
 Mathew W. Moore1
Mathew W. Moore1
- 1Genetics, ResearchDx, Irvine, CA, USA
- 2Department of Pediatrics, University of California San Francisco, San Francisco, CA, USA
The phosphatase and tensin homolog gene (PTEN) on chromosome 10q23.3 is a negative regulator of the PIK3/Akt survival pathway and is the most frequently deleted tumor suppressor gene in prostate cancer. Monoallelic loss of PTEN is present in up to 60% of localized prostate cancers and complete loss of PTEN in prostate cancer is linked to metastasis and androgen-independent progression. Studies on the genomic status of PTEN in prostate cancer initially used a two-color fluorescence in situ hybridization (FISH) assay for PTEN copy number detection in formalin fixed paraffin embedded tissue preparations. More recently, a four-color FISH assay containing two additional control probes flanking the PTEN locus with a lower false-positive rate was reported. Combined with the detection of other critical genomic biomarkers for prostate cancer such as ERG, androgen receptor, and MYC, the evaluation of PTEN genomic status has proven to be invaluable for patient stratification and management. Although less frequent than allelic deletions, point mutations in the gene and epigenetic silencing are also known to contribute to loss of PTEN function, and ultimately to prostate cancer initiation. Overall, it is clear that PTEN is a powerful biomarker for prostate cancer. Used as a companion diagnostic for emerging therapeutic drugs, FISH analysis of PTEN is promisingly moving human prostate cancer closer to more effective cancer management and therapies.
Introduction
Prostate cancer is one of the leading causes of cancer mortality in men in the Western world. In the United States, it is the most commonly diagnosed cancer in men and second only to lung cancer in the number of male cancer deaths (1). Prostate cancers display a variable range of clinical behaviors, from slow-growing tumors of little clinical significance to aggressively metastatic and lethal diseases. Current prognostic tools, such as pre-operative prostate specific antigen (PSA) levels, histological Gleason grading, clinical tumor, node, and metastasis (TNM) staging are used to place men in low-, intermediate-, and high-risk prostate cancer risk groupings. However, these prognostic tools often fail to accurately stratify individual patients at early stages of the disease. Although <5% of patients exhibit advanced disease, up to 40% of patients will eventually develop metastatic disease despite local therapy (2). Localized cancers are usually treated with radical prostatectomy or radiation. For more advanced cancers that have either recurred or metastasized, the gold standard treatment is androgen ablation therapy. Androgens play a central role in the normal development and growth of the prostate gland as well as the abnormal growth of prostate cancer. Androgen ablation by either surgical castration, or luteinizing hormone-releasing hormone (LHRH) analog treatments strongly inhibit the growth of localized advanced cancer by eliminating circulating testosterone (3, 4). Although very efficient at reducing cancer growth, this treatment eventually selects for cells that are no longer responsive to such therapy, resulting in a recurring lethal cancer within 18–24 months. This recurrent cancer is often referred to as Castration Resistant Prostate Cancer (CRPC) (5).
Given the broad spectrum of clinical and molecular behaviors, the wide range of clinical outcomes and their associated treatments, it is clear that prostate cancer is a highly heterogeneous disease that presents great complexities in determining risk stratification and appropriate treatment strategies. The main challenge for physicians remains to distinguish indolent from clinically significant tumors. With the goal of improving clinical management of the disease, current efforts are focusing on identifying the genes and understanding the pathways involved in mediating disease progression and treatment resistance. A further short term goal of genetic testing of tumor samples is the identification of appropriate companion diagnostics, allowing stratification of patients for treatment and monitoring of treatment.
Genetic Alterations in the PI3K/Akt/mTOR Signaling Pathway in Prostate Tumorigenesis
One pathway with a prominent role in prostate cancer is the phosphatidylinositide 3-kinases (PI3K) signaling pathway. Current estimates suggest that this signaling pathway is up-regulated in 30–50% of prostate cancers (6–8). PI3K signaling is initiated by the activation of a number of receptor tyrosine kinases, including platelet-derived growth factor receptor (PDGFR), insulin-like growth factor receptor (IGFR), and epidermal growth factor receptor (EGFR) (Figure 1).
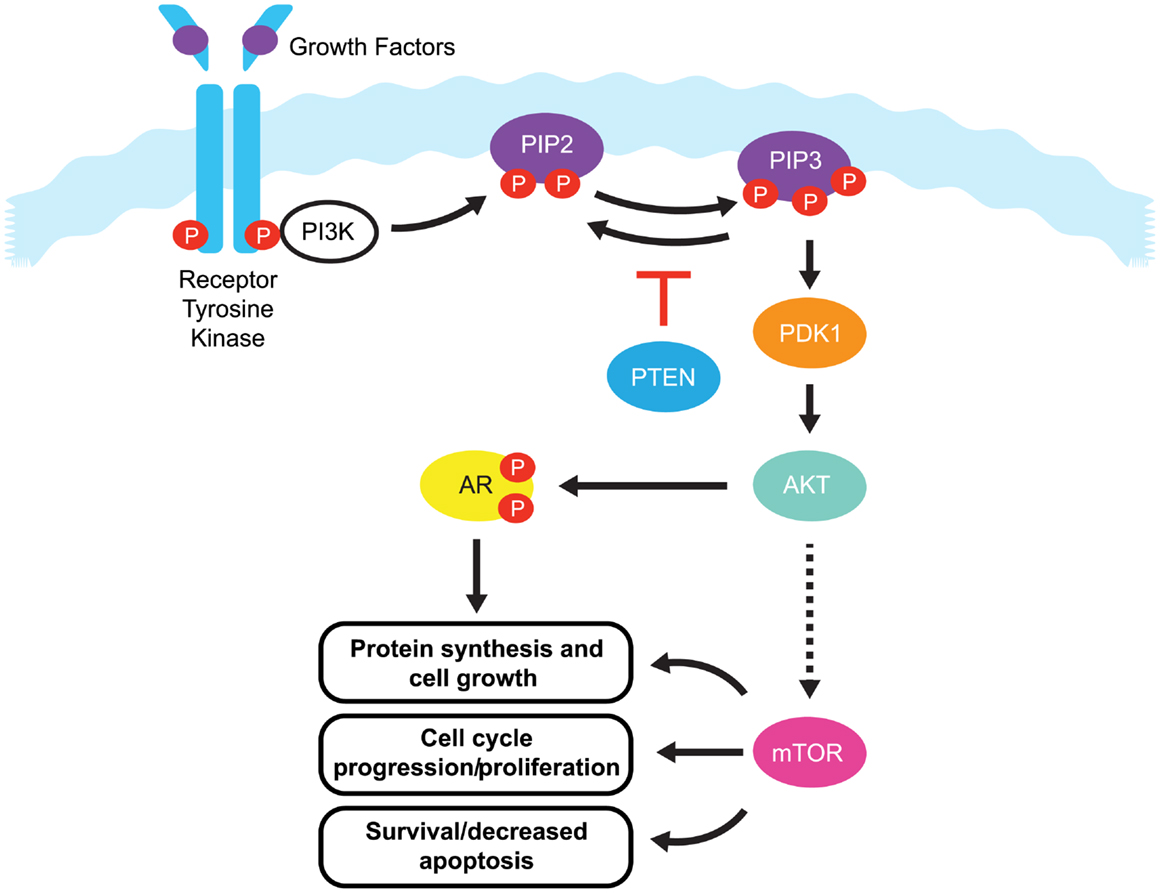
Figure 1. The PI3K/PTEN/Akt pathway. Binding of growth factors to the receptor tyrosine kinase activates the receptor complex, which in turn recruits and activates PI3K. Activated PI3K converts PIP2 to PIP3, which subsequently mediates the phosphorylation of Akt through PDK1. Phosphorylated Akt is active on a wide range of substrates, but one of its most important targets is mTOR, which is involved in cell growth, proliferation, and survival. Activated Akt also interacts with androgen receptor (AR) in an androgen-independent manner, leading to over-activation of the AR signaling pathway in castration resistant prostate cancer. PTEN is a tumor suppressor that negatively regulates the pathway by removing the 3-phosphate from PIP3, converting it back to PIP2. Loss of PTEN leads to over-activation of Akt which, in turn, is associated with uncontrolled cell proliferation, decreased apoptosis, and enhanced tumor angiogenesis.
