 Shannon Patricia Fortin Ensign1,2
Shannon Patricia Fortin Ensign1,2
- 1Cancer and Cell Biology Division, Translational Genomics Research Institute, Phoenix, AZ, USA
- 2Cancer Biology Graduate Interdisciplinary Program, University of Arizona, Tucson, AZ, USA
- 3Center for Oncology and Cell Biology, The Feinstein Institute for Medical Research at North Shore-LIJ, Manhasset, NY, USA
Glioblastoma (GB) is the most malignant of primary adult brain tumors, characterized by a highly locally invasive cell population, as well as abundant proliferative cells, neoangiogenesis, and necrosis. Clinical intervention with chemotherapy or radiation may either promote or establish an environment for manifestation of invasive behavior. Understanding the molecular drivers of invasion in the context of glioma progression may be insightful in directing new treatments for patients with GB. Here, we review current knowledge on Rho family GTPases, their aberrant regulation in GB, and their effect on GB cell invasion and tumor progression. Rho GTPases are modulators of cell migration through effects on actin cytoskeleton rearrangement; in non-neoplastic tissue, expression and activation of Rho GTPases are normally under tight regulation. In GB, Rho GTPases are deregulated, often via hyperactivity or overexpression of their activators, Rho GEFs. Downstream effectors of Rho GTPases have been shown to promote invasiveness and, importantly, glioma cell survival. The study of aberrant Rho GTPase signaling in GB is thus an important investigation of cell invasion as well as treatment resistance and disease progression.
Glioma Characterization
Gliomas comprise the most common group of primary brain tumors and are of neuroepithelial designation, named for a glial cell origin (1). These tumors include astrocytomas, oligodendrogliomas, and ependymomas, with infiltrating astrocytomas accounting for approximately 80% of adult primary brain tumors (2). Within each glial cell lineage, tumors are divided into grades based on their biologic behavior across one of four grades of increasing aggressiveness (grade I to grade IV); grading confers some prognostic guidance as well as informs treatment protocols. Clinically, glioblastoma (GB) (grade IV) represents the most malignant primary brain tumors, for which there is no cure (2).
Glioblastoma tumors arise in two scenarios: primary GB with new onset disease, or secondary GB with a previous history of lower grade astrocytoma. Gliomas rarely metastasize outside the CNS (2), but can be highly invasive within the brain parenchyma. Both lower grade as well as high-grade tumors display marked invasiveness, suggesting this malignant phenotype may be acquired early in tumorigenesis (3). Gliomas preferentially invade along white-matter tracts of the cerebrum, at times crossing the corpus callosum. Other patterns of cell spreading include perivascular growth, subpial spread, or perineuronal satellitosis (3).
The 5-year survival rate for GB is under 10% (4). Unfortunately, prognosis for GB patients has not significantly changed over the past several decades despite increased molecular understanding of the tumor. The problem of resistance to standard anti-proliferative treatment with concomitant radiotherapy and chemotherapy using the alkylating agent temozolomide (TMZ) is common, particularly manifest by the invasive cell population with subsequent tumor recurrence and death. The role of pro-invasive genes in GB progression remains understudied and the link between cell invasion and therapeutic insensitivity remains poorly characterized. Notably, treatments directed at impairing mediators of invasion have been shown to increase chemotherapeutic sensitivity (5–7).
Glioma cell invasion is promoted via the overexpression of receptors such as MET or EGFR, as well as downstream signaling through PI3K and MAPK pathways, and the Rho family of GTPases, among others (8, 9). This increased invasive potential arises from overexpression or mutation of drivers of cell motility, from interactions of glioma cells with pro-migratory signals within the brain tumor microenvironment, but also, notably, as a response to treatment (10). The Rho family of small GTPases, including primarily Rac1, Cdc42, and RhoG are key signaling mediators of GB cell invasion. Members of this signaling family as well as their regulators and effectors represent novel molecular targets against which therapeutic intervention could be aimed, and the implications for their signaling deregulation in gliomas is the subject of this review.
