


- 1CNRS, UMR8104, Paris, France
- 2INSERM, U1016, Paris, France
- 3Sorbonne Paris Cite, Universite Paris Descartes, Paris, France
The endothelial barrier strictly maintains vascular and tissue homeostasis, and therefore modulates many physiological processes such as angiogenesis, immune responses, and dynamic exchanges throughout organs. Consequently, alteration of this finely tuned function may have devastating consequences for the organism. This is particularly obvious in cancers, where a disorganized and leaky blood vessel network irrigates solid tumors. In this context, vascular permeability drives tumor-induced angiogenesis, blood flow disturbances, inflammatory cell infiltration, and tumor cell extravasation. This can directly restrain the efficacy of conventional therapies by limiting intravenous drug delivery. Indeed, for more effective anti-angiogenic therapies, it is now accepted that not only should excessive angiogenesis be alleviated, but also that the tumor vasculature needs to be normalized. Recovery of normal state vasculature requires diminishing hyperpermeability, increasing pericyte coverage, and restoring the basement membrane, to subsequently reduce hypoxia, and interstitial fluid pressure. In this review, we will introduce how vascular permeability accompanies tumor progression and, as a collateral damage, impacts on efficient drug delivery. The molecular mechanisms involved in tumor-driven vascular permeability will next be detailed, with a particular focus on the main factors produced by tumor cells, especially the emblematic vascular endothelial growth factor. Finally, new perspectives in cancer therapy will be presented, centered on the use of anti-permeability factors and normalization agents.
Vascular Permeability in Cancers
Vascular Barrier Organization
Endothelial cells, pericytes, smooth muscle cells, and the basal membrane collectively form the blood vascular wall, which ensures selective exchanges between plasma and irrigated tissues. The passage of macromolecules, fluids, and cells through this endothelial barrier can occur either through (transcellular) or between cells (paracellular) (1). The ability to pass from the interstitial space to the blood compartment, and vice versa depends on charge, size, and binding characteristics.
Small molecules (inferior to 3 nm) are commonly transported by the transcellular route, which requires a system of trafficking vesicles, called vesicular vacuolar organelles (VVOs) (Figure 1). Several permeability factors, such as vascular endothelial growth factor (VEGF) and histamine have been demonstrated to activate VVOs and to orchestrate vascular homeostasis (2). These VVOs comprise, among other things, clustered caveolae, and rely on caveolin-1 protein function, that notably guarantees albumin transport. Interestingly, caveolin-1 plays a dual regulatory role in microvascular permeability by stabilizing caveolae structures and allowing caveolar transcytosis, while acting as a negative regulator through endothelial nitric oxide synthase (eNOS) (3, 4).
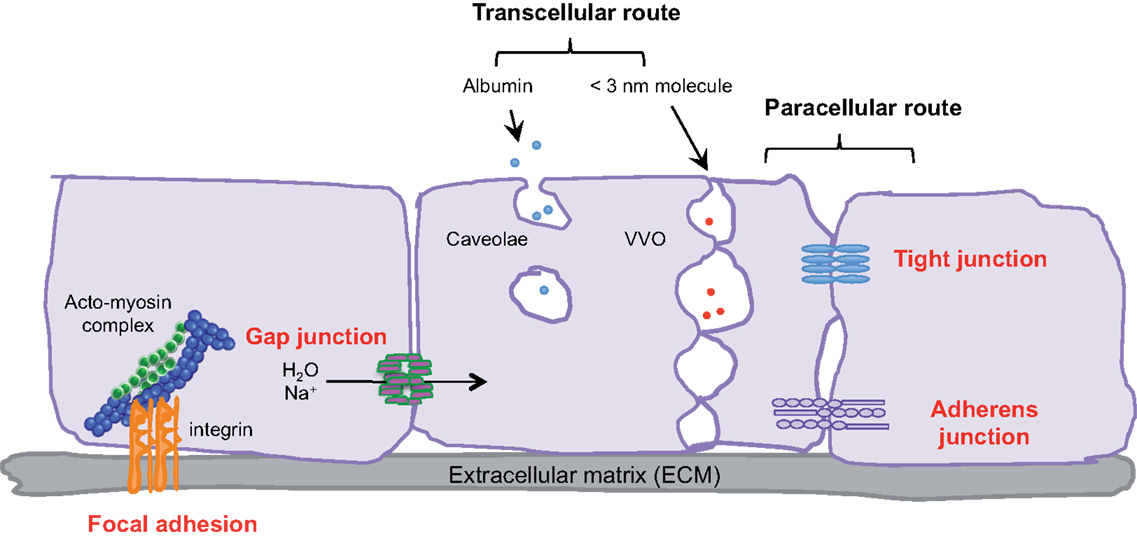
Figure 1. Transcellular and paracellular pathways in endothelial cells. The passage of cells and macromolecules through the endothelial barrier can occur through transcellular (vesicular vacuolar organelles) or paracellular (tight and adherens junctions) pathways. Gap junctions ensure water and ion transport. Moreover, endothelial cells are anchored and connected to the extracellular matrix (ECM) through integrin-based adhesion complexes, namely focal adhesions.
Cells and macromolecules larger than 3 nm use the paracellular pathway, which is mediated by the coordinated opening and closing of endothelial cell–cell junctions. Adherens (AJ) and tight (TJ) junctions maintain the restrictiveness of the barrier, while gap junctions connect adjacent endothelial cells. Gap junctions are responsible for water and ion transport but do not contribute significantly or directly to the establishment of vascular barriers (Figure 1). Among AJ proteins, the most important is vascular endothelial cadherin (VE-cadherin), which is exclusively expressed in vessels (1, 5). In mice, VE-cadherin gene deletion results in early embryonic lethality due to massive vascular defects, while loss of its function provokes a hyperpermeability phenotype in adults (6, 7). VE-cadherin comprises five immunoglobulin-like domain repeats in its extracellular region, one single-pass transmembrane domain and a short cytoplasmic tail. While the extracellular domain confers Ca++ dependency and allows homophilic interaction in trans (i.e., between cadherins on neighboring cells), the transmembrane domain participates in lateral clustering within the same cell (cis) (8). The cytoplasmic part of VE-cadherin binds to proteins from the armadillo-repeat gene family, namely p120-catenin and either β-catenin or plakoglobin (γ-catenin). This complex serves to strengthen adhesion forces and allows dynamic contacts (Figure 2). p120-catenin interacts with the juxtamembrane part of the VE-cadherin cytoplasmic domain and is involved in its retention at the cell surface, while β/γ-catenins, on the other hand, act as constitutive partners of VE-cadherin, bound to its carboxy-terminal part (9, 10). Importantly, VE-cadherin is also connected to the actin cytoskeleton through the actin-binding protein, α-catenin (5). Other adhesive proteins that accumulate in or close to AJ, include N-cadherin, platelet-endothelial cell adhesion molecule (PECAM-1), and junctional adhesion molecules (JAMs).
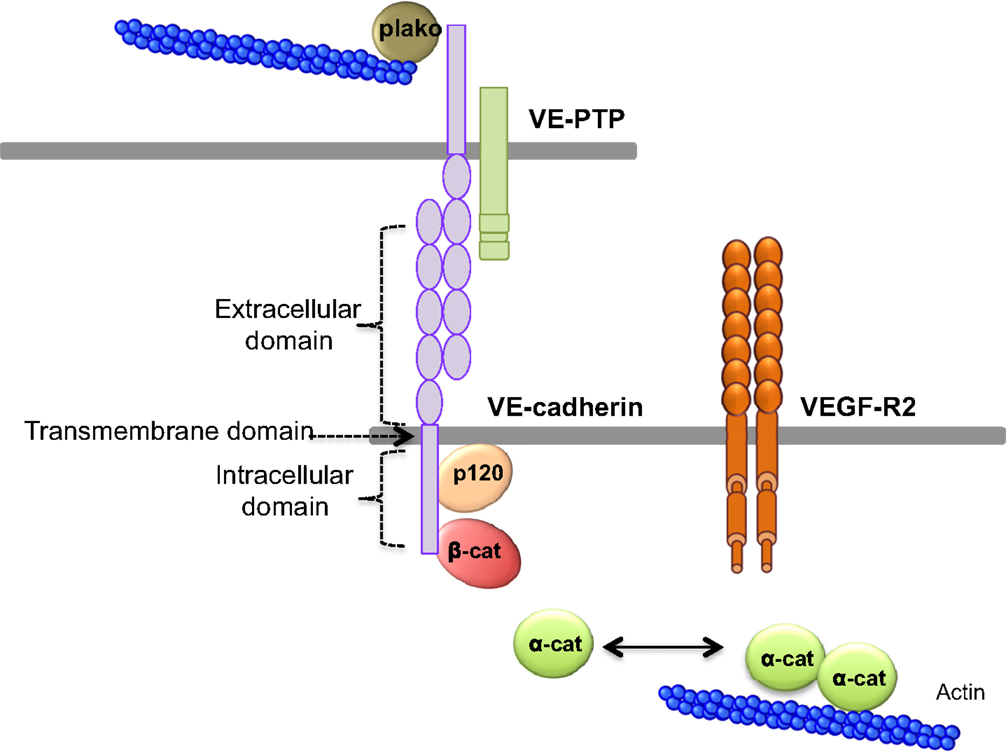
Figure 2. VE-cadherin adhesive complex. VE-cadherin mediates the adhesion between endothelial cells in calcium-dependent manner. VE-cadherin is constituted of an extracellular domain, which allows homophilic interaction in trans. The transmembrane domain participates to lateral clustering in cis. The intracellular domain of VE-cadherin binds p120-catenin (p120), and β-catenin (β-cat), which participates to VE-cadherin membrane retention. Actin cytoskeleton is anchored to VE-cadherin via α-catenin (α-cat) or plakoglobin (plako). In addition, VE-cadherin can bind VEGF-R2 (vascular endothelial growth factor receptor 2) and VE-PTP (vascular endothelial phosphotyrosine phosphatase).
