 Nikki A. Ford1
Nikki A. Ford1

- 1Department of Nutritional Sciences, University of Texas, Austin, TX, USA
- 2Department of Urologic Surgery, Duke University Medical Center, Durham, NC, USA
- 3Department of Molecular Carcinogenesis, University of Texas-MD Anderson Cancer Center, Smithville, TX, USA
The prevalence of obesity, an established risk and progression factor for many cancers, has increased dramatically in many countries over the past three decades. Worldwide, an estimated 600 million adults are currently obese. Thus, a better understanding of the mechanistic links between obesity and cancer is urgently needed to identify intervention targets and strategies to offset the procancer effects of obesity. This review synthesizes the evidence on key biological mechanisms underlying the obesity-cancer association, with particular emphasis on obesity-associated enhancements in growth factor signaling, inflammation, and perturbations in the tumor microenvironment. These interrelated pathways and processes that are aberrantly regulated in obese individuals represent mechanism-based targets for disrupting the obesity-cancer link using phytochemicals.
Introduction
An estimated 600 million adults throughout the world are obese, and the majority of these obese individuals meet the criteria for the metabolic syndrome, a state of metabolic dysregulation characterized by increased waist circumference, elevated fasting glucose levels, hypertension, and hypertriglyceridemia (1, 2). Increased circulating levels of insulin, bioavailable insulin-like growth factor (IGF)-1, leptin, inflammatory factors, and vascular integrity-related factors such as vascular endothelial growth factor (VEGF) and plasminogen activator inhibitor (PAI)-1, are typically observed in obese individuals (3–6). Through these, and likely other, mediators and their interacting pathways and processes, obesity increases the risk and/or worsens the outcome of several chronic diseases (3, 5) including cardiovascular disease, type II diabetes, and the focus of this review, cancer.
Obesity prevention or reversal is a major part of several evidence-based cancer prevention guidelines (7). Approximately 25% of cancer deaths in the US and United Kingdom are attributed to being overweight and obese (8), with the strongest evidence for endometrial, postmenopausal breast, colon, renal cell carcinoma, liver, gallbladder, esophageal adenocarcinoma, and pancreatic cancer, and mounting evidence for cervical, ovarian, prostate (prognosis, but not overall risk), and stomach cancer (7). This review focuses on possible mechanisms underlying the associations between obesity and cancer, with emphasis on obesity-associated enhancements in growth factor signaling, inflammatory processes, vascular perturbations, and microenvironmental disruptions, all linked with increased cancer susceptibility and poor prognosis. We will also discuss the potential for using phytochemicals known to modulate one or more of these energy balance-responsive pathways to contribute toward prevention or control of obesity-related cancers.
Growth Signal Dysregulation
Insulin and IGF-1, and Their Downstream Signals
Hyperinsulinemia and/or hyperglycemia are hallmarks of the obese state and are associated with insulin resistance, aberrant glucose metabolism, chronic inflammation, and the production of other metabolic hormones such as IGF-1, leptin, and adiponectin (9). Insulin is a peptide hormone produced by the beta cells of the pancreas and released in response to increased blood glucose. IGF-1 is a peptide growth factor that shares ∼50% sequence homology with insulin and is produced primarily by the liver following stimulation by growth hormone, although hyperinsulinemia and hyperglycemia can lead to increased hepatic IGF-1 production independent of growth hormone signaling. Circulating IGF-1 is typically bound to IGF binding proteins (IGFBPs) that regulate IGF-1 bioavailability and modulate growth and survival signals either directly or by repressing the IGF-1R (10). With obesity, the amount of bioavailable IGF-1 increases, possibly via hyperglycemia-induced suppression of IGFBP synthesis and/or hyperinsulinemia-induced promotion of hepatic growth hormone receptor expression and IGF-1 synthesis (10, 11). Elevated circulating IGF-1 is an established risk factor for many obesity-associated cancer types (11).