Rho GTPase Function and Regulation
The Rho family of GTPases belongs to the Ras superfamily; they are monomeric low molecular weight proteins. There are 20 known mammalian Rho protein members, divided in part into subfamilies including Rho, Rac, Cdc42, Rnd, RhoD, RhoF, RhoH, and RhoBTB (11–16). Rho GTPases exist in either an inactive GDP-bound state, or an active GTP-bound state during which the GTPase can interact with downstream effectors. Switching between the inactive and active conformations is mediated by three classes of proteins: Guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and Rho GDIs. GEFs are responsible for GTP loading. Stimulation of Rho GTPase intrinsic GTP-to-GDP hydrolysis by GAPs facilitates Rho GTPase inactivation (17). Rho GTPases undergo post-translational prenylation modifications, which are attached to the carboxyl-terminal cysteine residue of the protein, allowing for interactions with, and attachment at, phospholipid membranes where Rho GTPases are activated and interact with signaling complexes (18, 19). A third category of Rho GTPase family regulators are the guanine nucleotide dissociation inhibitors (Rho GDIs) which sequester Rho GTPases in their inactive conformation in the cytosol by shielding the hydrophobic tail (20). Rho GTPase activity can be regulated by nucleotide binding and subcellular localization (21). Recent studies have also shown Rho GTPase regulation via ubiquitin-proteasome degradation (22), as well as the importance of Rho GDIs in controlling the homeostasis of Rho proteins potentially by preventing protein degradation (23) (Figure 1).
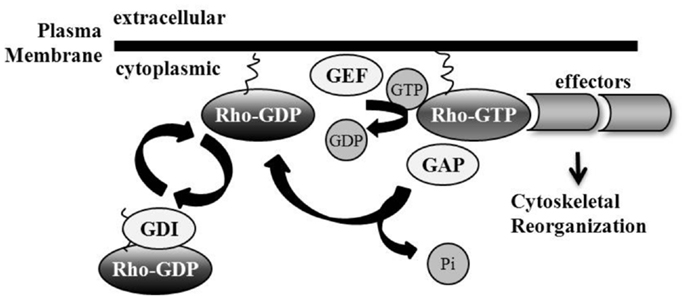
Figure 1. Regulation of Rho GTPases. Rho GTPase family members are active in their GTP-bound state, mediated by guanine nucleotide exchange factor (GEF) loading of GTP. GTPase-activating proteins (GAPs) catalyze intrinsic Rho GTPase GTP-to-GDP hydrolysis, including the removal of phosphate and the subsequent inactivation of the Rho GTPase. Guanine nucleotide dissociation inhibitors (GDIs) sequester Rho GTPases in their inactive GDP-bound conformation by shielding their hydrophobic tail. Membrane interaction and attachment is important for Rho GTPase activation and the promotion of subsequent downstream effector binding.
RhoA, Rac, and Cdc42 are well-characterized members of the Rho family of GTPases and have been described as key regulators of cell migration (17). RhoA was first described to promote the assembly of contractile actomyosin filaments, or stress fibers, and Rac promoted the assembly of a peripheral actin meshwork, including lamellipodia (17, 24, 25). In addition to inducing peripheral actin rich microspikes, or filopodia, Cdc42 has been described to activate Rac, demonstrating the existence of cross-talk within the Rho family of GTPases (17, 26). Furthermore, RhoA and Rac1 have been shown to mutually inhibit each other in some studies, while other reports postulate a positive feedback (27). Another Rho GTPase family member, RhoG, has been shown to act upstream of Rac1 to stimulate cell movement, but also signals to promote the robust formation of Rac1-independent lamellipodia (28–31).
During the process of cell migration, actin-driven protrusions at the leading edge of cell motility are driven by Rac activation, whereas actomyosin contractility at the cell body and rear are coordinated by active Rho (32), with Cdc42 regulating cell polarity through integrating extracellular directional cues (33, 34). Rac and Cdc42 share an overlapping set of downstream effectors to enact cytoskeletal changes. Signaling via Pak and MAPK activation, or via PI5K, Formin, or IQGAP leads to actin reorganization downstream of either Rac or Cdc42. In addition, Cdc42 further signals via WASP and Rac further signals via WAVE to enact cytoskeletal rearrangement (35). RhoG, which has been described to signal both in concert with or in parallel to Rac1 and Cdc42 also shares downstream effectors IQGAP2, MLK3, and PLD1 with Rac1 and Cdc42, but confers independent signaling as well (31). RhoA signaling leads to phosphorylation of myosin light chain (MLC) and is conferred by several downstream effectors including the serine/threonine kinase p160 ROCK (36–38). ROCK activity leads to increased MLC phosphorylation via the inhibition of MLC phosphatase as well as direct MLC phosphorylation (36, 38, 39). ROCK, together with the RhoA effector mDia, coordinates stress fiber formation (38, 40).
