed 13 different ERV (represented by 31 probe sets), L-428 cells expressed 10 ERV (36 probe sets), and L-540 cells expressed 11 ERV (28 probe sets). In normal blood cells, 13 ERV were expressed (43 probe sets with mean signal intensities above the threshold). Interestingly, the eight ERV that were expressed in all HL cell lines (ERVFC1-1, ERVH-1, ERVH-4, ERVH48-1, ERVK3-1, ERVK-7, ERVK-9/-4/-19, ERVK13-1), were also expressed in normal blood cells. In addition, normal blood cells expressed ERV3-1, ERVK-6, ERVV-1, ERVW-6, and ERVMER34-1. Isolated B cells expressed 15 ERV (45 probe sets with mean signal intensities above the threshold). In addition to all ERV that were found in normal blood, isolated B cells had high signal intensities for ERV9-1 and ERVK11-1. Additional ERV were found only in single HL cell lines: L-428 cells expressed ERVH-6; L-540 cells expressed ERVW-1 and ERVK3-2; HDLM-2 cells expressed ERVFRD-1 and ERVFRD-2. Taken all together, the number of expressed ERV in HL cell lines did not exceed the number of ERV expressed in normal blood or isolated B cells. Probe sets for ERV3-1 and ERVK13-1 showed significantly lower (p < 0.01) signal intensities in HL cells than in normal blood cells (Figure 2A). We found no ERV that were significantly up-regulated in HL cells. Mean signal intensities for ERV3-1 and ERVK13-1 in isolated B cells were lower than the signals in whole blood. It is known from the literature that ERV3 is up-regulated in cell cycle arrested differentiating cells (45). We tested whether ERV3 is also up-regulated in cell cycle arrested B cells. For this end, we analyzed DNA microarray data (41) from P493-6 cells that have been treated with tetracycline. Tetracycline switches off expression of MYC in this Burkitt lymphoma model cell line leading to cell cycle arrest (34–36). As shown in Figure 2B, we detected up-regulation of all probe sets from Figure 2A in arrested P493-6 cells.
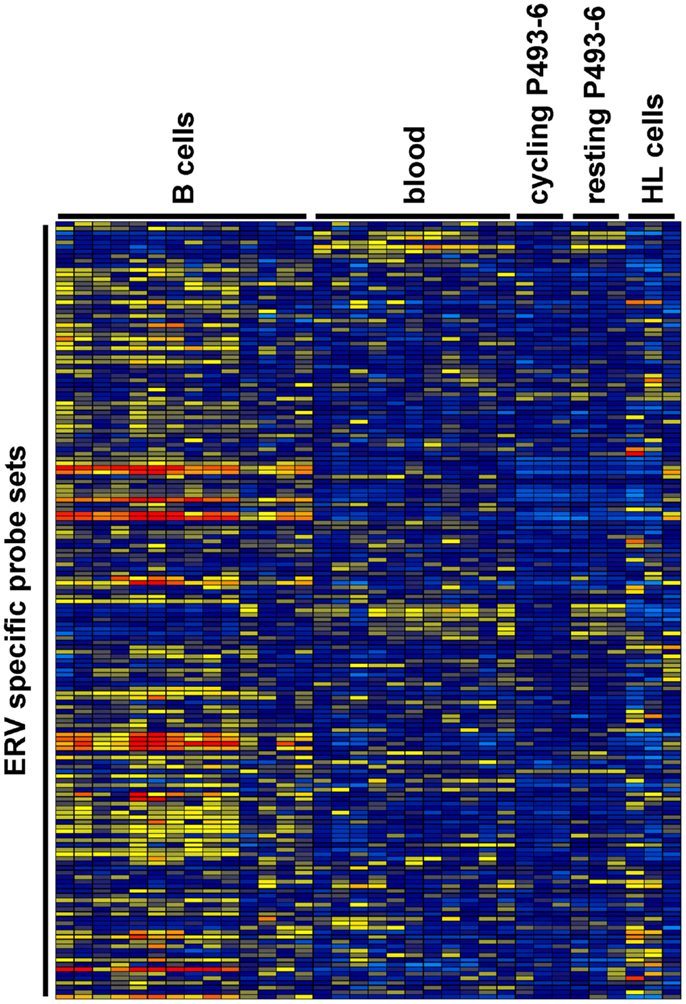
Figure 1. Heat map of ERV-specific probe sets in the investigated DNA microarray data. Probe sets with specificity for ERV-related sequences were selected from microarray data from normal blood cells [GEO data set GSE18838 (40)], normal B cells [GEO data sets GSE14352 and GSE5188 (42, 43)], P493-6 cells [GEO data set GSE32219 (41)], and HL cell lines. Signal intensities were visualized using the Genesis software (log2-transformed and mean centered data). The used arrays contain probe sets for the following ERV: ERV3-1, ERV3-2, ERV9-1, ERV18-1, ERVFC1-1, ERVFH21-1, ERVFRD-1, ERVFRD-2, ERVH-1, ERVH-4, ERVH-6, ERVH48-1, ERVI-1, ERVK3-1, ERVK3-2, ERVK-6, ERVK-7, ERVK-9/-4/-19, ERVK11-1, ERVK13-1, ERVMER34-1, ERVMER61-1, ERVV-1, ERVV-1/-2, ERVW-1, andERVW-6.
