

- Department of Medical Oncology, Harvard Medical School, Dana-Farber Cancer Institute, Boston, MA, USA
Recent developments in the study of epithelial ovarian cancer have called into question the traditional views regarding the site of tumor initiation. Histopathologic studies and genomic analyses suggest that extra-ovarian sites, like the fallopian tube, may harbor the coveted cell of origin and could therefore contribute significantly to the development of high-grade serous ovarian carcinoma (HG-SOC). Our ability to validate these emerging genomic and pathologic observations and characterize the early transformation events of HG-SOC hinges on the development of novel model systems. Currently, there are only a handful of new model systems that are addressing these concerns. This review will chronicle the convergent evolution of these ovarian cancer model systems in the context of the changing pathologic and genomic understanding of HG-SOC.
Introduction
In 2013, the American Cancer Society estimates that 22,240 women will receive a new diagnosis of ovarian cancer and that 14,030 women will die from this disease, making ovarian cancer the most lethal gynecological malignancy in the United States (1). Of these newly diagnosed cases, 80% of the serous ovarian carcinomas are diagnosed at late stage, for which the 5-year survival rate is only 9–35% (2). Despite advancements in technology, this poor survival rate has been consistent over the last 30 years, an indictment of the complexity of this disease. In order to combat this clinical challenge, it is imperative to generate robust early detection methods and novel treatment options.
Many of the characteristics confounding the study of ovarian cancer arise from the disease’s heterogeneity. Ovarian tumors can arise from three different cell types; epithelial, germ, and sex cord stromal cells, with epithelial accounting for approximately 90% of all ovarian cancers (1). Epithelial tumors are further grouped into different tumor types: Type I and Type II. Type I tumors include low-grade serous carcinoma, low-grade endometrioid carcinoma, mucinous carcinoma, and a subset of clear cell carcinomas, which develop in a stepwise fashion from well-recognized precursors, in most cases, borderline tumors (3–5) (Figure 1). These tumors are slow to develop and are generally confined to the ovary (6). Type I tumors are also genetically stable, with each histologic subtype corresponding to a distinct genetic profile (4–6). In contrast, Type II tumors encompass high-grade serous carcinoma, undifferentiated carcinoma, malignant mixed mesodermal tumor (carcinosarcoma), and some clear cell carcinomas (3) (Figure 1). High-grade serous carcinomas are the most common Type II tumor. These tumors progress rapidly, harbor TP53 mutations, and exhibit widespread DNA copy number alterations (3–7).
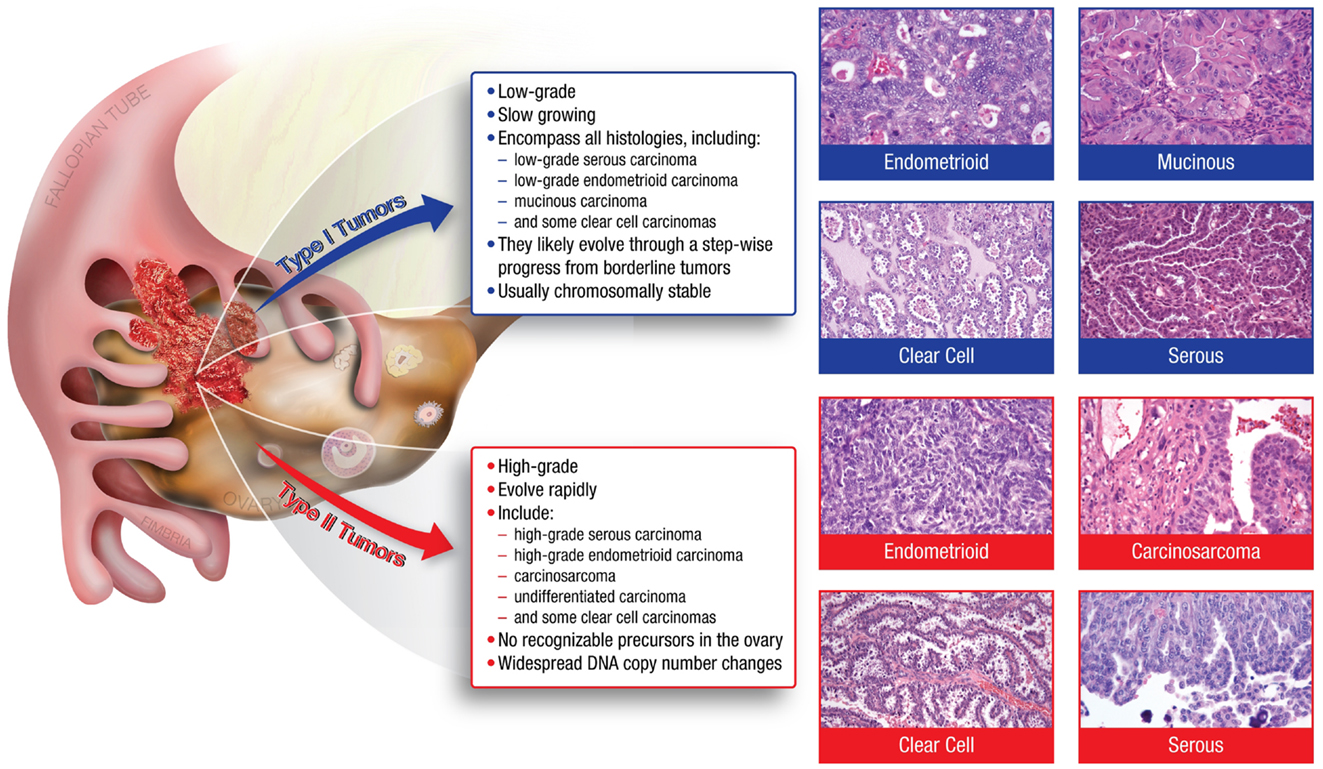
Figure 1. The major histologic subtypes of ovarian cancer fall into two subclassifications. Type I tumors are low-grade, slow growing carcinomas that typically arise from well recognized precursors lesions (borderline tumors) that themselves develop from the ovarian surface epithelium, inclusion cysts, or endometriosis. In contrast, Type II tumors are high-grade and rapidly growing carcinomas. Typically, they have spread well beyond the ovary at the time of diagnosis.
This new appreciation of tumor diversity and the rapid development of genomic technologies have helped redefine “ovarian cancer.” As the field grapples with these emerging concepts, experimental model systems will likely play a vital role in defining new opportunities for early detection and therapeutic intervention. This review will highlight the recent advancements in ovarian cancer genetics and pathology, and explore the past and present model systems employed to study high-grade serous ovarian carcinoma (HG-SOC).
Genetics
Until recently, neoplastic transformation was thought to be driven by the sequential acquisition of mutations in critical genes. For many epithelial cancers, including Type I ovarian cancer, this is true. The most prominent mutations present in Type I tumors include alterations to KRAS, BRAF, PTEN, CTNNB1, and TGFBR2 (3, 6, 8). However, besides mutations in the TP53 tumor suppressor gene and the BRCA1 or BRCA2 genes, very few recurrent somatic mutations have been associated with the more aggressive Type II tumors (6). This inability to systematically characterize Type II tumors was addressed by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) in the creation of The Cancer Genome Atlas (TCGA). In the TCGA’s pilot study of HG-SOC, microarray analyses and new sequencing technology were used to publish the largest and most comprehensive genetic analysis of HG-SOC. The study encompassed mRNA expression, microRNA expression, DNA copy number, and DNA promoter region methylation for 489 HG-SOC and whole exome DNA sequence information for 316 of these samples (7).