Tight junctions participate in endothelial cell cohesion and block molecule diffusion along the apical and basolateral poles (11). They rely on transmembrane adhesion proteins (occludin and claudins), JAM family proteins, and intracellular connectors, including ZO-1, -2, -3 (Zonula Occludens). First, occludin and claudins contain four transmembrane domains with N- and C-terminal intracellular parts. Second, JAM-A belongs to the immunoglobulin superfamily with one intracellular short domain, one single transmembrane domain, and two extracellular immunoglobulin-like domains. Third, the ZO proteins contain three PDZ (post synaptic density protein PSD95, Drosophila disk large tumor suppressor Dlg1, and zonula occludens-1 protein zo-1), one SH3 (SRC homology 3) and one guanylyl kinase-like domains (11). Contrary to VE-cadherin, deletion of the claudin-5 gene does not impair mouse embryo development, but rather leads to post-natal death caused by a defective blood-brain barrier (12). Thus, VE-cadherin is instrumental in vascular barrier integrity, while claudins may have a more restrictive role (13). Nevertheless AJ and TJ are functionally and structurally linked and can influence each other (14, 15).
Within blood vessels, endothelial cells are interactively anchored to the extracellular matrix (ECM) through integrin-based adhesion complexes, namely focal adhesions (Figure 1). Indeed, integrins bridge the ECM to the acto-myosin contractility apparatus (16), and allow endothelial cells to adapt to extracellular signals and cues (e.g., shear stress and secreted molecules). From a molecular standpoint, Rho-GTPase activation, stress fiber formation, and acto-myosin contraction are finely tuned through integrin adhesion and collectively contribute to the modulation of endothelial junction integrity (17, 18). More recently, it was demonstrated that the integrin-associated focal adhesion tyrosine kinase (FAK) contributes to the impairment of vascular barrier function (19). Indeed, VEGF-induced FAK activation was shown to lead to VE-cadherin/FAK interaction in association with β-catenin phosphorylation on tyrosine Y142, resulting in VE-cadherin/β-catenin dissociation, junction opening, and endothelial barrier disruption.
Hence, vascular barrier properties depend on both structural (basal membrane, smooth muscle cells, endothelial cells) and functional (VVO, AJ, TJ) features. To endorse this role, endothelial cell adhesion has to be tightly regulated. Indeed, aberrant and uncontrolled increase of vascular permeability can participate in the progression of many pathological states, such as chronic inflammatory diseases, diabetes, and tumor angiogenesis.
Vascular Leakage in the Tumor Microenvironment
Compared to normal tissues, tumor vasculature is immature and exhibits structural abnormalities, such as dilatation, saccular formation, and a hyper-branched and twisted pattern. Moreover, solid tumors usually present few to none functional lymphatic vessels (20, 21). Many molecular and cellular factors contribute to this morphological and functional failure, in which vascular permeability is central. Rapidly growing tumors secrete an abundance of different factors (VEGF, chemokines, and others) that govern uncontrolled angiogenesis. In such microenvironments, most of the criteria that define the endothelial barrier properties are circumvented.
First, tumor vessels are characterized by extensive angiogenesis, i.e., neovessel formation from pre-existing vascular networks. In this scenario, tumor endothelial cells have a proliferation rate 50–200 times faster than that of normal quiescent endothelial cells (22). They also have to migrate and rearrange into vascular tubules, dedicated to fuel the tumor mass. This high endothelial plasticity in the constantly remodeled vascular wall is accompanied by elevated permeability. Tumor vessel hyperpermeability correlates with faint VE-cadherin expression, opening of paracellular junctions, and transcellular holes formation (23). In the course of tumor growth, the direct consequence of hyperpermeable vessels is plasma membrane protein extravasation and formation of a provisory matrix to allow endothelial cell sprouting and formation of new vessels (24).
Morphologically, the pericytes surrounding tumor vessels are abnormally shaped and are weakly associated with endothelial cells (25). In addition, tumor blood vessels lack smooth muscles (Figure 3). Similarly, the basal membrane can be either unusually thick or totally absent (26). In these conditions, resistance to blood flow is increased, and thereby the efficacy in tumor blood supply is reduced. As a consequence, despite a high microvessel density, tumors are poorly vascularized with hyperpermeable vasculature. This could lead in fine to the accumulation of metabolic products (lactic and carbonic acids) and extracellular pH decrease (27). Tumor vessel defects also quell oxygen supply, frequently causing hypoxia in the tumor microenvironment. Hypoxia, in turn, supports tumor angiogenesis through the hypoxia-inducible transcription factors (HIF), and further elevates the expression of pro-angiogenic molecules, such as VEGF, TNF (tumor necrosis factor), and PDGF (platelet-derived growth factor). Interestingly, because of its involvement in chemo- and radio-resistance, as well as metastasis, hypoxia has been suggested as an adverse prognostic factor (28).
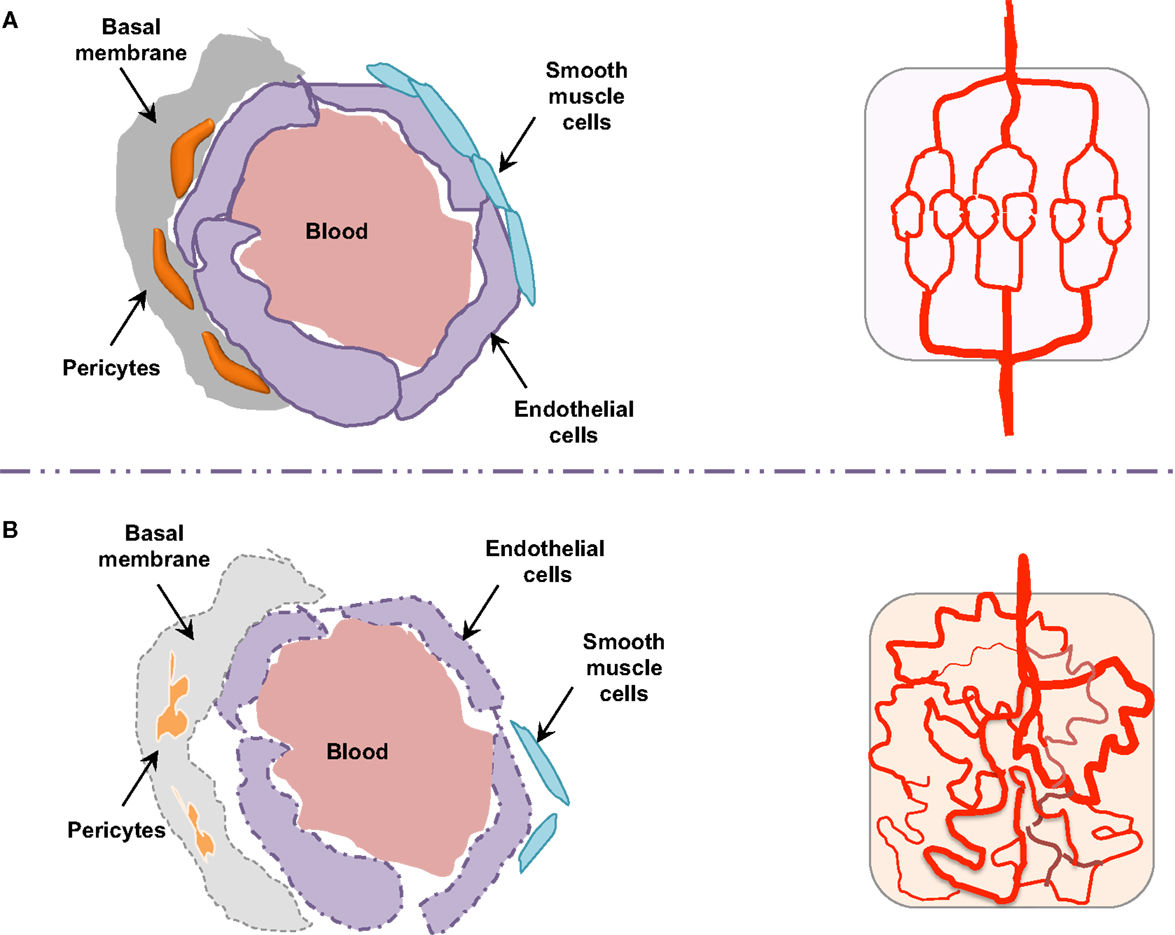
Figure 3. Endothelial barrier in normal and tumor vessels. The endothelial barrier structure differs in normal (A) and tumor (B) blood vessels. Contrary to normal vessels, the tumor vasculature pattern is extremely disorganized and anarchic, presents morphological and structural difference, i.e., weak association between endothelial cells, abnormal shapes of pericytes, lack of smooth muscles, as well as basal membrane modification.
Within the tumor microenvironment, the ECM undergoes significant compositional modifications most notably by increasing the levels of expression of collagen-1, matrix metalloproteases (MMP)-1 and -2, and laminin-5 (29). For instance, collagen-1 deposit increases ECM stiffness, and this is related with poor prognosis and higher metastasis potential (30). In addition, ECM stiffness enhances integrin expression and promotes focal adhesion signaling, and consequently influences tumor cell malignancy (31).