In contrast, the enhanced insulin sensitivity and normalized glucose levels in response to a calorie restriction (CR) regimen, relative to a control or diet-induced obesity (DIO) regimen, results in lowered serum insulin and IGF-1, and increased IGFBP production, particularly IGFBP1 and 3 (and hence low levels of bioavailable IGF-1) (11). The CR-induced reduction in glucose may also have direct anticancer effects. In cancer cells, mitochondrial metabolism of glucose is reprogrammed to meet the demands of macromolecular synthesis required for cellular proliferation. This metabolic switch of glucose metabolism from oxidative phosphorylation to oxidative glycolysis (called the Warburg effect) is now understood to be necessary to supply sufficient nucleotides, lipids, and proteins for daughter cell production (10). Cancer cells do this, however, at the expense of substrate inflexibility relative to normal cells, as the increased proliferation rate associated with most cancer cells can only be sustained by a constant supply of the necessary building blocks derived from the flux of glucose carbons through glycolysis. Thus, it is possible that precancerous or cancer cells undergoing this metabolic reprogram, and hence developing a glucose addiction, may have heightened sensitivity to alterations in glucose levels, as occurs with obesity and CR.
The phospatidylinositol-3-kinase (PI3K)/Akt pathway, downstream of the insulin receptor and IGF-1R, comprises a signaling network that regulates and integrates cellular growth, survival, and metabolism. Cantley and colleagues (12) established that this signaling cascade is one of the most commonly altered pathways in human epithelial tumors. Engagement of the PI3K/Akt pathway results in production of both intracellular and extracellular cues concerning substrate availability, growth factor supply, and levels of other factors that impact cell survival, growth, proliferation, and metabolism. Activation of receptor tyrosine kinases, such as the insulin receptor or IGF-1R, stimulates PI3K to produce lipid messengers that facilitate activation of the Akt cascade. Akt regulates the mammalian target of rapamycin (mTOR) (13), thus regulating cell growth, proliferation, and survival through downstream mediators. mTOR activation is inhibited by increased AMP-activated kinase (AMPK) under low nutrient conditions (14, 15). Increased activation of mTOR is common in tumors and many normal tissues from obese and/or diabetic mice, and specific mTOR inhibitors block the tumor-enhancing effects of obesity in mouse models (15–17).
Leptin, Adiponectin, and Their Ratio
Leptin, a peptide hormone produced predominantly by adipocytes, functions as an energy sensor to signal to the hypothalamus to reduce appetite. Insulin, glucocorticoids, tumor necrosis factor-alpha (TNF-α), and estrogens all stimulate leptin release (18). In the obese state, adipose tissue overproduces leptin, and the brain eventually becomes resistant to this satiety signal. In addition to its role in regulating appetite, leptin has direct pro-tumorigenic effects on peripheral tissues, as well as a role in modulating immune function, cytokine production, angiogenesis, carcinogenesis, and other biological processes (18). The leptin receptor has similar homology to class I cytokines that signal through the janus kinase and signal transducer activator of transcription (JAK/STAT) pathway that is often dysregulated in cancer (19).
Although adiponectin is a hormone mainly secreted from visceral adipose tissue, its serum levels, in contrast with serum leptin, negatively correlate with adiposity. Adiponectin functions to counter the metabolic program associated with obesity and hyperleptinemia by modulating glucose metabolism, increasing fatty acid oxidation and insulin sensitivity, and decreasing production of inflammatory cytokines (20). The possible mechanisms through which adiponectin exerts anticancer effects may include increasing insulin sensitivity, and decreasing insulin/IGF-1 and mTOR signaling via activation of AMPK. Adiponectin also reduces pro-inflammatory cytokine expression via inhibition of the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) (20–22).
In vitro, animal and epidemiologic evidence linking leptin (21, 23–26) or adiponectin (21, 27–31) individually to cancer risk is mixed. Associations between the adiponectin-to-leptin ratio and the metabolic syndrome and several cancers (32–34) have also been reported, but there is insufficient data thus far to assess the strength of this relationship.