Results from the initial TCGA study characterized HG-SOC as having TP53 mutations in nearly 100% of tumors and identified low prevalence but statistically significant recurrent somatic mutations in nine additional genes including NF1, BRCA1, BRCA2, RB1, and CDK12 (7). TCGA also described 113 DNA copy number alterations and implicated 168 genes involved in promoter methylation events (7). Considering the widespread DNA copy number aberrations observed across HG-SOC, it has been suggested that disruption of DNA repair pathways followed by chromosome instability is a viable model for the early progression of HG-SOC (9, 10). The TCGA provides an expanding database that is useful in identify high impact genes. However, because the TCGA studies the advanced state of HG-SOC, determining whether these genes are important to transformation, or instead are related to tumor maintenance, immune evasion, anti-apoptosis, and/or chemoresistance, requires further investigation.
Pathogenesis
Historically, ovarian cancer was believed to originate from the ovarian surface epithelium (OSE), where ovulation, follicular rupture, oocyte release, cytokine exposure, and reactive oxygen species introduce DNA damage into the ovarian epithelial layer (11, 12). Proposed back in 1971, the Fathalla “incessant ovulation” hypothesis (13) suggests that over a woman’s lifespan, the accrual of DNA damage and the development of cortical inclusion cysts (CICs) results in Mullerian metaplasia of the coelomic epithelium followed by neoplastic transformation (14, 15). This hypothesis attempts to explain the presentation of coexisting serous and non-serous tumor subtypes within ovarian tumors and incorporates the epidemiological data linking ovulatory activity with risk of ovarian cancer (16). However, while precursor lesions have been identified in the OSE that are linked to Type I tumors (17), reproducible pre-malignant lesions have been difficult to identify in the OSE for the high-grade Type II tumors.
A more recent analysis compares the major subtypes of ovarian carcinomas to tumors arising in the fallopian tube, endometrium, and endocervix. Evidence suggests that benign structures derived from these anatomic locations may serve as sites of origin for all tumors that have traditionally been regarded as of primary ovarian origin. Such epithelial structures, which include endosalpingiosis, endometriosis, and endocervicosis, represent non-neoplastic counterparts of serous, endometrioid/clear cell, and mucinous ovarian carcinomas, respectively, and are referred to as extra-uterine Müllerian epithelium (EUME) (15).
The most significant studies supporting the concept of EUME are those implicating the fallopian tube fimbria as the site of origin for high-grade serous carcinomas. Early studies of fallopian tube carcinomas noted TP53 and interleukin 6 (IL-6) mutations (18, 19). However, a link to ovarian cancer was not proposed until pathologists systematically analyzed fallopian tubes from women carrying mutations in the BRCA1 tumor suppressor gene. These studies identified preneoplastic lesions localized to the tubal fimbria (20–22), where they displayed secretory cell histology, DNA damage, mutations in TP53, and stable p53 protein expression (20, 23). This evidence suggests that HG-SOC tumor progression within the fallopian tube fimbria begins with TP53 mutations (p53 signatures), evolves to serous tubal intraepithelial carcinoma (STIC), and eventually transforms and metastasizes to the ovary presenting as HG-SOC (20, 24) (Figure 2).
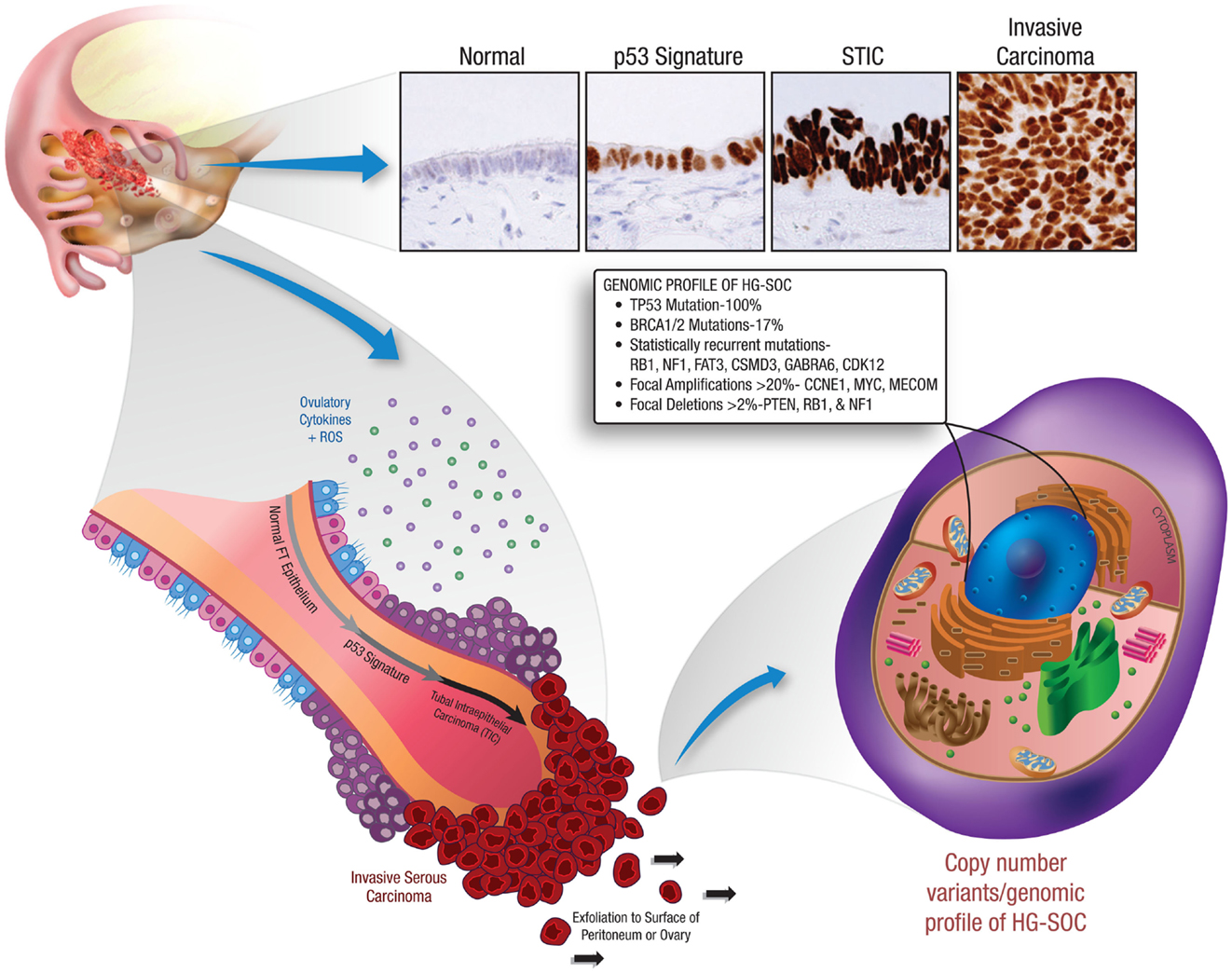
Figure 2. Early tumor progression within the fallopian tube and the resultant genetic profile of HG-SOC. This illustration depicts the recently identified precursor lesions of HG-SOC that are present in the fallopian tube. Mutations in the TP53 tumor suppressor gene are a very early event in the pathogenesis of HG-SOC, occurring exclusively in benign-appearing secretory cells. These preneoplastic lesions are referred to as ‘p53 signatures’. Acquisition of a neoplastic phenotype and proliferative capacity results in the development of serous tubal intraepithelial carcinoma (STIC). Breaching of the basement membrane and localized dissemination to the ovary and/or peritoneal cavity heralds the development of invasive HG-SOC and the associated clinical scenario. HG-SOCs that involve the ovary or peritoneum are characterized by mutations in TP53 (and BRCA1 in familial cases) and display a complex genomic terrain with widespread copy number alterations throughout the genome.
