
 Madonna M. McManus
Madonna M. McManus
- Department of Pediatrics – Research, UT MD Anderson Cancer Center, Houston, TX, USA
The two most common primary bone malignancies, osteosarcoma (OS), and Ewing sarcoma (ES), are both aggressive, highly metastatic cancers that most often strike teens, though both can be found in younger children and adults. Despite distinct origins and pathogenesis, both diseases share several mechanisms of progression and metastasis, including neovascularization, invasion, anoikis resistance, chemoresistance, and evasion of the immune response. Some of these processes are well-studies in more common carcinoma models, and the observation from adult diseases may be readily applied to pediatric bone sarcomas. Neovascularization, which includes angiogenesis and vasculogenesis, is a clear example of a process that is likely to be similar between carcinomas and sarcomas, since the responding cells are the same in each case. Chemoresistance mechanisms also may be similar between other cancers and the bone sarcomas. Since OS and ES are mesenchymal in origin, the process of epithelial-to-mesenchymal transition is largely absent in bone sarcomas, necessitating different approaches to study progression and metastasis in these diseases. One process that is less well-studied in bone sarcomas is dormancy, which allows micrometastatic disease to remain viable but not growing in distant sites – typically the lungs – for months or years before renewing growth to become overt metastatic disease. By understanding the basic biology of these processes, novel therapeutic strategies may be developed that could improve survival in children with OS or ES.
Basics of Pediatric Bone Sarcomas
Osteosarcoma (OS) derives from primitive bone-forming mesenchymal cells and is the most common primary bone cancer (1). It occurs predominantly in growing adolescents and young adults, with a peak incidence at the age of 15–19 years. OS accounts for approximately 900 new diagnoses each year in the US, of which 15–20% patients present with overt lung metastases at initial diagnosis and about 40% patients develop metastases at a later stage (2, 3). Based upon the clinical outcomes of patients without overt metastasis at diagnosis during the pre-chemotherapy area, in which approximately 90% of patients developed lung metastasis 6–36 months later, it is presumed that the vast majority of apparently non-metastatic patients actually have micrometastatic disease at diagnosis. Current treatment including surgery, neoadjuvant, and adjuvant chemotherapy, has increased the overall survival rate for OS to around 70%. However, the clinical outcome for metastatic OS remains poor: fewer than 30% of patients presenting with metastases survive 5 years after initial diagnosis (4). More intensive chemotherapy protocols have not substantially increased long-term survival for patients with resistant or recurrent disease. Our knowledge of the mechanisms underlying OS metastasis is still limited. In order to improve the clinical outcomes for patients with poor prognosis, it is urgent to find new approaches to block metastasis in this disease.
Ewing’s sarcoma (ES) is the second most common bone malignancy in adolescents and young adults, after OS. This disease is most often characterized by a chromosomal translocation between chromosome 11 and 22, generating the EWS-FLI1 fusion gene (5). The protein encoded by this and other related EWS translocations acts as an aberrant transcription factor, driving the malignant behaviors of the transformed cell. ES has a peak incidence in the second decade of life, with a slightly higher rate in males (6, 7). Each year over 200 new cases are diagnosed in the United States. ES can develop at any site of the body, but it arises most frequently in bones and occasionally in soft tissues of the leg, pelvis, and chest wall (8). Similar to other pediatric sarcomas, nearly all mortality in patients with ES is caused by metastatic disease, not the primary tumor. Current therapy is directed toward treating both the primary tumor, since reducing tumor bulk prior to local control can result in better control, a less morbid procedure and better functional outcomes, and also at treating presumed microscopic metastasis. The intensive multimodal treatment with combination chemotherapy, surgery, and radiation has increased the 5-year event-free survival rate from less than 10% to over 70% (9–14).
The third most common type of primary bone malignancy is chondrosarcoma, which occurs mostly in adults and older teens. Chondrosarcoma is less well-studied than OS or ES, and there are few chondrosarcoma laboratory models in wide use. Since this condition is extremely rare in children, we will not address the biology of chondrosarcoma in this review.
Bone Sarcoma Tumorigenesis
Initiating Events
Consistent with its high incidence in adolescents, OS preferentially develops in the growth plates of the most rapidly growing bones such as the distal femur and proximal tibia (15). A key driver of OS pathogenesis is thought to be over-activation of signal transduction pathways initiated by various growth factors such as insulin-like growth factor (IGF), transforming growth factor (TGF), and connective tissue growth factor (CTGF). Previous studies indicated that IGF-I, IGF-II, and their receptors are of vital importance to malignant growth of OS (16– 18). High expression levels of TGF-β1 in OS samples has been associated with a worse prognosis, and inhibition of TGF-β signaling severely hindered OS cell proliferation (19, 20). CTGF has been shown to exert its pro-tumorigenic effects on OS cells via integrin signaling (21).
A variety of genetic and chromosomal alterations also contribute to the generation of OS. Germline and somatic mutations of the retinoblastoma tumor suppressor gene (Rb) and p53 pathways are among the most common genetic changes found in OS (22, 23). Inactivation of the Rb pathway is observed in up to 70% of primary OS tumors (24). Retinoblastoma patients who have inherited Rb mutations are predisposed to OS (25). Experimental mice with both p53 and Rb inactivated in osteoblast progenitors or in limb-bud tissues develop OS (26, 27). While cyclin-dependent kinase inhibitor p16INK4a acts upstream to promote Rb dephosphorylation and activation, deletion of this protein is detected in more than 10% of OS (28–31). The negative regulators of Rb, including cyclin-dependent kinase 4 (CDK4) and cyclin D1, have been found to be overexpressed in a small portion of high-grade OS (28, 30). The frequency of p53 mutations in OS is between 20 and 50% according to different reports and patients with Li–Fraumeni syndrome who carry inactivating mutations in p53 are at a much higher risk for developing OS than is the general population (32–34). MDM2, a major negative regulator of p53, is frequently amplified in OS (35–37). The p14ARF protein which activates p53 function by inhibiting MDM2-mediated p53 degradation is absent in about 10% of OS cases (38). In addition to the alterations of major tumor suppressor pathways, oncogene activation is also centrally involved in OS tumorigenesis. Elevated expression of proto-oncogenes c-myc and c-fos could be detected in the majority of OS and is associated with poor prognosis (36, 39, 40). Forced overexpression of c-myc in bone marrow stromal cells derived from Ink4a/Arf null mice leads to genesis of OS (41). Coexpression of c-fos and c-jun enhanced OS formation in transgenic mice (42). The activator protein-1 (AP-1) is a heterodimeric transcription factor complex composed of c-jun and c-fos. Papachistou et al. displayed that AP-1 activity is associated with the pathogenesis and progression of OS (43). In addition, upregulation of Notch signaling components has been shown to contribute to the pathogenesis and invasiveness of OS (44, 45). Other molecular events that have been indicated in the tumor initiation of OS include the RECQ helicase pathways and the telomere maintenance mechanisms (46). These genetic alterations are summarized in Table 1.
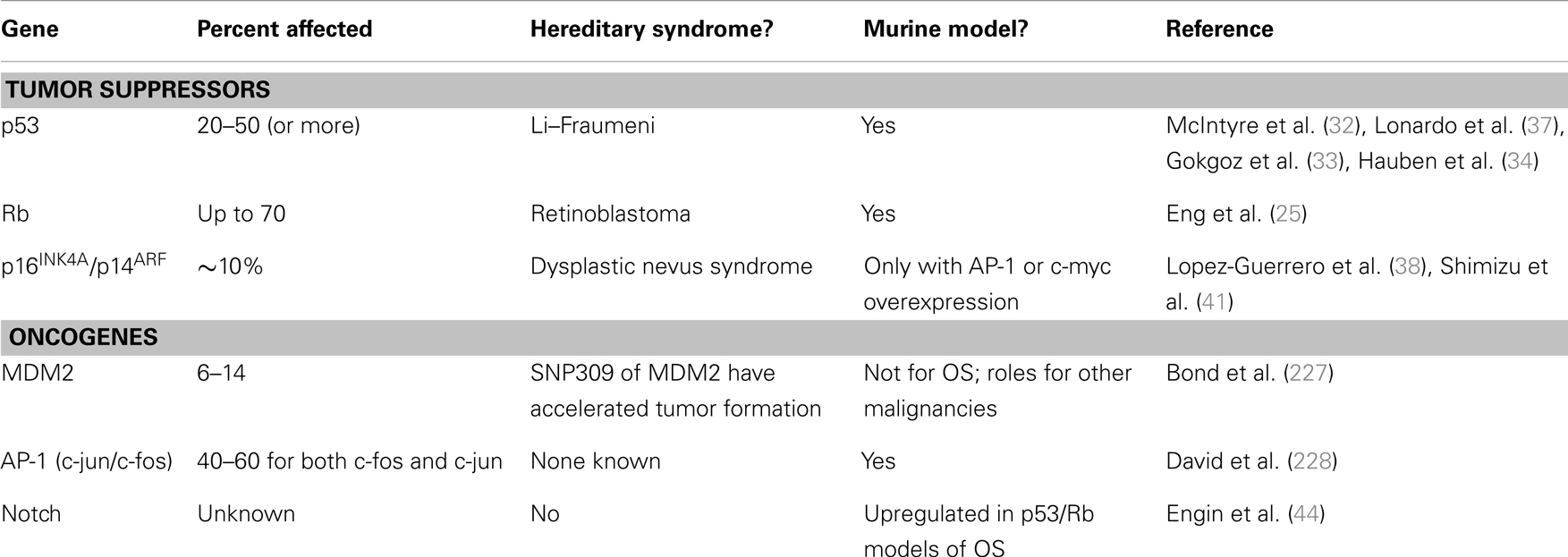
Table 1. Genetic alterations in osteosarcoma.
The vast majority of ES is characterized by the specific chromosomal translocation which fuses the EWSR1 gene on chromosome 22 and the FLI1 gene on chromosome 11 to form the chimeric EWS-FLI1 oncogene. It is believed that the EWS-FLI1 fusion protein is the master regulator of tumorigenesis in ES. It functions as a transcription factor and modulates multiple signaling pathways including Hedgehog/GLI, Wnt/β-catenin, IGF1/IGF1R and TGF-β, and Notch/p53 (47–52). According to previous studies, c-Myc, GLI1, cyclin D1, Cav-1, VEGFA, IGF1, NKX2-2, AURKA, EZH2, and NR0B1 are among the downstream targets of EWS-FLI1 that are upregulated in ES to promote cell survival and proliferation (53–56). Tumor suppressor genes such as NOTCH, p53, p21WAF/Cip1, p27Kip1, p57z, TGFBR2, IGFBP3 are downregulated by EWS-FLI1 to protect cells from growth arrest, senescence, and apoptosis (55). In addition to the formation of EWS-FLI1, other genetic alterations also contribute to pathogenesis of ES. Like OS and many other human cancers, p53 mutations have been detected in a small fraction of ES cases (57, 58). Amplification of MDM2 and Ras are also commonly found in ES tumor samples (59, 60). The CDKN2A locus where p14ARF and p16INK4a are located is lost in 15–30% of ES and is associated with poor clinical outcomes (60, 61).
Neovascularization
The process of blood vessel formation, or neovascularization, is comprised of angiogenesis and vasculogenesis. Tumor angiogenesis is the extension of blood vessels from preexisting vascular structures, while vasculogenesis is the de novo formation of vessel networks through the recruitment of bone marrow-derived precursor cells. Neovascularization is essential for sustained tumor growth and provides the systemic network that stimulates metastasis. Without the formation of supporting vasculature, tumor cells would be unable to obtain the nutrients and oxygen necessary for proliferation and would not be able to mediate metastatic spread. A delicately controlled balance between pro- and anti-angiogenic factors typically regulates angiogenesis; environmental stressors or genetic changes like hypoxia, acidosis, oncogene activation, and loss of tumor suppressor genes lead to dysfunction of this balance and result in angiogenesis.
Hypoxia-inducible factor-1 (HIF-1) is a key transcription factor that regulates the expression of genes responsible for the survival and adaptation of cells as they move from normoxia (∼21% O2) to hypoxia (∼1% O2). HIF-1 is made up of an oxygen related α subunit (HIF-1α) and a constitutive β subunit (HIF-1β) (62). The stability of HIF-1α is regulated by prolyl hydroxylase domain proteins (PHDs), while its transcription is regulated by factor inhibiting HIF (FIH). In normoxic and mildly hypoxic conditions, PHDs hydroxylate HIF-1α, resulting its association with von Hipper–Lindau (pVHL) ubiquitin E3 ligase complex allowing for rapid proteasomal degradation of HIF-1α (63–65). Under the extreme hypoxic conditions within a tumor, HIF-1α is stabilized and binds to the promoter region of VEGF where it mediates its upregulation (66). This signaling cascade can take place in both tumor cells and the non-malignant cells – tumor associated endothelial cells, etc. – that are found in the hypoxic center of tumors (67).
VEGF has been shown to be upregulated by a number of other factors that are released in response to the rapid proliferation of tumor cells; these include transforming growth factor α (TGF-α), fibroblast growth factor 2 (FGF-2), and hepatocyte growth factor (HGF) (68). Upregulation of VEGF also can be mediated by the transcription factor Wilms tumor protein 1 (WT1) (69). As the activation of growth factor receptors like EGFR and Integrin lead to Src activation, Ras/MAPK signaling and activation of the transcription factor STAT3 are initiated, allowing for cell cycle progression and proliferation (70, 71). STAT3 signaling is required for VEGF production and its activation results in a positive feedback loop that further increases the production of FGF and VEGF, leading to the increased induction of vascular permeability and neovascularization (72). Thus signaling by EGFR or other ERBB family kinases is upstream of VEGF release in most cases.