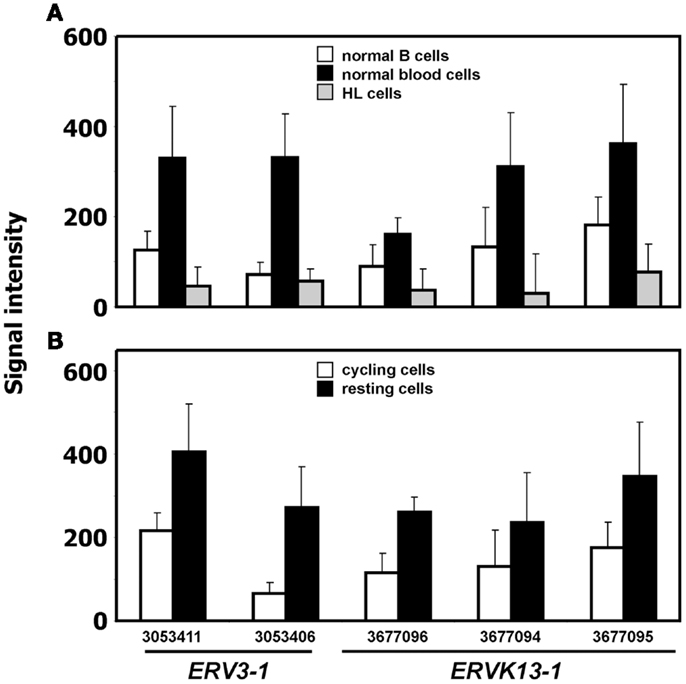
Figure 2. ERV with significant difference between HL and normal blood samples. (A) Presented are signal intensities (means and standard deviations) of the indicated probe sets (ERV3-1: probe sets 303411 and 3053406; ERVK13-1: probe sets 3677096, 3677094, and 3677095) in normal blood cells [GEO data set GSE18838 (40)], normal B cells [GEO data sets GSE14352 and GSE5188 (42, 43)], and HL cell lines. (B) Signal intensities of the same probe sets in P493-6 cells after treatment with tetracycline (resting cells) or medium without tetracycline [cycling cells; raw data from GEO data set GSE32219 (41)].
Validation of ERV3 Expression in Cell Cycle Arrested B Cells
To validate the observation of cell cycle dependent regulation of ERV3 we performed quantitative RT-PCR with P439-6 cells that had been cultured under different conditions. Cells were grown in medium (cycling cells) or after addition of tetracycline (arrested cells). In addition, the same cells were cultured in the presence of estrogen. Estrogen switches on functional EBNA2 in these cells and allows the proliferation in the presence of tetracycline. As shown in Figure 3, incubation of P493-6 cells with tetracycline resulted in marked up-regulation of ERV3. Switching on the EBV transformation program by addition of estrogen to tetracycline-treated P493-6 cells inhibited this up-regulation. Similar results were obtained with the conditional EBV-immortalized cell line EREB2-5 (46). This cell line is the parental cell line of P493-6 cells (without exogenous MYC) and proliferates only in the presence of estrogen in the culture medium. In this cell line we observed up-regulation of ERV3 after withdrawal of estrogen (data not shown).
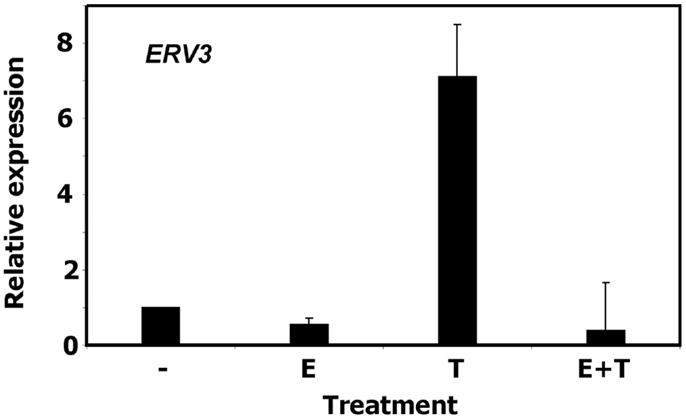
Figure 3. Expression of ERV3 in P439-6 cells. Expression of ERV3 was analyzed in P439-6 cells under different culture conditions by qRT-PCR. Cells were cultured in the absence or presence of estrogen (E) and/or tetracycline (T). Presented are means and standard errors from duplicate determinations. For comparative analysis, beta actin was used as housekeeping control and the mean of P439-6 cells cultured in medium was set as 1.