Experimental Models of HG-SOC
Experimental models systems in ovarian cancer biology have evolved significantly over the past 10–15 years. Today there exist a number of useful models that continue to advance translational research in ovarian cancer (Figure 3). It is beyond the scope of this mini-review to address all available experimental models. However, in order to demonstrate the utility and evolution of these research tools, a select few of these models will be discussed.
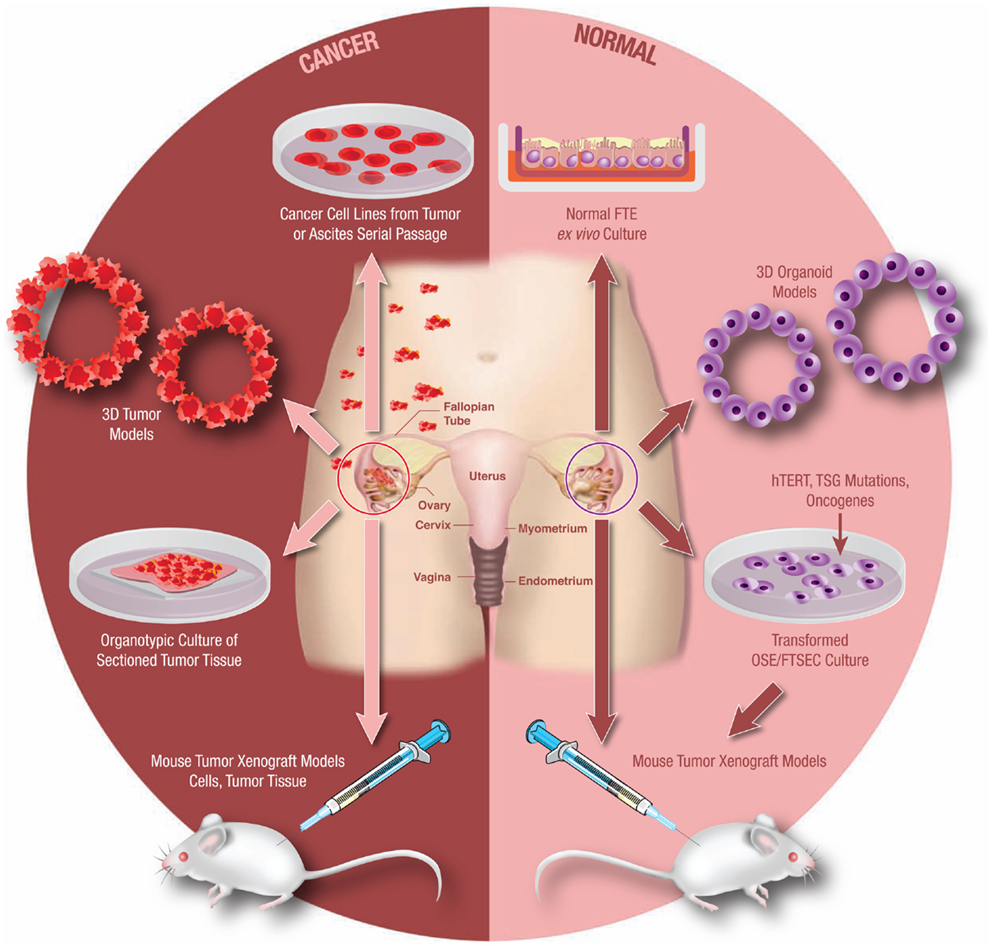
Figure 3. Model systems from primary human tissues. An array of experimental model systems spanning in vitro and in vivo approaches have been developed to study ovarian cancer. The models include platforms to interrogate the biology of cancer cells as well as for the study of benign epithelium. The expansion beyond traditional two-dimensional (2D) cell culture into 3D and organoid cultures has yielded important insights into the biology of this disease, as has the development of unique animal models. Development of these models is critical as our understanding of this cancer continues to evolve.
Xenografts
Xenograft models are possibly the most utilized experimental platform in the field of cancer research. Early developments of this model were reported in the late 1960s when Rygaard et al. found that mice suffering from recessive thymic aplasia could grow mammary and colon xenografted carcinomas (25). This effectively spawned the immunocompromised rodent model, which, due to its ease of application and histological insights, provided an extensive tool to study ovarian cancer tumorigenesis, chemotherapeutics, and biomarkers (26–29).
Despite its utility, questions still remain whether compromising the rodent immune system affects the tumor microenvironment. Various studies have shown that cell lines implanted in immune-compromised mice can lose their histological fidelity (30–33). Likewise, monitoring disease formation and progress is also complicated with immune-compromised mice, as disease and infection rates increase when mice are handled outside their protective environment. To combat this, a small number of syngeneic models have been developed (34, 35). However, this digression from human disease presents its own complications when translating experimental results into the clinical setting.
The location of tumor formation and its histologic fidelity to the human disease is also a concern when using xenograft models. The bursal membrane in rodents encapsulates the ovary and creates a unique microenvironment unlike the human equivalent. By acting as a barrier to the peritoneal cavity, the bursal membrane could hinder the development of these tumors (36). In addition, the anatomy of the murine reproductive system departs from that of humans and contains a bicornuate uterus with the fallopian tubes embedded in the aforementioned bursa. Furthermore, the intermittent ovulatory cycle of the mouse corresponds to its rare development of spontaneous ovarian carcinomas (37).
Even with these limitations, xenograft models are still important in translational research and have broad utility. All drug treatments must show promise in animal studies prior to investigation in human clinical trials. In addition, because a high priority has been placed on characterizing the early events of HG-SOC, xenograft models can be effectively paired with in vitro transformation studies to characterize preneoplastic and metastatic events (Figure 3). Karst et al. demonstrated this by confirming the transformative and metastatic potential of fallopian tube secretory epithelial cells (FT-SECs) in nude mice (38). Considering this versatility and practicality, the future use of xenograft models in ovarian cancer research is a certainty.
Cell Culture Models
OSE models
Prior to 1981, the isolation of untransformed primary ovarian tissue within the laboratory was unprecedented, making it difficult to discern molecular events related to transformation. This changed in 1981 when Adams and Auersperg isolated and transformed rat OSE (ROSE) cells with the Kirsten murine sarcoma virus (Ki-MSV) (39). The impact of this initial study led to the optimization of cell culture techniques (40, 41) and prompted investigators to start creating a vast cell bank for future studies.