Chronic Inflammation
Cytokines and Crosstalk between Epithelial Cells, Adipocytes, and Macrophages
Obesity and metabolic syndrome are associated with a low-grade, chronic state of inflammation characterized by increased circulating free fatty acids and chemoattraction of immune cells (such as macrophages that also produce inflammatory mediators) into the local milieu of expanded adipose tissue (35–37). These effects are further amplified by the release of inflammatory cytokines such as interleukin (IL)-1β, IL-6, TNF-α, and monocyte chemoattractant protein (MCP)-1. Hypertrophic adipocytes can enlarge past the point of effective oxygen diffusion, which results in hypoxia and eventually necrosis leading to further infiltration of scavenging macrophages and formation of crown-like structures. Free fatty acids escape the engorged/necrotic adipocytes and deposit in other tissues, which in turn promotes insulin resistance and diabetes (through downregulation of insulin receptors and glucose transporters), hypertension, and fatty liver disease and also activates signaling molecules involved in epithelial carcinogenesis, such as NF-κB (35–37).
NF-κB is a transcription factor that is activated in response to bacterial and viral stimuli, growth factors, and inflammatory molecules (e.g., TNF-α, IL-6, and IL-1β), and is responsible for inducing gene expression associated with cell proliferation, apoptosis, inflammation, metastasis, and angiogenesis (35–37). Activation of NF-κB is a common characteristic of many tumors and is associated with insulin resistance and elevated circulating levels of leptin, insulin, and/or IGF-1 (37–40).
Inflammation and Cancer
The link between chronic inflammation and cancer development was first reported more than 100 years ago by Rudolph Virchow, who observed an abundance of leukocytes in neoplastic tissue (41). Now, several tissue-specific inflammatory lesions are established neoplastic precursors for invasive cancer, including gastritis for gastric cancer, inflammatory bowel disease for colon cancer, and pancreatitis for pancreatic cancer (42, 43).
Tumor microenvironments are composed of multiple cell types including epithelial cells, fibroblasts, mast cells, and cells of the innate and adaptive immune system (43, 44). As discussed previously, macrophages, which are classically activated in the obese state, infiltrate tumors and amplify the inflammatory tumor microenvironment through production of pro-inflammatory cytokines, prostaglandins, and angiogenic factors (37, 44). Another important cancer-related inflammatory mediator is cyclooxygenase (COX)-2, an enzyme that is upregulated in most tumors and catalyzes the synthesis of the potent inflammatory lipid metabolite, prostaglandin E2. COX-2 overexpression is an indicator of poor prognosis in multiple cancer types (45).
Vascular Integrity-Related Factors
Plasminogen Activator Inhibitor-1
Plasminogen activator inhibitor-1 is a serine protease inhibitor produced by endothelial cells, stromal cells, and adipocytes in visceral white adipose tissue (46). Increased circulating PAI-1 levels, frequently found in obese subjects, are associated with increased risk of atherogenesis and cardiovascular disease, diabetes, and several cancers (4, 46). PAI-1, through its inhibition of urokinase-type and tissue-type plasminogen activators, regulates fibrinolysis and integrity of the extracellular matrix (ECM). Furthermore, PAI-1 can modulate cell adhesion through decreasing cell binding to the ECM protein, vitronectin, thus promoting tumor cell detachment from the ECM (46). PAI-1 is also involved in angiogenesis and thus may contribute to obesity-driven tumor cell growth, invasion, and metastasis (4).
Vascular Endothelial Growth Factor
Vascular endothelial growth factor, a heparin-binding glycoprotein produced by adipocytes and tumor cells, has angiogenic, mitogenic, and vascular permeability-enhancing activities specific for endothelial cells (47). Circulating levels of VEGF are increased in obese, relative to lean humans and animals, and increased tumoral expression of VEGF is associated with poor prognosis in several obesity-related cancers (48). The need for nutrients and oxygen triggers tumor cells to produce VEGF, which leads to the formation of new blood vessels to nourish the rapidly growing tumor and facilitate the metastatic spread of tumor cells (49).