In summary, abnormal blood vessels and lack of lymphatic vessels in tumors, as well as increased ECM stiffness and relatively high interstitial fluid pressure (IFP) collectively contribute to the functional outcome called enhanced permeability and retention (EPR). This phenomenon facilitates both macromolecule extravasation and retention. Whereas normal vessels form a selective barrier, limiting cell and macromolecule passage, the tumor vasculature is extremely leaky and not restrictive. Consequently, although these features could benefit to tumor angiogenesis and growth, anti-tumor drug delivery is rather limited.
Impact on Drug Delivery
To gain in bioavailability and selectivity toward tumor cells, therapeutic molecules must counteract biological and physical barriers, among which are endothelial transport and blood flow. Drug efflux pumps are one of the main obstacles that anti-cancer drugs must overcome. These transporters are highly expressed in a large panel of cancer cells, as well as in the blood-brain barrier, where they ensure drug detoxification (32, 33).
However, tumor vessels cannot ensure correct tumor blood perfusion, since they are structurally aberrant and hyperpermeable (24). The pressure difference between vessels and surrounding tissues constitutes also an important physical barrier. Upon vascular leakage, transcapillary interstitial fluid flow decreases and IFP increases resulting in poor drug penetration through tumor vessels (21). In addition to blood vessel leakage, both the absence of a functional lymphatic system and increased ECM-frictional resistance also lead to tumor IFP increase (34). This ultimately provokes disruption in blood flow directions, again limiting drug delivery.
Importantly, tumor drug delivery can be tailored by changing the size and charge of the delivered molecule. Of interest, molecules larger than 40 kDa cannot be passively eliminated through renal clearance and are unable to cross normal blood vessels through endothelial junctions; however, they could easily penetrate tumors through leaky vessels. EPR of tumor vessels permits the passage of molecules ranging from 40 to 70 kDa, thus, in association with other properties such as the ability to traverse relatively long distances, prolonged plasma half-life and slow clearance, these larger molecule have been proposed to be the most appropriate for specific tumor delivery (35, 36). In addition, due to the negative charges of the vessel luminal face, the use of cationic therapeutic molecules may also favor vascular accumulation, which in turn can elevate tumor drug concentration (37).
Thus, although drug delivery is strongly impaired in tumors because of structural and functional vascular defects, some of these constraints, such as vessel leakiness, can be exploited for curative purposes.
Molecular Mechanisms Involved in Vascular Permeability
As presented above, endothelial barrier integrity ensures vascular and tissue homeostasis. In cancers, deregulation of this fine-tuned function leads to the formation of a chaotic blood vessel network associated with elevated permeability. We will now detail the molecular mechanisms involved in tumor-driven vascular permeability, focusing on the main factors produced by tumor cells, such as VEGF and chemokines. This knowledge could open new avenues for drug design.
Vascular Endothelial Growth Factor
Vascular endothelial growth factor belongs to the family of platelet-derived growth factors and was originally referred to as vascular permeability factor (38). It is a homodimeric glycoprotein of which several forms have been described in mammals, these are: VEGF-A, B, C, D, and the placenta growth factor PlGF. Among these, VEGF-A is the most commonly studied and better described in literature. Various cell types, such as endothelial cells, smooth muscle cells, fibroblasts, and immune cells (macrophages, lymphocytes, neutrophils, and eosinophils) can produce and release VEGF within the environment. In turn, VEGF can act in both an autocrine and paracrine manner. In cancers, tumor cells constitute an important source of VEGF. VEGF stimulates endothelial cell growth and promotes vasculogenesis and angiogenesis. It also increases vascular permeability, its first described function, in many tissues, and plays a crucial role in tumor vasculature development (22). VEGF intracellular signaling is mediated by three tyrosine kinase-receptors, namely VEGF-R1, -R2, and -R3, as well as co-receptors such as neuropilins. The binding of the ligand to its cognate receptors induces their dimerization, autophosphorylation, and subsequent signal transduction (39). VEGF-A interacts with both VEGF-R1 and -R2, but only VEGF-R2 is directly involved in normal and pathological vascular permeability (40). However, VEGF-R1 is reported to act as a regulator of VEGF-R2 signaling, and thus might indirectly regulate vascular permeability.
VEGF-A promotes vascular permeability by disruption of AJ and TJ, resulting in transient opening of endothelial cell–cell contacts (5, 14) (Figure 4). Indeed, VEGF-A promotes tyrosine phosphorylation of VE-cadherin and of its binding partners β-catenin, plakoglobin, and p120, in a Src-dependent mechanism (41). Consistent with this, VE-cadherin phosphorylation is inhibited in Src-deficient mice (41). VE-cadherin can also associate with VEGF-R2 and inhibit its phosphorylation and subsequent internalization (42). This association potentiates the phosphorylation of AJ components by Src, thus impairing endothelial barrier integrity and favoring tumor cell extravasation and dissemination in pathological models (43). The VE-cadherin/VEGF-R2 association also contributes to VE-cadherin-induced contact inhibition of cell growth and requires the β-, but not p120-catenin, binding domain of VE-cadherin (42, 44). In addition, VEGF-A mediates VE-cadherin phosphorylation and internalization via the sequential activation of Src, the guanine nucleotide exchange factor Vav2, the Rho-GTPase Rac, and its downstream effector, the serine-threonine kinase PAK (Figure 4). This signaling pathway culminates in the PAK-dependent phosphorylation of VE-cadherin, which directs its internalization (45). Moreover, VEGF signaling decreases VE-cadherin/p120-catenin association promoting clathrin-dependent VE-cadherin endocytosis (46). Indeed, p120 binding to VE-cadherin prevents its internalization, while its silencing by siRNA leads to VE-cadherin degradation, and loss of cell–cell contacts (10, 47). The expression of VE-cadherin mutants that compete with the endogenous molecule for binding with p120, triggers VE-cadherin degradation, suggesting that p120 might act as plasma membrane retention signal. More recently, a motif within VE-cadherin was identified to be responsible for VE-cadherin/p120 coupling and endocytosis sorting (48). In this context, VE-cadherin-mediated cell–cell contacts are stabilized by the small GTPase Rap1 and its effector, the cyclic adenosine monophosphate (cAMP)-activated guanidine exchange factor Epac (49, 50). Small GTPases also regulate myosin light chain (MLC) phosphorylation, acto-myosin contractility, and endothelial permeability (51). Indeed, VEGF induces the phosphorylation of MLC that results in the formation of stress fibers which exert centripetal tension on intercellular junctions (52).
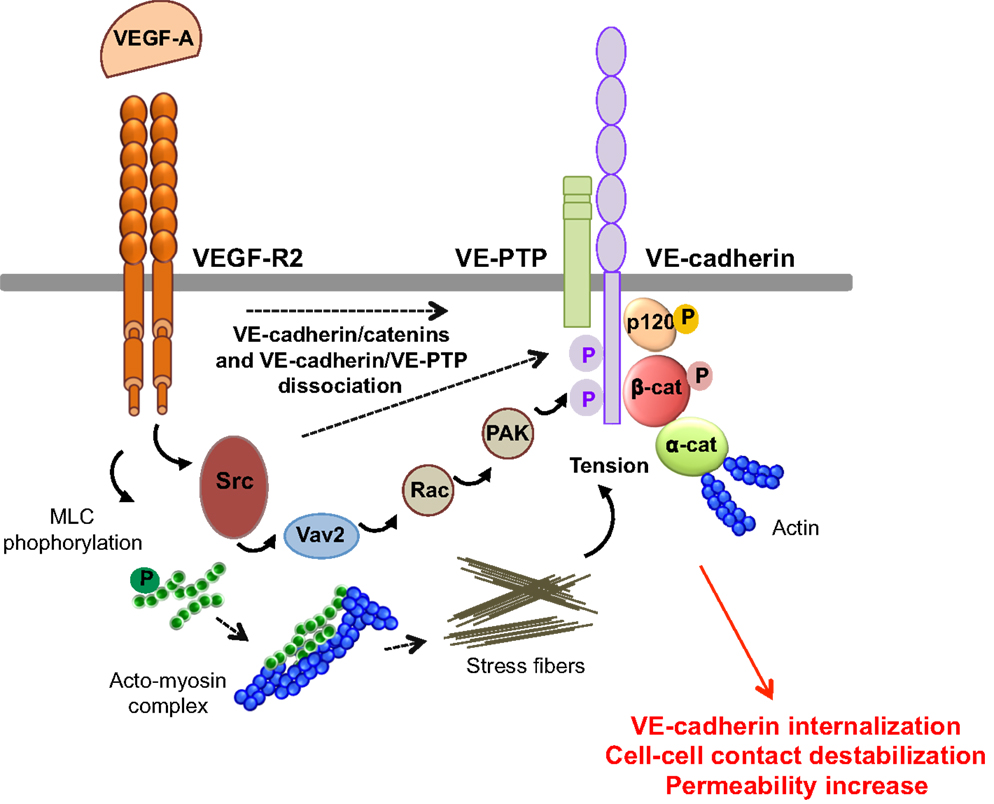
Figure 4. Molecular pathways involved in VEGF-endothelial permeability. VEGF-A stimulation induces VEGF-R2 dimerization and the sequential activation of Vav2, Rac, and PAK, through Src. This results in the serine phosphorylation of VE-cadherin by PAK, and its subsequent internalization into clathrin-coated pits. VEGF can also trigger the tyrosine phosphorylation of VE-cadherin and of its binding partners β-catenin (β-cat) and p120, in a Src-dependent fashion. In addition, VEGF-A decreases the VE-cadherin/p120-catenin association and promotes VE-cadherin endocytosis. VEGF-A also induces the phosphorylation of myosin light chains (MLC), which produces stress fibers that exert tension on intercellular junctions, thus weakening cell–cell contacts. Finally, VEGF-A stimulation causes the dissociation of VE-PTP/VE-cadherin and triggers loss of adhesion and permeability increase.