Investigators took advantage of this new technology in the early 1990s when a series of studies simulated incessant ovulation through repeated in vitro passaging of rodent OSE cells. Investigators found that primary ROSE and mouse OSE (MOSE) cells that had undergone serial propagation exhibited increased proliferative and tumorigenic properties (35, 42, 43). Further analyses indicated that these transformed cell lines displayed similar proliferative and genomic patterns observed in human tumors. This was the first comparative analysis between a transformed cell line and its primary parental line and provided supporting evidence for the Fathalla Hypothesis.
While these studies were limited to rodent OSE cells, studies involving isolated human OSE (HOSE) cells were also being attempted (44–46). However, unlike rodent OSE cells, HOSE cells have a limited growth potential in vitro and require genetic perturbations to increase cellular lifespan (Figure 3). In order to achieve immortalization, two important questions require constant attention; what are the pathways critical to immortalization and how can one alter those pathways without disrupting the normal function of the cell? Initially, these genetic perturbations were achieved via retroviral transduction of either the human papilloma virus E6/E7 oncogenes or the simian virus 40 T antigen (SV40-TAg) (46, 47). Cell lines generated through this method displayed increased proliferation without tumorigenicity and remained proliferative after multiple passages (46, 47). Additional retroviral constructs targeting hTERT, TP53, and RB have all been shown to be successful in the immortalization of primary HOSE cells (48–52).
The recent development of small interfering RNAs (siRNAs) has had an impact on ovarian cancer research as well. Primarily used to silence genes through the RNA interference pathway (RNAi), Yang et al. used siRNAs to immortalize OSE cells by targeting p53 (53) and Rb (54), while others have used siRNAs to explore the roles of PTTG (55), CD44 (56), and STAT3 (57). Certain investigators have even looked into siRNAs as a therapeutic agent. Huang et al. showed that by using a lipidoid-mediated delivery of siRNAs targeting CLDN3, OVCAR-3 xenografts showed reduced proliferation, metastasis, and tumor growth (58). The benefits of siRNAs include ease of application and more rapid results. However, specificity and cell toxicity have been a concern.
Fallopian tube models
The first fallopian tube epithelial cell (FTEC) culture system, developed for the purpose of studying the susceptibility of this epithelium to neoplastic transformation, was described in 2010 (59). Unlike traditional two-dimensional (2D) submerged cultures, this “ex vivo” system allows FTECs to grow at the air-surface liquid interface (Figure 3). This in-turn preserves the natural orientation, architecture, polarity, extracellular features, and biological functions of in vivo FTECs, including the retention of ciliated and secretory cells (59). Considering these advantages, this model is ideal to explore the stresses of hormone exposure, ovulation, and inflammatory response. In fact, Levanon et al. reported that in response to DNA damage the FT-SECs display delayed DNA repair kinetics compared to their ciliated cell neighbors (59). This makes secretory cells more sensitive to DNA damage and could explain why FT-SEC are susceptible to neoplastic transformation, especially in the absence of key DNA repair proteins like BRCA1 or BRCA2 (9). Despite the strengths of this model, it has two major limitations. First, it is limited by the dependence on fresh primary FT tissue. Second, the ex vivo cultures cannot be further propagated in culture. While they remain viable for weeks, they are not a renewable resource.
To alleviate the need for fresh tissue samples, and to create a long term self-propagating cell population, Karst et al. utilized fresh fallopian tube samples to create the first FT-SEC line (38). By transducing hTERT and either SV40-TAg or an shRNA targeting p53 and mutant CDK4R24C, FT-SECs were able to overcome senescence and apoptosis (38). Further transduction of either HRAS or an shRNA targeting the B56γ subunit of protein phosphatase 2A (PP2A-B56γ) and c-Myc resulted in an increase in proliferation, anchorage independent growth, and tumor formation in implanted nude mice (38).
Jazaeri and colleagues reported similar results by administering an oncogenic retroviral cocktail containing a myriad of known oncogenes to primary FT-SEC (60). After a period positive selection due to proliferative advantages, the genetic profile of transformed FT-SECs was determined. Increased c-Myc, HRAS, hTERT, and SV40-TAg transgene expression and protein accumulation was observed. Further experimentation showed that hTERT and SV40-TAg expression was sufficient to overcome senescence without tumor formation in nude mice (60). This confirmed the findings of Karst et al. showing that FT-SECs are a possible source for HG-SOC.
Further confirmation of these initial results was reported by Shan et al. These investigators immortalized human FT-SECs by overexpressing hTERT and SV40-TAg (61). However, when they transduced the cells with oncogenic HRAS and implanted them into nude mice, they observed tumor formation that resembled poorly differentiated mucinous adenocarcinomas rather than HG-SOC (61). This is consistent with recent reports showing that Type I, low-grade tumors can emerge from the fallopian tube as well (62, 63).
Recently, FTEC models have even stepped outside traditional human cultures and expanded to baboons and pigs. A recent study used baboon FTECs immortalized with SV40-TAg to study the effect of ovulation on FTEC proliferation (64). Likewise, porcine oviductal epithelial cells were used to optimize in vitro cell culture conditions to maintain de novo FTEC morphological features, i.e., secretory and ciliated cells (65). These new methods could prove useful as investigations into the FTEC continue to increase.
Conditionally reprogramed cells
An alternative to transgene immortalization is a newly developed technique where epithelial cells are “reprogramed” into a stem cell state through conditioned media. Schlegel et al. has been able to show that primary human prostate, liver, lung, and breast epithelial cells, when co-cultured with irradiated fibroblast feeder cells in the presence of the rho-kinase inhibitor Y-27632, can undergo unlimited expansion without senescence or apoptosis (66). This increase in cell proliferation is accompanied by the up regulation of stem cell markers and a decrease in Notch signaling (66). Even more intriguing is that this phenotype is reversible. The removal of Y-27632 and feeders results in the re-differentiation of cells accompanied with their natural polarity and orientation (66). Similarly, Ince et al. showed that human mammary epithelial cells (HMECs) are able to grow indefinitely in a serum-free, chemically defined medium termed WIT (67). The optimization of these techniques for either OSE cells or FT cells would be ideal and may eliminate transgene manipulations and reduce potential off-target effects.
Genome engineering
Despite its successes, certain drawbacks to retroviral transduction and RNAi systems, like oncogenic effects, toxicity, and off-target effects, have prompted investigators to develop targeted genome editing systems. The application of custom DNA-binding proteins, like transcription activator-like effector nuclease (TALENs) and the clustered regularly interspaced short palindromic repeats (CRISPR)/cas genome editing systems, have produced a flurry of papers within the last few years. TALENs use a restriction enzyme engineered to recognize specific DNA sequences through the fusion of a TAL effector DNA-binding domain (68). Once a gene is targeted, double strand breaks (DSBs) are introduced and non-homologous end joining occurs (68). However, TALENs are expensive to develop and suffer from off-target effects. The CRISPR/cas system provides a cheaper alternative and works in a similar manner. By utilizing endonucleases that use dual-RNAs for site-specific DNA cleavage, investigators are able to exploit the CRISPR/cas system for RNA-programmable genome editing (69). This had been shown to be very effective and site-specific in controlling gene expression and introducing genetic mutations (70). Overall, despite its lack of validation and limited use with ovarian cancer models, promising results should spark interest and new avenues of investigation.