Adipocytes communicate with endothelial cells by producing a variety of proangiogenic and vascular permeability-enhancing factors. These include VEGF, IGF-1, PAI-1, leptin, hepatocyte growth factor, and fibroblast growth factor-2 (49). In the obese, non-tumor setting, these factors stimulate neovascularization in support of the expanding fat mass. These adipose-derived factors may also contribute to obesity-associated enhancement of tumor angiogenesis. However, the relative contributions of tumor-derived, versus adipocyte-derived, proangiogenic factors in tumor development, progression, and metastasis remain unclear.
Phytochemical Modulation of Obesity-Cancer Links
Since the biological effects of a broad spectrum of phytochemicals have been reviewed in other articles in this special issue, we will focus here on some specific examples of phytochemicals that target the key pathways underlying obesity-cancer associations.
Resveratrol
Resveratrol, chemically known as 3,5,4′-trihydroxystilbene, is a naturally occurring polyphenolic compound present in grapes, berries, peanuts, and red wine. It is believed to be responsible for the so called “French paradox,” in which the consumption of red wine has been shown to reduce the mortality rates from cardiovascular disease and certain cancers. Resveratrol is an established anti-inflammatory agent that inhibits initiation and growth of numerous cancer types including breast, prostate, colon, and liver (50). Recent clinical studies demonstrate resveratrol improves glycemic control in diabetic patients (51) and reduces inflammatory signaling through TNF-α, IL-6, C-reactive protein, and NFκB pathways (52). Furthermore, at physiologic concentrations resveratrol (possibly by activating the sirtuin pathway) counteracts activation of tumoral COX-2 and NFκB (53). Recently published reports suggest that resveratrol mimics some of the effects of CR on lifespan in worms and other model organisms, apparently by inhibiting inflammation and the Akt/mTOR pathway (54, 55).
Ursolic Acid
Ursolic acid is a triterpenoid found in fairly high levels in rosemary and in smaller amounts in apples and other fruits and vegetables. Ursolic acid has been shown to inhibit carcinogen and tumor promoter-induced inflammation, hyperplasia, and tumor formation in multiple models (56–59). Ursolic acid is a very potent anti-inflammatory and insulin sensitizing agent with reported activities against the effects of obesity, such as the ability to counteract NF-κB, COX-2, and Akt activity (53, 59). Furthermore, we reported that a diet containing 0.1% (w/w) ursolic acid suppressed MMTV-Wnt-1 murine mammary tumor growth, decreased insulin and IGF-1, and decreased activation of the mTOR pathway (59). Thus, like resveratrol, ursolic acid is thought to mimic some of the effects of CR. Ursolic acid was also shown to inhibit tumor-associated capillary formation in mice through inhibition of VEGF and other inflammatory growth factors (60).
Curcumin
Curcumin, also known as diferuloylmethane, is an anti-inflammatory agent that gives yellow color to turmeric used in curry powder. Curcumin has been shown to have chemopreventative properties across several cancer types (61). These anticancer effects are modulated through suppression of several inflammatory and growth signaling pathways including NFκB, Stat3, COX-2, Akt, and mTOR (53, 62). Furthermore, inhibition of COX-2 by curcumin also results in suppression of VEGF-mediated angiogenesis (63). By targeting these pathways, curcumin has been shown to reverse hyperglycemia, hyperlipidemia, and other symptoms related to obesity and therefore reduce the growth and development of obesity-associated cancers (64, 65).
Quercetin
Quercetin is found commonly in citrus fruits, onions, tea, and red wine and possesses significant antioxidant activity, like other polyphenols (curcumin and resveratrol). Quercetin targets many of the key pathways involved in tumor initiation, development, growth, and metastases. Specifically, it can modulate the Akt/mTOR pathway, COX-2 expression, NFκB signaling, TNFα expression, and VEGF expression (53, 66–68). A phase I clinical study in cancer patients showed that quercetin effectively inhibits tyrosine kinases involved in growth, inflammation, and cell signaling (69).