Once activated, these receptors phosphorylate PI3K at the cell membrane. Phosphorylated PI3K in turn phosphorylates phosphatidylinositol-4,5-diphosphate (PIP2), leading to the accumulation of phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 recruits Akt (also known as protein kinase B) and phosphoinositide dependent protein kinase 1 (PDK1) to the cell membrane, where Akt is phosphorylated by PDK1. Phosphorylated Akt activity extends over a wide range of substrates, but most importantly activates the mammalian target of rapamycin (mTOR), which play a significant role in tumorigenesis (9). mTOR is a serine/threonine kinase that plays critical roles in the regulation of cell growth, survival, and division. Interaction between Akt and androgen receptor (AR) can lead to AR activation in a ligand-independent manner, ultimately up-regulating genes involved in CRPC tumorigenesis (9).
The primary negative regulator of the PI3K pathway is the tumor suppressor phosphatase and tensin homolog gene (PTEN). PTEN is a dual specificity protein and lipid phosphatase that not only targets acidic residues in protein substrates, but more importantly, the 3-phosphate from PIP3, converting it back to PIP2 (10). PTEN signaling regulates cell division and can also direct cells to enter a natural cell death pathway when sufficient growth has taken place by inducing G1-phase cell cycle arrest through the retinoblastoma protein (11, 12). As a regulator of PI3K signaling, loss of PTEN leads to over-activation of Akt, which, in turn, is associated with uncontrolled cell proliferation, decreased apoptosis, and enhanced tumor angiogenesis (13). The PTEN tumor suppressor gene maps to human chromosome 10q23.3, and this region is known to exhibit high rates of loss of heterozygosity in a variety of human malignancies, including kidney, lung, breast, and prostate cancer (14). In prostate cancer, early reports on the PTEN gene focused on small changes of DNA sequence or point mutations that led to inactivation of PTEN protein function (15). In addition, the PTEN gene may also be inactivated by epigenetic events such as promoter methylation (16, 17). However in recent years, it has become evident that relatively large deletions and genomic rearrangements affecting PTEN are most prevalent in prostate cancer (8, 18). Mechanisms including transcriptional repression, microRNA (miRNA) regulation (19), disruption of competitive endogenous RNA (CeRNA) networks (20), and post translational modifications have also been implicated in the loss of PTEN function and in the initiation of tumorigenesis (21, 22). However, this review will mainly focus on PTEN alterations at the genomic level.
PTEN Deletion Analyses by FISH
Fluorescence in situ hybridization (FISH) analyses have provided a robust evaluation of the genomic status of PTEN in prostate cancer. This assay design has a centromere probe as a chromosome copy number control, and the PTEN locus probe labeled in a different fluorochrome. Using this FISH method, early studies by Yoshimoto et al. (23) analyzing 35 radical prostatectomy specimens showed no PTEN deletion in benign glandular epithelium or low-grade Prostatic Intra-epithelial Neoplasia (PIN), while PTEN deletions were found in 23% of High-Grade Prostatic Intra-epithelial Neoplasia (HGPIN), a pre-malignant stage of prostate carcinoma, and 68% of overt prostate cancer. The authors concluded that acquisition of a PTEN deletion is an important step toward prostatic tumorigenesis (23). Subsequent studies have demonstrated an association between PTEN loss and poor clinical outcomes in cohorts ranging from 59 to 322 tumors samples, confirming that PTEN alterations confer substantial malignant potential to prostate cancer cells (24–26). Utilizing FISH to determine the hemizygous or homozygous PTEN deletion status, Yoshimoto et al. (8) analyzed paired primary adenocarcinomas and regional lymph node metastasis derived from 10 patients and determined that only 1 of the 10 patients retained both copies of the PTEN locus in his matched pair biopsy. Hemizygous PTEN deletion was found in both the primary and the metastatic nodal tumor samples in 4 of 10 patients, while homozygous PTEN deletion was found in both the primary tumor and their metastatic lymph nodes in 3 of the 10 patients. Interestingly, 2 of the 10 patients with a hemizygous PTEN deletion in their primary adenocarcinomas, had positive lymph node biopsies that had acquired a homozygous PTEN deletion (8). These findings suggest that the transition from one-copy loss to two-copy loss may be associated with metastasis (8). Subsequent supporting studies demonstrated that hemizygous PTEN deletions were associated with increased risk and earlier biochemical relapse after radical prostatectomy, whereas homozygous deletions were strongly linked to metastasis and androgen-independent progression (24, 26, 27).