3D cultures
In addition to conventional 2D culture systems, three-dimensional (3D) culture systems have become increasingly common. There are five major types of 3D culture systems: scaffold free for spheroid growth, scaffolds, gels, bioreactors, and microchips. In these settings, investigators concentrate on creating a more realistic environment where cells of interest can interact with surrounding tissues (71, 72). This seems relevant as studies show that differences in chemosensitivity, cell invasion, and protein expression exist when epithelial ovarian cancer cells are cultured in either 2D or 3D conditions (73–76). Difficulties associated with these models include cell removal, gelling variations, cost, and commercial availability, although the further optimization of these techniques should yield a host of useful tools.
Animal Models of Ovarian Cancer
Genetically engineered mouse models
In contrast to cell culture platforms, which rely on an artificial environment, genetically engineered mouse models are an efficient alternative for genetic modification and tumor observations in vivo. This is important, as questions regarding the identity of cell lines and the selective pressures of cell culture systems continue to surface (77, 78). In addition, investigators have a broad range of techniques to introduce genetic alterations in a temporal or spatial-dependent manner. These methodologies employ transgenic elements, RNAi technologies, and viruses to create both loss of function and gain of function traits within mice.
Limitations to these models include random integration of transgenic elements, limited tissue specific promoters, and difficulties achieving both spatial and temporal control simultaneously. In addition to experimental difficulties, it is also challenging to accurately mimic the human disease in rodents. For example, mice require fewer genetic alterations for tumor induction compared to humans (79–81). Furthermore, rodent tumors that are produced from defined genetic mutations do not always resemble their human counterparts (79–81). The HRAS oncogene is a prime example of this anomaly. Hamad et al. showed that the mechanisms of Ras-induced transformation in mice differ when compared to the mechanism of Ras-induced transformation in humans (79). By systematically comparing the murine and mammalian transformation pathways investigators highlighted a critical disadvantage to non-human model systems; the genetic and molecular disconnect between animal models and human disease. However, the ability to validate gene function and test novel therapeutics in a relevant microenvironment, when paired with relevant human studies, still makes these models especially useful (82).
Mouse OSE
Like other model systems, the initial ovarian cancer mouse models focused on the OSE (5, 83–85). The first ovarian cancer transgenic mouse model was developed in 2002 (86). By inducing the expression of the avian tumor virus receptor A (TVA) through the control of the keratin-5 promoter, these investigators were able to create a cell population within the mouse that was vulnerable to avian retrovirus infection (86). However, the transient expression of keratin-5 required OSE viral infection to occur in vitro with subsequent transplantation. Despite this drawback, infection with different combinations of c-Myc, AKT, and KRas, produced tumors in OSE cells harvested from TVAp53−/−mice and provided the first successful transgenic analysis of ovarian cancer in mice (86).
A more specific promoter, the Mullerian Inhibitory Substance Type II Receptor (MISIIR), was later identified and used by Connolly et al. to drive gynecological tissue specific transgene expression of SV40-TAg in mice resulting in the formation of ovarian carcinoma in 50% of the transgenic founders (87). However, aggressive tumor formation prohibited the study of early stage tumors and prevented reproduction. Additional studies utilizing the MISIIR promoter explored the oncogenic properties of PTTG and PIK3CA, however both had difficulties producing tumors (88, 89).
Rather than identify a specific promoter, some investigators have employed the Cre-loxP method to deliver specific genetic alterations (90). Administration of the Cre recombinase can be achieved either through injection of a viral vector (AdCre) or by crossing with a mouse generated to express the protein. This model is a clever way to circumvent problems inherent to typical transgenic models and has been used study TP53 and Rb (91), KRas and PTEN (92), PTEN and APC (93), and BRCA1 (94) within the context of ovarian cancer.
Most recently, Flesken-Nikitin and colleagues applied the AdCre system to perturb p53 and Rb in a stem cell niche in the transitional zone of the bursal cavity of mice. With p53 and Rb inactivated, these stem cells in the hilum region showed the earliest signs of transformation (95). However, perhaps the more interesting aspect of this study was the reporter mouse developed to characterize the fate of the hilum stem cells. A stem cell marker (LRG5), specific to the hilum region, was used to drive specific expression of CreERT2. In turn, subsequent tamoxifen (TAM) administration created a traceable knocked-in fluorescent probe. Results indicated that hilum cells do have the potential to repopulate the ovarian surface and suggest that stem cell niches could contribute to HG-SOC (95). Whether the hilar cells are OSE cells, or a different cell type altogether, remains to be seen. It is also worth noting that, since there is no bursa surrounding the human ovaries, it is not clear whether there is an equivalent structure or cell type in humans.
Overall, the mouse models developed thus far have focused on the OSE and some have exhibited difficulties with tumorigenicity, female reproduction, anatomical anomalies, and transient expression. Likewise, while these models have offered insight into genes that are important to transformation, they have not provided insight into HG-SOC preneoplastic lesions as such lesions have yet to be identified in the ovary. We anticipate that animal models that target the fallopian tube secretory cell will provide additional insights.
Mouse fallopian tube
The first mouse model targeting the extra-ovarian Mullerian epithelium was developed by Miyoshi et al. By exploiting the promoter of the murine oviduct-specific glycoprotein, Miyoshi was able to drive expression of the SV40-TAg in the oviduct, uterus, vagina, and ovary. Except for the ovary, subsequent tumor formation throughout the female reproductive tract was observed (96). Tumor formation was reduced in ovariectomized mice, but when estradiol was injected subcutaneously a dramatic increase in hyperplasia of the extra-ovarian Mullerian epithelia was observed (96). This suggests that ovarian cancer could originate outside the ovary, and that these preneoplastic lesions are highly reliant on hormone regulation pathways involving the ovary.
More recently, Kim et al. disabled DICER and PTEN using the anti-Mullerian hormone receptor type 2-directed Cre (Amhr2-Cre) (97). HG-SOC with aggressive metastasis was observed in these mice resulting in 100% death. In addition, the fallopian tube displayed the earliest lesions and cancer was prevented when the fallopian tube was removed at an early age (97). However, the first signs of increased proliferation within the fallopian tube appear to reside in the stromal compartment, counterintuitive to the epithelial properties presented in the advanced HG-SOC. Equally vexing was the low p53 expression in mouse tumors, a protein known to be mutated and highly expressed in almost 100% of human tumors (7).
Other animal models
An alternative to the mouse model, which has dominated the field since its initial use, is the domestic laying hen. The hen is the only animal identified to spontaneously develop HG-SOC that is histologically and morphologically similar to human HG-SOC (98). Likewise, because ovarian cancer of the hen presents so many similarities to human ovarian cancer, there are many opportunities to explore early preneoplastic lesions, chemopreventive trials, and perform genomic analyses (99). Disadvantages include a lack of reagents and genetic manipulation technologies that target the hen, as well as anatomical discrepancies (99).