Of note, endothelial permeability can also be regulated through changes in the expression of AJ and TJ components (15, 53). For example, VEGF signaling through VEGF-R2 induces the expression of SRF (serum response factor), which is important for VE-cadherin expression (54). Indeed, SRF knockdown in mice reduces VE-cadherin expression and angiogenesis. Furthermore, claudin-5 expression is regulated by VE-cadherin, confirming that the latter is instrumental in controlling endothelial barrier function (15). Recently, it has been described that VEGF is involved in claudin-5 down-regulation in peritoneal endothelium, inducing ascites in ovarian cancer patients (55).
Interleukin-8
Cytokines are key drivers of immune responses and play important roles in cancer progression. Among these, the chemokine IL-8 (CXCL8, CXC chemokine ligand 8) is overexpressed and secreted by cancerous cells. Of note, their cognate G-protein-coupled receptors (GPCR), CXCR1 and CXCR2, are expressed on endothelial cells, tumor cells, and neutrophils/tumor-associated-macrophages, indicative of pleiotropic activities of IL-8. Activation of IL-8 endothelial receptors is known to promote angiogenic responses, through enhanced proliferation, survival, and migration (56). Furthermore, intratumoral IL-8 concentration is proposed to chiefly cause neutrophil recruitment into the tumor microenvironment and to promote metastasis (57). Besides its chemotactic role, IL-8 arose as an essential factor of angiogenesis and increased vascular permeability (58). Indeed, IL-8 can provoke VEGF-R2 phosphorylation and transactivation, which in turn result in both Src and RhoA activation, leading to endothelial gap formation, and elevated permeability (59). IL-8 can also increase permeability in mouse and human endothelial cells via a VEGF-R2 independent mechanism (60). IL-8 initiates a signaling route through CXCR2/Rac/PI3Kγ that triggers the phosphorylation and subsequent internalization of VE-cadherin, thereby promoting increased permeability. Moreover, blockade of CXCR2 and PI3Kγ with pharmacological inhibitors or by RNA interference (RNAi), limits IL-8-induced neovascularization and vessel leakage (60). In glioblastoma, cancer cells were found to secrete high concentrations of IL-8, which was further demonstrated to function as a key factor involved in tumor-induced permeability in vitro, and to signal to brain microvascular endothelial cells via CXCR2, promoting VE-cadherin cell–cell junction remodeling, and elevated permeability (61). Similarly, in prostate cancers, IL-8 secretion is associated with increased Akt expression and activation, which impacts on endothelial cell survival, angiogenesis, and cell migration (62).
Transforming Growth Factor-β1
TGF-β1 is a multifunctional polypeptide member of the transforming growth factor beta superfamily. It regulates the production of cytokines and ECM components, and is involved in diverse biological processes, such as proliferation and differentiation in many cell types (63– 65). Within the tumor microenvironment, macrophages, mesenchymal, and cancer cells secrete TGF-β1 under hypoxic and inflammatory conditions. TGF-β1 was suggested to act as a potent inducer of angiogenesis, since its increased expression correlates with high microvessel density and poor prognosis in various types of cancers (66). TGF-β1 also augments vascular permeability by altering cell–cell contacts. This is thought to involve p38 mitogen-activated protein kinase (MAPK) and RhoA signaling cascades, which in turn modulate ECM adhesion and lead to the loss of endothelial-barrier integrity and function (67). In primary breast tumors, TGF-β1 activity is associated with an increased risk of lung metastasis. Indeed, angiopoietin-related protein 4 (ANGPTL4), a TGF-β1 target gene, disrupts endothelial cell–cell junctions, and facilitates the extravasation of breast cancer cells (68). Moreover, TGF-β1 induces the expression of VEGF in fibroblasts (69), whereas it inhibits angiopoietin-1, an anti-permeability factor, therefore exacerbating tumor-associated vascular leakage (70). In addition, TGF-β1 potentiates the secretion and activation of MMPs (71).
Stromal Cell-Derived Factor 1
Stromal cell-derived factor 1, also known as CXCL12, is a member of the α-chemokine subfamily and the ligand for the GPCR CXCR4. In adulthood, SDF-1 was implicated in angiogenesis by recruiting endothelial progenitor cells from the bone marrow (72). SDF-1 is highly expressed in a number of cancers and is associated with tumor extravasation and increased metastases (73, 74). CXCR4 expression also corroborates with metastatic properties of breast cancer cells (75). Indeed, CXCR4 levels were found to be higher in malignant breast tumors in comparison to those of normal healthy counterparts. In vivo, neutralizing CXCR4/SDF-1 signaling axis significantly impaired breast cancer cell extravasation and propagation (75, 76). SDF-1 can also mediate endothelial permeability via CXCR4, as for instance, SDF-1 stimulation of breast cancer cells in vitro increased their passage across the endothelial barrier. This effect is dependent on both PI3K/Akt and calcium signaling in endothelial cells (77). Inhibiting this pathway with anti-CXCR4 antibodies, on the other hand, decreased vascular leakage (77). Moreover, SDF-1 is involved in macrophage recruitment to breast tumors in mice, in response to chemotherapy (78). This action is believed to stimulate tumor blood vessel growth, counteracting the effects of the drug. Finally, it is to be noted that VEGF stimulates SDF-1 secretion and vice versa (79, 80). However, VEGF implication in SDF-1-induced permeability remains to be elucidated.
Interleukin-10
IL-10, also known as human cytokine synthesis inhibitory factor, is an anti-inflammatory cytokine. The role of IL-10 in cancers, though well accepted, is vaguely understood (81). Indeed, IL-10 is suspected to exert both pro- and anti-tumor activities, and contradictory results have been reported regarding its involvement in tumor angiogenesis. On one hand, IL-10 could hamper angiogenesis and tumor growth in mice bearing VEGF-producing ovarian cancer (82), and suppress tumor growth and metastasis of human melanoma cells (83). On the other hand, other studies have suggested that IL-10 may promote angiogenesis in a melanoma cell model, by inhibiting macrophage functions and inducing tumor and vascular cell proliferation (84).
Matrix Metalloproteinases
Metalloproteinases are a large family of proteases that include MMP and ADAM (a disintegrin and metalloproteinase). MMPs belong to a family of zinc-containing endopeptidases that degrade various components of the ECM. Their aberrant over-expression correlates with cancer progression, cell invasion, and metastasis (85). Tumor-associated macrophages secrete VEGF and MMP-9, which are directly involved in both breast cancer and colorectal cancer cell invasion and metastasis (86). In addition, MMPs promote tumor progression by rearrangement of the ECM. Indeed, they trim cell adhesion molecules and degrade matrix proteins, favoring cell proliferation, and angiogenesis. MMP-7 can shed VE-cadherin, while MMP-2 and MMP-9 are involved in occludin proteolysis, thus enhancing endothelial permeability (87, 88). MMPs can also potentiate vascular leakage in a more indirect fashion, via cleavage and activation of chemokines such as IL-8, which is processed by MMP-9 (89, 90). Moreover, in vitro experiments have demonstrated a role for ADAM10 and ADAM17 in endothelial gap formation in response to cytokines. This is probably mediated by the cleavage of adhesion molecules within cell–cell junctions, including VE-cadherin and JAM-A (91).
Semaphorins
Semaphorins correspond to a family of secreted and membrane-bound proteins that can act as both attractive and repulsive guidance molecules (92, 93). Besides their role in neural development, some of these molecules can modulate endothelial plasticity (94). Indeed, semaphorin 4D plays a positive role in endothelial migration and tumor angiogenesis (95, 96). In contrast, class 3 semaphorins, notably semaphorin 3A (S3A), and semaphorin 3E, are reported to operate as selective inhibitors of VEGF-induced angiogenesis (97– 100). However, S3A and VEGF can also cooperate to induce vascular permeability (101). Indeed, S3A induces Akt phosphorylation through PI3K signaling, thus enhancing vascular permeability (101). In glioblastoma, the cancer stem-like cell sub-population expresses and secretes S3A ex vivo (102). In this context, S3A mediates endothelial cell–cell junction destabilization and elevates endothelial permeability (102). On a molecular level, S3A disrupts the VE-cadherin/PP2A complex, allowing VE-cadherin serine phosphorylation and subsequent internalization (45, 102). Consistent with this, inhibition of S3A by blocking antibody or by silencing RNAs has been demonstrated to abrogate these effects.