Despite being the method of choice for detecting genomic status in FFPE sections, the reported frequency of PTEN deletion in prostate cancer tissue using a two-color FISH assay varies widely. To date, reports of PTEN deletions ranged from 20 to 60% of localized prostate cancers (24–26). This large range in frequency most likely results from a difference in tissue preparation, stage of disease, and the methodology used to detect the aberration. Several of the studies reported high cutoff values using the two-color technology, and attributed this to artifacts generated during sectioning. During tissue sectioning, part of the cell and nucleus can be sliced out, leading to a “truncation effect” where loss of PTEN signal from the nucleus could be scored falsely as gene deletion. It is therefore essential that the false-positives likely to come from truncation be determined by comparison with normal nuclei for all deletion FISH assays (28). One solution for minimizing truncation effects is the use of a four-color FISH assay incorporating additional control loci. Recently, Yoshimoto et al. (29) reported a meta-analysis of 311 published human genome array datasets determined that the minimal prostate cancer-associated PTEN deletion at 10q23.3 corresponds to a ∼2.06 Mb region flanked by the BMPR1A and FAS genes. A four-color FISH assay was designed to include BMPR1A and FAS probes flanking either side of the minimal deleted region, a PTEN probe, and a chromosome 10 centromere copy control probe (29) (Figure 2).
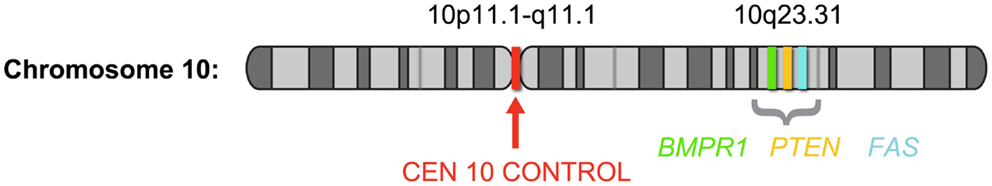
Figure 2. Schematic depiction of chromosome 10 showing the probe locations of the PTEN four-color FISH assay; based on Yoshimoto et al. (29). The four-color FISH assay was designed to include BMPR1A (green) and FAS (aqua) probes flanking either side of the 2.06-Mb minimal deleted region, a PTEN probe (orange) at 10q23.3 and a chromosome 10 centromere (red) copy control probe.
In this four-color FISH assay design, loss of PTEN signal, but presence of BMPR1A and FAS signals, indicates with a higher degree of accuracy that a PTEN deletion is present rather than a false-positive resulting from truncation effects. Loss of PTEN signal along with loss of flanking probe signals could suggest the presence of an artifact truncation loss. A recent comparison study between a four-color PTEN FISH assay and a two-color PTEN FISH assay using benign prostatic hyperplasia as a control tissue source for prostate cancer showed that two-color PTEN FISH has a mean of 12% of false-positive cells due to truncation losses whereas four-color PTEN FISH has a mean false-positive rate of only 4%. Thus, the addition of these control flanking probes provides three-dimensional information in FFPE sections that increases specificity and sensitivity of the assay (29). Representative deleted and normal PTEN FISH images are presented (Figure 3).
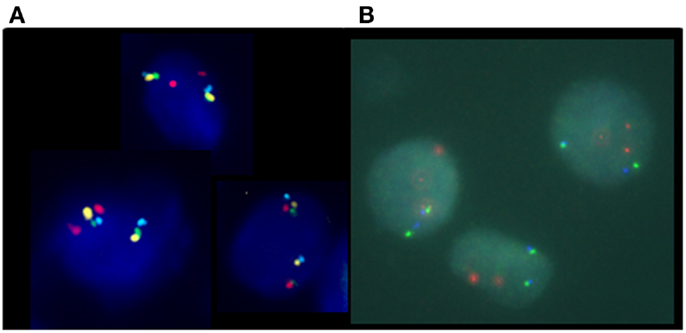
Figure 3. Phosphatase and tensin homolog gene four-color FISH assay. (A) Normal, non-deleted tissue with red signals showing chromosome 10 centromere regions, green signals showing the presence of BMPR1, blue signal showing the presence of FAS, and yellow signal indicating the presence of the PTEN gene; and (B) deleted PTEN locus, the yellow signals corresponding to the PTEN gene are absent indicating homozygous deletion of PTEN.
Furthermore, using this four-color FISH assay, breakpoints between PTEN and BMPR1A or FAS were mapped in 100 homozygous and 82 homozygous PTEN losses. The results revealed that 69% of the samples had one breakpoint within the 940-kb interval between BMPR1A and PTEN, suggesting that this interval was a “breakpoint cluster hotspot” (Figure 4).
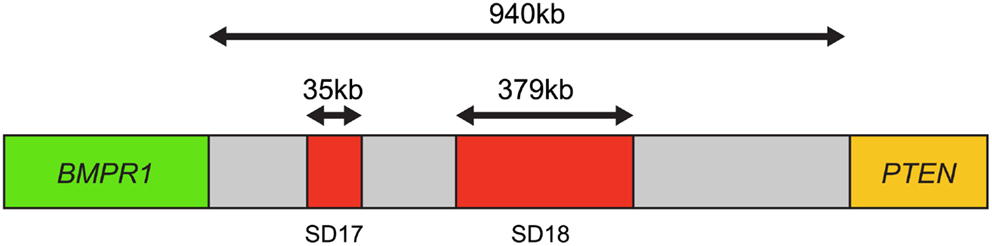
Figure 4. Map of the breakpoint cluster region between BMPR1 and PTEN. The 940-kb interval between BMPR1 and PTEN includes the segmental duplication clusters SD17 and SD18, which are 35 kb and 379 kb in length, respectively. Both contain clusters of homologous inverted repeat sequences thought to cause genomic instability and mediate PTEN deletion. Based on Yoshimoto et al. (29).
This region appeared to coincide with segmental duplication sites SD17 and SD18, containing at least 13 homologous inverted repeat sequences over 10 kb in length, which could promote intrachromosomal mis-pairing during homologous DNA repair or DNA replication. This genomic instability is the most likely mechanism by which PTEN deletion is mediated in prostate cancer (29).
PTEN Deletions Detected in Circulating Tumor Cells
Circulating prostate tumor cells are cells that have broken free from the tumor and circulate in the peripheral circulation. Most of these circulating tumor cells (CTCs) that depart from the primary tumor will die, whereas an estimated 0.01% of CTCs are likely to give rise to metastases (30). Despite having important prognostic and therapeutic implications, CTCs have not been widely studied in prostate cancer because of the technical challenges of their detection. Early studies have shown that PSA-positive CTCs from the peripheral blood of patients suffering from multifocal cancers could be subjected to genetic profiling along with tissue isolated from each cancer focus by multiplex PCR-based microsatellite analysis (31–33). Results showed that the mutations detected in CTCs were most likely derived from only one distinct focus, while other foci from the same tumor sometimes had additional LOH. Interestingly, the highest LOH in the PSA-positive CTCs was observed at the PTEN locus, suggesting that PTEN genetic imbalances might be associated with the occurrence of CTCs in the peripheral blood of patients and with early biochemical recurrence (33). An additional study using FISH analysis of PTEN showed heterogeneous loss of PTEN in prostate cancer CTCs (34). Their data demonstrated that 6 of 13 patients had homogenous homozygous loss of PTEN in all their CTCs, while 7 patients had heterogeneous PTEN loss with combinations ranging from a mixture of heterozygous loss/normal PTEN to heterozygous/homozygous PTEN loss (34). With emerging technology such as the CellSearch detection method, isolation of CTCs from the peripheral blood of patients is now becoming a valid alternative source of tumor tissue that can be subjected to molecular characterization, FISH analyses, and cancer monitoring (35).