VEGF is the best characterized pro-angiogenic factor and is considered the most important factor involved in the development of the vasculature. There are a number of different VEGF molecules (VEGFA through VEGFE) that bind to VEGF receptors (VEGFR1-3). VEGFA binds to VEGFR2 and initiates a number of divergent signaling pathways (73). Among the proteins that are upregulated upon VEGF activation are the matrix metalloproteinase (MMP) and plasmin proteases (74), which act on the vascular network by breaking down the extracellular matrix (ECM) and allow for tumor cell invasion, as well as the migration of the precursor cells that give rise to vascular structures: pericytes and endothelial cells. These events are depicted in Figure 1. Additionally, VEGF signaling also induces the expression of the anti-apoptotic factors Bcl-2 and survivin (75), as well as the ERK/NF-kB and PI3K pathways (76). These effectors promote tumor cell proliferation and survival.
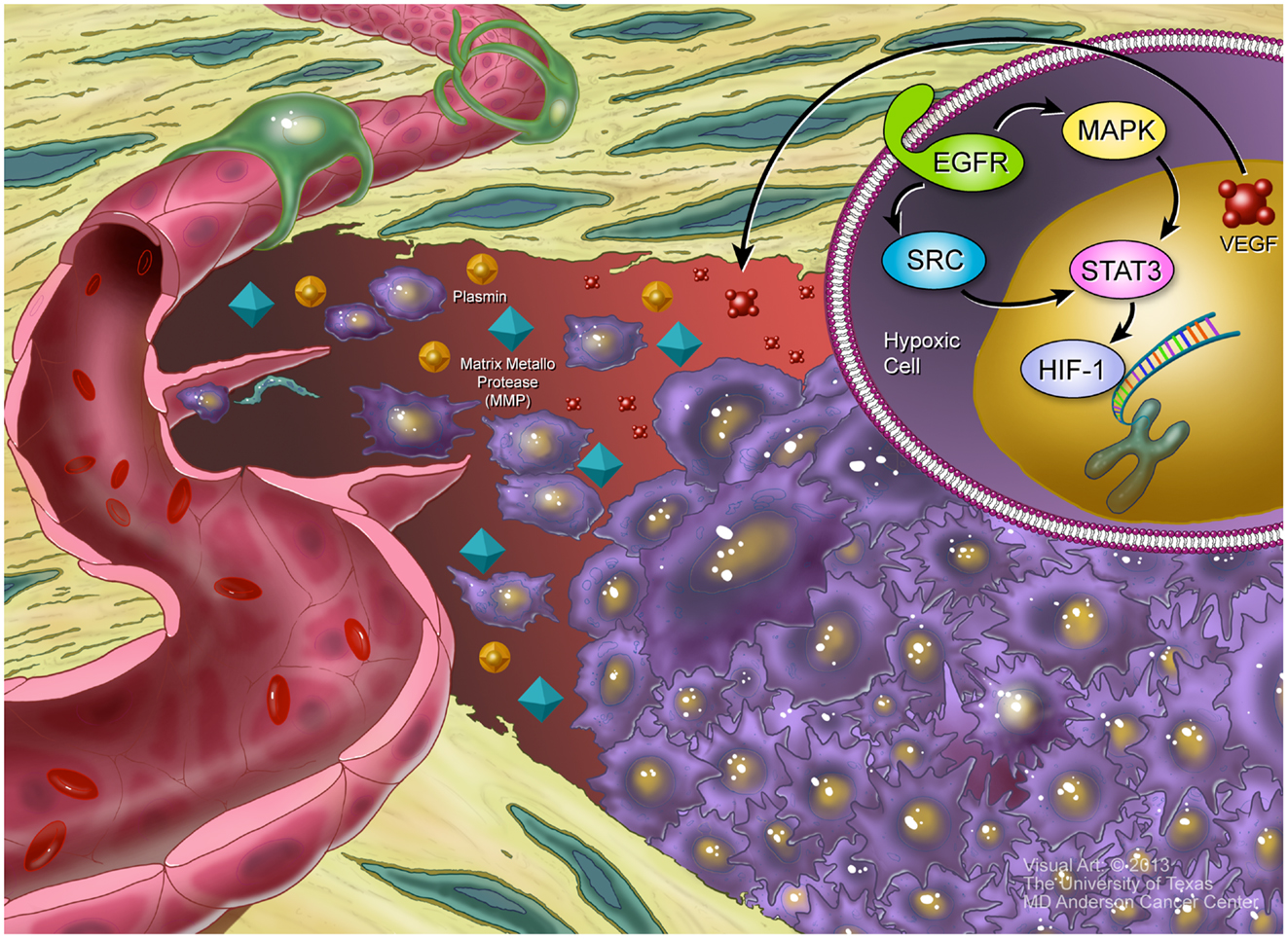
Figure 1. Hypoxia and angiogenesis. Inset, upper right: HIF1a has been stabilized in the hypoxic center of a tumor, allowing it to bind to the VEGF promotor. Additionally, EGFR signaling, acting through SRC and the MAPK cascade, induce STAT3, which helps promote VEGF transcription. Larger picture: the hypoxic tumor, in purple, releases VEGF that establishes a gradient from the tumor to the nearby blood vessel. VEGF stimulates release of MMPs and plasmin, which help mediate digestion of extracellular matrix proteins, facilitating migration of both endothelial cells and tumor cells. In response to the VEGF gradient, some endothelial cells acquire a “tip cell” phenotype, pushing long extensions toward the tumor. These tip cells will eventually form the new blood vessels that provide a blood supply to the growing tumor.
VEGF has been shown to be amplified in human OS (77). Elevated VEGF expression in OS has been associated with the development of lung metastases, compared to patients with VEGF-negative tumors (82 vs. 10% respectively), and VEGF-positive OS tumors have been shown to have significantly worse overall and disease-free survival rates (78–80). Patients with ES have increased circulating VEGF levels compared to controls (79, 81, 82). Interestingly, EWS-ETS fusion oncoproteins drive the expression of VEGF in an SP1 dependent manner (83) and may contribute to the increased VEGF levels observed in patients. Bone marrow driven vasculogenesis is essential for tumor growth in ES (84). The upregulation of pro-angiogenic factors like VEGF, FGF, TGF-α, HGF, platelet-derived growth factor (PDGF), angiopoietin 1 (Ang1), and ephrin-B2 combined with the down-regulation of anti-angiogenic proteins like thrombospondin-1, TGF-β, troponin I, pigment epithelial-derived factor (PEDF), and reversion-inducing cysteine-rich protein with Kazal motifs (RECK) allows for rapid neovascularization (68, 85–88).
Intravasation
The first step in metastasis is migration from the primary tumor site and invasion through the basement membrane, allowing metastatic cells to enter circulation and disseminate distantly. These events are depicted in Figure 2. Since pulmonary metastasis is the major cause of death in both OS and ES, elucidating the mechanisms controlling metastasis is vital for improving patient outcomes. By identifying the molecular alterations associated with cell migration and invasion, it is hoped that novel therapies could be developed to prevent metastases.
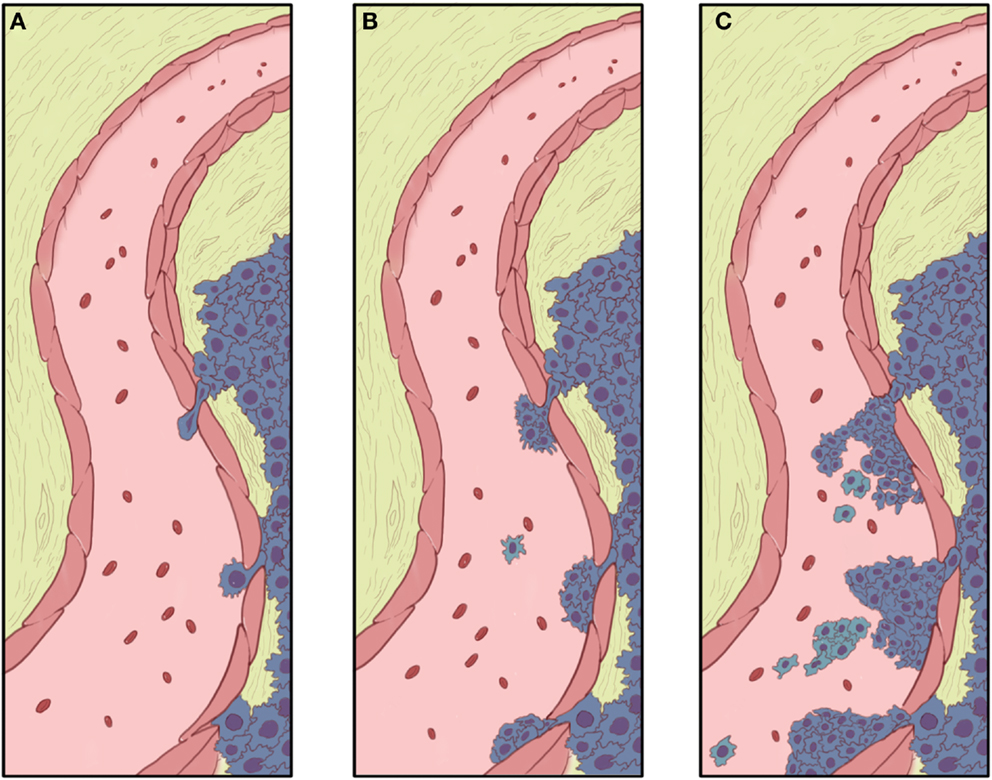
Figure 2. Invasion and intravasation. (A) The growing tumor advances to the basement membrane of a nearby blood vessel, with the proteolytic functions of MMPs and plasmins clearing a pathway and allowing the leading cells to approach (bottom) and eventually push between (middle and top) the endothelial cells. With mass migration, other tumor cells may follow behind the leading cells. These trailing cells do not necessarily have as much invasive capacity as the lead cell, but may have their invasion and intravasation facilitated by the more invasive lead cells. (B) Migrating tumor cells effectively form a small “beachhead” in the spaces where lead cells have pushed between and through gaps in the endothelial cells. (C) With further mass migration, tumor cells begin obstructing the flow of blood through the vessel, and encounter red cells, platelets, and other blood components. A tumor thrombus may occlude blood flow and, is the vessel is large enough, cause symptoms of deep venous thrombosis. Clumps of tumor and individual tumor cells detach from the main mass of tumors and are carried away in the blood stream. Individual cells would be subject to anoikis when loose in the blood stream. Some cells in tumor clumps may be protected from anoikis by their attachment to other cells in the clump.
Degradation of the ECM, facilitated by the action of MMPs, is a prerequisite of tumor invasion and metastasis in a variety of human cancers including OS and ES. MMP-2 and MMP-9 in particular have been repetitively implicated in OS cell invasion (89–91). In addition, m-calpain, an intracellular protease that modulates cell adhesion and motility, plays a pivotal role in promoting cell invasion in OS. Fan et al. demonstrated that the inhibition of m-calpain and the concomitant blockade of MMP-2 secretion led to a strong decrease in OS cell adhesive and invasive ability (92). The urokinase plasminogen activator (uPA) and its receptor uPAR are important mediators of OS cell invasion through activation of plasminogen and pro-MMPs (93). Down-regulation of uPAR prohibited cell adhesion, migration, and invasion without affecting cell proliferation in vitro, resulting in a marked reduction of pulmonary metastases in vivo.
The Wnt/β-catenin pathway and its antagonists are involved in multiple facets of tumor progression including invasion and metastasis. High levels of β-catenin and aberrant activation of Wnt signaling are frequently observed in OS samples and such molecular alterations are correlated with the incidence of metastasis (94, 95). Evidence from multiple sources suggests that the Wnt/β-catenin pathway promotes metastasis in OS through modulation of Wnt target genes including MMP-9, cyclin D, c-myc, and survivin (96). Various approaches to disrupt Wnt signaling have been proven effective in inhibiting cell migration and invasion in laboratory models (96, 97). Wnt signaling also may mediate activation of Notch pathway signaling, which also has been implicated in OS pathogenesis and metastasis. The Notch receptor 1, 2, and the downstream target gene Hes1 is upregulated in highly metastatic OS cells (44, 98), and inhibition of Notch signaling by either molecular or pharmacologic means reduced both OS proliferation and metastasis (44).
Src signaling is another pivotal pathway that contributes to the aggressive phenotype of OS cells. Pharmacologic inhibition of the c-Src-mediated signaling pathway suppressed phosphorylation of focal adhesion kinase (FAK) and activation of other downstream proteins, resulting in significantly decreased cell migration and invasion in a panel of OS cells (70, 99). However, the therapeutic effect of Src inhibition is very limited in animal models (70), suggesting that Src is not an essential mediator of metastasis in OS, or that “rescue pathways” exist to activate FAK and other Src targets.
Recent studies in OS have uncovered some novel targets for anti-metastatic strategies. Parathyroid hormone (PTH), PTH peptides, as well as the PTH receptor (PTHR) have been shown by multiple reports to enhance OS cell migration and invasion (100, 101). Autocrine motility factor (AMF), a major cell motility-stimulating factor secreted by tumor cells, is associated with tumor metastasis in various human cancers including OS (102). Suppression of AMF expression induced mesenchymal-to-epithelial transition (MET) in OS and reduced cell motility and invasiveness (103). Tissue microarray analysis revealed that cysteine-rich protein 61 (CCN1/Cyr61) is overexpressed in most cases of OS when compared to normal bone tissue (104). The same study showed that genetic manipulation of Cyr61 expression levels in OS cells concomitantly influenced cell invasion and migration in vitro and metastatic potential in vivo. Interleukin-6 (IL-6) is a multipotent cytokine which has been implicated in the progression of multiple human malignancies including OS. Lin et al. demonstrated through a series of IL-6 knockdown experiments that interaction between IL-6 and its receptor IL-6R is required for OS cell migration via the activation of intercellular adhesion molecule-1 (ICAM-1) expression (105).