Conclusion
As summarized in Figure 1, multiple hormones, growth factors, cytokines, and other mediators associated with the metabolic perturbations of the obese state enable crosstalk between macrophages, adipocytes, endothelial cells, and epithelial cells. These obesity-associated factors contribute to cancer-related processes (including growth signaling, inflammation, and vascular alterations). Components of these interrelated processes and pathways represent promising mechanism-based targets for phytochemical interventions, with the goal of breaking the links between obesity (and its metabolic dysregulation) and cancer.
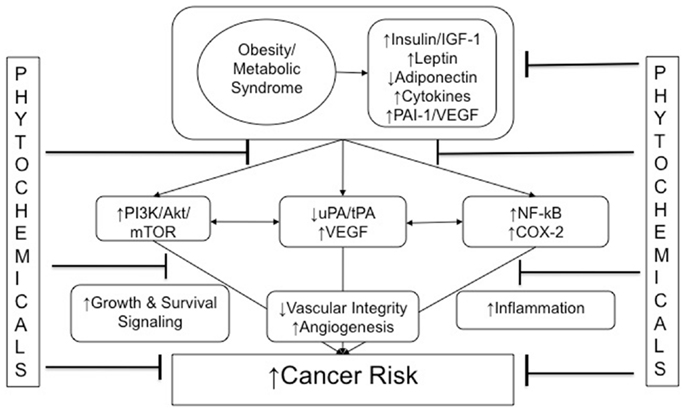
Figure 1. Obesity and cancer: mechanistic targets for phytochemical interventions. An arrow preceding text denotes a directional effect (e.g., activity or concentration). Abbreviations: IGF-1, insulin-like growth factor-1; ApN, adiponectin; PAI-1, plasminogen activator inhibitor-1; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator; VEGF, vascular endothelial growth factor; PI3K, phosphoinositide-3-kinase; NF-κB, nuclear factor κB; COX-2, cyclooxygenase-2; EMT, epithelial-to-mesenchymal transition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Stephen D. Hursting is funded, in part, by grants from the National Cancer Institute (R01CA129409), the Breast Cancer Research Foundation (UTA09-001068). Dr. Nikki A. Ford is funded by an American Institute for Cancer Research Postdoctoral Fellowship, and Dr. Emma H. Allott is funded by an R25 Fellowship from the National Cancer Institute.
References
1. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA (2010) 303:235–41. doi:10.1001/jama.2009.2014
2. Ervin RB. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race, and ethnicity, and body mass index: United States, 2003-2006. Natl Health Stat Report (2009) 13:1–7.
3. Hursting SD, Berger NA. Energy balance, host-related factors, and cancer progression. J Clin Oncol (2010) 28:4058–65. doi:10.1200/JCO.2010.27.9935
4. Carter JC, Church FC. Obesity and breast cancer: the roles of peroxisome proliferator-activated receptor-gamma and plasminogen activator inhibitor-1. PPAR Res (2009) 2009:345320.
5. Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol (2006) 26:968–76. doi:10.1161/01.ATV.0000216787.85457.f3
6. Dunlap SM, Chiao LJ, Nogueira L, Usary J, Perou CM, Varticovski L, et al. Obesity enhances epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prev Res (Phila) (2012) 5:930–42. doi:10.1158/1940-6207.CAPR-12-0034
7. World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity and the Prevention of Cancer: A Global Perspective. Washington, DC: AICR (2007).
8. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospective studied cohort of U.S. adults. N Engl J Med (2003) 348:1625–38. doi:10.1056/NEJMoa021423
9. Braun S, Bitton-Worms K, LeRoith D. The link between the metabolic syndrome and cancer. Int J Biol Sci (2011) 7:1003–15. doi:10.7150/ijbs.7.1003
10. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer (2012) 12:159–69. doi:10.1038/nrc3215
11. Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis (2010) 31:83–9. doi:10.1093/carcin/bgp280
12. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev (2010) 20:87–90. doi:10.1016/j.gde.2009.11.002
13. Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal (2009) 21:656–64. doi:10.1016/j.cellsig.2009.01.004
14. Lindsley JE, Rutter J. Nutrient sensing and metabolic decisions. Comp Biochem Physiol B Biochem Mol Biol (2004) 139:543–59. doi:10.1016/j.cbpc.2004.06.014
15. Moore T, Beltran LD, Carbajal S, Strom S, Traag J, Hursting SD, et al. Dietary energy balance modulates signaling through the Akt/mammalian target of rapamycin pathway in multiple epithelial tissues. Cancer Prev Res (Phila) (2008) 1:65–76. doi:10.1158/1940-6207.CAPR-08-0022
16. De Angel RE, Conti CJ, Wheatley KE, Brenner AJ, deGraffenried LA, Hursting SD. The enhancing effects of obesity on mammary tumor growth and Akt/mTOR pathway activation persist after weight loss and are reversed by Rad001. Mol Carcinog (2012) 52:446–58. doi:10.1002/mc.21878
17. Nogueira LM, Dunlap SM, Ford NA, Hursting SD. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr Relat Cancer (2012) 19:57–68. doi:10.1530/ERC-11-0213
18. Gautron L, Elmquist JK. Sixteen years and counting: an update on leptin in energy balance. J Clin Invest (2011) 121:2087–93. doi:10.1172/JCI45888
19. Villanueva EC, Myers MG. Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (2008) 32:S8–12. doi:10.1038/ijo.2008.232
20. Vaiopoulos AG, Marinou K, Christodoulides C, Koutsilieris M. The role of adiponectin in human vascular physiology. Int J Cardiol (2012) 155:188–93. doi:10.1016/j.ijcard.2011.07.047
21. Barb D, Williams CJ, Neuwirth AK, Mantzoros CS. Adiponectin in relation to malignancies: a review of existing basic research and clinical evidence. Am J Clin Nutr (2007) 86:s858–66.
22. Stofkova A. Leptin and adiponectin: from energy and metabolic dysbalance to inflammation and autoimmunity. Endocr Regul (2009) 43:157–68.
23. Stattin P, Lukanova A, Biessy C, Soderberg S, Palmqvist R, Kaaks R, et al. Obesity and colon cancer: does leptin provide a link? Int J Cancer (2004) 109:149–52. doi:10.1002/ijc.11668
24. Chang S, Hursting SD, Contois JH, Strom SS, Yamamura Y, Babaian RJ, et al. Leptin and prostate cancer. Prostate (2001) 46:62–7. doi:10.1002/1097-0045(200101)46:1<62::AID-PROS1009>3.0.CO;2-V
25. Wu MH, Chou YC, Chou WY, Hsu GC, Chu CH, Yu CP, et al. Circulating levels of leptin, adiposity and breast cancer risk. Br J Cancer (2009) 100:578–82. doi:10.1038/sj.bjc.6604913
26. Fenton JI, Hord NG, Lavigne JA, Perkins SN, Hursting SD. Leptin, insulin-like growth factor-1, and insulin-like growth factor-2 are mitogens in ApcMin/+ but not Apc+/+ colonic epithelial cells. Cancer Epidemiol Biomarkers Prev (2005) 14:1646–52. doi:10.1158/1055-9965.EPI-04-0916
27. Grossmann ME, Nkhata KJ, Mizuno NK, Ray A, Cleary MP. Effects of adiponectin on breast cancer cell growth and signaling. Br J Cancer (2008) 98:370–9. doi:10.1038/sj.bjc.6604166
28. Rzepka-Gorska I, Bedner R, Cymbaluk-Ploska A, Chudecka-Glaz A. Serum adiponectin in relation to endometrial cancer and endometrial hyperplasia with atypia in obese women. Eur J Gynaecol Oncol (2008) 29:594–7.