Regulation of ERV3 Expression in HL Cell Lines
In our previous work we observed that treatment of HL cells with the histone deacetylase inhibitor vorinostat induces cell cycle arrest (37). Therefore, we asked whether this cell cycle arrest is also accompanied by up-regulation of ERV3 in HL cells. Figure 4 show the results of this analysis. Incubation of all tested HL cell lines with vorinostat resulted in an increased expression of ERV3. We observed that treatment of HL cells with the hypoxia-mimetic CoCl2 led to a pronounced inhibition of proliferation (Figure 5). As shown in Figure 6, the expression of ERV3 again increased when cells were cultured under conditions of inhibited proliferation.
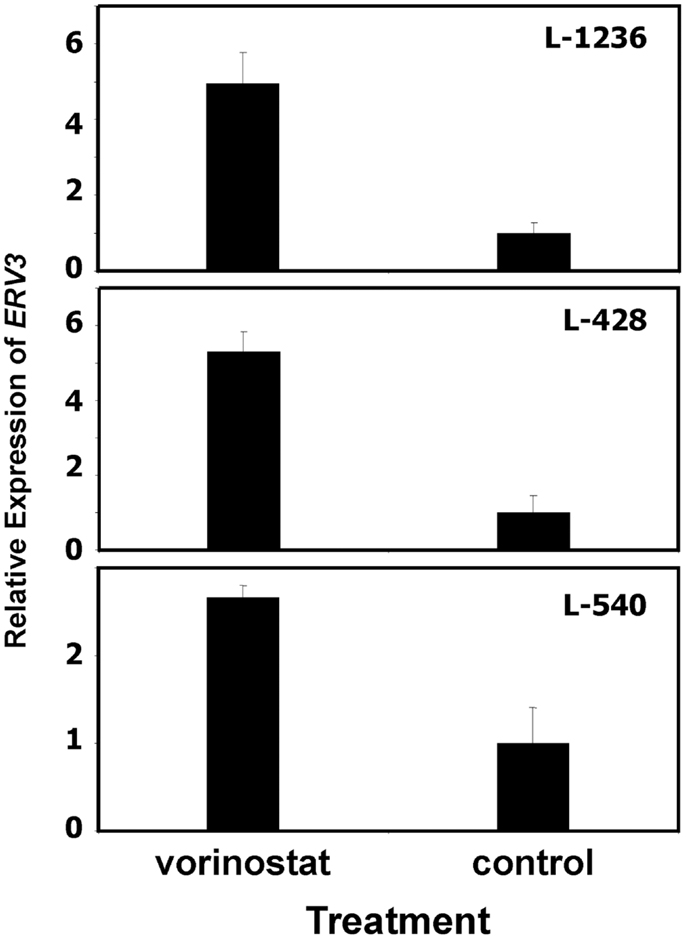
Figure 4. Incubation of HL cells with the histone deacetylase inhibitor vorinostat leads to increased expression of ERV3. Expression of ERV3 was analyzed in HL cell lines with and without 1 μM vorinostat by quantitative RT-PCR. Presented are means and standard errors from triplicates. Beta actin was used as housekeeping control and the mean of the HL cells without vorinostat was set as 1.
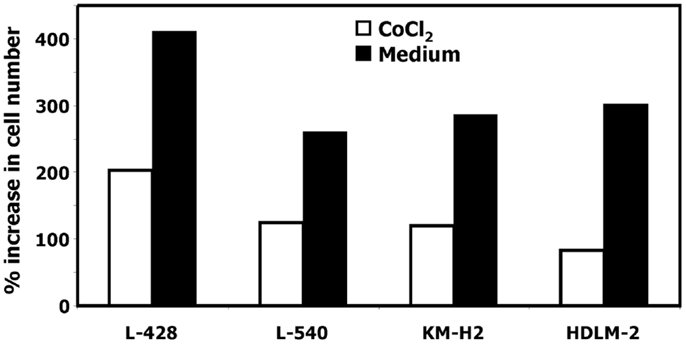
Figure 5. Incubation of HL cells with cobalt(II) chloride inhibits proliferation. Cells of the indicated cell lines were cultured in the presence or absence of 200 μM cobalt(II) chloride for 4 days. Thereafter the cell number was determined. Percentage of increase in cell number was calculated as [(final cell number)/(starting cell number) × 100].