Similar to their role in OS, MMPs are essential mediators of ES cell migration and invasion as they are responsible for the localized degradation of ECM components, which serve to clear a pathway for the invading ES cells. Caveolin-1 (CAV-1) controls ES cell migration, invasion, and lung colonization by upregulation of secreted protein acidic and rich in cysteine (SPARC), which in turn activates MMP-2 and MMP-9 (106). Studies on ES patient samples have established a link between the expression of CCN-3 which is a secreted, ECM-associated signaling protein, and a higher risk to develop lung metastases (107, 108). Ectopic expression of CCN-3 in ES cells reduced cell proliferation rate and anchorage-independent growth while promoting cell migratory and invasive capabilities, which could be due to the decreased expression of α2β4 integrin receptor and increased cell surface localization of MMP-9 (109). Hauer et al. identified DKK-2 as a pro-metastatic gene highly overexpressed in a panel of ES cell lines (110). By regulating the activity of MMP-1, DKK-2 increased ES cell invasiveness and metastatic potential. The same study also demonstrated that DKK-2 overexpression enhanced bone invasiveness and osteolysis, both of which facilitate the metastatic spread of ES. A DNA microarray analysis of 11 ES samples and 133 normal tissues revealed that the six-transmembrane epithelial antigen of the prostate 1 (STEAP1), a membrane-bound mesenchymal stem cell marker, is a signature gene highly expressed in ES (111). Further study on this gene demonstrated that STEAP1 promotes proliferation, anchorage-independent growth, tumorigenicity, and metastasis in ES cells (112). In addition, STEAP1 overexpression is associated with elevated reactive oxygen species (ROS) level in ES, which could also promote tumor aggressiveness. The histone methyltransferase enhancer of Zeste, Drosophila, Homolog 2 (EZH2) is another important mediator of ES tumor growth and metastasis, driven by EWS/FLI1 (54). Down-regulation of EZH2 significantly impeded tumor growth and inhibited lung and liver metastasis in vivo.
Other signaling pathways, such as Src and Wnt signaling, are also of great importance in ES cell migration and intravasation. Constitutive activation of Src kinase has been observed in ES metastasis (99, 113). Shor et al. demonstrated that inhibition of Src phosphorylation and its downstream targets, including FAK and Crk-associated substrate (CAS), by dasatinib blocked in vitro cell migration and invasion (99). Overexpression of C-X-C chemokine receptor type 4 (CXCR4) correlates with ES metastases (114, 115). Upregulation of CXCR4 by the non-canonical Wnt family member Wnt5a has been shown to promote ES cell migration in absence of Wnt antagonist SFRP5 (115). Landuzzi et al. reported a positive role of stem cell factor (SCF) and its receptor c-kit in promoting ES metastasis (116). They have demonstrated in multiple ES cell lines that SCF, a growth factor abundantly expressed in ES and the tumor microenvironment, induced a strong increase in cell motility via activation of c-kit. A recent publication also indicated that overexpression of ERBB-4, which is observed in a panel of ES cell lines and metastatic tumor samples, led to increased invasion and migration in vitro, as well as enhanced metastatic capacity in vivo through activation of PI3K-Akt, FAK, and the Rac1 GTPase (117).
Specific regulation of microRNA expression also may play a role in regulating intravasation and metastasis. MicroRNAs (miRNAs) are a class of endogenous non-coding small RNAs that contains about 18–25 nucleotides. miRNAs regulate translational suppression or cleavage of their target mRNAs through perfect or imperfect binding to the 3′-untranslated region (3′-UTR) of their target mRNA. Deregulation of miRNAs has been observed in many human cancers, and recent reports indicate the involvement of miRNAs in development and metastasis of bone sarcomas. Ziyan et al. identified a pro-metastatic function of miR-21 by comparing tumor samples and matched normal bone tissues (118). miR-21 promotes the activity of matrix MMPs by negatively regulating the tumor suppressor gene reversion-inducing cysteine-RECK, and knockdown of miR-21 decreased cell migration and invasion. miR-27a is another pro-metastatic miRNA validated by Jones and colleagues after they surveyed a panel of OS cell lines together with normal osteoblast cells (119). On the contrary, miR-143 stood out as an anti-metastatic miRNA when the miRNA expression profiles were compared between HOS cells and its metastatic subclone 143B cells (120). Reintroduction of miR-143 into the highly metastatic cells suppressed invasiveness and impeded pulmonary metastasis through inhibition of MMP-13. Expression of miR-183 also correlates with OS pulmonary metastasis (121). Suppression of miR-183 led to elevated ezrin level and activation of p-p44/42, resulting in enhanced metastatic ability of OS cells. Mao et al. found that miR-195 plays a similar role in OS metastasis (122). With the fatty acid synthase (FASN) as a direct target, miR-195 significantly inhibited OS invasion and migration in USOS cells.
Anoikis Resistance
After tumor cells detach from their primary site and enter into the bloodstream, the absence of cell–cell adhesion, and cell-ECM interaction can trigger specific cellular apoptosis, a process termed “anoikis.” Therefore, acquisition of resistance to anoikis is a critical step in survival and expansion of metastatic cells. Multiple molecular pathways contribute to anoikis evasion in OS and ES, including integrin signaling, PI3K/Akt, Src, Wnt/β-catenin, and the BcL family (123). Since anoikis resistance is an essential step in tumor progression, elucidation of the molecular mechanisms underlying this process may provide additional therapeutic strategies for targeting metastasis.
Integrins, a family of cell adhesion receptors that can activate multiple signal transduction pathways, play a key role in the survival of tumor cells in anchorage-independent environments. Several studies have indicated a correlation between integrin expression and OS metastasis (124, 125). Wan et al. demonstrated that β4 integrin is highly expressed in OS cell lines and tumor samples (125). Knockdown of β4 integrin suppressed cell proliferation in anchorage-independent conditions without affecting cell growth in adherent cultures in vitro and effectively inhibited pulmonary metastasis in vivo. In addition, β4 integrin has been found to interact with ezrin as a way to maintain its expression level in OS cells. Marco et al. reported that α4 integrin expression also confers resistance to anoikis in OS cells (124). They demonstrated that α4 integrin is abundantly expressed in metastatic OS lesions. Blocking α4 integrin with a monoclonal antibody increased cell death in suspended cells but not in adherent cells.
Another way for tumor cell to avoid anoikis is to alter the expression pattern of integrin subunits, which leads to elevated PI3K/Akt signaling and decreased cell apoptosis (126). One study revealed that Src-dependent activation of the PI3K/Akt pathway, which is independent of FAK phosphorylation, is essential for OS cells to gain anoikis resistance (127). Pharmacological inhibition of Src or PI3K activity restored sensitivity to anoikis in OS cells. Cantiani et al. also confirmed the role of Src in mediating anoikis resistance by showing that Cav-1, a tumor suppressor gene significantly downregulated in OS cell lines and tumor samples, inhibited migration, invasion, and anchorage-independent growth of OS cells by blocking Src family kinase activity and Met signaling (128). Beristain et al. indicated that the receptor activator of NF-κB (RANK) signaling pathway also protects OS cells from anoikis (129). Activation of RANK enhanced cell survival in anchorage-independent environments while knockdown of endogenous RANK inhibited the tumorigenic ability of several mouse OS cell lines. Other survival mechanisms adopted by solitary OS cells to evade anoikis include activation of the Wnt/β-catenin pathway and overexpression of anti-apoptotic genes such as Bcl-2. Lin et al. demonstrated that upregulation of Bcl-2 protected detached OS cells from anoikis by inhibiting activation of caspase-8, which can induce apoptosis when cell adherence is disrupted (130). High levels of Wnt family proteins have been reported in OS patient samples, and constitutive Wnt activation is associated with high metastatic potential (94). Rubin et al. showed that overexpression of the Wnt inhibitory factor-1 (WIF-1) significantly decreased anchorage-independent growth and cell motility in 143B cells and inhibited in vivo tumorigenesis and metastasis (97).
Some of the molecular pathways mediating anoikis resistance in ES are similar to that of OS. Sustained activation of PI3K/Akt and Ras/ERK pathways in ES cells has been indicated as an essential process for anchorage-independent cell survival (131). PI3K inhibition markedly reduced anchorage-independent growth of TC32 cells through regulation of the cyclin D1 level. Aberrant activation of the MEK/MAPK pathway contributes to the survival of detached ES cells (132). Blockade of the MEK/MAPK pathway by specific inhibitors impaired cell motility and colony formation ability in vitro. The insulin-like growth factor receptor (IGF-1R), a receptor tyrosine kinase upstream of PI3K/Akt and MAPK pathways, is another important signaling pathway involved in anoikis resistance in ES (133). IGF-1R is highly expressed in both ES cell lines and patient tumor samples and inhibition of the IGF-1R signaling resulted in profoundly reduced cell migratory ability and increased anoikis-induced apoptosis in several ES cell lines. Additional pathways also may mediate anoikis resistance in ES. Douglas et al. demonstrated that the polycomb gene BMI-1 is highly expressed in ES cells and it promotes anchorage-independent growth and tumorigenesis by activating downstream targets which modulate cell adhesion pathways (134). Tirado et al. showed that, unlike its tumor suppressive role in OS, Cav-1 is highly expressed as an oncogene in ES cell lines and tumor samples (135). As the direct transcriptional target of EWS-FLI1, Cav-1 promotes the malignant phenotype of ES cells. Knockdown of Cav-1 impeded anchorage-independent growth in vitro and tumorigenesis in vivo, which is associated with suppression of E-cadherin and upregulation of Snail. E-cadherin belongs to a class of transmembrane proteins that regulate cell–cell adhesion. As ES cells readily form multicellular spheroids under detachment conditions, a concomitant upregulation of E-cadherin has been observed. Kang et al. reported that E-cadherin protects ES cells from anoikis-induced apoptosis through downstream activation of Akt in an ERBB-4 dependent manner (136). Several pathways relevant to anoikis resistance are depicted in Figure 3.
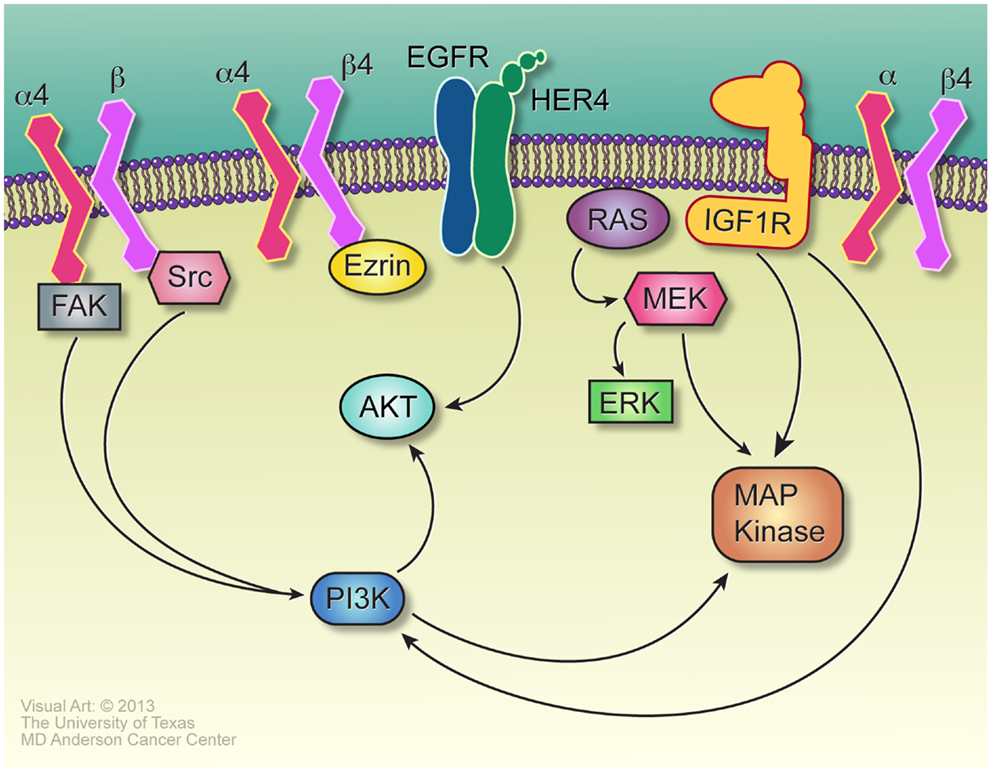
Figure 3. Signaling in anoikis resistance. The α4 and β4 integrins, paired either with each other or with other integrins, initiate signals vital to anoikis resistance, through interaction with Ezrin in the case of β4, and Src for α4. Src-mediated activation of PI3K may be independent of its well-described role in activating FAK. Her-4, induced by E-cadherin, signals strongly through AKT and mediates anoikis resistance, certainly for Ewing sarcoma and probably in osteosarcoma as well. While many receptor tyrosine kinases may mediate Ras activation and Ras-dependent and – independent activation of the MAP kinase cascade, IGF1R is an especially important source of these signals, especially for anoikis resistance in Ewing sarcoma.
Extravasation and Attachment
To form metastases, disseminated tumor cells (DTCs) that survive circulation must extravagate into foreign tissues by attaching and adapting to the metastatic microenvironment. DTCs are significantly larger than normal blood cells; this causes the formation of microembolisms that can get trapped in capillary beds throughout the body (137). Interestingly, these microembolism do not spread indiscriminately. Rather, they tend to appear in a small number of highly specific target organs, which can vary depending on the histology of the original tumor. Over 80% of all metastases in OS occur in the lungs (138). Most ES metastases also occur in the lungs, but ES also spreads frequently to the bone and bone marrow (139). This suggests that there are specific environmental cues that allow for the survival and growth of DTCs. These environmental factors that make specific organ sites favorable to DTC metastases rely on the interactions between specific molecules expressed on both the DTCs and the endothelium of the target organ (76).