Conclusion
Our understanding of ovarian cancer has dramatically changed in the last 10 years. In our search for a cell of origin, our evolving knowledge about the pathogenesis of the disease has led us to sites neighboring the ovary. At the same time, we now appreciate that this is a heterogeneous disease with a complex genomic landscape. In particular, HG-SOCs are marked by surprisingly few recurrent somatic mutations. Instead, this tumor exhibits a complex genome marked by copy number alterations so widespread that few other cancer types mirror its complexity. The challenge now is to elucidate the key alterations related to tumorigenesis, tumor viability, and chemotherapy resistance. In order to achieve this goal, experimental model systems must take center stage and continue to evolve to meet the demanding needs of the scientific community.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. Kevin Elias and Marian Novak for helpful comments on the manuscript, Dr. Michelle Hirsch for histologic images, Michael Cooper for medical illustrations (Cooper Graphics: www.Cooper247.com), and the members of the Drapkin laboratory for fruitful discussions. This work was supported by grants from the National Cancer Institute at the NIH P50-CA105009 (Drapkin Ronny), NIH U01 CA-152990 (Drapkin Ronny), NIH R21 CA-156021 (Drapkin Ronny); the Honorable Tina Brozman “Tina’s Wish” Foundation (Drapkin Ronny), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (Drapkin Ronny), The Robert and Debra First Fund (Drapkin Ronny), and The Gamel Family Fund (Drapkin Ronny). Paul Michael Jones was supported by a grant from Kaleidoscope of Hope Foundation.
References
1. Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi:10.3322/caac.21166
2. Karst AM, Drapkin R. The new face of ovarian cancer modeling: better prospects for detection and treatment. F1000 Med Rep (2011) 3:22. doi:10.3410/M3-22
3. Shih IeM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol (2004) 164(5):1511–8. doi:10.1016/S0002-9440(10)63708-X
4. Landen CN Jr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol (2008) 26(6):995–1005. doi:10.1200/JCO.2006.07.9970
5. Cho KR, Shih IeM. Ovarian cancer. Annu Rev Pathol (2009) 4:287–313. doi:10.1146/annurev.pathol.4.110807.092246
6. Kurman RJ, Shih IeM. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol (2010) 34(3):433–43. doi:10.1097/PAS.0b013e3181cf3d79
7. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474(7353):609–15. doi:10.1038/nature10166
8. Bast RC Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer (2009) 9(6):415–28. doi:10.1038/nrc2644
9. Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer (2010) 10(11):803–8. doi:10.1038/nrc2946
10. Berns EM, Bowtell DD. The changing view of high-grade serous ovarian cancer. Cancer Res (2012) 72(11):2701–4. doi:10.1158/0008-5472.CAN-11-3911
11. Murdoch WJ, Martinchick JF. Oxidative damage to DNA of ovarian surface epithelial cells affected by ovulation: carcinogenic implication and chemoprevention. Exp Biol Med (Maywood) (2004) 229(6):546–52.
12. Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol (2008) 26(32):5284–93. doi:10.1200/JCO.2008.18.1107
13. Fathalla MF. Incessant ovulation – a factor in ovarian neoplasia? Lancet (1971) 2(7716):163. doi:10.1016/S0140-6736(71)92335-X
14. Resta L, Russo S, Colucci GA, Prat J. Morphologic precursors of ovarian epithelial tumors. Obstet Gynecol (1993) 82(2):181–6.
15. Dubeau L. The cell of origin of ovarian epithelial tumours. Lancet Oncol (2008) 9(12):1191–7. doi:10.1016/S1470-2045(08)70308-5
16. Casagrande JT, Louie EW, Pike MC, Roy S, Ross RK, Henderson BE. “Incessant ovulation” and ovarian cancer. Lancet (1979) 2(8135):170–3. doi:10.1016/S0140-6736(79)91435-1
17. Ho CL, Kurman RJ, Dehari R, Wang TL, Shih IeM. Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer Res (2004) 64(19):6915–8. doi:10.1158/0008-5472.CAN-04-2067
18. Costa MJ, Vogelsan J, Young LJ. p53 gene mutation in female genital tract carcinosarcomas (malignant mixed Müllerian tumors): a clinicopathologic study of 74 cases. Mod Pathol (1994) 7(6):619–27.
19. Runnebaum IB, Tong XW, Möbus VJ, Kieback DG, Rosenthal HE, Kreienberg R. p53 mutant His175 identified in a newly established fallopian tube carcinoma cell line secreting interleukin 6. FEBS Lett (1994) 353(1):29–32. doi:10.1016/0014-5793(94)00953-8
20. Crum CP, Drapkin R, Miron A, Ince TA, Muto M, Kindelberger DW, et al. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol (2007) 19(1):3–9. doi:10.1097/GCO.0b013e328011a21f
21. Lee Y, Miron A, Drapkin R, Nucci MR, Medeiros F, Saleemuddin A, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol (2007) 211(1):26–35. doi:10.1002/path.2091
22. Crum CP. Intercepting pelvic cancer in the distal fallopian tube: theories and realities. Mol Oncol (2009) 3(2):165–70. doi:10.1016/j.molonc.2009.01.004
23. Salvador S, Rempel A, Soslow RA, Gilks B, Huntsman D, Miller D. Chromosomal instability in fallopian tube precursor lesions of serous carcinoma and frequent monoclonality of synchronous ovarian and fallopian tube mucosal serous carcinoma. Gynecol Oncol (2008) 110(3):408–17. doi:10.1016/j.ygyno.2008.05.010
24. Karst AM, Drapkin R. Ovarian cancer pathogenesis: a model in evolution. J Oncol (2010). doi:10.1155/2010/932371 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2739011/,
25. Rygaard J, Povsen CO. Heterotransplantation of a human malignant tumour to ‘nude’ mice. 1969. APMIS (2007) 115(5):604–6. discussion 607–8. doi:10.1111/j.1600-0463.2007.apm_689a.x
26. Ward BG, Wallace K. Localization of the monoclonal antibody HMFG2 after intravenous and intraperitoneal injection into nude mice bearing subcutaneous and intraperitoneal human ovarian cancer xenografts. Cancer Res (1987) 47(17):4714–8.
27. Hamilton TC, Young RC, Louie KG, Behrens BC, McKoy WM, Grotzinger KR, et al. Characterization of a xenograft model of human ovarian carcinoma which produces ascites and intraabdominal carcinomatosis in mice. Cancer Res (1984) 44(11):5286–90.
28. Massazza G, Tomasoni A, Lucchini V, Allavena P, Erba E, Colombo N, et al. Intraperitoneal and subcutaneous xenografts of human ovarian carcinoma in nude mice and their potential in experimental therapy. Int J Cancer (1989) 44(3):494–500. doi:10.1002/ijc.2910440320
29. Kelland LR, Jones M, Abel G, Valenti M, Gwynne J, Harrap KR. Human ovarian-carcinoma cell lines and companion xenografts: a disease-oriented approach to new platinum anticancer drug discovery. Cancer Chemother Pharmacol (1992) 30(1):43–50. doi:10.1007/BF00686484