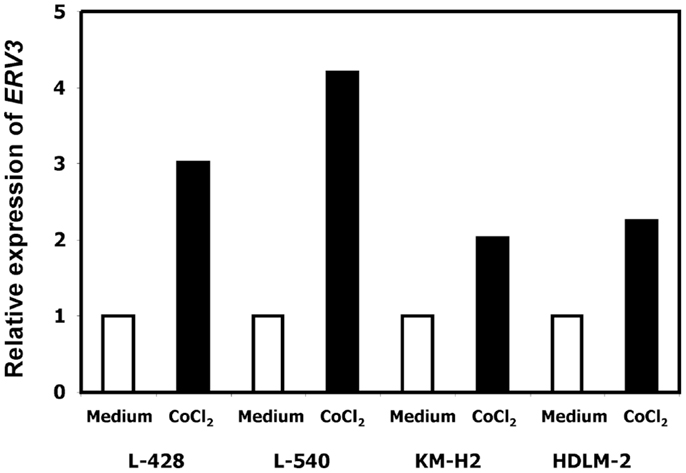
Figure 6. Incubation of HL cells with cobalt(II) chloride leads to an increased expression of ERV3. Expression of ERV3 was analyzed in HL cell lines L-428, L-540, KM-H2, and HDLM-2 after treatment with 200 μM cobalt(II) chloride or without cobalt(II) chloride by quantitative RT-PCR. Beta actin was used as housekeeping control and the mean of HL cells without cobalt(II) chloride was set as 1.
Discussion
Increased expression of ERV derived sequences has been observed in cancer cells (14, 15, 47–51) and in patients with autoimmune diseases or neurodegenerative diseases (52–54). ERV can act as alternative promoters for adjacent genes (7, 55–58). The resulting fusion transcripts can result in new protein isoforms, or the ERV component of these fusion transcripts can inhibit translation (58). In addition, ERV expression can interfere with the expression of adjacent genes at the level of transcription (59). In cancer cells such interference may lead to the inactivation of tumor suppressor genes. Some ERV-encoded proteins can directly bind and inactivate tumor suppressor genes (60). In our present study we provide preliminary evidence for a differential expression of ERV3 in HL cells under conditions of growth arrest. The method used for analysis of ERV expression in HL has several limitations. Not all human ERV loci are represented on the arrays and it might be that other ERV are differentially expressed in the investigated cells. In addition, the comparability of our data sets from HL cells and published data sets from normal blood and B cells might also be sub-optimal. However, our results gave no evidence for a general up-regulation of the investigated ERV loci in HL cells. The ERV3 signals in normal B cells are relatively low. Therefore and based on the limitations of the study, we cannot conclude that the low ERV3 expression is a specific feature of HL cells. However, up-regulation of ERV3 in HL cells under conditions of growth arrest suggests that ERV3 might be an interesting gene for further studies. ERV3 is unique among ERV as it is considered to be a tumor suppressor (61). ERV3 is abundantly expressed in the placenta and it is expressed in most other tissues at lower levels (62). Absence of expression in choriocarcinoma was observed (62), and transgenic expression of ERV3 in choriocarcinoma cells inhibits cell proliferation (63). This growth inhibition is associated with down-regulation of cyclin B and up-regulation of the cyclin dependent kinase inhibitor p21 (63). Expression of ERV3 is up-regulated during terminal differentiation of leukemia cells and highest in cell cycle arrested cells (45, 64). ERV3 is a member of an ERV family with more than 40 members, but only ERV3 has intact ORF for viral proteins (65). The chromosomal location of human ERV3 is characterized by a high number of pseudogenes (data not shown), and the complete ERV3 locus is present only in Old World primates with the exception of gorillas (66). Surprisingly, approximately 1% of Caucasians with normal phenotype have mutations in ERV3 which interrupt the ORF of the ERV3 envelope (67). This observation suggests that the ERV3 encoded envelope protein is not critically involved in the physiological function of this gene. Interestingly, read-through transcript between ERV3 and the neighboring zinc finger protein 117 (ZNF117) have been described (68). Lower expression of these transcripts has been observed in patients with multiple sclerosis (69). The human zinc finger proteins ZNF107, ZNF138, and ZNF92 have high homology with ZNF117. Together with other zinc finger proteins these genes form a cluster on human chromosome 7. The physiological function of ZNF117 has not been clarified, but it seems possible that this gene contributes to the biological effects of ERV3.
Endogenous retrovirus reactivation might occur only in transcriptionally active regions of the genome. In such cases, ERV reactivation might be only an epiphenomenon of chromatin opening and depends on the presence of adequate competence factors allowing transcription of the ERV. In such a model one would expect that several ERV loci are activated at the same time point. Our results show no evidence for such a general activation of ERV loci in HL. However, our analysis includes only well characterized ERV loci which are detectable by the used microarrays. Reactivation of ERV associated alternative promoter in the CSF1R gene seems to be involved in the pathophysiology of HL (7). The identification of such alternative transcription start sites by means of DNA exon microarray analysis requires new bioinformatics tools which are currently being developed in our lab.