While these signaling pathways are well-characterized, the understanding of Rho GTPase regulation of cell movement has become more complex. Tumor cell motility has been characterized to occur not only in a mesenchymal pattern with a spindle like shape and an obvious leading cell edge, but also in an amoeboid fashion with cycles of expansion and contraction of the cell body, potentially dependent upon the environment through which the cells move (41, 42). Moreover, biosensors capable of visualizing active Rho family members have implicated that both Rho and Rac can be active at leading edge protrusions (43). Thus the environmental regulation and spatiotemporal control of Rho GTPases are key factors in the regulation of cytoskeletal dynamics toward cell locomotion.
The activity of Rho GTPases is tightly regulated. Rho GEFs belong either to the Dbl or Dock families. Dbl proteins contain a Dbl homology (DH) domain responsible for catalyzing the exchange of GDP for GTP (12); DH domain binding to the Rho GTPase switch region induces conformational remodeling of the nucleotide-binding pocket (44). Pleckstrin homology (PH) domains are C-terminal adjacent associated regions to the DH domain that regulate GEF activity (45). PH domains bind phosphoinositides which facilitates plasma membrane localization, although it has also been suggested that other domains are necessary for directing subcellular localization (12). Furthermore, there is evidence that GEFs may be negatively regulated by sites in their N-terminus. The constitutive activation of many Rho GEFs occurs following removal of N-terminal sequences with in vivo protein expression (45); thus, the N-terminal region may act to auto-inhibit the DH domain, the control of which can be relieved by phosphorylation (12). Within the Dock family there are 11 members that are characterized by the presence of two conserved domains, termed Dock-homology region-1 and -2 (DHR1 and DHR2). For some Dock proteins, DHR2 has been shown to be sufficient for catalytic activity (12, 46, 47). The mechanisms of signal activation, including GEF localization and protein interaction, relief of auto-inhibition, and alteration of activity for Rho GEFs remain poorly characterized.
Deregulation of RhoGTPases in Gliomas
Although Rac1-activating mutations are newly discovered and described via exome sequencing in melanoma tissue (48, 49), to date there are no reports of these mutations in other tumor types including GB. Increased activity of Rac1 has been reported in GB, and data supporting the role of additional Rho GTPases including Cdc42, RhoG, and RhoA in GB progression have been detailed as well, the findings of which are described below.
Rac
The levels of Rac1 protein correlate with tumor grade in astrocytomas. In GB, Rac1 prominent plasma membrane staining is observed, indicating a potential hyper-activation status (50). Additionally, Rac1 promotes invasive glioma cell behavior (50, 51). While most data supports the role of Rac1 in GB progression, the Rac3 GTPase, which has high homology to Rac1, has also been described to play a role in GB cell invasion; the siRNA-mediated depletion of Rac3 led to strong inhibition of GB cell invasion in vitro (52).
Rac1 facilitation of glioma cell invasion occurs via signaling through several receptors and effectors. The tumor necrosis factor receptor superfamily (TNFRSF) contains two members known to utilize Rac1 in GB. Downstream of the fibroblast growth factor-inducible 14 receptor (Fn14), the Rac1 protein is important in promoting the TNF-like weak inducer of apoptosis (TWEAK) ligand-induced activation of the Akt and NF-κB-pathways, and Fn14 signaling through Rac1 promoted increased cell invasion and resistance to cytotoxic therapy-induced apoptosis (51, 53, 54). Additionally, Fn14-induced Rac1 activation is mediated by Cdc42. TWEAK-Fn14-induced Rac1 activation was dependent upon the presence of Cdc42 protein, while Rac1 depletion had no effect on TWEAK-induced Cdc42 activity (55). Moreover, TWEAK-Fn14 signaling has been demonstrated to induce Rac1 activation through TNF receptor associated factor 2 (TRAF2)-dependent activation of SGEF and RhoG (56). Another member of the TNFRSF family, TROY, is overexpressed in glioma cells and activates Rac1 signaling in a Pyk-2 dependent fashion, leading to enhanced GB cell motility (57).