30. Buick RN, Pullano R, Trent JM. Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Res (1985) 45(8):3668–76.
31. Möbus V, Gerharz CD, Press U, Moll R, Beck T, Mellin W, et al. Morphological, immunohistochemical and biochemical characterization of 6 newly established human ovarian carcinoma cell lines. Int J Cancer (1992) 52(1):76–84. doi:10.1002/ijc.2910520115
32. Molthoff CF, Calame JJ, Pinedo HM, Boven E. Human ovarian cancer xenografts in nude mice: characterization and analysis of antigen expression. Int J Cancer (1991) 47(1):72–9. doi:10.1002/ijc.2910470114
33. Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther (2004) 10(6):1032–42. doi:10.1016/j.ymthe.2004.08.013
34. Rose GS, Tocco LM, Granger GA, DiSaia PJ, Hamilton TC, Santin AD, et al. Development and characterization of a clinically useful animal model of epithelial ovarian cancer in the Fischer 344 rat. Am J Obstet Gynecol (1996) 175(3 Pt 1):593–9. doi:10.1053/ob.1996.v175.a73595
35. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis (2000) 21(4):585–91. doi:10.1093/carcin/21.4.585
36. Sale S, Orsulic S. Models of ovarian cancer metastasis: murine models. Drug Discov Today Dis Models (2006) 3(2):149–54. doi:10.1016/j.ddmod.2006.05.006
37. Cline JM. Neoplasms of the reproductive tract: the role of hormone exposure. ILAR J (2004) 45(2):179–88. doi:10.1093/ilar.45.2.179
38. Karst AM, Levanon K, Drapkin R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc Natl Acad Sci U S A (2011) 108(18):7547–52. doi:10.1073/pnas.1017300108
39. Adams AT, Auersperg N. Transformation of cultured rat ovarian surface epithelial cells by Kirsten murine sarcoma virus. Cancer Res (1981) 41(6):2063–72.
40. Siemens CH, Auersperg N. Serial propagation of human ovarian surface epithelium in tissue culture. J Cell Physiol (1988) 134(3):347–56. doi:10.1002/jcp.1041340305
41. Kruk PA, Maines-Bandiera SL, Auersperg N. A simplified method to culture human ovarian surface epithelium. Lab Invest (1990) 63(1):132–6.
42. Godwin AK, Testa JR, Handel LM, Liu Z, Vanderveer LA, Tracey PA, et al. Spontaneous transformation of rat ovarian surface epithelial cells: association with cytogenetic changes and implications of repeated ovulation in the etiology of ovarian cancer. J Natl Cancer Inst (1992) 84(8):592–601. doi:10.1093/jnci/84.8.592
43. Testa JR, Getts LA, Salazar H, Liu Z, Handel LM, Godwin AK, et al. Spontaneous transformation of rat ovarian surface epithelial cells results in well to poorly differentiated tumors with a parallel range of cytogenetic complexity. Cancer Res (1994) 54(10):2778–84.
44. Gregoire L, Munkarah A, Rabah R, Morris RT, Lancaster WD. Organotypic culture of human ovarian surface epithelial cells: a potential model for ovarian carcinogenesis. In vitro Cell Dev Biol Anim (1998) 34(8):636–9. doi:10.1007/s11626-996-0012-z
45. Nitta M, Katabuchi H, Ohtake H, Tashiro H, Yamaizumi M, Okamura H. Characterization and tumorigenicity of human ovarian surface epithelial cells immortalized by SV40 large T antigen. Gynecol Oncol (2001) 81(1):10–7. doi:10.1006/gyno.2000.6084
46. Tsao SW, Mok SC, Fey EG, Fletcher JA, Wan TS, Chew EC, et al. Characterization of human ovarian surface epithelial cells immortalized by human papilloma viral oncogenes (HPV-E6E7 ORFs). Exp Cell Res (1995) 218(2):499–507. doi:10.1006/excr.1995.1184
47. Maines-Bandiera SL, Kruk PA, Auersperg N. Simian virus 40-transformed human ovarian surface epithelial cells escape normal growth controls but retain morphogenetic responses to extracellular matrix. Am J Obstet Gynecol (1992) 167(3):729–35. doi:10.1016/S0002-9378(11)91579-8
48. Li NF, Kocher HM, Salako MA, Obermueller E, Sandle J, Balkwill F. A novel function of colony-stimulating factor 1 receptor in hTERT immortalization of human epithelial cells. Oncogene (2009) 28(5):773–80. doi:10.1038/onc.2008.412
49. Li NF, Broad S, Lu YJ, Yang JS, Watson R, Hagemann T, et al. Human ovarian surface epithelial cells immortalized with hTERT maintain functional pRb and p53 expression. Cell Prolif (2007) 40(5):780–94. doi:10.1111/j.1365-2184.2007.00462.x
50. Davies BR, Steele IA, Edmondson RJ, Zwolinski SA, Saretzki G, von Zglinicki T, et al. Immortalisation of human ovarian surface epithelium with telomerase and temperature-sensitive SV40 large T antigen. Exp Cell Res (2003) 288(2):390–402. doi:10.1016/S0014-4827(03)00218-0
51. Kusakari T, Kariya M, Mandai M, Tsuruta Y, Hamid AA, Fukuhara K, et al. C-erbB-2 or mutant Ha-ras induced malignant transformation of immortalized human ovarian surface epithelial cells in vitro. Br J Cancer (2003) 89(12):2293–8. doi:10.1038/sj.bjc.6601423
52. Liu J, Yang G, Thompson-Lanza JA, Glassman A, Hayes K, Patterson A, et al. A genetically defined model for human ovarian cancer. Cancer Res (2004) 64(5):1655–63. doi:10.1158/0008-5472.CAN-03-3380
53. Yang G, Rosen DG, Mercado-Uribe I, Colacino JA, Mills GB, Bast RC Jr, et al. Knockdown of p53 combined with expression of the catalytic subunit of telomerase is sufficient to immortalize primary human ovarian surface epithelial cells. Carcinogenesis (2007) 28(1):174–82. doi:10.1093/carcin/bgl115
54. Yang G, Rosen DG, Colacino JA, Mercado-Uribe I, Liu J. Disruption of the retinoblastoma pathway by small interfering RNA and ectopic expression of the catalytic subunit of telomerase lead to immortalization of human ovarian surface epithelial cells. Oncogene (2007) 26(10):1492–8. doi:10.1038/sj.onc.1209905
55. El-Naggar SM, Malik MT, Kakar SS. Small interfering RNA against PTTG: a novel therapy for ovarian cancer. Int J Oncol (2007) 31(1):137–43.
56. Li CZ, Liu B, Wen ZQ, Li HY. Inhibition of CD44 expression by small interfering RNA to suppress the growth and metastasis of ovarian cancer cells in vitro and in vivo. Folia Biol (2008) 54(6):180–6.
57. Cai L, Zhang G, Tong X, You Q, An Y, Wang Y, et al. Growth inhibition of human ovarian cancer cells by blocking STAT3 activation with small interfering RNA. Eur J Obstet Gynecol Reprod Biol (2010) 148(1):73–80. doi:10.1016/j.ejogrb.2009.09.018
58. Huang YH, Bao Y, Peng W, Goldberg M, Love K, Bumcrot DA, et al. Claudin-3 gene silencing with siRNA suppresses ovarian tumor growth and metastasis. Proc Natl Acad Sci U S A (2009) 106(9):3426–30. doi:10.1073/pnas.0813348106
59. Levanon K, Ng V, Piao HY, Zhang Y, Chang MC, Roh MH, et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene (2010) 29(8):1103–13. doi:10.1038/onc.2009.402
60. Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, et al. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia (2011) 13(10):899–911.