The up-regulation of ERV3 under conditions of cell cycle inhibition and/or terminal differentiation is not specific for HL. Whether such up-regulation occurs only in transformed hematopoietic cells or also in other cell types has to be determined. Up-regulation of ERV3 in HL cells occurred under conditions which are characterized by increased apoptosis. CoCl2 can induce apoptosis in hematopoietic and non-hematopoietic tumor cells (70–73). Similarly, vorinostat and other histone deacetylase inhibitors induce apoptosis in HL cells (74–76). If ERV3 is a tumor suppressor gene (as suggested by the choriocarcinoma data discussed above), expression of ERV3 in HL cells and other hematopoietic cells under pro-apoptotic and anti-proliferative conditions might indicate a tumor suppressing activity of ERV3 also in these cell types. The elucidation of ERV3 activities in the context of growth inhibition and apoptosis might help the identification of new targets for the treatment of HL and other malignant diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank I. Volkmer for grateful technical assistance. We thank Vera Marks (communication skills) for critically reading and copy editing of the manuscript. This work was supported by a fellowship from the Konrad-Adenauer-Stiftung (Stefanie Kewitz) and the Wilhelm-Roux-Program of the Martin-Luther-University Halle-Wittenberg (FKZ 25/22).
References
1. Tzankov A, and Dirnhofer S. Pathobiology of classical Hodgkin lymphoma. Pathobiology (2006) 73:107–25. doi:10.1159/000095558
2. Drexler HG, and Minowada J. Hodgkin’s disease derived cell lines: a review. Hum Cell (2009) 5:42–53.
3. Schwering I, Bräuninger A, Klein U, Jungnickel B, Tinguely M, Diehl V, et al. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood (2003) 101:1505–12. doi:10.1182/blood-2002-03-0839
4. Staege MS, Banning-Eichenseer U, Weissflog G, Volkmer I, Burdach S, Richter G, et al. Gene expression profiles of Hodgkin’s lymphoma cell lines with different sensitivity to cytotoxic drugs. Exp Hematol (2008) 36:886–96. doi:10.1016/j.exphem.2008.02.014
5. Körholz D, Claviez A, Hasenclever D, Kluge R, Hirsch W, Kamprad F, et al. The concept of the GPOH-HD 2003 therapy study for pediatric Hodgkin’s disease: evolution in the tradition of the DAL/GPOH studies. Klin Padiatr (2004) 216:150–6. doi:10.1055/s-2004-822627
6. Mauz-Körholz C, Hasenclever D, Dörffel W, Ruschke K, Pelz T, Voigt A, et al. Procarbazine-free OEPA-COPDAC chemotherapy in boys and standard OPPA-COPP in girls have comparable effectiveness in pediatric Hodgkin’s lymphoma: the GPOH-HD-2002 study. J Clin Oncol (2010) 28:3680–6. doi:10.1200/JCO.2009.26.9381
7. Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med (2010) 16:571–9. doi:10.1038/nm.2129
8. Weiss RA. The discovery of endogenous retroviruses. Retrovirology (2006) 3:67. doi:10.1186/1742-4690-3-S1-S67
9. Buzdin A. Human-specific endogenous retroviruses. ScientificWorldJournal (2007) 7:1848–68. doi:10.1100/tsw.2007.270
10. Basta HA, Cleveland SB, Clinton RA, Dimitrov AG, and McClure MA. Evolution of teleost fish retroviruses: characterization of new retroviruses with cellular genes. J Virol (2009) 83:10152–62. doi:10.1128/JVI.02546-08
11. Du J, Tian Z, Hans CS, Laten HM, Cannon SB, Jackson SA, et al. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J (2010) 63:584–98. doi:10.1111/j.1365-313X.2010.04263.x
12. Cui J, and Holmes EC. Endogenous RNA viruses of plants in insect genomes. Virology (2012) 427:77–9. doi:10.1016/j.virol.2012.02.014
13. Harzmann R, Löwer J, Löwer R, Bichler KH, and Kurth R. Synthesis of retrovirus-like particles in testicular teratocarcinomas. J Urol (1982) 128:1055–9.
14. Stefanov Y, Salenko V, and Glukhov I. Drosophila errantiviruses. Mob Genet Elements (2012) 2:36–45. doi:10.4161/mge.19234
15. Bieda K, Hoffmann A, and Boller K. Phenotypic heterogeneity of human endogenous retrovirus particles produced by teratocarcinoma cell lines. J Gen Virol (2001) 82:591–6.