Chemokines and proteinases mediate distant colonization (76, 140). Chemokines and proteinases are responsible for the organ-specific localization of DTCs and the extravasation of DTCs into foreign tissues, respectively (140–145). In sarcomas, chemokines bind to G-protein coupled receptors on the plasma membrane of tumor cells in the lungs (115, 141, 142, 144, 146, 147). CXCR4 is a highly expressed chemokine in OS and is implicated as the most important contributor in the development of site-specific metastases. Binding of CXCR4 to C-X-C motif chemokine ligand 12 (CXCL12), which is abundantly expressed in the lungs, allows for the adhesion and extravasation of OS cells into the lung (140–144, 146, 148, 149). High CXCR4 expression in OS patient samples is inversely correlated with event-free, overall, and metastasis-free survival (142). Although inhibition of CXCR4 binding to CXCL12 did not consistently inhibit the development of tumor metastases (148), targeting CXCR3 in vivo to inhibit its binding to ligands CXCL9, 10, and 11 did inhibit OS lung metastases (149). In ES CXCR4 expression correlated with Wnt5a expression and was significantly higher in patients that presented with metastasis at diagnosis, compared with patients without metastases, and also correlated with poor survival (115). Selectins and integrins also facilitate the adhesion of malignant sarcoma cells to the endothelial lining of the lungs (150).
Ezrin is a membrane cytoskeleton linker protein that mediates membrane organization and cell microenvironment interactions (151) and is also believed to facilitate OS-DTC anchorage to the lung tissue. High Ezrin expression is correlated with: metastasis in animal models of OS (152–154), higher risk of metastatic relapse, and poor survival in pediatric OS patients (153, 154). Ezrin is thought to promote the formation of metastases through β4 Integrin mediated activation of the PI3K, Akt, and MAPK pathways and thus Ezrin stimulates survival and proliferation of the DTC in the lung (76, 125, 146, 153, 154). Inhibition of Ezrin-activated MAPK/Akt signaling with Sorafenib suppresses the development of lung metastases in laboratory models (155).
The nuclear factor-kappa B (NF-κB) is known to be involved in the development of lung metastases (156–159). The linear ubiquitin chain assembly complex (LUBAC) activates both NF-κB and intercellular adhesion molecule-1 (ICAM-1), and is necessary for the extravasation and retention of DTCs in the lungs (160). Therefore, the knockdown of LUBAC is able to decrease both the number and the size of the metastatic nodules within the lungs of mice injected with OS cells (160).
Dormancy
For patients who present without radiographic evidence of metastasis, pulmonary metastases often become evident only 6 months to 3 years after diagnosis, long after the resection of the primary site. This suggests that as cells disseminate, extravagate, and attach to the lung environment, they undergo growth arrest and become dormant. Despite the clinical significance of this dormancy, the mechanisms underlying it and the outgrowth of macrometastatic lesions from these dormant micro-metastases remain poorly understood. Some evidence suggests that a subpopulation of cancer cells exhibit stem-like properties which are capable of disseminating to distant organs (161–165). These cancer stem-like cells (CSCs) are purported to have the ability to self-renew and populate a growing tumor, an increased capacity for DNA repair, and higher expression levels of anti-apoptotic proteins than differentiated cells (166–170). These properties provide CSCs with the ability to survive for long periods of time under metabolic and environmental stresses (e.g., hypoxia) while they arm tumor cells with ATP-binding cassette (ABC) transporters that actively efflux chemotherapeutics from target cells, thus providing tumor cells with increased drug resistance (171–176).
Two additional explanations for tumor dormancy focus on the senescence of either multiple cells (tumor mass dormancy) or individual cells (cellular dormancy). Tumor mass dormancy is the process by which the cells that make up micro-metastases are balanced between proliferation and apoptosis, and accordingly the mass does not grow (177, 178). This process purportedly relates to the lack of nutrients and oxygen from the vasculature (179–183). Cellular dormancy cites the process by which individual tumor cells enter a quiescent state and do not divide anymore (177, 184, 185). Such cells are typically more resistant to conventional drugs because current treatments tend to target dividing cells. The two processes, of course, are not mutually exclusive, as small foci of tumors may contain individual cells that are growth arrested but not terminally differentiated.
In order to elucidate the cellular mechanisms that define metastatic dormancy, Almog et al. designed an OS in vivo model for dormancy and performed gene expression analysis of cells in the dormant state versus cells in the proliferative state (186). Since tumor growth is dependent on vascularization, metastatic dormancy is generally associated with the upregulation of anti-angiogenic proteins such as angiomotin, which suppresses tumor growth while maintaining the dormant state of DTCs (179). Almog et al. observed a marked increase in the upregulation of anti-angiogenic proteins in dormant cells (186). Administration of recombinant angiogenic factors was shown to allow the transition from anti-angiogenic dormancy, to pro-angiogenic tumor growth (187). Additionally, an upregulation of endocan in rapidly proliferating cells was observed, suggesting that poor connection to the ECM of the pulmonary environment may also be associated with dormant micro-metastases (186) while proper anchorage to the ECM would stimulate cells to proliferate via β1-integrin signaling (183). The anti-apoptotic protein Bcl-xL, α5β1-integrin mediated activation of NF-κB, and the ratio between ERK and p38-MAPK proteins all appear to be involved in the regulation of tumor dormancy (76, 146, 182, 183). The role of anti-angiogenic signals in maintaining dormancy and pro-angiogenic signals in promoting renewed growth of pulmonary micro-metastases may explain the clinical observation, often repeated by patients and families, that pulmonary metastases are often identified within a few months after a major operation. Patients often tell us that “operations spread the sarcoma.” The more probable truth is that major operations, and the cytokines released as a result of them, awaken dormant micrometastatic lesions.
As of now, we are unaware of publications that describe dormancy in ES. Given that ES patients do experience recurrence after long periods of disease-free survival, it seems probable that systems of dormancy do occur for ES. Whether these systems are mechanistically similar to dormancy in other sarcomas remains to be shown.
Chemoresistance
Tumor resistance to chemotherapy has always been a major obstacle for treating patients with bone sarcomas. Intrinsic resistance to chemotherapy has been detected in a small portion of patients. More commonly, however, patients present with disease that initially is responsive to cytotoxic therapy, but acquires resistance that is evident when tumors recur. Thus, a better understanding of the molecular mechanisms underlying the development of chemoresistance will facilitate the discoveries of novel treatment strategies to overcome this resistance and improve patient outcomes.
Most studies of chemoresistance in bone sarcomas thus far have focused on molecules and pathways already known to effect sensitivity to chemotherapy in more common tumors. Increased levels of P-glycoprotein, a transmembrane ATP-dependent efflux pump which plays a key role in multidrug resistance, is strongly associated with poor response to chemotherapy and reduced survival in both OS and ES patients (188–191). As an important predictive factor for chemoresponse in many other cancers, Her-2/ERBB2 has been extensively studied in bone tumors, yet the role of Her-2 overexpression in the development of chemoresistance in OS and ES remains controversial and ambiguous. Some studies suggested that Her-2 expression is significantly correlated with histologic responses to preoperative chemotherapy while other studies indicated that Her-2 expression in OS and ES has no prognostic significance (191–195). Our group has shown that expression of Her-4/ERBB-4 confers a worse prognosis for neuroblastoma by mediating reduced cell proliferation and anoikis resistance, as well as conferring direct resistance to cisplatin, doxorubicin, and ifosfamide (196). Similar studies to evaluate the role of ERBB-4 in OS are ongoing in our lab, and a role for ERBB-4 in ES has been established (136). The Bcl-2 family proteins, which mediate various steps of apoptosis, are important regulators of chemotherapy-induced cell death. The anti-apoptotic proteins Bcl-2 and Bcl-xL are frequently overexpressed in OS and ES and are associated with development of resistance to a variety of chemotherapeutic agents or radiation (197–201). Up- and down-regulation of these proteins can significantly reduce or enhance the chemosensitivity of OS and ES cells. Overexpression of EWS/FLI-1 direct target genes, CAV-1 and glutathione S-transferase M4 (GSTM4), also help modulate resistance to chemotherapy in ES cells (202, 203). In addition, miRNA has been found to play a vital role in conferring chemoresistance in OS and ES cells. Song et al. indicated that miR-140 and miR-215 are both involved in OS chemoresistance by inducing a decreased cell proliferation through cell cycle arrest at G1 and G2 phase respectively (204, 205). The miRNAs miR-34a, miR-708, and miR-125b are essential miRNAs that regulate cell survival and chemosensitivity of ES cells (206–208). Analysis of 34 ES tumor samples revealed that reduced or absent expression of miR-34a, which is associated with p53 inactivation, predicted a worse clinical outcome and restoration of miR-34 activity in vitro markedly increased ES cell sensitivity to doxorubicin and vincristine (206). Similarly, repression of miR-708 in ES tumor samples is associated with up-regulation of the DNA repair protein and transcriptional cofactor EYA3, which directly led to enhanced cell survival and resistance to DNA-damaging chemotherapeutics (207). On the contrary, miR-125b has been shown to promote multidrug resistance in ES cells by suppressing the expression of pro-apoptotic proteins, p53 and Bak. Knocking down miR-125b in the chemo-resistant ES cells increased their sensitivity to doxorubicin, etoposide, and vincristine (208).
Evasion of Immune Surveillance
The survival of DTCs and, to a lesser extent primary tumors, depends on the evasion of the host immune system. Faulty regulation of a number of key genes that control the immune system allow DTCs to achieve a survival advantage. The down-regulation of HLA class 1, a cell surface receptor that impairs the recognition of tumor cells by the host cytotoxic T-lymphocytes, is one such mechanism (209, 210), and expression of HLA class I has prognostic significance for OS (209). DTCs can also induce the production of IL-10 expression in OS, resulting in immunosuppression (146, 211). Additionally, corruption of the Fas pathway or low expression of Fas on the cell surface of OS and ES cells inhibits the ability of cytotoxic natural killer cells to detect and clear DTCs, thus yielding an increase in metastatic potential. OS metastatic tissue samples are Fas negative (212, 213), and low Fas expression correlates with disease progression and poor survival (212, 214–217). Despite these observations, adoptively transferred T cells did show some clinical benefit in one pilot study (218). The use of interferon in OS patients has been studied, though the results of the Euramos study – the largest clinical evaluation of interferon for OS to date, have still not been reported as of the time of this review.
The innate immune system may offer greater promise for treating OS and ES. The macrophage-activating agent L-MTP-PE, first evaluated by Kleinerman and colleagues, reduced metastatic tumor burden in experimental laboratory models (219), and had proven benefit in dogs with spontaneously occurring OS (220). After a series of smaller trials, a cooperative group study of L-MTP-PE demonstrated an 8% improvement in overall survival in patients treated with L-MTP-PE (221), though the initial studies were difficult to interpret due to an interaction with ifosfamide that has not been fully characterized (222). The benefit was greatest for patients who presented with metastatic disease that were able to achieve a complete remission through surgical resection (26). This drug, now with the generic name mifamurtide, has been given regulatory approval by the EMA, Mexico, Japan, Israel, and several other countries, but has yet to be accepted by the FDA. A similar agent, Imm-Ther, was studied in ES, but the data from that small study have not been reported.
Natural killer cells (NK cells) may be another option for harnessing immune responses against OS and ES. Multiple laboratory studies demonstrated the ability of NK cells to recognize and kill OS cells (223–226). The regulation of NK cell cytolytic activity is complex, and includes both activating and inhibitory receptors, as well as induced tolerance to specific self-receptors. These mechanisms have been reviewed recently (227). One aspect on NK recognition, however, can be described relatively simply: the presence of “self-HLA Class I” has an inhibiting function for NK cells. Thus the down-regulation of HLA that facilitates evasion of T cell recognition actually facilitates NK recognition. Clinical trials using autologous and haplo-identical (i.e., parent or sibling donor) NK cells have been conducted for several tumor types, with some promising early results. These events are depicted graphically in Figure 4.