Nitric Oxide and Peroxynitrite
Nitric oxide (NO) is a highly reactive free radical, which mediates a myriad of cellular reactions (103). NO is produced from l-arginine and oxygen by NO synthases (NOS). There are three major NOS isoforms: inducible NO synthase (NOS2/iNOS), eNOS (NOS3/eNOS), and neuronal NO synthase (NOS1/nNOS). NOS3 is constitutively expressed in endothelial cells, cardiac myocytes, and hippocampal pyramidal cells and is involved in suppressing platelet aggregation, maintaining vascular tone, inhibiting smooth muscle cell proliferation, and prompting angiogenesis (104). In cancers, NOS3 generates NO in blood vessels, which can favor endothelial proliferation, migration, and tumor progression (105, 106). Of note, NOS3 can be induced by VEGF in a MAPK/PLC-γ-dependent manner (107). NOS3 may also be involved in modulating vascular leakage. Indeed, it has been reported that eNOS translocation to the cytosol, but not to the Golgi, is associated with hyperpermeability in vitro and in vivo (108, 109). Stimulation of endothelial cells with platelet-activating factor (PAF) induces S-nitrosylation of β-catenin and p120 and significantly diminishes their association with VE-cadherin (110). Furthermore, VEGF treatment elicited S-nitrosylation of β-catenin at the Cys619 residue, within the VE-cadherin interaction site (111). Inhibition of β-catenin S-nitrosylation prevents NO-dependent dissociation of β-catenin from VE-cadherin and disassembly of AJ complexes, thereby inhibiting VEGF-mediated endothelial permeability (111). Moreover, oxidized products of NO, such as peroxynitrite (ONOO-), activate MMPs, which favor matrix rearrangement and endothelial permeability as discussed above. However, NO can induce cytotoxic effects on cancer cells. The balance between NO-mediated permeability and angiogenesis or apoptosis should thus be considered in tumor-targeted therapy (112).
In conclusion, endothelial permeability-mediated signaling pathways converge at the disruption and destabilization of cell–cell contacts, promoting AJ and TJ restructuration and subsequent opening of endothelial cell–cell junctions. We will now present the anti-permeability factors and normalization agents that may represent new perspectives in cancer therapy.
Perspectives in Cancer Therapy
It is now well accepted that vascular permeability limits drug delivery thus restraining the efficacy of conventional therapies. New approaches aim now at diminishing both excessive angiogenesis and hyperpermeability. In this paragraph, perspectives in cancer therapy, such as the use of anti-permeability factors and blood vessel normalization agents will be discussed.
Anti-Permeability Factors
The most relevant anti-permeability factors are angiopoietin-1 and its cognate receptor Tie2, sphingosine-1-phosphate (S1P), and fibroblast growth factor (FGF) (Figure 5).
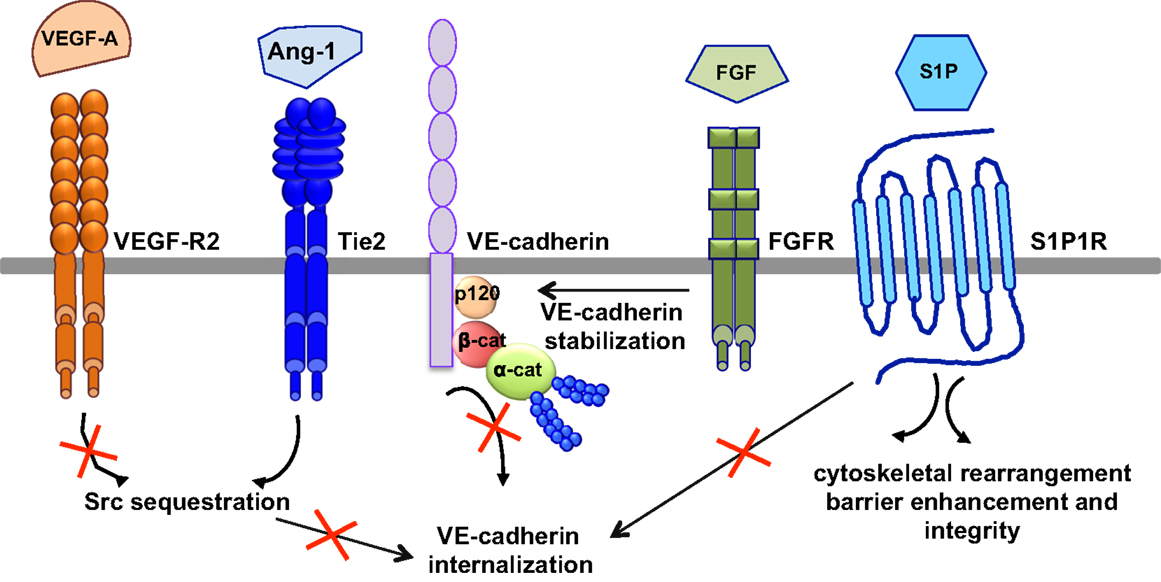
Figure 5. Signaling pathways of anti-permeability factors. The most relevant anti-permeability factors are angiopoietin-1 (Ang1), sphingosine-1-phosphate (S1P), and fibroblast growth factor (FGF). Ang1 activation of its cognate receptor, Tie2, elicits a signaling pathway and promotes Src sequestration; thus hindering VEGF-A signaling and VE-cadherin internalization. S1P signaling maintains vascular integrity by modulating VE-cadherin internalization, cytoskeletal rearrangement, barrier enhancement and integrity, through its cognate receptor S1P1R. FGF maintains the integrity of the VE-cadherin/p120-catenin complex, thus stabilizing VE-cadherin at the membrane.
Angiopoeitin-1 is a potent pro-angiogenic factor with the particularity of stabilizing blood vessels and counteracting VEGF-induced vascular permeability (63, 113). As mentioned above, VEGF elevates endothelial permeability via VE-cadherin adhesion destabilization in a Src-dependent mechanism (41, 45). Angiopoietin-1 elicits a signaling pathway through Tie2 that promotes the sequestration of Src though mammalian diaphanous (mDia) (114), thus hindering VEGF signaling and VE-cadherin internalization. In an intact endothelial monolayer, angiopoietin-1 promotes the interaction of Tie2 with the vascular endothelial protein tyrosine phosphatase VE-PTP (115, 116). VE-PTP associates with VE-cadherin and stabilizes it at the plasma membrane by blocking its tyrosine phosphorylation in response to VEGF-R2 activation (117). Angiopoeitin-1 also maintains the barrier integrity by increasing the association between VE-cadherin and plakoglobin (118), thus strengthening cell–cell contacts and limiting endothelial permeability. Consistently, VEGF signaling in vivo triggers the dissociation of VE-PTP from VE-cadherin, facilitating leukocyte extravasation and vessel leakage (119). Furthermore, angiopoeitin-1 balances VEGF pro-permeability actions by controlling NO release from endothelial cells. Indeed, it increases eNOS phosphorylation on Thr497, and subsequently reduces NO release and transendothelial permeability (120).
In addition to angiopoeitin-1, S1P, a biologically active phosphorylated lipid growth factor released from activated platelets, has emerged as an endothelial barrier protective agent. In both pulmonary artery and lung microvascular endothelial cells, S1P was able to reverse barrier dysfunctions elicited by thrombin (121). At the molecular level, S1P signaling maintains vascular integrity by cytoskeletal rearrangement and barrier enhancement through Rac activation (121). Moreover, high-density lipoproteins, acting as major plasma carriers for S1P, promote endothelial-barrier integrity via the Akt signaling pathway (122). Plasma-derived S1P also plays an essential role in maintaining vascular integrity. Indeed, mutant mice engineered to selectively lack S1P in plasma show increased vascular leak and impaired survival after administration of permeability inducing factors (123). Elevated leak was associated with interendothelial cell gaps in venules and was reversed by acute treatment with an agonist for the S1P receptor 1 (S1PR1) (123). Furthermore, recent works present S1PR1, as a key component of vascular stability (124, 125). These two studies elegantly showed that S1PR1 inhibits VEGF-induced VE-cadherin destabilization and internalization, and thereby enhances cell–cell adhesion (124, 125).
Other factors, such as FGF, can act as VE-cadherin stabilizing agents. Indeed, the inhibition of FGF signaling results in the dissociation of the VE-cadherin/p120-catenin complex, and subsequent VE-cadherin internalization, disassembly of AJ and TJ, and loss of vascular barrier integrity (126).
Thus, since they counteract VEGF-induced permeability and contribute to the maintenance of vascular barrier function, anti-permeability factors appear as potential therapeutic candidates. Another promising approach is the use of anti-VEGF/VEGF-R drugs to promote normalization of the vascular wall and its microenvironment.
Normalization Agents
From 1950 to the 2000s, the only existing non-invasive treatment for solid tumors has been chemotherapy, which is mainly based on reducing tumor cell proliferation. Because its lack of selectivity causes a large panel of side effects, new strategies, such as molecular and personalized therapies, attempt to focus on molecules overexpressed in cancers. Unfortunately, the results from clinical trials targeting such molecules in anti-cancer therapy have been quite disappointing with an overall low extension of survival, with the exception of imatinib, a tyrosine kinase inhibitor (TKI) used in chronic myelogenous leukemia treatment (127). In addition, anti-angiogenic molecules have been suggested to improve anti-cancer therapy, not only because they reduce tumor vascularization, but also thanks to their “normalization” action, which improves drug delivery.
Bevacizumab (commercialized as Avastin) was one of the first clinically available anti-angiogenic drugs. This humanized mouse antibody targeting VEGF was FDA-approved about a decade ago for combination use with standard chemotherapy in colorectal and non-small cell lung cancers. In addition, bevacizumab alone can significantly curb disease progression in patients with metastatic renal cell cancer (128). Recently published data suggest promising clinical efficacy of bevacizumab monotherapy in metastatic melanoma (129). Beside anti-VEGF, broad-spectrum multi-target TKI prolong cancer-free survival by collectively decreasing tumor vessel diameter, density, and permeability, even when administered in the absence of conventional therapies. For instance, sunitinib and sorafenib monotherapies appear particularly efficient in gastrointestinal and renal cancers (130). Nevertheless, both bevacizumab and TKI can cause serious adverse effects, such as gastrointestinal perforations.