29. Tian YF, Chu CH, Wh MH, Chang CL, Yang T, Chou YC, et al. Anthropometric measures, plasma adiponectin, and breast cancer risk. Endocr Relat Cancer (2007) 14:669–77. doi:10.1677/ERC-06-0089
30. Wei EK, Giovannucci E, Fuch CS, Willett WC, Mantzoros CS. Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. J Natl Cancer Inst (2005) 97:1688–94. doi:10.1093/jnci/dji376
31. Sher DJ, Oh WH, Jacobus S, Regan MM, Lee GS, Mantzoros C. Relationship between serum adiponectin and prostate cancer grade. Prostate (2008) 68:1592–8. doi:10.1002/pros.20823
32. Cleary MP, Ray A, Rogozina OP, Dogan S, Grossman ME. Targeting the adiponectin:leptin ratio for postmenopausal breast cancer prevention. Front Biosci (2009) 1:329–57. doi:10.2741/e30
33. Li H, Stampfer MJ, Mucci L, Rifai N, Qui W, Kurth T, et al. A 25-year prospective study of plasma adiponectin and leptin concentrations and prostate cancer risk and survival. Clin Chem (2010) 56:34–43. doi:10.1373/clinchem.2009.133272
34. Yamaji T, Iwasaki M, Sasazuki S, Tsugane S. Interaction between adiponectin and leptin influences the risk of colorectal adenoma. Cancer Res (2010) 70:5430–7. doi:10.1158/0008-5472.CAN-10-0178
35. Gupta SC, Kim JH, Kannappan R, Reuter S, Doughtery PM, Aggarwal BB. Role of nuclear factorkB-mediated inflammatory pathways in cancer-related symptoms and their regulation by nutritional agents. Exp Biol Med (2012) 236:658–71.
36. Subbaramaiah K, Howe LR, Bhardway P, Du B, Gravaghi C, Yantiss RK, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila) (2011) 4:329–46. doi:10.1158/1940-6207.CAPR-10-0381
37. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
38. O’Rourke RW. Inflammation in obesity-related diseases. Surgery (2009) 145:255–9. doi:10.1016/j.surg.2008.08.038
39. Renehan AG, Roberts DL, Dive C. Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem (2008) 114:71–83. doi:10.1080/13813450801954303
40. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature (2006) 441:431–6. doi:10.1038/nature04870
41. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
42. Foltz CJ, Fox JG, Cahill R, Murphy JC, Yan L, Shames B, et al. Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter (1998) 3:69–78. doi:10.1046/j.1523-5378.1998.08006.x
44. Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev (2008) 222:155–61. doi:10.1111/j.1600-065X.2008.00607.x
45. Koki A, Khan NK, Woerner BM, Dannenberg AJ, Olson L, Seibert K, et al. Cyclooxygenase-2 in human pathological disease. Adv Exp Med Biol (2002) 507:177–84. doi:10.1007/978-1-4615-0193-0_28
46. Iwaki T, Urano T, Umemura K. PAI-1, progress in understanding the clinical problem and its aetiology. Br J Haematol (2012) 157:291–8. doi:10.1111/j.1365-2141.2012.09074.x
47. Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J Cell Mol Med (2005) 9:777–94. doi:10.1111/j.1582-4934.2005.tb00379.x
48. Liu Y, Tamimi RM, Collins LC, Schnitt SJ, Gilmore HL, Connolly JL, et al. The association between vascular endothelial growth factor expression in invasive breast cancer and survival varies with intrinsic subtypes and use of adjuvant systemic therapy: results from the Nurses’ Health Study. Breast Cancer Res Treat (2011) 129:175–84. doi:10.1007/s10549-011-1432-3
49. Cao Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest (2007) 117:2362–8. doi:10.1172/JCI32239
50. Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res (2004) 5A:2783–840.
51. Bhatt JK, Thomas S, Nanjan MJ. Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr Res (2012) 32:537–41. doi:10.1016/j.nutres.2012.06.003
52. Ghanim H, Sia CL, Abuaysheh S, Korzeniewski K, Patnaik P, Marumganti A, et al. An antiinflammatory and reactive oxygen species suppressive effects of an extract of Polygonum cuspidatum containing resveratrol. J Clin Endocrinol Metab (2010) 95:E1–8. doi:10.1210/jc.2010-0482
53. Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol (2006) 71:1397–421. doi:10.1016/j.bcp.2006.02.009
54. Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy (2010) 6:186–8. doi:10.4161/auto.6.1.10817
55. Lanzilli G, Fuggetta MP, Tricarico M, Cottarelli A, Serafino A, Falchetti R, et al. Resveratrol down-regulates the growth and telomerase activity of breast cancer cells in vitro. Int J Oncol (2006) 28:641–8.