PTEN Deletions and Associated Biomarkers in Prostate Cancer
The importance of PTEN genomic status for prostate cancer prognosis is compelling but understanding its co-operation with other genetic aberrations in the context of this highly heterogeneous disease is crucial for accurately predicting clinical outcomes and developing targeted therapies. One of the most pivotal interactions in both human and murine prostate cancer is that between PTEN and the ETS-related gene, ERG. Genomic alterations of ERG resulting from a fusion with the androgen responsive trans-membrane protease, serine 2 (TMPRSS2) gene are highly pervasive in prostate cancer and can be detected in about 40–70% of clinically significant diseases. Several studies have confirmed that tumors with TMPRSS2:ERG gene rearrangements are enriched for PTEN genomic deletions in localized prostate cancer and CRPC (18, 24, 25, 36). Bismar et al. (37) reported that these two genetic alterations showed significant concurrence within the same tumor focus, ranging between 44 and 71% of hemizygous and homozygous PTEN deletions, respectively. King et al. (38) and Carver et al. (39), confirmed this association in a murine model, both documenting that the development of invasive prostate cancer in double-transgenic models occurs at a higher rate than in ERG rearrangements or PTEN genomic deletions alone. Furthermore, Bismar et al. (24) proposed that the synergistic co-operation between PTEN deletion and ERG rearrangement could be a significant driver for PCA development and tumor progression. A subset of PCA may be driven initially by PTEN genomic instability, which may facilitate the chromosomal rearrangement leading to gene fusion formation and progression of cancer. Subsequent PTEN deregulation by homozygous deletion could then occur to induce invasive PCA possibly through Akt activation (Figure 5). In a recent study using a Tissue MicroArray (TMA), Krohn et al. (40) analyzed PTEN status in 2217 hormone naive and 49 hormone-refractory tumors. Their data showed that PTEN deletion was strongly associated with ERG fusion-positive tumors (29.1 versus 10.7%), suggesting a selective advantage for tumor cells harboring both PTEN deletion and ERG fusion (40). However, their findings also suggested that ERG fusion is not required for PTEN loss to determine aggressive tumor behavior, because PTEN deletion in both ERG fusion-positive and fusion-negative cancers was independently linked to poor prognosis, while the presence of ERG fusion was unrelated to patient prognosis (40). Collectively, these FISH studies of PTEN gene loss and ERG gene rearrangements could be pursued for patient stratification and patient management.
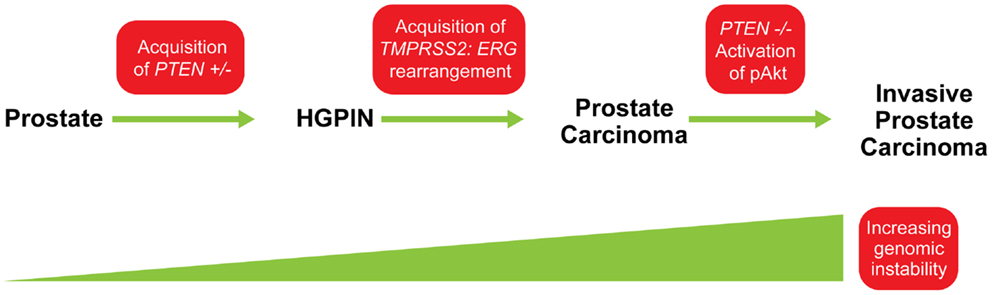
Figure 5. Model for possible sequence of genomic events in PCA progression. The acquisition of PTEN haplo-insufficiency in prostatic precursor results in a decrease in PTEN protein level, which could lead to genomic instability and the formation of high-grade prostatic intra-epithelial neoplasia (HGPIN). This genomic instability could facilitate ERG rearrangement, and together, synergistic co-operation between PTEN and ERG abnormalities is associated with early steps of PCA formation. Continuing genetic instability generates “PTEN null” subclones and allows the selective advantage for further tumor progression through activation of the Akt pathway. Modified from (24).
The AR is a steroid receptor member of the larger nuclear receptor superfamily, and plays a central role in normal prostate development as well as in prostate cancer initiation and progression. Androgen deprivation is currently the standard therapy for metastatic prostate cancer, but patients invariably relapse with a more aggressive CRPC. It has been widely recognized that AR signaling remains important even in the presence of reduced androgen levels and thus remains a major target for targeted therapeutic interventions. In 43 primary prostate cancer samples, Choucair et al. (41) determined that AR expression was significantly lower in prostate cancer tumors harboring a PTEN deletion compared to those with no PTEN deletion. However, the underlying mechanism associating decreased AR expression in the presence of PTEN deletion is still unclear (41). El Sheikh et al. (42) also showed in 47 samples of hormone naive prostate cancers that reduced expression of either biomarker was associated with the development of androgen independence and reduced survival in patients whose tumors remained androgen dependent (42). In contrast, Sircar et al. (26) reported a positive correlation between PTEN deletion status and up-regulation of AR expression in CRPC patients. These results suggest that AR expression levels differ according to the stage of the disease, with genomic amplification of AR likely to occur in CRPC, but rarely in untreated prostate cancer (43, 44). Therefore, AR gene amplification may play a direct role in prostate cancer progression and may be associated with failure of androgen deprivation therapy (ADT). Since AR expression levels are low in hormone naive tumors, addition of an inhibitor of PI3K/mTOR to the standard ADT of advanced prostate cancer may therefore be beneficial to patients with PTEN deleted tumor. A number of other studies have implicated differential expression of AR and PTEN deletions/Akt activation as significant predictors for disease progression and the development of CRPC (37, 44, 45). Their data suggest that assessment of PTEN/AR co-expression proves useful in distinguishing prostate cancer with a more favorable prognosis from those with a high likelihood of developing recurrent disease.