Rac1 activation and promotion of cell migration and invasion in glioma is also seen downstream of signaling networks known to be utilized in neuronal signaling and development. The neuropeptide neurotensin induced activation of Rac1 in U373 GB cells which express three subtypes of neurotensin receptors; neurotensin enhances specifically the migration of cells cultured on laminin, with neurotensin-treated cells migrating more slowly when cultured on plastic (58). In addition, neuropilin-1 is a receptor for the semaphorin family of axon guidance molecules, and signaling through its ligand semaphorin3A promotes Rac1 activity and GB cell migration (59). Semaphorin 5A and its receptor plexin-B3, however, have been shown to significantly inhibit glioma cell migration and invasion, with concomitant inactivation of Rac1 through RhoGDIα and the promotion of glioma cell differentiation; semaphorin5A protein expression was significantly reduced in high-grade astrocytomas (60, 61). The axon guidance ligand Ephrin-B3 is overexpressed in GB cells and expression correlates with invading cells, with Ephrin-B3 co-localizing with Rac1 at areas of lamellipodia formation (62). The expression of Ephrin-B3 induced Rac1 activation in GB cells (62).
Rac1 has also been shown to regulate the formation of invadopodia, which are specialized formations of the plasma membrane that promote degradation of the extracellular matrix, an action critical in glioma cell invasion (52, 63). The depletion of either Rac1 or synaptojanin 2, a Rac1 effector with phosphatidylinositol phosphatase activity, decreases invadopodia formation, and glioma cell invasion (64). Synaptojanin 2 is also enriched in invadopodia (64). Additionally, Rac1 activation at the plasma membrane in glioma is regulated by Geranylgeranyltransferase I as well as RLIP76 modulation of ubiquitination (65, 66).
Rac1 signaling has also been reported to be activated downstream of several known receptor drivers of glioma malignancy. For example, Rac1 was activated subsequent to EGFRvIII-Src family kinase-dependent or protein kinase A-dependent phosphorylation of Dock180 (67, 68). Over half of “Classical” GB tumors, a genetically defined GB subtype, contain either point or vIII EGFR activating mutations (69), which results in a gain of function in-frame deletion of the extracellular portion of the receptor protein (70). Rac1 activation may be a key event in tumors of this subtype. Similarly, the PDGFRα receptor, which is amplified in a significant proportion of gliomas (71), induced Src-dependent Dock180 phosphorylation, with subsequent increased Rac1-GTP activity and GB cell growth and invasion (72). Lastly, Rac1 activation has been described downstream of several other Ras or Rho superfamily GTPases, including Rac1 activation subsequent to IQ-domain GTPase-activating protein 1 (IQGAP1)-dependent ADP-ribosylation factor 6 (ARF6) signaling (73). Rac1 activation downstream of additional Rho GTPase family members in GB is detailed below.
Cdc42 and RhoG
There have been multiple described mechanisms of Rac1 activation in GB. Cdc42 and RhoG both share an overlapping set of activators and effectors with Rac1, and have been shown to function upstream of Rac1 in the regulation of several biological functions, including cell polarity and cell migration (29, 30, 35, 74–77). In GB, there is data to support the activity of Cdc42 and RhoG upstream of Rac1 activation; however the overall data on the status of these GTPases in GB is limited. The TWEAK-Fn14 ligand receptor axis-induced activation of Rac1 is one mechanism to enhance Rac1-GTP that is dependent upon a functional and activated Cdc42 protein; the depletion of Cdc42 abrogated glioma cell migration in vitro and invasion ex vivo (55). Cdc42 activation has also been demonstrated alongside Rac1 activation in GB cells downstream of PDGFRα association with SHP-2 non-receptor protein tyrosine phosphatase and Dynamin 2 (Dyn2) to promote glioma cell migration (78). The activation of Rac1 in neurotensin neuropeptide treated glioma cells was described in parallel with Cdc42 activity, although the dependence of one GTPase on the other for activation was not explored (58). In addition, biosensor studies have shown that both Cdc42 and Rac1 display high activity in the leading GB cells in the process of penetrating the brain parenchyma (79).