16. Dupressoir A, Lavialle C, and Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta (2012) 33:663–71. doi:10.1016/j.placenta.2012.05.005
17. Beyer U, Moll-Rocek J, Moll UM, and Dobbelstein M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc Natl Acad Sci U S A (2011) 108:3624–9. doi:10.1073/pnas.1016201108
18. Meylan F, De Smedt M, Leclercq G, Plum J, Leupin O, Marguerat S, et al. Negative thymocyte selection to HERV-K18 superantigens in humans. Blood (2005) 105:4377–82. doi:10.1182/blood-2004-07-2596
19. Sutkowski N, Conrad B, Thorley-Lawson DA, and Huber BT. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity (2001) 15:579–89. doi:10.1016/S1074-7613(01)00210-2
20. Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, et al. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res (2008) 68:5869–77. doi:10.1158/0008-5472.CAN-07-6838
21. Hahn S, Ugurel S, Hanschmann KM, Strobel H, Tondera C, Schadendorf D, et al. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res Hum Retroviruses (2008) 4:717–23. doi:10.1089/aid.2007.0286
22. Schiavetti F, Thonnard J, Colau D, Boon T, and Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res (2002) 62:5510–6.
23. Mullins CS, and Linnebacher M. Endogenous retrovirus sequences as a novel class of tumor-specific antigens: an example of HERV-H env encoding strong CTL epitopes. Cancer Immunol Immunother (2012) 61:1093–100. doi:10.1007/s00262-011-1183-3
24. Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest (2008) 118:1099–109. doi:10.1172/JCI34409
25. Wang-Johanning F, Rycaj K, Plummer JB, Li M, Yin B, Frerich K, et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst (2012) 104:189–210. doi:10.1093/jnci/djr540
26. Sacha JB, Kim IJ, Chen L, Ullah JH, Goodwin DA, Simmons HA, et al. Vaccination with cancer- and HIV infection-associated endogenous retrotransposable elements is safe and immunogenic. J Immunol (2012) 189:1467–79. doi:10.4049/jimmunol.1200079
27. Peggs KS, Hunter A, Chopra R, Parker A, Mahendra P, Milligan D, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet (2005) 365:1934–41. doi:10.1016/S0140-6736(05)66659-7
28. Staege MS, Hattenhorst UE, Neumann UE, Hutter C, Foja S, and Burdach S. DNA-microarrays as tools for the identification of tumor specific gene expression profiles: applications in tumor biology, diagnosis and therapy. Klin Padiatr (2003) 215:135–9. doi:10.1055/s-2003-39371
29. Drexler HG, Gaedicke G, Lok MS, Diehl V, and Minowada J. Hodgkin’s disease derived cell lines HDLM-2 and L-428: comparison of morphology, immunological and isoenzyme profiles. Leuk Res (1986) 10:487–500. doi:10.1016/0145-2126(86)90084-6
30. Kamesaki H, Fukuhara S, Tatsumi E, Uchino H, Yamabe H, Miwa H, et al. Cytochemical, immunologic, chromosomal, and molecular genetic analysis of a novel cell line derived from Hodgkin’s disease. Blood (1986) 68:285–92.
31. Wolf J, Kapp U, Bohlen H, Kornacker M, Schoch C, Stahl B, et al. Peripheral blood mononuclear cells of a patient with advanced Hodgkin’s lymphoma give rise to permanently growing Hodgkin-Reed Sternberg cells. Blood (1996) 87:3418–28.
32. Schaadt M, Fonatsch C, Kirchner H, and Diehl V. Establishment of a malignant, Epstein-Barr-virus (EBV)-negative cell-line from the pleura effusion of a patient with Hodgkin’s disease. Blut (1979) 38:185–90. doi:10.1007/BF01007965
33. Diehl V, Kirchner HH, Burrichter H, Stein H, Fonatsch C, Gerdes J, et al. Characteristics of Hodgkin’s disease-derived cell lines. Cancer Treat Rep (1982) 66:615–32.
34. Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, et al. Control of cell growth by c-Myc in the absence of cell division. Curr Biol (1999) 9:1255–8. doi:10.1016/S0960-9822(99)80507-7
35. Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, et al. Cell cycle activation by c-myc in a Burkitt lymphoma model cell line. Int J Cancer (2000) 87:787–93. doi:10.1002/1097-0215(20000915)87:6<787::AID-IJC4>3.0.CO;2-6
36. Pajic A, Staege MS, Dudziak D, Schuhmacher M, Spitkovsky D, Eissner G, et al. Antagonistic effects of c-myc and Epstein-Barr virus latent genes on the phenotype of human B cells. Int J Cancer (2001) 93:810–6. doi:10.1002/ijc.1404
37. Kewitz S, Bernig T, and Staege MS. Histone deacetylase inhibition restores cisplatin sensitivity of Hodgkin’s lymphoma cells. Leuk Res (2012) 36:773–8. doi:10.1016/j.leukres.2012.02.021
38. Livak KJ, and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) method. Methods (2001) 25:402–8. doi:10.1006/meth.2001.1262
39. Kewitz S, Müller J, Winkler C, and Staege MS. Retinoic acid signaling and immunotherapy of cancer. In: LH Cheng and Y Ito editors. Retinoic Acid: Structure, Metabolism and Roles in Disease. Hauppauge: Nova Science Publishers (2012). p. 87–104.