The proto-oncogene MYC has been associated with cell transformation. It is known that overexpression of MYC can immortalize human prostatic epithelial cells, so gain of function of MYC is clearly an oncogenic factor in human prostate cancer. Comparative Genomic Hybridization (CGH) studies have showed that gain of 8q, including 8q24 involving the MYC gene, is one of the most frequent alterations in prostate cancer (46). In a comprehensive study using FISH with probes for MYC (8q24), ∼40% of primary tumors and >90% of metastases showed varying levels of MYC copy number gains (47). Whereas primary tumors typically have simple gain of MYC due to an extra copy of 8q, metastases have more frequent regional MYC amplification, suggesting that MYC is more commonly involved in prostate cancer progression (15). Recently, Zafarana et al. (48) reported for the first time the combined role of PTEN loss and MYC gain using global high-resolution CGH analyses validated by FISH in a cohort of 126 intermediate-risk prostate cancer patients after receiving radiotherapy. They reported that copy number alterations in PTEN (allelic loss) and MYC (allelic gain) were associated with significantly increased genetic instability and biochemical relapse compared to tumors with normal PTEN and MYC status. In light of these results, triaging patients by the use of PTEN and MYC copy number changes within pre-treatment biopsies may allow for better use of systemic therapies to target sub-clinical metastases or locally recurrent disease and improve clinical outcomes (48).
PTEN Mutation and Methylation Analysis
In addition to allelic deletion, functional loss of PTEN can also be caused by mutations and epigenetic modifications. Some of the most frequent mutations identified in PTEN are frameshift or non-sense mutations that lead to inactivation of the protein (Table 1). In localized prostate cancer, the incidence of PTEN mutations has been found to occur at <20%, a frequency notably lower than that of PTEN deletions (∼40% of localized cancers) (2, 49–53). However, in advanced and metastatic disease, PTEN appears to be more frequently mutated in up to 60% of patients who had multiple metastases as well as in cell lines and xenografts derived from metastatic prostate cancer (7, 54). PTEN appears to be more frequently mutated in metastases of prostate cancer, providing further evidence for the idea that PTEN plays a crucial role in the progression of prostate cancer (55). Recent advances in massively parallel DNA sequencing technologies, allowing for the identification of base substitutions as well as indels and copy number alterations at greater sensitivity and cost effectiveness than screening by traditional Sanger sequencing, have great promises to offer a new strategy for personalized cancer care. Several studies have shown that targeted mutational and copy number analyses in advance prostate cancer using Next-Generation Sequencing (NGS) were able to detect PTEN somatic point mutations as well as genomic deletion in up to 44% of analyzed patient samples (2, 56, 57). The comprehensive genomic profiling that NGS technologies offer is an exciting new tool that will not only help in the discovery of new biomarkers but also in the design of personalized therapies in the context of such a heterogeneous disease. Epigenetic inactivation of the PTEN promoter has been identified in multiple types of cancers such as thyroid, lung, cervical, and breast cancer (58). Even though several genes such as APC, GSTP1, and MGMT, are known to be highly methylated in prostate cancer (59–61), to date, limited data has shown the occurrence of PTEN methylation in prostate cancer. One study using prostate cancer xenografts, demonstrated by Northern analysis and RT-PCR that the level of PTEN mRNA expression was reduced or absent in five of the nine xenografts studied. The mRNA expression was then restored with the demethylating agent 5-azadeoxycytidine in at least one of the five xenografts suggesting that methylation of CpG islands in the PTEN genomic locus was responsible for decreased PTEN expression. However, it is also possible that the effect of 5-azadeoxycytidine on PTEN is mediated indirectly through a second gene, such as a transcription factor that may be the true target of methylation. Direct evidence that the PTEN gene is methylated in prostate cancer is still required (17, 62).
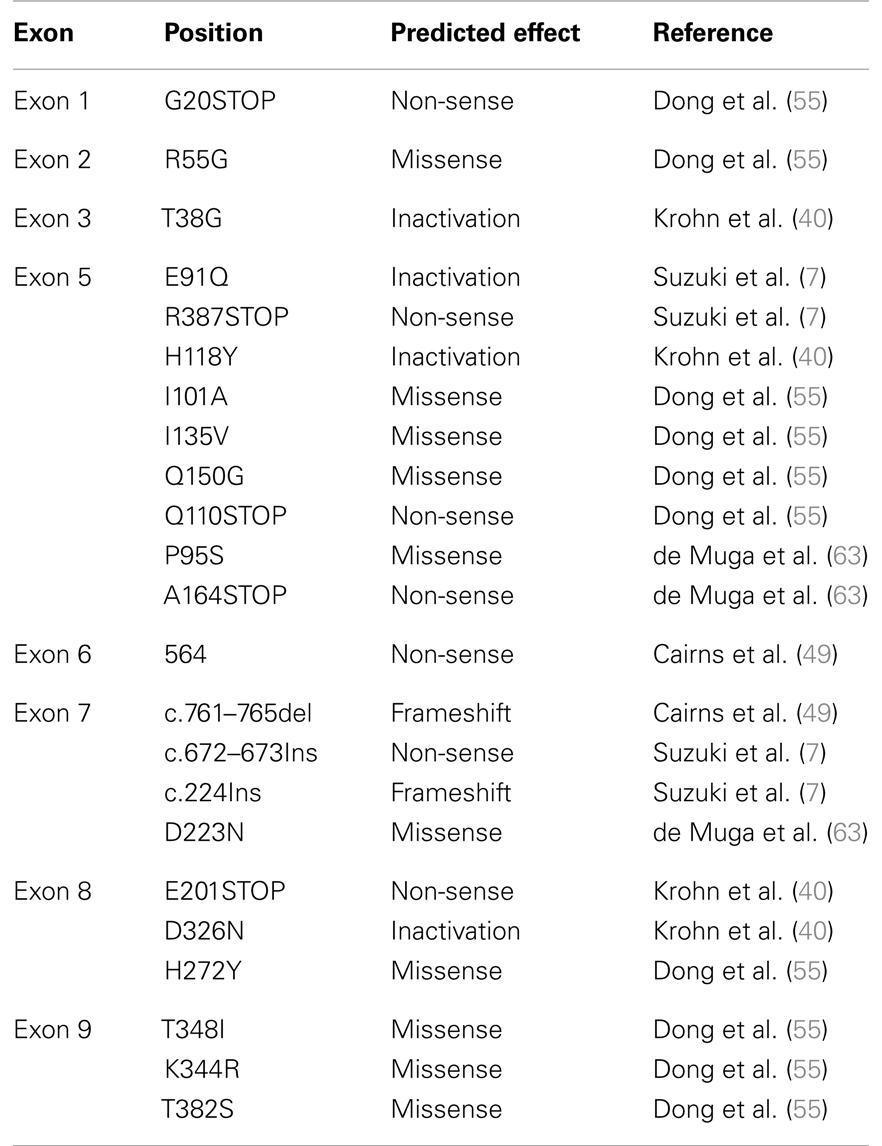
Table 1. Mutations in the PTEN gene identified in prostate cancer.