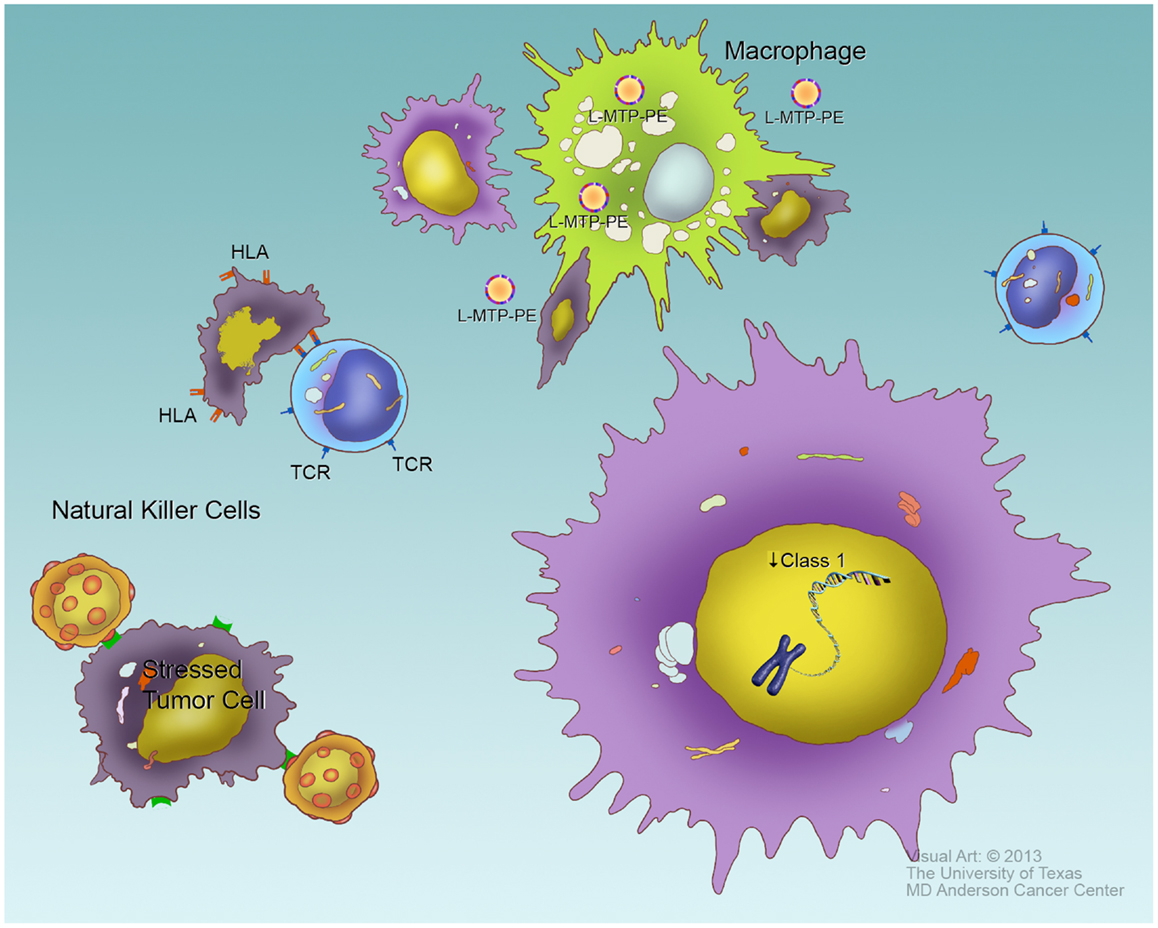
Figure 4. Evasion of immune response. To survive, tumor cells must evade several components of cellular immunity. Cytotoxic T cells, through their T cell receptors (TCR), recognize foreign antigens, and kill cells bearing these antigens presented by class I HLA. Tumor-specific antigens can potentially be treated as foreign by the immune system. Tumor cells, such as the one in the lower right, typically downregulate HLA expression as a means of evading T cell response. Natural killer (NK) cells (left side of figure) recognize specific surface molecules expressed on “stressed” cells, especially tumor cells. One requirement for full NK response is the absence of self-HLA, so the down-regulation of HLA that allows tumors to evade cytotoxic T cells may facilitate their recognition by NK cells. Macrophages may ingest and destroy tumor cells in an antigen non-specific manner, though this process may require macrophage activation such as typically occurs in the sites of infection. The macrophage-activating drug L-MTP-PE has been shown to induce macrophage activation (top of figure) and improves survival in high-risk osteosarcoma patients when given in first complete remission or at a time of minimal residual disease, presumably by increasing phagocytosis on tumor cells.
Once factor related to the efficacy of any immune-mediated therapy that must be considered is the tumor burden at the time of treatment. Whether the intervention is T cells, NK cells, cytokines, or immune-modulating agents such as L-MTP-PE, the greatest efficacy would be expected if the intervention begins in a minimal residual disease state. All available data regarding immune therapies suggests that bulky tumors are unlikely to respond, while microscopic disease may be controlled. This fact about immune therapies needs to be considered carefully, both when clinicians and investigators are evaluating outcomes from early-phase studies and when clinician-investigators are designing phase II and phase III studies incorporating immune-based treatments. If we design trials evaluating immune approaches in advanced-disease patients, we are quite likely to discard potentially beneficial treatments because we evaluated them in the wrong cohort of patients.
Summary and Next Steps
The desperate need for better therapies for both OS and ES is clear, given the numbers of children and young adults each year who develop treatment-resistant disease that follows a relentless, downward course. A better understanding of the biology related to the several steps of disease development and progression, especially the biology of metastasis, immune evasion, and latency/reactivation, is needed to foster new approaches. While our investigations can sometimes be informed by following the literature for common adult carcinomas, one must bear in mind the important differences between carcinomas and either OS or ES. For example, since OS is derived from mesenchymal tissues, studies of epithelial-to-mesenchymal transition (EMT) and the reverse –MET – seem unlikely to be very informative, since the tumor is mesenchymal in the first place, and the data for state changes such as EMT or MET as a part of OS and ES metastasis is not very clear. The specific signals involved in tumor dormancy in particular also are not well-characterized and may offer a new therapeutic entry point for changing the landscape of OS and ES. Three distinct parts of dormancy should be considered, as each could be its own novel therapy. First, an active signal causes OS tumor cells to arrest growth while metastasizing, which helps contribute to chemoresistance. At the same time, dormant tumor cells have an active developmental block that prevents them from pursuing a differentiation cascade. Finally, something later causes individual tumors in the metastatic niche to resume growth. The latter may be connected to angiogenesis and the cytokines of wound healing, given the association, more frequently commented upon than observed, of metastatic tumor growth beginning a few weeks after major surgery. Better understanding of these signals and pathways will enable novel therapies to be used in the manner most likely to lead to improved outcomes, such as employing immune therapies in a minimal residual disease state.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res (2009) 152:3–13. doi:10.1007/978-1-4419-0284-9_1
2. Provisor AJ, Ettinger LJ, Nachman JB, Krailo MD, Makley JT, Yunis EJ, et al. Treatment of nonmetastatic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: a report from the Children’s Cancer Group. J Clin Oncol (1997) 15:76–84.
3. Ferguson WS, Goorin AM. Current treatment of osteosarcoma. Cancer Invest (2001) 19:292–315. doi:10.1081/CNV-100102557
4. Sung L, Anderson JR, Donaldson SS, Spunt SL, Crist WM, Pappo AS. Late events occurring five years or more after successful therapy for childhood rhabdomyosarcoma: a report from the soft tissue sarcoma committee of the Children’s Oncology Group. Eur J Cancer (2004) 40:1878–85. doi:10.1016/j.ejca.2004.04.005
5. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature (1992) 359:162–5. doi:10.1038/359162a0
6. Hense HW, Ahrens S, Paulussen M, Lehnert M, Jurgens H. Descriptive epidemiology of Ewing’s tumor – analysis of German patients from (EI)CESS 1980-1997. Klin Padiatr (1999) 211:271–5. doi:10.1055/s-2008-1043799
7. Ries LG, SEER Program-National Cancer Institute (US). Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995, Lynn A, Ries G, editor. Bethesda, MD: National Cancer Institute, SEER Program (1999).
8. Wilkins RM, Pritchard DJ, Burgert EO Jr, Unni KK. Ewing’s sarcoma of bone. Experience with 140 patients. Cancer (1986) 58:2551–5. doi:10.1002/1097-0142(19861201)58:11<2551::AID-CNCR2820581132>3.0.CO;2-Y
9. Nesbit ME Jr, Perez CA, Tefft M, Burgert EO Jr, Vietti TJ, Kissane J, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: an Intergroup Study. Natl Cancer Inst Monogr (1981):255–62.
10. Nesbit ME Jr, Gehan EA, Burgert EO Jr, Vietti TJ, Cangir A, Tefft M, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol (1990) 8:1664–74.
11. Paulussen M, Ahrens S, Dunst J, Winkelmann W, Exner GU, Kotz R, et al. Localized Ewing tumor of bone: final results of the cooperative Ewing’s sarcoma study CESS 86. J Clin Oncol (2001) 19:1818–29.
12. Bacci G, Forni C, Longhi A, Ferrari S, Donati D, De Paolis M, et al. Long-term outcome for patients with non-metastatic Ewing’s sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer (2004) 40:73–83. doi:10.1016/j.ejca.2003.08.022
13. Granowetter L, Womer R, Devidas M, Krailo M, Wang C, Bernstein M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group Study. J Clin Oncol (2009) 27:2536–41. doi:10.1200/JCO.2008.19.1478
14. Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol (2012) 30:4148–54. doi:10.1200/JCO.2011.41.5703
15. Gelberg KH, Fitzgerald EF, Hwang S, Dubrow R. Growth and development and other risk factors for osteosarcoma in children and young adults. Int J Epidemiol (1997) 26:272–8. doi:10.1093/ije/26.2.272
16. Rikhof B, De Jong S, Suurmeijer AJ, Meijer C, Van Der Graaf WT. The insulin-like growth factor system and sarcomas. J Pathol (2009) 217:469–82. doi:10.1002/path.2499
17. Dong J, Demarest SJ, Sereno A, Tamraz S, Langley E, Doern A, et al. Combination of two insulin-like growth factor-I receptor inhibitory antibodies targeting distinct epitopes leads to an enhanced antitumor response. Mol Cancer Ther (2010) 9:2593–604. doi:10.1158/1535-7163.MCT-09-1018
18. Wang YH, Xiong J, Wang SF, Yu Y, Wang B, Chen YX, et al. Lentivirus-mediated shRNA targeting insulin-like growth factor-1 receptor (IGF-1R) enhances chemosensitivity of osteosarcoma cells in vitro and in vivo. Mol Cell Biochem (2010) 341:225–33. doi:10.1007/s11010-010-0453-2
19. Franchi A, Arganini L, Baroni G, Calzolari A, Capanna R, Campanacci D, et al. Expression of transforming growth factor beta isoforms in osteosarcoma variants: association of TGF beta 1 with high-grade osteosarcomas. J Pathol (1998) 185:284–9. doi:10.1002/(SICI)1096-9896(199807)185:3<284::AID-PATH94>3.0.CO;2-Z
20. Navid F, Letterio JJ, Yeung CL, Pegtel M, Helman LJ. Autocrine transforming growth factor-beta growth pathway in murine osteosarcoma cell lines associated with inability to affect phosphorylation of retinoblastoma protein. Sarcoma (2000) 4:93–102. doi:10.1080/13577140020008057
21. Nishida T, Nakanishi T, Asano M, Shimo T, Takigawa M. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J Cell Physiol (2000) 184:197–206. doi:10.1002/1097-4652(200008)184:2<197::AID-JCP7>3.0.CO;2-R
22. Scholz RB, Kabisch H, Weber B, Roser K, Delling G, Winkler K. Studies of the RB1 gene and the p53 gene in human osteosarcomas. Pediatr Hematol Oncol (1992) 9:125–37. doi:10.3109/08880019209018328
23. Marina N, Gebhardt M, Teot L, Gorlick R. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist (2004) 9:422–41. doi:10.1634/theoncologist.9-4-422
24. Wadayama B, Toguchida J, Shimizu T, Ishizaki K, Sasaki MS, Kotoura Y, et al. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res (1994) 54:3042–8.
25. Eng C, Li FP, Abramson DH, Ellsworth RM, Wong FL, Goldman MB, et al. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst (1993) 85:1121–8. doi:10.1093/jnci/85.14.1121
26. Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A (2008) 105:11851–6. doi:10.1073/pnas.0805462105
27. Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev (2008) 22:1662–76. doi:10.1101/gad.1656808
28. Maelandsmo GM, Berner JM, Florenes VA, Forus A, Hovig E, Fodstad O, et al. Homozygous deletion frequency and expression levels of the CDKN2 gene in human sarcomas – relationship to amplification and mRNA levels of CDK4 and CCND1. Br J Cancer (1995) 72:393–8. doi:10.1038/bjc.1995.344
29. Miller CW, Aslo A, Campbell MJ, Kawamata N, Lampkin BC, Koeffler HP. Alterations of the p15, p16,and p18 genes in osteosarcoma. Cancer Genet Cytogenet (1996) 86:136–42. doi:10.1016/0165-4608(95)00216-2
30. Wei G, Lonardo F, Ueda T, Kim T, Huvos AG, Healey JH, et al. CDK4 gene amplification in osteosarcoma: reciprocal relationship with INK4A gene alterations and mapping of 12q13 amplicons. Int J Cancer (1999) 80:199–204. doi:10.1002/(SICI)1097-0215(19990118)80:2<199::AID-IJC7>3.0.CO;2-4
31. Sharpless NE. Ink4a/Arf links senescence and aging. Exp Gerontol (2004) 39:1751–9. doi:10.1016/j.exger.2004.06.025
32. McIntyre JF, Smith-Sorensen B, Friend SH, Kassell J, Borresen AL, Yan YX, et al. Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol (1994) 12:925–30.
33. Gokgoz N, Wunder JS, Mousses S, Eskandarian S, Bell RS, Andrulis IL. Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer (2001) 92:2181–9. doi:10.1002/1097-0142(20011015)92:8<2181::AID-CNCR1561>3.0.CO;2-3
34. Hauben EI, Arends J, Vandenbroucke JP, Van Asperen CJ, Van Marck E, Hogendoorn PC. Multiple primary malignancies in osteosarcoma patients. Incidence and predictive value of osteosarcoma subtype for cancer syndromes related with osteosarcoma. Eur J Hum Genet (2003) 11:611–8. doi:10.1038/sj.ejhg.5201012
35. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature (1992) 358:80–3. doi:10.1038/358080a0
36. Ladanyi M, Cha C, Lewis R, Jhanwar SC, Huvos AG, Healey JH. MDM2 gene amplification in metastatic osteosarcoma. Cancer Res (1993) 53:16–8.
37. Lonardo F, Ueda T, Huvos AG, Healey J, Ladanyi M. p53 and MDM2 alterations in osteosarcomas: correlation with clinicopathologic features and proliferative rate. Cancer (1997) 79:1541–7. doi:10.1002/(SICI)1097-0142(19970415)79:8<1541::AID-CNCR15>3.3.CO;2-5
38. Lopez-Guerrero JA, Lopez-Gines C, Pellin A, Carda C, Llombart-Bosch A. Deregulation of the G1 to S-phase cell cycle checkpoint is involved in the pathogenesis of human osteosarcoma. Diagn Mol Pathol (2004) 13:81–91. doi:10.1097/00019606-200406000-00004
39. Wu JX, Carpenter PM, Gresens C, Keh R, Niman H, Morris JW, et al. The proto-oncogene c-fos is over-expressed in the majority of human osteosarcomas. Oncogene (1990) 5:989–1000.
40. Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR, et al. C-myc and c-fos in human osteosarcoma: prognostic value of mRNA and protein expression. Oncology (1998) 55:556–63. doi:10.1159/000011912
41. Shimizu T, Ishikawa T, Sugihara E, Kuninaka S, Miyamoto T, Mabuchi Y, et al. c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene (2010) 29:5687–99. doi:10.1038/onc.2010.312
42. Wang ZQ, Liang J, Schellander K, Wagner EF, Grigoriadis AE. c-fos-induced osteosarcoma formation in transgenic mice: cooperativity with c-jun and the role of endogenous c-fos. Cancer Res (1995) 55:6244–51.
43. Papachristou DJ, Batistatou A, Sykiotis GP, Varakis I, Papavassiliou AG. Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone (2003) 32:364–71. doi:10.1016/S8756-3282(03)00026-7
44. Engin F, Bertin T, Ma O, Jiang MM, Wang L, Sutton RE, et al. Notch signaling contributes to the pathogenesis of human osteosarcomas. Hum Mol Genet (2009) 18:1464–70. doi:10.1093/hmg/ddp057
45. Zhang P, Yang Y, Nolo R, Zweidler-McKay PA, Hughes DP. Regulation of NOTCH signaling by reciprocal inhibition of HES1 and Deltex 1 and its role in osteosarcoma invasiveness. Oncogene (2010) 29:2916–26. doi:10.1038/onc.2010.62
46. Wang LL. Biology of osteogenic sarcoma. Cancer J (2005) 11:294–305. doi:10.1097/00130404-200507000-00005
47. Hahm KB. Repression of the gene encoding the TGF-beta type II receptor is a major target of the EWS-FLI1 oncoprotein. Nat Genet (1999) 23:481. doi:10.1038/70611
48. Im YH, Kim HT, Lee C, Poulin D, Welford S, Sorensen PH, et al. EWS-FLI1, EWS-ERG, and EWS-ETV1 oncoproteins of Ewing tumor family all suppress transcription of transforming growth factor beta type II receptor gene. Cancer Res (2000) 60:1536–40.
49. Ban J, Bennani-Baiti IM, Kauer M, Schaefer KL, Poremba C, Jug G, et al. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res (2008) 68:7100–9. doi:10.1158/0008-5472.CAN-07-6145
50. Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, et al. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem (2009) 284:9074–82. doi:10.1074/jbc.M806233200
51. Herrero-Martin D, Osuna D, Ordonez JL, Sevillano V, Martins AS, MacKintosh C, et al. Stable interference of EWS-FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target. Br J Cancer (2009) 101:80–90. doi:10.1038/sj.bjc.6605104
52. Navarro D, Agra N, Pestana A, Alonso J, Gonzalez-Sancho JM. The EWS/FLI1 oncogenic protein inhibits expression of the Wnt inhibitor DICKKOPF-1 gene and antagonizes beta-catenin/TCF-mediated transcription. Carcinogenesis (2010) 31:394–401. doi:10.1093/carcin/bgp317
53. Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res (2009) 69:9047–55. doi:10.1158/0008-5472.CAN-09-1540
54. Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A (2009) 106:5324–9. doi:10.1073/pnas.0810759106
55. MacKintosh C, Madoz-Gurpide J, Ordonez JL, Osuna D, Herrero-Martin D. The molecular pathogenesis of Ewing’s sarcoma. Cancer Biol Ther (2010) 9:655–67. doi:10.4161/cbt.9.9.11511
56. Simmons O, Maples PB, Senzer N, Nemunaitis J. Ewing’s sarcoma: development of RNA interference-based therapy for advanced disease. ISRN Oncol (2012) 2012:247657. doi:10.5402/2012/247657
57. de Alava E, Antonescu CR, Panizo A, Leung D, Meyers PA, Huvos AG, et al. Prognostic impact of P53 status in Ewing sarcoma. Cancer (2000) 89:783–92. doi:10.1002/1097-0142(20000815)89:4<783::AID-CNCR10>3.3.CO;2-H
58. Park YK, Chi SG, Kim YW, Park HR, Unni KK. P53 mutations in Ewing’s sarcoma. Oncol Rep (2001) 8:533–7.
59. Radig K, Schneider-Stock R, Rose I, Mittler U, Oda Y, Roessner A. p53 and ras mutations in Ewing’s sarcoma. Pathol Res Pract (1998) 194:157–62. doi:10.1016/S0344-0338(98)80016-2
60. Tsuchiya T, Sekine K, Hinohara S, Namiki T, Nobori T, Kaneko Y. Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet Cytogenet (2000) 120:91–8. doi:10.1016/S0165-4608(99)00255-1
61. Wei G, Antonescu CR, De Alava E, Leung D, Huvos AG, Meyers PA, et al. Prognostic impact of INK4A deletion in Ewing sarcoma. Cancer (2000) 89:793–9. doi:10.1002/1097-0142(20000815)89:4<793::AID-CNCR11>3.0.CO;2-M
62. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol (2006) 70:1469–80. doi:10.1124/mol.106.027029
63. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi:10.1126/science.1059796
64. Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem (2005) 74:115–28. doi:10.1146/annurev.biochem.74.082803.133142
65. Kuzmanov A, Wielockx B, Rezaei M, Kettelhake A, Breier G. Overexpression of factor inhibiting HIF-1 enhances vessel maturation and tumor growth via platelet-derived growth factor-C. Int J Cancer (2012) 131:E603–13. doi:10.1002/ijc.27360
66. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol (2005) 23:1011–27. doi:10.1200/JCO.2005.06.081
67. Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, et al. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell (2004) 6:485–95. doi:10.1016/j.ccr.2004.09.026
68. Dvorak HF. Angiogenesis: update 2005. J Thromb Haemost (2005) 3:1835–42. doi:10.1111/j.1538-7836.2005.01361.x
69. McCarty G, Awad O, Loeb DM. WT1 protein directly regulates expression of vascular endothelial growth factor and is a mediator of tumor response to hypoxia. J Biol Chem (2011) 286: 43634–43. doi:10.1074/jbc.M111.310128
70. Hingorani P, Zhang W, Gorlick R, Kolb EA. Inhibition of Src phosphorylation alters metastatic potential of osteosarcoma in vitro but not in vivo. Clin Cancer Res (2009) 15:3416–22. doi:10.1158/1078-0432.CCR-08-1657
71. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol (2009) 6:587–95. doi:10.1038/nrclinonc.2009.129
72. Bid HK, Oswald D, Li C, London CA, Lin J, Houghton PJ. Anti-angiogenic activity of a small molecule STAT3 inhibitor LLL12. PLoS ONE (2012) 7:e35513. doi:10.1371/journal.pone.0035513
73. Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res (2006) 312:549–60. doi:10.1016/j.yexcr.2005.11.012
74. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology (2005) 69(Suppl 3):4–10. doi:10.1159/000088478
75. Tran J, Rak J, Sheehan C, Saibil SD, Lacasse E, Korneluk RG, et al. Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem Biophys Res Commun (1999) 264:781–8. doi:10.1006/bbrc.1999.1589
76. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med (2006) 12:895–904. doi:10.1038/nm1469
77. Yang J, Yang D, Sun Y, Sun B, Wang G, Trent JC, et al. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer (2011) 117:4925–38. doi:10.1002/cncr.26116
78. Kaya M, Wada T, Akatsuka T, Kawaguchi S, Nagoya S, Shindoh M, et al. Vascular endothelial growth factor expression in untreated osteosarcoma is predictive of pulmonary metastasis and poor prognosis. Clin Cancer Res (2000) 6:572–7.
79. DuBois S, Demetri G. Markers of angiogenesis and clinical features in patients with sarcoma. Cancer (2007) 109:813–9. doi:10.1002/cncr.22455
80. Lammli J, Fan M, Rosenthal HG, Patni M, Rinehart E, Vergara G, et al. Expression of vascular endothelial growth factor correlates with the advance of clinical osteosarcoma. Int Orthop (2012) 36:2307–13. doi:10.1007/s00264-012-1629-z
81. Holzer G, Obermair A, Koschat M, Preyer O, Kotz R, Trieb K. Concentration of vascular endothelial growth factor (VEGF) in the serum of patients with malignant bone tumors. Med Pediatr Oncol (2001) 36:601–4. doi:10.1002/mpo.1136
82. Pavlakovic H, Von Schutz V, Rossler J, Koscielniak E, Havers W, Schweigerer L. Quantification of angiogenesis stimulators in children with solid malignancies. Int J Cancer (2001) 92:756–60. doi:10.1002/1097-0215(20010601)92:5<756::AID-IJC1253>3.0.CO;2-F
83. Fuchs B, Inwards CY, Janknecht R. Vascular endothelial growth factor expression is up-regulated by EWS-ETS oncoproteins and Sp1 and may represent an independent predictor of survival in Ewing’s sarcoma. Clin Cancer Res (2004) 10:1344–53. doi:10.1158/1078-0432.CCR-03-0038
84. Stewart KS, Kleinerman ES. Tumor vessel development and expansion in Ewing’s sarcoma: a review of the vasculogenesis process and clinical trials with vascular-targeting agents. Sarcoma (2011) 2011:165837. doi:10.1155/2011/165837
85. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature (2000) 407:242–8. doi:10.1038/35025215
86. Cai J, Parr C, Watkins G, Jiang WG, Boulton M. Decreased pigment epithelium-derived factor expression in human breast cancer progression. Clin Cancer Res (2006) 12:3510–7. doi:10.1158/1078-0432.CCR-06-0094
87. Ren B, Yee KO, Lawler J, Khosravi-Far R. Regulation of tumor angiogenesis by thrombospondin-1. Biochim Biophys Acta (2006) 1765:178–88.
88. Clark JC, Akiyama T, Thomas DM, Labrinidis A, Evdokiou A, Galloway SJ, et al. RECK in osteosarcoma: a novel role in tumour vasculature and inhibition of tumorigenesis in an orthotopic model. Cancer (2011) 117:3517–28. doi:10.1002/cncr.25757
89. Cheng YY, Huang L, Lee KM, Li K, Kumta SM. Alendronate regulates cell invasion and MMP-2 secretion in human osteosarcoma cell lines. Pediatr Blood Cancer (2004) 42:410–5. doi:10.1002/pbc.20019
90. Cho HJ, Lee TS, Park JB, Park KK, Choe JY, Sin DI, et al. Disulfiram suppresses invasive ability of osteosarcoma cells via the inhibition of MMP-2 and MMP-9 expression. J Biochem Mol Biol (2007) 40:1069–76. doi:10.5483/BMBRep.2007.40.6.1069
91. Xin ZF, Kim YK, Jung ST. Risedronate inhibits human osteosarcoma cell invasion. J Exp Clin Cancer Res (2009) 28:105. doi:10.1186/1756-9966-28-105
92. Fan DG, Dai JY, Tang J, Wu MM, Sun SG, Jiang JL, et al. Silencing of calpain expression reduces the metastatic potential of human osteosarcoma cells. Cell Biol Int (2009) 33:1263–7. doi:10.1016/j.cellbi.2009.08.014
93. Dass CR, Nadesapillai AP, Robin D, Howard ML, Fisher JL, Zhou H, et al. Downregulation of uPAR confirms link in growth and metastasis of osteosarcoma. Clin Exp Metastasis (2005) 22:643–52. doi:10.1007/s10585-006-9004-3
94. Haydon RC, Deyrup A, Ishikawa A, Heck R, Jiang W, Zhou L, et al. Cytoplasmic and/or nuclear accumulation of the beta-catenin protein is a frequent event in human osteosarcoma. Int J Cancer (2002) 102:338–42. doi:10.1002/ijc.10719
95. Iwaya K, Ogawa H, Kuroda M, Izumi M, Ishida T, Mukai K. Cytoplasmic and/or nuclear staining of beta-catenin is associated with lung metastasis. Clin Exp Metastasis (2003) 20:525–9. doi:10.1023/A:1025821229013
96. Leow PC, Tian Q, Ong ZY, Yang Z, Ee PL. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/beta-catenin antagonists against human osteosarcoma cells. Invest New Drugs (2010) 28:766–82. doi:10.1007/s10637-009-9311-z
97. Rubin EM, Guo Y, Tu K, Xie J, Zi X, Hoang BH. Wnt inhibitory factor 1 decreases tumorigenesis and metastasis in osteosarcoma. Mol Cancer Ther (2010) 9:731–41. doi:10.1158/1535-7163.MCT-09-0147
98. Hughes DP. How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat Res (2009) 152:479–96. doi:10.1007/978-1-4419-0284-9_28
99. Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res (2007) 67:2800–8. doi:10.1158/0008-5472.CAN-06-3469
100. Yang R, Hoang BH, Kubo T, Kawano H, Chou A, Sowers R, et al. Over-expression of parathyroid hormone Type 1 receptor confers an aggressive phenotype in osteosarcoma. Int J Cancer (2007) 121:943–54. doi:10.1002/ijc.22749
101. Berdiaki A, Datsis GA, Nikitovic D, Tsatsakis A, Katonis P, Karamanos NK, et al. Parathyroid hormone (PTH) peptides through the regulation of hyaluronan metabolism affect osteosarcoma cell migration. IUBMB Life (2010) 62:377–86.