RhoG protein levels are elevated in GB (77), and RhoG has been reported to stimulate lamellipodia formation and confer downstream activation of Rac1 with a subsequent increase in cell migration (28–30, 77). Furthermore, TWEAK-Fn14 signaling in GB induces the rapid activation of RhoG dependent upon the receptor recruitment of TRAF2 and SGEF-mediated guanine nucleotide exchange, leading to increased levels of active Rac1 (56). RhoG can also activate Rac1 and Cdc42 in the regulation of neuronal process plasticity (80, 81), thus it remains a possibility that deregulated neuronal signaling and development pathways may also utilize RhoG to promote glioma malignancy. In addition, RhoG promoted caveolar endocytosis of growth factor receptors, including the regulation of EGF receptor internalization (82, 83). In GB, RhoG is activated downstream of EGF signaling and promotes GB cell migration (77); RhoG, therefore, may play a yet unidentified role in receptor trafficking within GB tumors.
There have also been several Rac1-independent functions of Cdc42 and RhoG described in other signaling systems (31, 84–86), although the data in GB is limited. Depletion of the Cdc42-specific GAP ARHGAP21 altered GB cell morphology and increased Cdc42 activity, FAK phosphorylation, MMP-2 production, and the rate of cell migration (87). Moreover, the Slit2 axon guidance molecule, which has been characterized to have lower expression in primary glioma specimens and invasive glioma cells relative to non-neoplastic brain, was described as part of the Slit2-Robo1 axis that inhibited glioma invasion through attenuation of Cdc42 activity (88). Evidence for RhoG regulation of GB cell invasion in a Rac1-independent fashion has been demonstrated downstream of HGF-induced invasion. RhoG depletion abolished HGF-induced invasion, but only partially inhibited HGF-stimulated Rac1 activity (77), suggesting RhoG also can utilize Rac1-independent mechanisms to promote HGF-induced cell motility.
RhoA
To date, much of the data on RhoA function in glioma remains largely correlative. Decreased RhoA activity occurred in correlation with increased glioma cell migration (51, 89–91). Functional evidence for the role of RhoA has been demonstrated under the inhibition of the RhoA effector ROCK, which led to activation of Rac1 in glioma cells and promoted invasion. Of note, this inhibition led to the cellular morphological changes including an increase in the number and length of cell processes, increased membrane ruffling, and collapse of actin stress fibers (92). In addition, the inhibition of Rho/ROCK downstream of LPA induced glioma cell chemotaxis also led to cells that displayed long thin morphologies with extension of processes (93). RhoA activation in astrocytoma cells has been demonstrated to confer cell process retraction and rounding in the presence of decreased Rac1 activity (94). Also, pharmacologic inhibition of Ras in GB resulted in decreased Rac1 activity with coincident increased RhoA activation and stress fiber formation leading to rigid matrix attachment and cell immobility (95). Similarly, pharmacologic EGFR inhibition led to the Rho/ROCK-dependent formation of stress fibers with consequent decreased glioma cell invasion (96). Additionally, treatment of GB cells with the plant growth modulator Narciclasine led to increased ROCK activity and stress fiber formation, and resulted in increased survival of orthotopic xenograft-bearing mice (97). One recent report links RhoA inhibition with decreased mesenchymal type invasion, however the inhibitor utilized also targets RhoB and RhoC, thus excluding a definitive conclusion on the involvement of RhoA (98).
The RhoA and Rac1 pathways are increasingly associated with the promotion of divergent modes of cell migration in other systems; amoeboid type motility, characterized by cell rounding has been demonstrated to be regulated by the Rho/ROCK pathway, whereas prominent Rac signaling regulates mesenchymal type invasion with long morphological process extension (99). These modes of migration have been demonstrated to occur on differing substrate rigidity and size (42). In GB there is insufficient functional evidence for the role of RhoA in promoting amoeboid cell migration.
It also is interesting to note that substrate preference in the mode of GB cell invasion may be influenced by RhoA signaling. FRET studies have shown that while activated Rac1 and Cdc42 was high in rat GB cells invading brain parenchyma, RhoA activity was high in the perivascular region where there was coincident lower activity of Rac1 and Cdc42 (79). Furthermore, the downstream Rho effector ROCK signaling has been demonstrated to be an important modulator of substrate preference for GB cell route of invasion into brain parenchyma (100).