The Notch ligand delta-like 4 (Dll4) has recently emerged as a critical regulator of tumor angiogenesis (131). Activation of the Notch pathway in neighboring endothelial cells causes inhibition of tip cell formation, an early event in sprouting angiogenesis. Mechanistically, this is believed to occur through the down-regulation of VEGF-R2/3 pro-angiogenic pathway and the up-regulation of VEGF-R1 anti-angiogenic pathway (132). Interestingly, VEGF can also operate upstream of Dll4 to potentiate its effects (133). However, the exact role of Dll4 in tumor growth and its potential in anti-cancer therapy remain unclear. Indeed, Dll4/Notch activation reduces overall tumor angiogenesis, while tumor vascular function was improved and tumor growth was heightened (134). This supports the notion that further strategy in anti-cancer therapy could be based on Dll4/Notch signaling blockade. On the other hand, Dll4-driven Notch activation might reduce both tumor-induced angiogenesis and endothelial cell responsiveness to VEGF (135), and therefore argue rather for the use of Dll4 as an effective therapeutic approach in cancers.
Combination Therapy and Future Strategies
Recently, based on general hallmarks of the tumor vasculature, i.e., poor blood flow, leakage, and reduced drug uptake, a new trend in anti-cancer treatment has emerged, that involves combining vascular normalization agents with traditional therapies to improve treatment response.
To re-establish an efficient tumor vascularization, assistant molecules, namely those bearing anti-angiogenic or anti-permeability properties, have been designed and tested in clinical trials, in parallel with cytotoxic drugs. In this scenario, the use of either the vasoconstrictor Angiotensin II (136) or the vasodilator Bradykinin B2 receptor agonist (137) improves tumor treatment uptake, through an increase in transcapillary pressure. Similarly, to facilitate stromal barrier crossing, the ECM-degrading enzyme collagenase (138) was shown to exert favorable changes in the transcapillary pressure gradient and thereby enhance anti-cancer drug penetration.
Alternatively, bevacizumab, the anti-VEGF drug, impinges on both microvascular density and tumor IFP, and improves drug uptake in colorectal carcinoma patients (139). Moreover, pazopanib, an inhibitor of VEGF and PDGF receptors, induces better tumor liposomal drug delivery (140). Likewise, radiotherapy combined with anti-integrin antibody (intetumumab) reduces tumor vessel density, while increasing tumor cell apoptosis and hindering metastasis (141). Interestingly, apart from its role in permeability, high levels of VEGF have been reported to promote T-reg proliferation, inhibit antigen-presenting cell maturation and as a consequence, decrease immune responses (142, 143). Therefore, anti-angiogenic drugs, especially those targeting VEGF actions, could improve cancer immunotherapy by stimulating tumor microenvironment immune responses (142). Although significant evidence has demonstrated the benefits of anti-VEGF therapies in cancer treatment, its general use is still controversial. First, significant increase in overall survival is observed only when bevacizumab is combined with standard chemotherapies. In addition, many patients exhibit resistance to anti-VEGF treatments, while timing and doses to be administered, cost and relapse effects raise some major concerns. Finally, vascular regrowth remains highly problematic. Indeed, a second wave of angiogenesis orchestrated by pro-angiogenic ligands of the FGF family could account for the short-term efficacy of VEGF-based anti-angiogenic therapies (144). Unlike bevacizumab, combination of TKI with conventional chemotherapy does not improve the outcome of anti-cancer treatment. In this scenario, use of erlotinib, a potent inhibitor of the epidermal growth factor receptor tyrosine kinase, with standard chemotherapy has failed to enhance tumor response or survival in lung carcinoma (145). Highly efficient TKI monotherapy could be combined with chemotherapy only when tumor cells become resistant to TKIs.
Alternatively, dose, schedule, and decreased toxicity may amend tumor responses. Contrary to standard chemotherapy, i.e., high doses with prolonged drug-free breaks, metronomic chemotherapy refers to chronic and equally spaced administration of low doses of cytotoxic drugs, without pauses. For example, reduced but continuous doses of cyclophosphamide suppressed tumor growth more effectively than canonical chemotherapy scheduling, even in drug-resistant tumors (146). Interestingly, metronomic chemotherapy exerts anti-tumor and anti-metastatic actions by decreasing VEGF serum concentration and increasing apoptosis of cancer cells (147). For those reasons, metronomic chemotherapy could be considered as an anti-angiogenic chemotherapy.
Importantly, the efficacy of anti-angiogenic therapy combined with cytotoxic conventional therapies (chemo- and/or radio-therapies) depends on optimal treatment scheduling. Indeed for each anti-cancer therapy, a “normalization window,” has to be determined to define period and doses necessary for tumor vessel normalization (148).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are thankful to the members from the Julie Gavard laboratory, especially Dr. J. Dwyer for comments on the manuscript. This research was funded by Ligue nationale contre le cancer comite de Paris, Fondation ARC, Fondation pour la Recherche Medicale, and by a Marie Curie International Reintegration Grant within The Seventh Framework Programme. Jagoda K. Hebda is supported by doctoral fellowship from Universite Paris Descartes and Sandy Azzi by a post-doctoral fellowship from Fondation ARC.
References
1. Gavard J. Breaking the VE-cadherin bonds. FEBS Lett (2009) 583:1–6. doi:10.1016/j.febslet.2008.11.032
2. Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med (1996) 183:1981–6. doi:10.1084/jem.183.5.1981
3. Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, et al. Microvascular hyperpermeability in caveolin-1 (−/−) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem (2002) 277:40091–8. doi:10.1074/jbc.M205948200
4. Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol (2003) 285:L1179–83.
5. Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol (2004) 5:261–70. doi:10.1038/nrm1357
6. Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell (1999) 98:147–57. doi:10.1016/S0092-674(00)81010-7
7. Crosby CV, Fleming PA, Argraves WS, Corada M, Zanetta L, Dejana E, et al. VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood (2005) 105:2771–6. doi:10.1182/blood-2004-06-2244
8. Koch AW, Bozic D, Pertz O, Engel J. Homophilic adhesion by cadherins. Curr Opin Struct Biol (1999) 9:275–81. doi:10.1016/S0959-440X(99)80038-4
9. Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI. The cadherin cytoplasmic domain is unstructured in the absence of beta-catenin. A possible mechanism for regulating cadherin turnover. J Biol Chem (2001) 276:12301–9. doi:10.1074/jbc.M010377200
10. Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, et al. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol (2003) 163:535–45. doi:10.1083/jcb.200306001
11. Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol (2001) 2:285–93. doi:10.1038/35067088
12. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol (2003) 161:653–60. doi:10.1083/jcb.200302070
13. Kluger MS, Clark PR, Tellides G, Gerke V, Pober JS. Claudin-5 controls intercellular barriers of human dermal microvascular but not human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol (2013) 33:489–500. doi:10.1161/ATVBAHA.112.300893
14. Gavard J, Gutkind JS. VE-cadherin and claudin-5: it takes two to tango. Nat Cell Biol (2008) 10:883–5. doi:10.1038/ncb0808-883
15. Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol (2008) 10:923–34. doi:10.1038/ncb1752
16. Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol (2009) 10:21–33. doi:10.1038/nrm2593
17. Schwartz MA, Shattil SJ. Signaling networks linking integrins and rho family GTPases. Trends Biochem Sci (2000) 25:388–91. doi:10.1016/S0968-0004(00)01605-4
18. Huveneers S, Oldenburg J, Spanjaard E, Van Der Krogt G, Grigoriev I, Akhmanova A, et al. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol (2012) 196:641–52. doi:10.1083/jcb.201108120
19. Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell (2012) 22:146–57. doi:10.1016/j.devcel.2011.11.002
20. Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res (2000) 60:4324–7.
21. Wu M, Frieboes HB, Mcdougall SR, Chaplain MA, Cristini V, Lowengrub J. The effect of interstitial pressure on tumor growth: coupling with the blood and lymphatic vascular systems. J Theor Biol (2013) 320:131–51. doi:10.1016/j.jtbi.2012.11.031
23. Hashizume H, Baluk P, Morikawa S, Mclean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol (2000) 156:1363–80. doi:10.1016/S0002-9440(10)65006-7
24. Le Guelte A, Dwyer J, Gavard J. Jumping the barrier: VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biol Cell (2011) 103:593–605. doi:10.1042/BC20110069
25. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, Mcdonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol (2002) 160:985–1000. doi:10.1016/S0002-9440(10)64920-6
26. Baluk P, Morikawa S, Haskell A, Mancuso M, Mcdonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol (2003) 163:1801–15. doi:10.1016/S0002-9440(10)63540-7
27. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst (2007) 99:1441–54. doi:10.1093/jnci/djm135
28. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2012) 12:9–22.
29. Seftor RE, Seftor EA, Koshikawa N, Meltzer PS, Gardner LM, Bilban M, et al. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res (2001) 61:6322–7.
30. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer (2010) 46:1181–8. doi:10.1016/j.ejca.2010.02.027
31. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139:891–906. doi:10.1016/j.cell.2009.10.027
32. Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A (1989) 86:695–8. doi:10.1073/pnas.86.2.695
33. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med (2002) 53:615–27. doi:10.1146/annurev.med.53.082901.103929
34. Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res (2000) 60:2497–503.
35. Seymour LW, Miyamoto Y, Maeda H, Brereton M, Strohalm J, Ulbrich K, et al. Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur J Cancer (1995) 31A:766–70. doi:10.1016/0959-8049(94)00514-6
36. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev (2011) 63:136–51. doi:10.1016/j.addr.2010.04.009
37. Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res (2002) 62:6831–6.
38. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science (1983) 219:983–5. doi:10.1126/science.6823562
39. Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem (1994) 269:26988–95.
40. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol (2006) 7:359–71. doi:10.1038/nrm1911
41. Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell (1999) 4:915–24. doi:10.1016/S1097-2765(00)80221-X
42. Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol (2006) 174:593–604. doi:10.1083/jcb.200602080
43. Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol (2004) 167:223–9. doi:10.1083/jcb.200408130
44. Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol (2003) 161:793–804. doi:10.1083/jcb.200209019
45. Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol (2006) 8:1223–34. doi:10.1038/ncb1486
46. Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, et al. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell (2005) 16:5141–51. doi:10.1091/mbc.E05-05-0440
47. Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol (2003) 163:525–34. doi:10.1083/jcb.200307111
48. Nanes BA, Chiasson-Mackenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, et al. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol (2012) 199:365–80. doi:10.1083/jcb.201205029
49. Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood (2005) 105:1950–5. doi:10.1182/blood-2004-05-1987
50. Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol (2005) 25:136–46. doi:10.1128/MCB.25.1.136-146.2005
51. Stockton RA, Schaefer E, Schwartz MA. p21-activated kinase regulates endothelial permeability through modulation of contractility. J Biol Chem (2004) 279:46621–30. doi:10.1074/jbc.M408877200
52. Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol (2002) 39:187–99. doi:10.1016/S1537-1891(03)00008-9
53. Hebda JK, Leclair HM, Azzi S, Roussel C, Scott MG, Bidere N, et al. The C-terminus region of beta-arrestin1 modulates VE-cadherin expression and endothelial cell permeability. Cell Commun Signal (2013) 11:37. doi:10.1186/1478-811X-11-37
54. Franco CA, Mericskay M, Parlakian A, Gary-Bobo G, Gao-Li J, Paulin D, et al. Serum response factor is required for sprouting angiogenesis and vascular integrity. Dev Cell (2008) 15:448–61. doi:10.1016/j.devcel.2008.07.019
55. Herr D, Sallmann A, Bekes I, Konrad R, Holzheu I, Kreienberg R, et al. VEGF induces ascites in ovarian cancer patients via increasing peritoneal permeability by downregulation of Claudin 5. Gynecol Oncol (2012) 127:210–6. doi:10.1016/j.ygyno.2012.05.002
56. Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol (2003) 170:3369–76.
57. De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin Cancer Res (2004) 10:4895–900. doi:10.1158/1078-0432.CCR-03-0760
58. Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol (2001) 280:L1094–103.
59. Petreaca ML, Yao M, Liu Y, Defea K, Martins-Green M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol Biol Cell (2007) 18:5014–23. doi:10.1091/mbc.E07-01-0004
60. Gavard J, Hou X, Qu Y, Masedunskas A, Martin D, Weigert R, et al. A role for a CXCR2/phosphatidylinositol 3-kinase gamma signaling axis in acute and chronic vascular permeability. Mol Cell Biol (2009) 29:2469–80. doi:10.1128/MCB.01304-08
61. Dwyer J, Hebda JK, Le Guelte A, Galan-Moya EM, Smith SS, Azzi S, et al. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLoS One (2012) 7:e45562. doi:10.1371/journal.pone.0045562
62. MacManus CF, Pettigrew J, Seaton A, Wilson C, Maxwell PJ, Berlingeri S, et al. Interleukin-8 signaling promotes translational regulation of cyclin D in androgen-independent prostate cancer cells. Mol Cancer Res (2007) 5:737–48. doi:10.1158/1541-7786.MCR-07-0032
63. Dumont N, Arteaga CL. A kinase-inactive type II TGFbeta receptor impairs BMP signaling in human breast cancer cells. Biochem Biophys Res Commun (2003) 301:108–12. doi:10.1016/S0006-291X(02)02977-7
64. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer (2003) 3:807–21. doi:10.1038/nrc1208
65. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer (2006) 6:506–20. doi:10.1038/nrc1926
66. Hasegawa Y, Takanashi S, Kanehira Y, Tsushima T, Imai T, Okumura K. Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer (2001) 91:964–71. doi:10.1002/1097-0142(20010301)91:5<964::AID-CNCR1086>3.3.CO;2-F
67. Goldberg PL, Macnaughton DE, Clements RT, Minnear FL, Vincent PA. p38 MAPK activation by TGF-beta1 increases MLC phosphorylation and endothelial monolayer permeability. Am J Physiol Lung Cell Mol Physiol (2002) 282:L146–54.
68. Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell (2008) 133:66–77. doi:10.1016/j.cell.2008.01.046
69. Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, et al. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem (1994) 269:6271–4.
70. Enholm B, Paavonen K, Ristimaki A, Kumar V, Gunji Y, Klefstrom J, et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene (1997) 14:2475–83. doi:10.1038/sj.onc.1201090
71. Hagedorn HG, Bachmeier BE, Nerlich AG. Synthesis and degradation of basement membranes and extracellular matrix and their regulation by TGF-beta in invasive carcinomas (Review). Int J Oncol (2001) 18:669–81.
72. Salcedo R, Oppenheim JJ. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation (2003) 10:359–70. doi:10.1080/mic.10.3-4.359.370
73. Dewan MZ, Ahmed S, Iwasaki Y, Ohba K, Toi M, Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother (2006) 60:273–6. doi:10.1016/j.biopha.2006.06.004
74. Gassmann P, Haier J, Schluter K, Domikowsky B, Wendel C, Wiesner U, et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia (2009) 11:651–61.
75. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410:50–6. doi:10.1038/35065016
76. Yagi H, Tan W, Dillenburg-Pilla P, Armando S, Amornphimoltham P, Simaan M, et al. A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci Signal (2011) 4:ra60. doi:10.1126/scisignal.2002221
77. Lee BC, Lee TH, Avraham S, Avraham HK. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol Cancer Res (2004) 2:327–38.
78. Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest (2011) 121:1969–73. doi:10.1172/JCI44562
79. Kijowski J, Baj-Krzyworzeka M, Majka M, Reca R, Marquez LA, Christofidou-Solomidou M, et al. The SDF-1-CXCR4 axis stimulates VEGF secretion and activates integrins but does not affect proliferation and survival in lymphohematopoietic cells. Stem Cells (2001) 19:453–66. doi:10.1634/stemcells.19-5-453
80. Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood (2002) 99:2703–11. doi:10.1182/blood.V99.8.2703
81. Hamidullah, Changkija B, Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat (2012) 133:11–21. doi:10.1007/s10549-011-1855-x
82. Kohno T, Mizukami H, Suzuki M, Saga Y, Takei Y, Shimpo M, et al. Interleukin-10-mediated inhibition of angiogenesis and tumor growth in mice bearing VEGF-producing ovarian cancer. Cancer Res (2003) 63:5091–4.
83. Huang S, Xie K, Bucana CD, Ullrich SE, Bar-Eli M. Interleukin 10 suppresses tumor growth and metastasis of human melanoma cells: potential inhibition of angiogenesis. Clin Cancer Res (1996) 2:1969–79.
84. Garcia-Hernandez ML, Hernandez-Pando R, Gariglio P, Berumen J. Interleukin-10 promotes B16-melanoma growth by inhibition of macrophage functions and induction of tumour and vascular cell proliferation. Immunology (2002) 105:231–43. doi:10.1046/j.1365-2567.2002.01363.x
85. Rundhaug JE. Matrix metalloproteinases, angiogenesis, and cancer: commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin. Cancer Res., 9: 00-00, 2003. Clin Cancer Res (2003) 9:551–4.
86. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi:10.1038/nrc1256
87. Ichikawa Y, Ishikawa T, Momiyama N, Kamiyama M, Sakurada H, Matsuyama R, et al. Matrilysin (MMP-7) degrades VE-cadherin and accelerates accumulation of beta-catenin in the nucleus of human umbilical vein endothelial cells. Oncol Rep (2006) 15:311–5.
88. Reijerkerk A, Kooij G, Van Der Pol SM, Khazen S, Dijkstra CD, De Vries HE. Diapedesis of monocytes is associated with MMP-mediated occludin disappearance in brain endothelial cells. FASEB J (2006) 20:2550–2. doi:10.1096/fj.06-6099fje
89. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood (2000) 96:2673–81.