Emerging Anticancer Therapies for PTEN-Deficient Prostate Cancers
Because PTEN is a tumor suppressor that negatively regulates the PI3K pathway, up-regulation of this pathway’s downstream targets is commonly observed in PTEN-deficient prostate cancers. Both Akt and mTOR are two important PI3K targets that are frequently activated in human primary prostate cancer specimens, as evidenced by increased phosphorylation of both Akt and S6RP, a downstream target of mTOR (64, 65). The development of specific inhibitors for either one of these kinases has thus emerged as important cancer therapeutic targets. It was reported that inhibition of mTOR by rapamycin analogs such as ridaforolimus or temsirolimus showed antitumor effects in clinical studies (65–68). MK-2206 has also recently emerged as a potent allosteric inhibitor of Akt and is currently in Phase 1 trial studies for the treatment of prostate cancer (69). Recently, Zhang et al. (70) showed that inhibition of either mTOR with ridaforolimus or Akt with MK-2206 alone had no effect on the status of the other kinase in castrate-sensitive settings. However, simultaneous inhibition of both mTOR and Akt demonstrated additive antitumor effects (70). These findings strongly indicate that the mTOR signaling network in the PTEN-null tumor is independent of Akt activity and that both Akt and mTOR downstream signaling pathways play a part in PTEN-deficient prostate tumors. These results strongly support the rationale for using Akt and mTOR inhibitors in combination therapy (ClinicalTrials.gov identifier NCT01295632).
Most recently, the mTOR inhibitor drug Everolimus was evaluated in a Phase 2 trial as a first-line treatment in patients with mCRPC (ClinicalTrials.gov identifier NCT00976755) (71). Everolimus has been reported to inhibit tumor growth in human prostate cancer cell lines as well as in transgenic mice expressing human Akt (72). Everolimus treatment was also shown to increase sensitivity to mTOR inhibition in tumors cells with PTEN loss in vitro (73, 74). In this study 35% of chemotherapy-naïve patients with mCRPC treated with a daily dose of 10 mg of single-agent Everolimus showed progression-free survival (PFS) at the primary end point of 12 weeks. Thirty percent of analyzable tumor samples were found to have a PTEN deletion determined using FISH, and those in particular showed a trend toward longer PFS after treatment. Despite modest activity in some patients with mCRPC, Everolimus is a promising candidate drug for treating PTEN-deleted prostate tumors, and further investigations of this drug in combination with other therapies are warranted.
Targeting AR through androgen ablation therapy is the mainstay of prostate cancer treatment. However, these cancers often progress and, as a result, treatment options become limited. While often termed “androgen-independent,” recent work has shown that AR signaling remains critical throughout the course of the disease (75). This hypothesis has been validated by the clinical successes of a new generation of potent AR antagonists such as MDV3100 or antiandrogen agents such as bicalutamide and abiraterone (76, 77). Recently, it has also been demonstrated that the AR and PI3K signaling pathways are feedback regulated in a reciprocal manner (78). That is, androgen independence, commonly associated with PTEN loss, is associated with the activation of PI3K signaling; conversely, PI3K inhibitors can reactivate androgen signaling and sensitivity to antiandrogen therapy. In particular, recent studies have identified several mechanisms for the crosstalk between AR by mTOR (79–81). In light of these findings, Squillace et al. (82) recently demonstrated that dual inhibition of AR and mTOR with bicalutamide and ridaforolimus, respectively, had synergistic antiproliferative effect in prostate cancer cells in vitro when compared with each agent alone (82).
The combination therapy resulted in full inhibition of each pathway and exhibited potent antitumor activity with parallel reductions in plasma PSA levels in xenograft models. Taken together, ridaforolimus and bicalutamide represent a potentially effective combination strategy for PTEN-deficient prostate cancer therapy. Considering the success of these new compounds and their mechanisms of action, the identification of PTEN deletions has the potential to be a useful companion diagnostic assay for therapeutics targeting the PIK3/mTOR pathway.
Conclusion
Over the past decade, extensive research has led to a more detailed understanding of the molecular mechanism(s) governing the initiation and progression of prostate cancer. Although significant progress has been made in our ability to forecast outcomes for prostate cancer after therapy using clinical and histological variables, the ability to accurately predict response to a specific treatment remains elusive. Molecular and cytogenetic assays such as FISH analyses of PTEN have paved the way to a much clearer understanding of cancer status and disease progression. With the improved design of the four-color FISH assay, PTEN genomic status can be used as a reliable diagnostic tool and potential companion diagnostic for emerging anticancer drugs. Overall it is clear that the status of PTEN is a powerful biomarker that promise effective diagnosis and improved patient stratification and management.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. Cancer J Clin (2012) 62:10–29. doi:10.3322/caac.21149 OPMID
2. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, Macdonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol (2012) 63(5):920–6. doi:10.1016/j.eururo.2012.08.053
3. Lu-Yao GL, Albertsen PC, Moore DF, Shih W, Lin Y, DiPaola RS, et al. Survival following primary androgen deprivation therapy among men with localized prostate cancer. JAMA (2008) 300:173–81. doi:10.1001/jama.300.2.173
4. Lu-Yao GL, Albertsen PC, Stanford JL, Stukel TA, Walker-Corkery E, Barry MJ. Screening, treatment, and prostate cancer mortality in the Seattle area and Connecticut: fifteen-year follow-up. J Gen Intern Med (2008) 23:1809–14. doi:10.1007/s11606-008-0785-8
5. Pomerantz M, Kantoff P. Advances in the treatment of prostate cancer. Annu Rev Med (2007) 58:205–20. doi:10.1146/annurev.med.58.101505.115650
6. De Velasco MA, Uemura H. Preclinical remodeling of human prostate cancer through the PTEN/AKT pathway. Adv Urol (2012) 2012:419348. doi:10.1155/2012/419348
7. Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, Sidransky D, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res (1998) 58:204–9.
8. Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, Soares FA, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer (2007) 97:678–85. doi:10.1038/sj.bjc.6603924
9. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer (2001) 1:34–45. doi:10.1038/35094009
10. Chia JY, Gajewski JE, Xiao Y, Zhu HJ, Cheng HC. Unique biochemical properties of the protein tyrosine phosphatase activity of PTEN-demonstration of different active site structural requirements for phosphopeptide and phospholipid phosphatase activities of PTEN. Biochim Biophys Acta (2010) 1804:1785–95. doi:10.1016/j.bbapap.2010.05.009
11. Paramio JM, Navarro M, Segrelles C, Gomez-Casero E, Jorcano JL. PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene (1999) 18:7462–8. doi:10.1038/sj.onc.1203151
12. Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol (2003) 23:6139–49. doi:10.1128/MCB.23.17.6139-6149.2003
13. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets (2008) 8:187–98. doi:10.2174/156800908784293659
14. Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, et al. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A (2001) 98:11563–8. doi:10.1073/pnas.201167798
15. Dong JT. Prevalent mutations in prostate cancer. J Cell Biochem (2006) 97:433–47. doi:10.1002/jcb.20696
16. Konishi N, Nakamura M, Kishi M, Nishimine M, Ishida E, Shimada K. Heterogeneous methylation and deletion patterns of the INK4a/ARF locus within prostate carcinomas. Am J Pathol (2002) 160:1207–14. doi:10.1016/S0002-9440(10)62547-3
17. Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A (1998) 95:5246–50. doi:10.1073/pnas.95.9.5246
18. Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, Ludkovski O, et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol (2008) 21:1451–60. doi:10.1038/modpathol.2008.96
19. Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b ∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal (2010) 3:ra29. doi:10.1126/scisignal.2000594
20. Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell (2011) 147:344–57. doi:10.1016/j.cell.2011.09.029
21. Singh G, Chan AM. Post-translational modifications of PTEN and their potential therapeutic implications. Curr Cancer Drug Targets (2011) 11:536–47. doi:10.2174/156800911795655930
22. Wang X, Jiang X. Post-translational regulation of PTEN. Oncogene (2008) 27:5454–63. doi:10.1038/onc.2008.242
23. Yoshimoto M, Cutz JC, Nuin PA, Joshua AM, Bayani J, Evans AJ, et al. Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet Cytogenet (2006) 169:128–37. doi:10.1016/j.cancergencyto.2006.04.003
24. Bismar TA, Yoshimoto M, Vollmer RT, Duan Q, Firszt M, Corcos J, et al. PTEN genomic deletion is an early event associated with ERG gene rearrangements in prostate cancer. BJU Int (2011) 107:477–85. doi:10.1111/j.1464-410X.2010.09470.x
25. Han B, Mehra R, Lonigro RJ, Wang L, Suleman K, Menon A, et al. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod Pathol (2009) 22:1083–93. doi:10.1038/modpathol.2009.69
26. Sircar K, Yoshimoto M, Monzon FA, Koumakpayi IH, Katz RL, Khanna A, et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J Pathol (2009) 218:505–13. doi:10.1002/path.2559
27. Squire JA. TMPRSS2-ERG and PTEN loss in prostate cancer. Nat Genet (2009) 41:509–10. doi:10.1038/ng0509-509
28. Zordan A. Fluorescence in situ hybridization on formalin-fixed, paraffin-embedded tissue sections. Methods Mol Biol (2011) 730:189–202. doi:10.1007/978-1-61779-074-4_14
29. Yoshimoto M, Ludkovski O, DeGrace D, Williams JL, Evans A, Sircar K, et al. PTEN genomic deletions that characterize aggressive prostate cancer originate close to segmental duplications. Genes Chromosomes Cancer (2012) 51:149–60. doi:10.1002/gcc.20939
30. Zhe X, Cher ML, Bonfil RD. Circulating tumor cells: finding the needle in the haystack. Am J Cancer Res (2011) 1:740–51.
31. Grasso YZ, Gupta MK, Levin HS, Zippe CD, Klein EA. Combined nested RT-PCR assay for prostate-specific antigen and prostate-specific membrane antigen in prostate cancer patients: correlation with pathological stage. Cancer Res (1998) 58:1456–9.
32. Kantoff PW, Halabi S, Farmer DA, Hayes DF, Vogelzang NA, Small EJ. Prognostic significance of reverse transcriptase polymerase chain reaction for prostate-specific antigen in men with hormone-refractory prostate cancer. J Clin Oncol (2001) 19:3025–8.
33. Schmidt H, DeAngelis G, Eltze E, Gockel I, Semjonow A, Brandt B. Asynchronous growth of prostate cancer is reflected by circulating tumor cells delivered from distinct, even small foci, harboring loss of heterozygosity of the PTEN gene. Cancer Res (2006) 66:8959–65. doi:10.1158/0008-5472.CAN-06-1722
34. Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, A’Hern R, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res (2009) 69:2912–8. doi:10.1158/0008-5472.CAN-08-3667
35. Thalgott M, Rack B, Maurer T, Souvatzoglou M, Eiber M, Kress V, et al. Detection of circulating tumor cells in different stages of prostate cancer. J Cancer Res Clin Oncol (2013) 139(5):755–63. doi:10.1007/s00432-013-1377-5
36. Yoshimoto M, Ding K, Sweet JM, Ludkovski O, Trottier G, Song KS, et al. PTEN losses exhibit heterogeneity in multifocal prostatic adenocarcinoma and are associated with higher Gleason grade. Mod Pathol (2012) 26(3):435–47. doi:10.1038/modpathol.2012.162
37. Bismar TA, Yoshimoto M, Duan Q, Liu S, Sircar K, Squire JA. Interactions and relationships of PTEN, ERG, SPINK1 and AR in castration-resistant prostate cancer. Histopathology (2012) 60:645–52. doi:10.1111/j.1365-2559.2011.04116.x
38. King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet (2009) 41:524–6. doi:10.1038/ng.371
39. Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet (2009) 41:619–24. doi:10.1038/ng.370
40. Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, Meyer-Kornblum M, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol (2012) 181:401–12. doi:10.1016/j.ajpath.2012.04.026
41. Choucair K, Ejdelman J, Brimo F, Aprikian A, Chevalier S, Lapointe J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer (2012) 12:543. doi:10.1186/1471-2407-12-543
42. El Sheikh SS, Romanska HM, Abel P, Domin J, Lalani el-N. Predictive value of PTEN and AR coexpression of sustained responsiveness to hormonal therapy in prostate cancer – a pilot study. Neoplasia (2008) 10:949–53.
43. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet (1995) 9:401–6. doi:10.1038/ng0495-401
44. Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urol Oncol (2009) 27:36–41. doi:10.1016/j.urolonc.2008.03.021
45. Burnstein KL. Regulation of androgen receptor levels: implications for prostate cancer progression and therapy. J Cell Biochem (2005) 95:657–69. doi:10.1002/jcb.20460
46. Knuutila S, Bjorkqvist AM, Autio K, Tarkkanen M, Wolf M, Monni O, et al. DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol (1998) 152:1107–23.
47. Jenkins RB, Qian J, Lieber MM, Bostwick DG. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res (1997) 57:524–31.
48. Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, Thoms J, et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer (2012) 118:4053–62. doi:10.1002/cncr.26729
49. Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res (1997) 57:4997–5000.