40. Shehadeh LA, Yu K, Wang L, Guevara A, Singer C, Vance J, et al. SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson’s disease. PLoS ONE (2010) 5:e9104. doi:10.1371/journal.pone.0009104
41. Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE (2011) 6:e26057. doi:10.1371/journal.pone.0026057
42. Grigoryev YA, Kurian SM, Nakorchevskiy AA, Burke JP, Campbell D, Head SR, et al. Genome-wide analysis of immune activation in human T and B cells reveals distinct classes of alternatively spliced genes. PLoS ONE (2009) 4:e7906. doi:10.1371/journal.pone.0007906
43. Price AM, Tourigny JP, Forte E, Salinas RE, Dave SS, and Luftig MA. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-κB activation. J Virol (2012) 86:11096–106. doi:10.1128/JVI.01069-12
44. Sturn A, Quackenbush J, and Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics (2002) 18:207–8. doi:10.1093/bioinformatics/18.1.207
45. Abrink M, Larsson E, and Hellman L. Demethylation of ERV3, an endogenous retrovirus regulating the Krüppel-related zinc finger gene H-plk, in several human cell lines arrested during early monocyte development. DNA Cell Biol (1998) 7:27–37. doi:10.1089/dna.1998.17.27
46. Kempkes B, Spitkovsky D, Jansen-Dürr P, Ellwart JW, Kremmer E, Delecluse HJ, et al. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J (1995) 14:88–96.
47. McClain K, and Wilkowski C. Activation of endogenous retroviral sequences in human leukemia. Biochem Biophys Res Commun (1985) 133:945–50. doi:10.1016/0006-291X(85)91227-6
48. Tomita N, Horii A, Doi S, Yokouchi H, Ogawa M, Mori T, et al. Transcription of human endogenous retroviral long terminal repeat (LTR) sequence in a lung cancer cell line. Biochem Biophys Res Commun (1990) 166:1–10. doi:10.1016/0006-291X(90)91904-7
49. Löwer R, Boller K, Hasenmaier B, Korbmacher C, Müller-Lantzsch N, Löwer J, et al. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc Natl Acad Sci U S A (1993) 90:4480–4. doi:10.1073/pnas.90.10.4480
50. Patience C, Simpson GR, Colletta AA, Welch HM, Weiss RA, and Boyd MT. Human endogenous retrovirus expression and reverse transcriptase activity in the T47D mammary carcinoma cell line. J Virol (1996) 70:2654–7.
51. Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR, et al. Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin Cancer Res (2001) 7:1553–60.
52. Balada E, Vilardell-Tarrés M, and Ordi-Ros J. Implication of human endogenous retroviruses in the development of autoimmune diseases. Int Rev Immunol (2010) 29:351–70. doi:10.3109/08830185.2010.485333
53. Antony JM, Deslauriers AM, Bhat RK, Ellestad KK, and Power C. Human endogenous retroviruses and multiple sclerosis: innocent bystanders or disease determinants? Biochim Biophys Acta (2011) 812:162–76. doi:10.1016/j.bbadis.2010.07.016
54. Douville R, Liu J, Rothstein J, and Nath A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol (2011) 69:141–51. doi:10.1002/ana.22149
55. Liu AY, and Abraham BA. Subtractive cloning of a hybrid human endogenous retrovirus and calbindin gene in the prostate cell line PC3. Cancer Res (1991) 51:4107–10.
56. Feuchter-Murthy AE, Freeman JD, and Mager DL. Splicing of a human endogenous retrovirus to a novel phospholipase A2 related gene. Nucleic Acids Res (1993) 21:135–43. doi:10.1093/nar/21.1.135
57. Landry JR, Rouhi A, Medstrand P, and Mager DL. The Opitz syndrome gene Mid1 is transcribed from a human endogenous retroviral promoter. Mol Biol Evol (2002) 19:1934–42. doi:10.1093/oxfordjournals.molbev.a004017
58. Kowalski PE, and Mager DL. A human endogenous retrovirus suppresses translation of an associated fusion transcript, PLA2L. J Virol (1998) 72:6164–8.