56. Liu W, Tan X, Shu L, Sun H, Song J, Jin P, et al. Ursolic acid inhibits cigarette smoke extract-induced human bronchial epithelial cell injury and prevents development of lung cancer. Molecules (2012) 17:9104–15. doi:10.3390/molecules17089104
57. Li J, Liang X, Yang X. Ursolic acid inhibits growth and induces apoptosis in gemcitabine-resistant human pancreatic cancer via the JNK and PI3K/Akt/NF-κB pathways. Oncol Rep (2012) 28:501–10. doi:10.3892/or.2012.1827
58. Prasad S, Yadav VR, Sung B, Reuter S, Kannappan R, Deorukhkar A, et al. Ursolic acid inhibits growth and metastasis of human colorectal cancer in an orthotopic nude mouse model by targeting multiple cell signaling pathways: chemosensitization with capecitabine. Clin Cancer Res (2012) 18:4942–53. doi:10.1158/1078-0432.CCR-11-2805
59. De Angel RE, Smith SM, Glickman RD, Perkins SN, Hursting SD. Antitumor effects of ursolic acid in a mouse model of postmenopausal breast cancer. Nutr Cancer (2010) 62:1074–86. doi:10.1080/01635581.2010.492092
60. Kanjoormana M, Kuttan G. Antiangiogenic activity of ursolic acid. Integr Cancer Ther (2010) 9:224–35. doi:10.1177/1534735410367647
61. Epstein J, Sanderson IR, Macdonald TT. Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr (2010) 103:1545–57. doi:10.1017/S0007114509993667
62. Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem (2010) 10:571–81. doi:10.2174/187152010793498663
63. Binion DG, Otterson MF, Rafiee P. Curcumin inhibits VEGF-mediated angiogenesis in human intestinal microvascular endothelial cells through COX-2 and MAPK inhibition. Gut (2008) 57:1509–17. doi:10.1136/gut.2008.152496
64. Shehzad A, Khan S, Sup Lee Y. Curcumin molecular targets in obesity and obesity-related cancers. Future Oncol (2012) 8:179–90. doi:10.2217/fon.11.145
65. Aggarwal BB. Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu Rev Nutr (2010) 30:173–99. doi:10.1146/annurev.nutr.012809.104755
66. Zhang X, Chen L-X, Ouyang L, Cheng Y, Liu B. Plant natural compounds: targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif (2012) 45:466–76. doi:10.1111/j.1365-2184.2012.00833.x
67. Vargas AJ, Burd R. Hormesis and synergy: pathways and mechanisms of quercetin in cancer prevention and management. Nutr Rev (2010) 68:418–28. doi:10.1111/j.1753-4887.2010.00301.x
Keywords: obesity, metabolism, cancer prevention, phytochemicals, inflammation, mTOR pathway
Citation: Ford NA, Lashinger LM, Allott EH and Hursting SD (2013) Mechanistic targets and phytochemical strategies for breaking the obesity-cancer link. Front. Oncol. 3:209. doi: 10.3389/fonc.2013.00209
Received: 28 May 2013; Paper pending published: 16 July 2013;
Accepted: 30 July 2013; Published online: 19 August 2013.
Edited by:
Zigang Dong, University of Minnesota, USAReviewed by:
Maria Felice Brizzi, University of Torino, ItalyXingzhi Xu, Capital Normal University, China
Copyright: © 2013 Ford, Lashinger, Allott and Hursting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen D. Hursting, Department of Nutritional Sciences, Dell Pediatric Research Institute, University of Texas at Austin, 1400 Barbara Jordan Blvd., Austin, TX 78723, USA e-mail:c2h1cnN0aW5nQGF1c3Rpbi51dGV4YXMuZWR1