50. Dong JT, Sipe TW, Hyytinen ER, Li CL, Heise C, McClintock DE, et al. PTEN/MMAC1 is infrequently mutated in pT2 and pT3 carcinomas of the prostate. Oncogene (1998) 17:1979–82. doi:10.1038/sj.onc.1202119
51. Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene (1998) 16:1743–8. doi:10.1038/sj.onc.1200205
52. Orikasa K, Fukushige S, Hoshi S, Orikasa S, Kondo K, Miyoshi Y, et al. Infrequent genetic alterations of the PTEN gene in Japanese patients with sporadic prostate cancer. J Hum Genet (1998) 43:228–30. doi:10.1007/s100380050078
53. Wang SI, Parsons R, Ittmann M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res (1998) 4:811–5.
54. Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res (1998) 58:2720–3.
55. Dong JT, Li CL, Sipe TW, Frierson HF Jr. Mutations of PTEN/MMAC1 in primary prostate cancers from Chinese patients. Clin Cancer Res (2001) 7:304–8.
56. Robbins CM, Tembe WA, Baker A, Sinari S, Moses TY, Beckstrom-Sternberg S, et al. Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors. Genome Res (2011) 21:47–55. doi:10.1101/gr.107961.110
57. Kovtun IV, Cheville JC, Murphy SJ, Johnson SH, Zarei S, Kosari F, et al. Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer. Cancer Res (2013) 73:3275–84. doi:10.1158/0008-5472.CAN-12-2803
58. Ongenaert M, Van Neste L, De Meyer T, Menschaert G, Bekaert S, Van Criekinge W. PubMeth: a cancer methylation database combining text-mining and expert annotation. Nucleic Acids Res (2008) 36:D842–6. doi:10.1093/nar/gkm788
59. Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol (2004) 202:233–40. doi:10.1002/path.1503
60. Roupret M, Hupertan V, Yates DR, Catto JW, Rehman I, Meuth M, et al. Molecular detection of localized prostate cancer using quantitative methylation-specific PCR on urinary cells obtained following prostate massage. Clin Cancer Res (2007) 13:1720–5. doi:10.1158/1078-0432.CCR-06-2467
61. Yamanaka M, Watanabe M, Yamada Y, Takagi A, Murata T, Takahashi H, et al. Altered methylation of multiple genes in carcinogenesis of the prostate. Int J Cancer (2003) 106:382–7. doi:10.1002/ijc.11227
62. Chin SP, Dickinson JL, Holloway AF. Epigenetic regulation of prostate cancer. Clin Epigenetics (2011) 2:151–69. doi:10.1007/s13148-011-0041-7
63. De Muga S, Hernandez S, Agell L, Salido M, Juanpere N, Lorenzo M, et al. Molecular alterations of EGFR and PTEN in prostate cancer: association with high-grade and advanced-stage carcinomas. Mod pathol (2010) 23:703–12. doi:10.1038/modpathol.2010.45
64. Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene (2005) 24:7465–74. doi:10.1038/sj.onc.1209096
65. Zhang W, Zhu J, Efferson CL, Ware C, Tammam J, Angagaw M, et al. Inhibition of tumor growth progression by antiandrogens and mTOR inhibitor in a Pten-deficient mouse model of prostate cancer. Cancer Res (2009) 69:7466–72. doi:10.1158/0008-5472.CAN-08-4385
66. Fagone P, Donia M, Mangano K, Quattrocchi C, Mammana S, Coco M, et al. Comparative study of rapamycin and temsirolimus demonstrates superimposable anti-tumour potency on prostate cancer cells. Basic Clin Pharmacol Toxicol (2013) 112:63–9. doi:10.1111/j.1742-7843.2012.00923.x
67. Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A (2001) 98:10314–9. doi:10.1073/pnas.171076798
68. Rivera VM, Squillace RM, Miller D, Berk L, Wardwell SD, Ning Y, et al. Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor, has broad antitumor activity and can be optimally administered using intermittent dosing regimens. Mol Cancer Ther (2011) 10:1059–71. doi:10.1158/1535-7163.MCT-10-0792
69. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther (2010) 9:1956–67. doi:10.1158/1535-7163.MCT-09-1012
70. Zhang W, Haines BB, Efferson C, Zhu J, Ware C, Kunii K, et al. Evidence of mTOR activation by an AKT-independent mechanism provides support for the combined treatment of PTEN-deficient prostate tumors with mTOR and AKT inhibitors. Transl Oncol (2012) 5:422–9.
71. Templeton AJ, Dutoit V, Cathomas R, Rothermundt C, Bartschi D, Droge C, et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur Urol (2013) 64:150–8. doi:10.1016/j.eururo.2013.03.040
72. Wu L, Birle DC, Tannock IF. Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res (2005) 65:2825–31. doi:10.1158/0008-5472.CAN-04-3137
73. Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res (2006) 66:10040–7. doi:10.1158/0008-5472.CAN-06-0802
74. Cao F, Jin TY, Zhou YF. Inhibitory effect of isoflavones on prostate cancer cells and PTEN gene. Biomed Environ Sci (2006) 19:35–41.
75. Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol (2008) 8:440–8. doi:10.1016/j.coph.2008.07.005
76. El-Amm J, Aragon-Ching JB. The changing landscape in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Med Oncol (2013) 5:25–40. doi:10.1177/1758834012458137
77. Fradet Y. Bicalutamide (Casodex) in the treatment of prostate cancer. Expert Rev Anticancer Ther (2004) 4:37–48. doi:10.1586/14737140.4.1.37
78. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell (2011) 19:575–86. doi:10.1016/j.ccr.2011.04.008
79. Cinar B, De Benedetti A, Freeman MR. Post-transcriptional regulation of the androgen receptor by mammalian target of rapamycin. Cancer Res (2005) 65:2547–53. doi:10.1158/0008-5472.CAN-04-3411
80. Wang Y, Mikhailova M, Bose S, Pan CX, deVere White RW, Ghosh PM. Regulation of androgen receptor transcriptional activity by rapamycin in prostate cancer cell proliferation and survival. Oncogene (2008) 27:7106–17. doi:10.1038/onc.2008.318
81. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res (2006) 66:7783–92. doi:10.1158/0008-5472.CAN-05-4472
Keywords: prostate cancer, PTEN, genomic rearrangements, fluorescence in situ hybridization
Citation: Phin S, Moore MW and Cotter PD (2013) Genomic rearrangements of PTEN in prostate cancer. Front. Oncol. 3:240. doi: 10.3389/fonc.2013.00240
Received: 24 June 2013; Accepted: 30 August 2013;
Published online: 17 September 2013.
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Marco Alessandro Pierotti, Fondazione IRCCS IStituto Nazionale dei Tumori, ItalyAngela Bononi, University of Hawaii, USA
Copyright: © 2013 Phin, Moore and Cotter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip D. Cotter, Genetics, ResearchDx, 5 Mason, Irvine, CA 92618, USA e-mail:cGNvdHRlckByZXNlYXJjaGR4LmNvbQ==