59. Gosenca D, Gabriel U, Steidler A, Mayer J, Diem O, Erben P, et al. HERV-E-mediated modulation of PLA2G4A transcription in urothelial carcinoma. PLoS ONE (2012) 7:e49341. doi:10.1371/journal.pone.0049341
60. Denne M, Sauter M, Armbruester V, Licht JD, Roemer K, and Mueller-Lantzsch N. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J Virol (2007) 81:5607–16. doi:10.1128/JVI.02771-06
61. Matsuda T, Sasaki M, Kato H, Yamada H, Cohen M, Barrett JC, et al. Human chromosome 7 carries a putative tumor suppressor gene(s) involved in choriocarcinoma. Oncogene (1997) 15:2773–81. doi:10.1038/sj.onc.1201461
62. Cohen M, Kato N, and Larsson E. ERV3 human endogenous provirus mRNAs are expressed in normal and malignant tissues and cells, but not in choriocarcinoma tumor cells. J Cell Biochem (1988) 36:121–8. doi:10.1002/jcb.240360203
63. Lin L, Xu B, and Rote NS. The cellular mechanism by which the human endogenous retrovirus ERV-3 env gene affects proliferation and differentiation in a human placental trophoblast model, BeWo. Placenta (2000) 21:73–8. doi:10.1053/plac.1999.0443
64. Larsson E, Venables P, Andersson AC, Fan W, Rigby S, Botling J, et al. Tissue and differentiation specific expression on the endogenous retrovirus ERV3 (HERV-R) in normal human tissues and during induced monocytic differentiation in the U-937 cell line. Leukemia (1997) 11(Suppl 3):142–4.
65. Andersson AC, Yun Z, Sperber GO, Larsson E, and Blomberg J. ERV3 and related sequences in humans: structure and RNA expression. J Virol (2005) 79:9270–84. doi:10.1128/JVI.79.14.9270-9284.2005
66. Hervé CA, Forrest G, Löwer R, Griffiths DJ, and Venables PJ. Conservation and loss of the ERV3 open reading frame in primates. Genomics (2004) 83:940–3. doi:10.1016/j.ygeno.2003.10.003
67. de Parseval N, and Heidmann T. Physiological knockout of the envelope gene of the single-copy ERV-3 human endogenous retrovirus in a fraction of the Caucasian population. J Virol (1998) 72:3442–5.
68. Kato N, Shimotohno K, VanLeeuwen D, and Cohen M. Human proviral mRNAs down regulated in choriocarcinoma encode a zinc finger protein related to Krüppel. Mol Cell Biol (1990) 10:4401–5.
69. Rasmussen HB, Geny C, Deforges L, Perron H, Tourtelotte W, Heltberg A, et al. Expression of endogenous retroviruses in blood mononuclear cells and brain tissue from multiple sclerosis patients. Mult Scler (1995) 1:82–7.
70. Bae S, Jeong HJ, Cha HJ, Kim K, Choi YM, An IS, et al. The hypoxia-mimetic agent cobalt chloride induces cell cycle arrest and alters gene expression in U266 multiple myeloma cells. Int J Mol Med (2012) 30:1180–6. doi:10.3892/ijmm.2012.1115
71. Guo M, Song LP, Jiang Y, Liu W, Yu Y, and Chen GQ. Hypoxia-mimetic agents desferrioxamine and cobalt chloride induce leukemic cell apoptosis through different hypoxia-inducible factor-1alpha independent mechanisms. Apoptosis (2006) 11:67–77. doi:10.1007/s10495-005-3085-3
72. Zeno S, Zaaroor M, Leschiner S, Veenman L, and Gavish M. CoCl(2) induces apoptosis via the 18 kDa translocator protein in U118MG human glioblastoma cells. Biochemistry (2009) 48:4652–61. doi:10.1021/bi900064t
73. Kim HJ, Yang SJ, Kim YS, and Kim TU. Cobalt chloride-induced apoptosis and extracellular signal-regulated protein kinase activation in human cervical cancer HeLa cells. J Biochem Mol Biol (2003) 36:468–74. doi:10.5483/BMBRep.2003.36.5.468
74. Buglio D, Georgakis GV, Hanabuchi S, Arima K, Khaskhely NM, Liu YJ, et al. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood (2008) 112:1424–33. doi:10.1182/blood-2008-01-133769
75. Hartlapp I, Pallasch C, Weibert G, Kemkers A, Hummel M, and Re D. Depsipeptide induces cell death in Hodgkin lymphoma-derived cell lines. Leuk Res (2009) 33:929–36. doi:10.1016/j.leukres.2008.12.013
Keywords: Hodgkin’s lymphoma, gene expression, endogenous retrovirus ERV3, histone deacetylase inhibitor, hypoxia-mimetic cobalt(II) chloride
Citation: Kewitz S and Staege MS (2013) Expression and regulation of the endogenous retrovirus 3 in Hodgkin’s lymphoma cells. Front. Oncol. 3:179. doi: 10.3389/fonc.2013.00179
Received: 27 March 2013; Accepted: 25 June 2013;
Published online: 10 July 2013.
Edited by:
Stephen Lessnick, University of Utah, USACopyright: © 2013 Kewitz and Staege. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Martin Sebastian Staege, Department of Pediatrics, Martin-Luther-University Halle-Wittenberg, Ernst-Grube-Str. 40, 06097 Halle, Germany e-mail:bWFydGluLnN0YWVnZUBtZWRpemluLnVuaS1oYWxsZS5kZQ==