102. Dobashi Y, Watanabe H, Matsubara M, Yanagawa T, Raz A, Shimamiya T, et al. Autocrine motility factor/glucose-6-phosphate isomerase is a possible predictor of metastasis in bone and soft tissue tumours. J Pathol (2006) 208:44–53. doi:10.1002/path.1878
103. Niinaka Y, Harada K, Fujimuro M, Oda M, Haga A, Hosoki M, et al. Silencing of autocrine motility factor induces mesenchymal-to-epithelial transition and suppression of osteosarcoma pulmonary metastasis. Cancer Res (2010) 70:9483–93. doi:10.1158/0008-5472.CAN-09-3880
104. Fromigue O, Hamidouche Z, Vaudin P, Lecanda F, Patino A, Barbry P, et al. CYR61 downregulation reduces osteosarcoma cell invasion, migration, and metastasis. J Bone Miner Res (2011) 26:1533–42. doi:10.1002/jbmr.343
105. Lin YM, Chang ZL, Liao YY, Chou MC, Tang CH. IL-6 promotes ICAM-1 expression and cell motility in human osteosarcoma. Cancer Lett (2013) 328:135–43. doi:10.1016/j.canlet.2012.08.029
106. Sainz-Jaspeado M, Lagares-Tena L, Lasheras J, Navid F, Rodriguez-Galindo C, Mateo-Lozano S, et al. Caveolin-1 modulates the ability of Ewing’s sarcoma to metastasize. Mol Cancer Res (2010) 8:1489–500. doi:10.1158/1541-7786.MCR-10-0060
107. Manara MC, Perbal B, Benini S, Strammiello R, Cerisano V, Perdichizzi S, et al. The expression of ccn3(nov) gene in musculoskeletal tumors. Am J Pathol (2002) 160:849–59. doi:10.1016/S0002-9440(10)64908-5
108. Perbal B, Lazar N, Zambelli D, Lopez-Guerrero JA, Llombart-Bosch A, Scotlandi K, et al. Prognostic relevance of CCN3 in Ewing sarcoma. Hum Pathol (2009) 40:1479–86. doi:10.1016/j.humpath.2009.05.008
109. Benini S, Perbal B, Zambelli D, Colombo MP, Manara MC, Serra M, et al. In Ewing’s sarcoma CCN3(NOV) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene (2005) 24:4349–61. doi:10.1038/sj.onc.1208620
110. Hauer K, Calzada-Wack J, Steiger K, Grunewald TG, Baumhoer D, Plehm S, et al. DKK2 mediates osteolysis, invasiveness, and metastatic spread in Ewing sarcoma. Cancer Res (2013) 73:967–77. doi:10.1158/0008-5472.CAN-12-1492
111. Staege MS, Hutter C, Neumann I, Foja S, Hattenhorst UE, Hansen G, et al. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res (2004) 64:8213–21. doi:10.1158/0008-5472.CAN-03-4059
112. Grunewald TG, Diebold I, Esposito I, Plehm S, Hauer K, Thiel U, et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res (2012) 10:52–65. doi:10.1158/1541-7786.MCR-11-0524
113. Guan H, Zhou Z, Gallick GE, Jia SF, Morales J, Sood AK, et al. Targeting Lyn inhibits tumor growth and metastasis in Ewing’s sarcoma. Mol Cancer Ther (2008) 7:1807–16. doi:10.1158/1535-7163.MCT-08-0058
114. Bennani-Baiti IM, Cooper A, Lawlor ER, Kauer M, Ban J, Aryee DN, et al. Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing’s sarcoma. Clin Cancer Res (2010) 16:3769–78. doi:10.1158/1078-0432.CCR-10-0558
115. Jin Z, Zhao C, Han X, Han Y. Wnt5a promotes Ewing sarcoma cell migration through upregulating CXCR4 expression. BMC Cancer (2012) 12:480. doi:10.1186/1471-2407-12-480
116. Landuzzi L, De Giovanni C, Nicoletti G, Rossi I, Ricci C, Astolfi A, et al. The metastatic ability of Ewing’s sarcoma cells is modulated by stem cell factor and by its receptor c-kit. Am J Pathol (2000) 157:2123–31. doi:10.1016/S0002-9440(10)64850-X
117. Mendoza-Naranjo A, El-Naggar A, Wai DH, Mistry P, Lazic N, Ayala FR, et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol Med (2013) 5:1087–102. doi:10.1002/emmm.201202343
118. Ziyan W, Shuhua Y, Xiufang W, Xiaoyun L. MicroRNA-21 is involved in osteosarcoma cell invasion and migration. Med Oncol (2011) 28. doi:10.1007/s12032-010-9563-7
119. Jones KB, Salah Z, Del Mare S, Galasso M, Gaudio E, Nuovo GJ, et al. miRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Res (2012) 72:1865–77. doi:10.1158/0008-5472.CAN-11-2663
120. Osaki M, Takeshita F, Sugimoto Y, Kosaka N, Yamamoto Y, Yoshioka Y, et al. MicroRNA-143 regulates human osteosarcoma metastasis by regulating matrix metalloprotease-13 expression. Mol Ther (2011) 19:1123–30. doi:10.1038/mt.2011.53
121. Zhu J, Feng Y, Ke Z, Yang Z, Zhou J, Huang X, et al. Down-regulation of miR-183 promotes migration and invasion of osteosarcoma by targeting Ezrin. Am J Pathol (2012) 180:2440–51. doi:10.1016/j.ajpath.2012.02.023
122. Mao JH, Zhou RP, Peng AF, Liu ZL, Huang SH, Long XH, et al. microRNA-195 suppresses osteosarcoma cell invasion and migration in vitro by targeting FASN. Oncol Lett (2012) 4:1125–9.
123. Ando K, Mori K, Verrecchia F, Marc B, Redini F, Heymann D. Molecular alterations associated with osteosarcoma development. Sarcoma (2012) 2012:523432. doi:10.1155/2012/523432
124. Marco RA, Diaz-Montero CM, Wygant JN, Kleinerman ES, Mcintyre BW. Alpha 4 integrin increases anoikis of human osteosarcoma cells. J Cell Biochem (2003) 88:1038–47. doi:10.1002/jcb.10465
125. Wan X, Kim SY, Guenther LM, Mendoza A, Briggs J, Yeung C, et al. Beta4 integrin promotes osteosarcoma metastasis and interacts with ezrin. Oncogene (2009) 28:3401–11. doi:10.1038/onc.2009.206
126. Janes SM, Watt FM. Switch from alphavbeta5 to alphavbeta6 integrin expression protects squamous cell carcinomas from anoikis. J Cell Biol (2004) 166:419–31. doi:10.1083/jcb.200312074
127. Diaz-Montero CM, Wygant JN, McIntyre BW. PI3-K/Akt-mediated anoikis resistance of human osteosarcoma cells requires Src activation. Eur J Cancer (2006) 42:1491–500. doi:10.1016/j.ejca.2006.03.007
128. Cantiani L, Manara MC, Zucchini C, De Sanctis P, Zuntini M, Valvassori L, et al. Caveolin-1 reduces osteosarcoma metastases by inhibiting c-Src activity and met signaling. Cancer Res (2007) 67:7675–85. doi:10.1158/0008-5472.CAN-06-4697
129. Beristain AG, Narala SR, Di Grappa MA, Khokha R. Homotypic RANK signaling differentially regulates proliferation, motility and cell survival in osteosarcoma and mammary epithelial cells. J Cell Sci (2012) 125:943–55. doi:10.1242/jcs.094029
130. Lin D, Feng J, Chen W. Bcl-2 and caspase-8 related anoikis resistance in human osteosarcoma MG-63 cells. Cell Biol Int (2008) 32:1199–206. doi:10.1016/j.cellbi.2008.07.002
131. Lawlor ER, Scheel C, Irving J, Sorensen PH. Anchorage-independent multi-cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene (2002) 21:307–18. doi:10.1038/sj.onc.1205053
132. Benini S, Manara MC, Cerisano V, Perdichizzi S, Strammiello R, Serra M, et al. Contribution of MEK/MAPK and PI3-K signaling pathway to the malignant behavior of Ewing’s sarcoma cells: therapeutic prospects. Int J Cancer (2004) 108:358–66. doi:10.1002/ijc.11576
133. Scotlandi K, Maini C, Manara MC, Benini S, Serra M, Cerisano V, et al. Effectiveness of insulin-like growth factor I receptor antisense strategy against Ewing’s sarcoma cells. Cancer Gene Ther (2002) 9:296–307. doi:10.1038/sj.cgt.7700442
134. Douglas D, Hsu JH, Hung L, Cooper A, Abdueva D, Van Doorninck J, et al. BMI-1 promotes Ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res (2008) 68:6507–15. doi:10.1158/0008-5472.CAN-07-6152
135. Tirado OM, Mateo-Lozano S, Villar J, Dettin LE, Llort A, Gallego S, et al. Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res (2006) 66:9937–47. doi:10.1158/0008-5472.CAN-06-0927
136. Kang HG, Jenabi JM, Zhang J, Keshelava N, Shimada H, May WA, et al. E-cadherin cell-cell adhesion in Ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res (2007) 67:3094–105. doi:10.1158/0008-5472.CAN-06-3259
137. Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med (2000) 6:100–2. doi:10.1038/71429
138. Jeffree GM, Price CH, Sissons HA. The metastatic patterns of osteosarcoma. Br J Cancer (1975) 32:87–107. doi:10.1038/bjc.1975.136
139. Cotterill SJ, Ahrens S, Paulussen M, Jurgens HF, Voute PA, Gadner H, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol (2000) 18:3108–14.
140. Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet (2007) 369:1742–57. doi:10.1016/S0140-6736(07)60781-8
141. Murphy PM. Chemokines and the molecular basis of cancer metastasis. N Engl J Med (2001) 345:833–5. doi:10.1056/NEJM200109133451113
142. Laverdiere C, Hoang BH, Yang R, Sowers R, Qin J, Meyers PA, et al. Messenger RNA expression levels of CXCR4 correlate with metastatic behavior and outcome in patients with osteosarcoma. Clin Cancer Res (2005) 11:2561–7. doi:10.1158/1078-0432.CCR-04-1089
143. de Nigris F, Rossiello R, Schiano C, Arra C, Williams-Ignarro S, Barbieri A, et al. Deletion of Yin Yang 1 protein in osteosarcoma cells on cell invasion and CXCR4/angiogenesis and metastasis. Cancer Res (2008) 68:1797–808. doi:10.1158/0008-5472.CAN-07-5582
144. Huang CY, Lee CY, Chen MY, Yang WH, Chen YH, Chang CH, et al. Stromal cell-derived factor-1/CXCR4 enhanced motility of human osteosarcoma cells involves MEK1/2, ERK and NF-kappaB-dependent pathways. J Cell Physiol (2009) 221:204–12. doi:10.1002/jcp.21846
145. Lin F, Zheng SE, Shen Z, Tang LN, Chen P, Sun YJ, et al. Relationships between levels of CXCR4 and VEGF and blood-borne metastasis and survival in patients with osteosarcoma. Med Oncol (2011) 28:649–53. doi:10.1007/s12032-010-9493-4
146. Krishnan K, Khanna C, Helman LJ. The biology of metastases in pediatric sarcomas. Cancer J (2005) 11:306–13. doi:10.1097/00130404-200507000-00006
147. Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev (2006) 25:521–9. doi:10.1007/s10555-006-9036-9
148. Kim SY, Lee CH, Midura BV, Yeung C, Mendoza A, Hong SH, et al. Inhibition of the CXCR4/CXCL12 chemokine pathway reduces the development of murine pulmonary metastases. Clin Exp Metastasis (2008) 25:201–11. doi:10.1007/s10585-007-9133-3
149. Pradelli E, Karimdjee-Soilihi B, Michiels JF, Ricci JE, Millet MA, Vandenbos F, et al. Antagonism of chemokine receptor CXCR3 inhibits osteosarcoma metastasis to lungs. Int J Cancer (2009) 125:2586–94. doi:10.1002/ijc.24665
150. Paavonen T, Tiisala S, Majuri ML, Bohling T, Renkonen R. In vivo evidence of the role of alpha 4 beta 1-VCAM-1 interaction in sarcoma, but not in carcinoma extravasation. Int J Cancer (1994) 58:298–302. doi:10.1002/ijc.2910580225
151. Mangeat P, Roy C, Martin M. ERM proteins in cell adhesion and membrane dynamics. Trends Cell Biol (1999) 9:187–92. doi:10.1016/S0962-8924(99)01544-5
152. Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, et al. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res (2001) 61:3750–9.
153. Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med (2004) 10:182–6. doi:10.1038/nm982
154. Ren L, Hong SH, Cassavaugh J, Osborne T, Chou AJ, Kim SY, et al. The actin-cytoskeleton linker protein ezrin is regulated during osteosarcoma metastasis by PKC. Oncogene (2009) 28:792–802. doi:10.1038/onc.2008.437
155. Pignochino Y, Grignani G, Cavalloni G, Motta M, Tapparo M, Bruno S, et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol Cancer (2009) 8:118. doi:10.1186/1476-4598-8-118
156. Huang S, Deguzman A, Bucana CD, Fidler IJ. Nuclear factor-kappaB activity correlates with growth, angiogenesis, and metastasis of human melanoma cells in nude mice. Clin Cancer Res (2000) 6:2573–81.
157. Futakuchi M, Ogawa K, Tamano S, Takahashi S, Shirai T. Suppression of metastasis by nuclear factor kappaB inhibitors in an in vivo lung metastasis model of chemically induced hepatocellular carcinoma. Cancer Sci (2004) 95:18–24. doi:10.1111/j.1349-7006.2004.tb03165.x
158. Batra S, Balamayooran G, Sahoo MK. Nuclear factor-kappaB: a key regulator in health and disease of lungs. Arch Immunol Ther Exp (Warsz) (2011) 59:335–51. doi:10.1007/s00005-011-0136-z
159. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res (2012) 72:1290–300. doi:10.1158/0008-5472.CAN-11-3123
160. Tomonaga M, Hashimoto N, Tokunaga F, Onishi M, Myoui A, Yoshikawa H, et al. Activation of nuclear factor-kappa B by linear ubiquitin chain assembly complex contributes to lung metastasis of osteosarcoma cells. Int J Oncol (2012) 40:409–17. doi:10.3892/ijo.2011.1209
161. van Breugel FM. The action of the Notch locus in Drosophila hydei. Genetica (1971) 42:25–41. doi:10.1007/BF00154837
162. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414:105–11. doi:10.1038/35102167
163. Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res (2007) 17:3–14. doi:10.1038/sj.cr.7310118
164. Marotta LL, Polyak K. Cancer stem cells: a model in the making. Curr Opin Genet Dev (2009) 19:44–50. doi:10.1016/j.gde.2008.12.003
165. Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A (2010) 107:18115–20. doi:10.1073/pnas.1006732107
166. Cairns J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc Natl Acad Sci U S A (2002) 99:10567–70. doi:10.1073/pnas.162369899
167. Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci (2002) 115:2381–8.