90. Van Den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur J Biochem (2003) 270:3739–49. doi:10.1046/j.1432-1033.2003.03760.x
91. Koenen RR, Pruessmeyer J, Soehnlein O, Fraemohs L, Zernecke A, Schwarz N, et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood (2009) 113:4799–809. doi:10.1182/blood-2008-04-152330
92. Kolodkin AL, Matthes DJ, O’Connor TP, Patel NH, Admon A, Bentley D, et al. Fasciclin IV: sequence, expression, and function during growth cone guidance in the grasshopper embryo. Neuron (1992) 9:831–45. doi:10.1016/0896-6273(92)90237-8
93. Kolodkin AL, Matthes DJ, Goodman CS. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell (1993) 75:1389–99. doi:10.1016/0092-8674(93)90625-Z
94. Treps L, Le Guelte A, Gavard J. Emerging roles of semaphorins in the regulation of epithelial and endothelial junctions. Tissue Barriers (2013) 1:e23272. doi:10.4161/tisb.23272
95. Basile JR, Holmbeck K, Bugge TH, Gutkind JS. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J Biol Chem (2007) 282:6899–905. doi:10.1074/jbc.M609570200
96. Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A, et al. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J Exp Med (2008) 205:1673–85. doi:10.1084/jem.20072602
97. Kessler O, Shraga-Heled N, Lange T, Gutmann-Raviv N, Sabo E, Baruch L, et al. Semaphorin-3F is an inhibitor of tumor angiogenesis. Cancer Res (2004) 64:1008–15. doi:10.1158/0008-5472.CAN-03-3090
98. Varshavsky A, Kessler O, Abramovitch S, Kigel B, Zaffryar S, Akiri G, et al. Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res (2008) 68:6922–31. doi:10.1158/0008-5472.CAN-07-5408
99. Sakurai A, Gavard J, Annas-Linhares Y, Basile JR, Amornphimoltham P, Palmby TR, et al. Semaphorin 3E initiates antiangiogenic signaling through plexin D1 by regulating Arf6 and R-Ras. Mol Cell Biol (2010) 30:3086–98. doi:10.1128/MCB.01652-09
100. Wu F, Zhou Q, Yang J, Duan GJ, Ou JJ, Zhang R, et al. Endogenous axon guiding chemorepulsant semaphorin-3F inhibits the growth and metastasis of colorectal carcinoma. Clin Cancer Res (2011) 17:2702–11. doi:10.1158/1078-0432.CCR-10-0839
101. Acevedo LM, Barillas S, Weis SM, Gothert JR, Cheresh DA. Semaphorin 3A suppresses VEGF-mediated angiogenesis yet acts as a vascular permeability factor. Blood (2008) 111:2674–80. doi:10.1182/blood-2007-08-110205
102. Le Guelte A, Galan-Moya EM, Dwyer J, Treps L, Kettler G, Hebda JK, et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J Cell Sci (2012) 125:4137–46. doi:10.1242/jcs.108282
103. Blaise GA, Gauvin D, Gangal M, Authier S. Nitric oxide, cell signaling and cell death. Toxicology (2005) 208:177–92. doi:10.1016/j.tox.2004.11.032
104. Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol Cancer Ther (2003) 2:815–23.
105. Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, et al. Roles of nitric oxide in tumor growth. Proc Natl Acad Sci U S A (1995) 92:4392–6. doi:10.1073/pnas.92.10.4392
106. Ambs S, Merriam WG, Bennett WP, Felley-Bosco E, Ogunfusika MO, Oser SM, et al. Frequent nitric oxide synthase-2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res (1998) 58:334–41.
107. Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol (2001) 2:149–56. doi:10.1016/S1470-2045(00)00256-4
108. Figueroa XF, Gonzalez DR, Martinez AD, Duran WN, Boric MP. ACh-induced endothelial NO synthase translocation, NO release and vasodilatation in the hamster microcirculation in vivo. J Physiol (2002) 544:883–96. doi:10.1113/jphysiol.2002.021972
109. Sanchez FA, Savalia NB, Duran RG, Lal BK, Boric MP, Duran WN. Functional significance of differential eNOS translocation. Am J Physiol Heart Circ Physiol (2006) 291:H1058–64. doi:10.1152/ajpheart.00370.2006
110. Marin N, Zamorano P, Carrasco R, Mujica P, Gonzalez FG, Quezada C, et al. S-Nitrosation of beta-catenin and p120 catenin: a novel regulatory mechanism in endothelial hyperpermeability. Circ Res (2012) 111:553–63. doi:10.1161/CIRCRESAHA.112.274548
111. Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, et al. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell (2010) 39:468–76. doi:10.1016/j.molcel.2010.07.013
112. Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer (2003) 3:276–85. doi:10.1038/nrc1046
113. Thurston G, Suri C, Smith K, Mcclain J, Sato TN, Yancopoulos GD, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science (1999) 286:2511–4. doi:10.1126/science.286.5449.2511
114. Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell (2008) 14:25–36. doi:10.1016/j.devcel.2007.10.019
115. Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, et al. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol (2008) 10:513–26. doi:10.1038/ncb1714
116. Saharinen P, Eklund L, Miettinen J, Wirkkala R, Anisimov A, Winderlich M, et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol (2008) 10:527–37. doi:10.1038/ncb1715
117. Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J (2002) 21:4885–95. doi:10.1093/emboj/cdf497
118. Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med (2008) 205:2929–45. doi:10.1084/jem.20080406
119. Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med (2011) 208:2393–401. doi:10.1084/jem.20110525
120. Oubaha M, Gratton JP. Phosphorylation of endothelial nitric oxide synthase by atypical PKC zeta contributes to angiopoietin-1-dependent inhibition of VEGF-induced endothelial permeability in vitro. Blood (2009) 114:3343–51. doi:10.1182/blood-2008-12-196584
121. Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest (2001) 108:689–701. doi:10.1172/JCI200112450
122. Argraves KM, Gazzolo PJ, Groh EM, Wilkerson BA, Matsuura BS, Twal WO, et al. High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function. J Biol Chem (2008) 283:25074–81. doi:10.1074/jbc.M801214200
123. Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest (2009) 119:1871–9.
124. Gaengel K, Niaudet C, Hagikura K, Lavina B, Muhl L, Hofmann JJ, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell (2012) 23:587–99. doi:10.1016/j.devcel.2012.08.005
125. Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell (2012) 23:600–10. doi:10.1016/j.devcel.2012.07.015
126. Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, et al. The FGF system has a key role in regulating vascular integrity. J Clin Invest (2008) 118:3355–66. doi:10.1172/JCI35298
127. Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med (2012) 2:a006593. doi:10.1101/cshperspect.a006593
128. Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med (2003) 349:427–34. doi:10.1056/NEJMoa021491
129. Schuster C, Eikesdal HP, Puntervoll H, Geisler J, Geisler S, Heinrich D, et al. Clinical efficacy and safety of bevacizumab monotherapy in patients with metastatic melanoma: predictive importance of induced early hypertension. PLoS ONE (2012) 7:e38364. doi:10.1371/journal.pone.0038364
130. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol (2006) 3:24–40. doi:10.1038/ncponc0403
131. Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature (2007) 445:776–80. doi:10.1038/nature05571
132. Phng LK, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell (2009) 16:70–82. doi:10.1016/j.devcel.2008.12.009
133. Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res (2005) 65:8690–7. doi:10.1158/0008-5472.CAN-05-1208
134. Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, et al. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res (2007) 67:11244–53. doi:10.1158/0008-5472.CAN-07-0969
135. Segarra M, Williams CK, Sierra Mde L, Bernardo M, Mccormick PJ, Maric D, et al. Dll4 activation of Notch signaling reduces tumor vascularity and inhibits tumor growth. Blood (2008) 112:1904–11. doi:10.1182/blood-2007-11-126045
136. Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol (2009) 39:756–66. doi:10.1093/jjco/hyp074
137. Sarin H, Kanevsky AS, Fung SH, Butman JA, Cox RW, Glen D, et al. Metabolically stable bradykinin B2 receptor agonists enhance transvascular drug delivery into malignant brain tumors by increasing drug half-life. J Transl Med (2009) 7:33. doi:10.1186/1479-5876-7-33
138. Eikenes L, Bruland OS, Brekken C, Davies Cde L. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res (2004) 64:4768–73. doi:10.1158/0008-5472.CAN-03-1472
139. Willett CG, Boucher Y, Di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med (2004) 10:145–7. doi:10.1038/nm988
140. Tailor TD, Hanna G, Yarmolenko PS, Dreher MR, Betof AS, Nixon AB, et al. Effect of pazopanib on tumor microenvironment and liposome delivery. Mol Cancer Ther (2010) 9:1798–808. doi:10.1158/1535-7163.MCT-09-0856
141. Ning S, Tian J, Marshall DJ, Knox SJ. Anti-alphav integrin monoclonal antibody intetumumab enhances the efficacy of radiation therapy and reduces metastasis of human cancer xenografts in nude rats. Cancer Res (2010) 70:7591–9. doi:10.1158/0008-5472.CAN-10-1639
142. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res (2013) 73:2943–8. doi:10.1158/0008-5472.CAN-12-4354
143. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res (2013) 73:539–49. doi:10.1158/0008-5472.CAN-12-2325
144. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell (2005) 8:299–309. doi:10.1016/j.ccr.2005.09.005
145. Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, De Rosa F, et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol (2007) 25:1545–52. doi:10.1200/JCO.2005.05.1474
146. Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res (2000) 60:1878–86.
147. Mainetti LE, Rico MJ, Fernandez-Zenobi MV, Perroud HA, Roggero EA, Rozados VR, et al. Therapeutic efficacy of metronomic chemotherapy with cyclophosphamide and doxorubicin on murine mammary adenocarcinomas. Ann Oncol (2013). doi:10.1093/annonc/mdt164. [Epub ahead of print].
Keywords: VEGF, permeability, VE-cadherin, endothelial barrier, tumor angiogenesis
Citation: Azzi S, Hebda JK and Gavard J (2013) Vascular permeability and drug delivery in cancers. Front. Oncol. 3:211. doi: 10.3389/fonc.2013.00211
Received: 29 May 2013; Paper pending published: 23 June 2013;
Accepted: 01 August 2013; Published online: 15 August 2013.
Edited by:
Fabrizio Marcucci, Istituto Superiore di Sanità, ItalyReviewed by:
Ronald Berenson, Compliment Corporation, USARaffaella Giavazzi, Istituto di Ricerche Farmacologiche “Mario Negri” – IRCCS, Italy
Copyright: © 2013 Azzi, Hebda and Gavard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie Gavard, Institut Cochin, 22 rue Mechain, Room 306, Paris 75014, France e-mail:anVsaWUuZ2F2YXJkQGluc2VybS5mcg==
†Sandy Azzi and Jagoda K. Hebda have contributed equally to this work.