61. Shan W, Mercado-Uribe I, Zhang J, Rosen D, Zhang S, Wei J, et al. Mucinous adenocarcinoma developed from human fallopian tube epithelial cells through defined genetic modifications. Cell Cycle (2012) 11(11):2107–13. doi:10.4161/cc.20544
62. Laury AR, Ning G, Quick CM, Bijron J, Parast MM, Betensky RA, et al. Fallopian tube correlates of ovarian serous borderline tumors. Am J Surg Pathol (2011) 35(12):1759–65. doi:10.1097/PAS.0b013e318233b0f7
63. Vang R, Shih IeM, Kurman RJ. Fallopian tube precursors of ovarian low-grade and high-grade serous neoplasms. Histopathology (2013) 62(1):44–58. doi:10.1111/his.12046
64. King SM, Hilliard TS, Wu LY, Jaffe RC, Fazleabas AT, Burdette JE. The impact of ovulation on fallopian tube epithelial cells: evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr Relat Cancer (2011) 18(5):627–42. doi:10.1530/ERC-11-0107
65. Miessen K, Sharbati S, Einspanier R, Schoen J. Modelling the porcine oviduct epithelium: a polarized in vitro system suitable for long-term cultivation. Theriogenology (2011) 76(5):900–10. doi:10.1016/j.theriogenology.2011.04.021
66. Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X, et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A (2012) 109(49):20035–40. doi:10.1073/pnas.1213241109
67. Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell (2007) 12(2):160–70. doi:10.1016/j.ccr.2007.06.013
69. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell (2013) 152(5):1173–83. doi:10.1016/j.cell.2013.02.022
70. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (2013) 339(6121):819–23. doi:10.1126/science.1231143
71. Xu F, Celli J, Rizvi I, Moon S, Hasan T, Demirci U. A three-dimensional in vitro ovarian cancer coculture model using a high-throughput cell patterning platform. Biotechnol J (2011) 6(2):204–12. doi:10.1002/biot.201000340
72. Lawrenson K, Benjamin E, Turmaine M, Jacobs I, Gayther S, Dafou D. In vitro three-dimensional modelling of human ovarian surface epithelial cells. Cell Prolif (2009) 42(3):385–93. doi:10.1111/j.1365-2184.2009.00604.x
73. Loessner D, Stok KS, Lutolf MP, Hutmacher DW, Clements JA, Rizzi SC. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials (2010) 31(32):8494–506. doi:10.1016/j.biomaterials.2010.07.064
74. Lee JM, Mhawech-Fauceglia P, Lee N, Parsanian LC, Lin YG, Gayther SA, et al. A three-dimensional microenvironment alters protein expression and chemosensitivity of epithelial ovarian cancer cells in vitro. Lab Invest (2013) 93(5):528–42. doi:10.1038/labinvest.2013.41
75. Touboul C, Lis R, Al Farsi H, Raynaud CM, Warfa M, Althawadi H, et al. Mesenchymal stem cells enhance ovarian cancer cell infiltration through IL6 secretion in an amniochorionic membrane based 3D model. J Transl Med (2013) 11:28. doi:10.1186/1479-5876-11-28
76. Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer (2007) 121(7):1463–72. doi:10.1002/ijc.22874
77. Korch C, Spillman MA, Jackson TA, Jacobsen BM, Murphy SK, Lessey BA, et al. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol Oncol (2012) 127(1):241–8. doi:10.1016/j.ygyno.2012.06.017
78. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun (2013) 4:2126. doi:10.1038/ncomms3126
79. Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev (2002) 16(16):2045–57. doi:10.1101/gad.993902
80. Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell (2005) 8(5):381–92. doi:10.1016/j.ccr.2005.10.014
81. Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell (2004) 6(2):171–83. doi:10.1016/j.ccr.2004.07.009
82. Cheon DJ, Orsulic S. Mouse models of cancer. Annu Rev Pathol (2011) 6:95–119. doi:10.1146/annurev.pathol.3.121806.154244
83. Garson K, Gamwell LF, Pitre EM, Vanderhyden BC. Technical challenges and limitations of current mouse models of ovarian cancer. J Ovarian Res (2012) 5(1):39. doi:10.1186/1757-2215-5-39
84. King SM, Burdette JE. Evaluating the progenitor cells of ovarian cancer: analysis of current animal models. BMB Rep (2011) 44(7):435–45. doi:10.5483/BMBRep.2011.44.7.435
85. Fong MY, Kakar SS. Ovarian cancer mouse models: a summary of current models and their limitations. J Ovarian Res (2009) 2:12. doi:10.1186/1757-2215-2-12
86. Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell (2002) 1(1):53–62. doi:10.1016/S1535-6108(01)00002-2
87. Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res (2003) 63(6):1389–97.
88. El-Naggar SM, Malik MT, Martin A, Moore JP, Proctor M, Hamid T, et al. Development of cystic glandular hyperplasia of the endometrium in Mullerian inhibitory substance type II receptor-pituitary tumor transforming gene transgenic mice. J Endocrinol (2007) 194(1):179–91. doi:10.1677/JOE-06-0036
89. Liang S, Yang N, Pan Y, Deng S, Lin X, Yang X, et al. Expression of activated PIK3CA in ovarian surface epithelium results in hyperplasia but not tumor formation. PLoS One (2009) 4(1):e4295. doi:10.1371/journal.pone.0004295
90. Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods (1998) 14(4):381–92. doi:10.1006/meth.1998.0593
91. Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of P53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res (2003) 63(13):3459–63.
92. Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med (2005) 11(1):63–70. doi:10.1038/nm1173
93. Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell (2007) 11(4):321–33. doi:10.1016/j.ccr.2007.02.016
94. Clark-Knowles KV, Garson K, Jonkers J, Vanderhyden BC. Conditional inactivation of Brca1 in the mouse ovarian surface epithelium results in an increase in preneoplastic changes. Exp Cell Res (2007) 313(1):133–45. doi:10.1016/j.yexcr.2006.09.026
95. Flesken-Nikitin A, Hwang CI, Cheng CY, Michurina TV, Enikolopov G, Nikitin AY. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature (2013) 495(7440):241–5. doi:10.1038/nature11979
96. Miyoshi I, Takahashi K, Kon Y, Okamura T, Mototani Y, Araki Y, et al. Mouse transgenic for murine oviduct-specific glycoprotein promoter-driven simian virus 40 large T-antigen: tumor formation and its hormonal regulation. Mol Reprod Dev (2002) 63(2):168–76. doi:10.1002/mrd.10175
97. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A (2012) 109(10):3921–6. doi:10.1073/pnas.1117135109
98. Fredrickson TN. Ovarian tumors of the hen. Environ Health Perspect (1987) 73:35–51. doi:10.1289/ehp.877335
Keywords: ovarian cancer, genetics, pathogenesis, model systems, OSE, fallopian tube
Citation: Jones PM and Drapkin R (2013) Modeling high-grade serous carcinoma: how converging insights into pathogenesis and genetics are driving better experimental platforms. Front. Oncol. 3:217. doi: 10.3389/fonc.2013.00217
Received: 30 June 2013; Paper pending published: 19 July 2013;
Accepted: 07 August 2013; Published online: 26 August 2013.
Edited by:
Viive Maarika Howell, University of Sydney, AustraliaCopyright: © 2013 Jones and Drapkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronny Drapkin, Dana-Farber Cancer Institute, Jimmy Fund Building, Room 215D, 450 Brookline Avenue, Boston, MA 02215, USA e-mail:cm9ubnlfZHJhcGtpbkBkZmNpLmhhcnZhcmQuZWR1