168. Wang S, Yang D, Lippman ME. Targeting Bcl-2 and Bcl-XL with nonpeptidic small-molecule antagonists. Semin Oncol (2003) 30:133–42. doi:10.1053/j.seminoncol.2003.08.015
169. Park Y, Gerson SL. DNA repair defects in stem cell function and aging. Annu Rev Med (2005) 56:495–508. doi:10.1146/annurev.med.56.082103.104546
170. Shinin V, Gayraud-Morel B, Gomes D, Tajbakhsh S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol (2006) 8:677–87. doi:10.1038/ncb1425
171. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer (2003) 3:895–902. doi:10.1038/nrc1232
172. Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene (2007) 26:1357–60. doi:10.1038/sj.onc.1210200
173. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia (2009) 14:3–9. doi:10.1007/s10911-009-9109-9
174. Donnenberg VS, Meyer EM, Donnenberg AD. Measurement of multiple drug resistance transporter activity in putative cancer stem/progenitor cells. Methods Mol Biol (2009) 568:261–79. doi:10.1007/978-1-59745-280-9_17
175. Ding XW, Wu JH, Jiang CP. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci (2010) 86:631–7. doi:10.1016/j.lfs.2010.02.012
176. Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther (2011) 89:491–502. doi:10.1038/clpt.2011.14
177. Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med (1995) 1:149–53. doi:10.1038/nm0295-149
178. Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS (2008) 116:754–70. doi:10.1111/j.1600-0463.2008.01033.x
179. Cao Y, O’Reilly MS, Marshall B, Flynn E, Ji RW, Folkman J. Expression of angiostatin cDNA in a murine fibrosarcoma suppresses primary tumor growth and produces long-term dormancy of metastases. J Clin Invest (1998) 101:1055–63. doi:10.1172/JCI1558
180. Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst (2006) 98:316–25. doi:10.1093/jnci/djj068
181. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature (2007) 450:903–7. doi:10.1038/nature06309
182. Favaro E, Amadori A, Indraccolo S. Cellular interactions in the vascular niche: implications in the regulation of tumor dormancy. APMIS (2008) 116:648–59. doi:10.1111/j.1600-0463.2008.01025.x
183. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer (2010) 46:1181–8. doi:10.1016/j.ejca.2010.02.027
184. Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat (2003) 82:199–206. doi:10.1023/B:BREA.0000004377.12288.3c
185. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer (2007) 7:834–46. doi:10.1038/nri2210
186. Almog N, Ma L, Raychowdhury R, Schwager C, Erber R, Short S, et al. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res (2009) 69:836–44. doi:10.1158/0008-5472.CAN-08-2590
187. Indraccolo S, Favaro E, Amadori A. Dormant tumors awaken by a short-term angiogenic burst: the spike hypothesis. Cell Cycle (2006) 5:1751–5. doi:10.4161/cc.5.16.2985
188. Roessner A, Ueda Y, Bockhorn-Dworniczak B, Blasius S, Peters A, Wuisman P, et al. Prognostic implication of immunodetection of P glycoprotein in Ewing’s sarcoma. J Cancer Res Clin Oncol (1993) 119:185–9. doi:10.1007/BF01624429
189. Baldini N, Scotlandi K, Barbanti-Brodano G, Manara MC, Maurici D, Bacci G, et al. Expression of P-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med (1995) 333:1380–5. doi:10.1056/NEJM199511233332103
190. Serra M, Scotlandi K, Manara MC, Maurici D, Benini S, Sarti M, et al. Analysis of P-glycoprotein expression in osteosarcoma. Eur J Cancer (1995) 31A:1998–2002. doi:10.1016/0959-8049(95)00335-5
191. Ferrari S, Bertoni F, Zanella L, Setola E, Bacchini P, Alberghini M, et al. Evaluation of P-glycoprotein, HER-2/ErbB-2, p53, and Bcl-2 in primary tumor and metachronous lung metastases in patients with high-grade osteosarcoma. Cancer (2004) 100:1936–42. doi:10.1002/cncr.20151
192. Gorlick R, Huvos AG, Heller G, Aledo A, Beardsley GP, Healey JH, et al. Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J Clin Oncol (1999) 17:2781–8.
193. Kilpatrick SE, Geisinger KR, King TS, Sciarrotta J, Ward WG, Gold SH, et al. Clinicopathologic analysis of HER-2/neu immunoexpression among various histologic subtypes and grades of osteosarcoma. Mod Pathol (2001) 14:1277–83. doi:10.1038/modpathol.3880474
194. Anninga JK, Van De Vijver MJ, Cleton-Jansen AM, Kristel PM, Taminiau AH, Nooij M, et al. Overexpression of the HER-2 oncogene does not play a role in high-grade osteosarcomas. Eur J Cancer (2004) 40:963–70. doi:10.1016/j.ejca.2003.10.025
195. Scotlandi K, Manara MC, Hattinger CM, Benini S, Perdichizzi S, Pasello M, et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur J Cancer (2005) 41:1349–61. doi:10.1016/j.ejca.2005.03.015
196. Hua Y, Gorshkov K, Yang Y, Wang W, Zhang N, Hughes D. Slow down to stay alive: HER4 protects against cellular stress and confers chemoresistance in neuroblastoma. Cancer (2012) 118:5140–54. doi:10.1002/cncr.27496
197. Posl M, Amling M, Werner M, Basler I, Salzer-Kuntschik M, Winkler K, et al. Osteosarcoma – apoptosis and proliferation. Study of bcl-2 expression. Pathologe (1994) 15:337–44. doi:10.1007/s002920050063
198. Soldatenkov V, Notario V, Dritschilo A. Expression of the human Bcl-2 increases resistance of Ewing’s sarcoma cells to apoptosis and inhibits poly(ADP-ribose) polymerase cleavage induced by radiation. Int J Oncol (1996) 9:547–51.
199. Zhao Y, Zhang CL, Zeng BF, Wu XS, Gao TT, Oda Y. Enhanced chemosensitivity of drug-resistant osteosarcoma cells by lentivirus-mediated Bcl-2 silencing. Biochem Biophys Res Commun (2009) 390:642–7. doi:10.1016/j.bbrc.2009.10.020
200. Wang ZX, Yang JS, Pan X, Wang JR, Li J, Yin YM, et al. Functional and biological analysis of Bcl-xL expression in human osteosarcoma. Bone (2010) 47:445–54. doi:10.1016/j.bone.2010.05.027
201. Fan DG, Zhao F, Ding Y, Wu MM, Fan QY, Shimizu K, et al. BLCAP induces apoptosis in human Ewing’s sarcoma cells. Exp Biol Med (Maywood) (2011) 236:1030–5. doi:10.1258/ebm.2011.010315
202. Luo W, Gangwal K, Sankar S, Boucher KM, Thomas D, Lessnick SL. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene (2009) 28:4126–32. doi:10.1038/onc.2009.262
203. Tirado OM, MacCarthy CM, Fatima N, Villar J, Mateo-Lozano S, Notario V. Caveolin-1 promotes resistance to chemotherapy-induced apoptosis in Ewing’s sarcoma cells by modulating PKCalpha phosphorylation. Int J Cancer (2010) 126:426–36. doi:10.1002/ijc.24754
204. Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene (2009) 28:4065–74. doi:10.1038/onc.2009.274
205. Song B, Wang Y, Titmus MA, Botchkina G, Formentini A, Kornmann M, et al. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol Cancer (2010) 9:96. doi:10.1186/1476-4598-9-96
206. Nakatani F, Ferracin M, Manara MC, Ventura S, Del Monaco V, Ferrari S, et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J Pathol (2012) 226:796–805. doi:10.1002/path.3007
207. Robin TP, Smith A, McKinsey E, Reaves L, Jedlicka P, Ford HL. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Mol Cancer Res (2012) 10:1098–108. doi:10.1158/1541-7786.MCR-12-0086
208. Iida K, Fukushi J, Matsumoto Y, Oda Y, Takahashi Y, Fujiwara T, et al. miR-125b develops chemoresistance in Ewing sarcoma/primitive neuroectodermal tumor. Cancer Cell Int (2013) 13:21. doi:10.1186/1475-2867-13-21
209. Tsukahara T, Kawaguchi S, Torigoe T, Asanuma H, Nakazawa E, Shimozawa K, et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci (2006) 97:1374–80. doi:10.1111/j.1349-7006.2006.00317.x
210. PosthumaDeBoer J, Witlox MA, Kaspers GJ, Van Royen BJ. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clin Exp Metastasis (2011) 28:493–503. doi:10.1007/s10585-011-9384-x
211. Whelan J, Patterson D, Perisoglou M, Bielack S, Marina N, Smeland S, et al. The role of interferons in the treatment of osteosarcoma. Pediatr Blood Cancer (2010) 54:350–4. doi:10.1002/pbc.22136
212. Lafleur EA, Jia SF, Worth LL, Zhou Z, Owen-Schaub LB, Kleinerman ES. Interleukin (IL)-12 and IL-12 gene transfer up-regulate Fas expression in human osteosarcoma and breast cancer cells. Cancer Res (2001) 61:4066–71.
213. Anderson P, Kopp L, Anderson N, Cornelius K, Herzog C, Hughes D, et al. Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma). Expert Opin Investig Drugs (2008) 17:1703–15. doi:10.1517/13543784.17.11.1703
214. Worth LL, Lafleur EA, Jia SF, Kleinerman ES. Fas expression inversely correlates with metastatic potential in osteosarcoma cells. Oncol Rep (2002) 9:823–7.
215. Lafleur EA, Koshkina NV, Stewart J, Jia SF, Worth LL, Duan X, et al. Increased Fas expression reduces the metastatic potential of human osteosarcoma cells. Clin Cancer Res (2004) 10:8114–9. doi:10.1158/1078-0432.CCR-04-0353
216. Gordon N, Koshkina NV, Jia SF, Khanna C, Mendoza A, Worth LL, et al. Corruption of the Fas pathway delays the pulmonary clearance of murine osteosarcoma cells, enhances their metastatic potential, and reduces the effect of aerosol gemcitabine. Clin Cancer Res (2007) 13:4503–10. doi:10.1158/1078-0432.CCR-07-0313
217. Alexander S, Friedl P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med (2012) 18:13–26. doi:10.1016/j.molmed.2011.11.003
218. MacKall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res (2008) 14:4850–8. doi:10.1158/1078-0432.CCR-07-4065
219. Kleinerman ES, Erickson KL, Schroit AJ, Fogler WE, Fidler IJ. Activation of tumoricidal properties in human blood monocytes by liposomes containing lipophilic muramyl tripeptide. Cancer Res (1983) 43:2010–4.
220. MacEwen EG, Kurzman ID, Rosenthal RC, Smith BW, Manley PA, Roush JK, et al. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J Natl Cancer Inst (1989) 81:935–8. doi:10.1093/jnci/81.12.935
221. Meyers PA, Schwartz CL, Krailo MD, Healey JH, Bernstein ML, Betcher D, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival – a report from the Children’s Oncology Group. J Clin Oncol (2008) 26:633–8. doi:10.1200/JCO.2008.14.0095
222. Meyers PA, Schwartz CL, Krailo M, Kleinerman ES, Betcher D, Bernstein ML, et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol (2005) 23:2004–11. doi:10.1200/JCO.2005.06.031
223. Tarozzi A, Mariani E, Facchini A. In vitro cytolytic activity of human NK cells against osteosarcoma cell lines. Boll Soc Ital Biol Sper (1995) 71:221–6.
224. Mariani E, Tarozzi A, Meneghetti A, Cattini L, Facchini A. Human osteosarcoma cell susceptibility to natural killer cell lysis depends on CD54 and increases after TNF alpha incubation. FEBS Lett (1997) 406:83–8. doi:10.1016/S0014-5793(97)00247-0
225. Honorati MC, Neri S, Cattini L, Facchini A. IL-17 enhances the susceptibility of U-2 OS osteosarcoma cells to NK cell lysis. Clin Exp Immunol (2003) 133:344–9. doi:10.1046/j.1365-2249.2003.02234.x
226. Nakashima Y, Deie M, Yanada S, Sharman P, Ochi M. Magnetically labeled human natural killer cells, accumulated in vitro by an external magnetic force, are effective against HOS osteosarcoma cells. Int J Oncol (2005) 27:965–71.
227. Bond G, Hu W, Bond E, Robins H, Lutzker S, Arva N, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell (2004) 119:591–602. doi:10.1016/j.cell.2004.11.022
Keywords: osteosarcoma, Ewing sarcoma, metastasis, intravasation, neovascularization, tumor dormancy, anoikis resistance, cancer signaling
Citation: Zhu L, McManus MM and Hughes DPM (2013) Understanding the biology of bone sarcoma from early initiating events through late events in metastasis and disease progression. Front. Oncol. 3:230. doi: 10.3389/fonc.2013.00230
Received: 23 May 2013; Paper pending published: 05 June 2013;
Accepted: 21 August 2013; Published online: 17 September 2013.
Edited by:
Stephen Lessnick, University of Utah, USAReviewed by:
Patrick Joseph Grohar, Vanderbilt University, USATimothy M Fan, University of Illinois, USA
Copyright: © 2013 Zhu, McManus and Hughes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dennis P. M. Hughes, Department of Pediatrics – Research, UT MD Anderson Cancer Center, Unit 853; MOD 1.021d, Houston, TX 77030, USA e-mail:ZHBodWdoZXNAbWRhbmRlcnNvbi5vcmc=