Role of GEFs in Glioma
Of the more than 80 characterized Rho GEFs, the contribution of GEFs to glioma pathobiology may be in large part due to their expression patterns. Five of the GEFs described to promote GB cell motility, including Ect2, Vav3, Trio, SGEF, and SWAP-70, are overexpressed in GB versus non-neoplastic brain, and Dock180 expression is higher in the tumor rim than in the tumor core (50, 56, 76, 101).
Ect2, Beta-PIX, SWAP-70, SGEF
It is likely that tumor cell invasion depends on the specific intracellular localization of Rho GEFs. Ect2, a GEF with RhoA, Cdc42, and possible Rac1 activity, was initially shown to regulate cytokinesis and actin cortex organization during mitosis (102, 103). While low-grade astrocytomas show predominantly nuclear Ect2 staining; GBs display prominent Ect2 staining in both the cytoplasm and nucleus (50, 104). In GB cells, Ect2 confers exchange for Cdc42, and subsequently Rac1, downstream of TWEAK-Fn14 (55). Moreover, Ect2 co-localizes with Rac1 and Cdc42 in the membrane ruffles of migratory astrocytoma cells, and inhibition of Ect2 led to decreased Rac1 and Cdc42 activity, with no change in Rho activity (104). Ect2 may additionally promote mesenchymal-amoeboid transition via interaction with the Rho-GAP RASAL2 (104). Overexpression of Ect2 in vivo promotes astrocyte migration in mice (55). ARHGEF7 (βPIX), a GEF that acts on both Rac1 and Cdc42, has additionally been studied in the context of cell migration (105). In GB, βPIX has been shown to be recruited to the leading edge of cell migration by the TIP-1 PDZ-scaffolding protein, where co-localization with Rac1 and Cdc42 promotes cell motility (106).
The Rho GEF SWAP-70 was discovered as a primarily nuclear protein with weak DNA-binding affinity in activated B-lymphocytes (107). Recently, it has been shown to localize at the leading edge of migrating glioma cells where it promoted membrane ruffling and migration and invasion of glioma cells, as well as the EGF-induced activation of Rac1 (101). In addition, the RhoG-specific exchange factor SGEF, which was initially characterized as an androgen-responsive gene in human prostate cells, has been shown to induce membrane ruffling and macropinocytosis in human fibroblasts (108, 109). SGEF localization in non-transformed human keratinocytes was demonstrated to be primarily nuclear, although HPV-mediated transformation was shown to be dependent upon SGEF and RhoG cytoplasmic activity, leading to increased membrane ruffling and cell invasion (110). In GB, SGEF is overexpressed relative to non-neoplastic brain tissue, and promotes the RhoG-mediated activation of Rac1 downstream of TWEAK-Fn14 signaling leading to increased cell migration and invasion (56). Thus, the specific localization of GEFs in brain tumors may promote cell invasion.
Vav
The Vav family of Rho GEFs has been shown to promote glioma malignancy, however their isoform-specific roles vary among tumor grade and cell type context dependence. Vav1 and Vav3 have independently been shown to be overexpressed in patients with high-grade gliomas; Vav1, however, is overexpressed in peritumoral and perivascular, non-neoplastic astrocytes and thus its role may be related to cross-talk between the microenvironment and glioma cells. In contrast, Vav3 expression is localized to the glioma cells and Vav3 was shown to mediate exchange for Rac1 in the promotion of glioma cell migration (50, 111). Vav2 has been shown to exchange for Rac1 and promote chemotactic cell migration via the G protein-coupled P2Y(2) nucleotide receptor [P2Y(2)R] in low-grade astrocytomas, but has not been reported to play a role in high-grade GB (112). Thus the Vav family of Rho GEFs may promote malignancy across varying grades of brain tumors.
Trio
Trio is defined by a unique structure containing two active GEF domains, one with Rac-specific activity and the second with Rho-specific activity, as well as a protein serine/threonine kinase (PSK) domain (113). Trio is overexpressed in high-grade gliomas and correlates with poor patient outcome; the depletion of Trio inhibits glioma cell invasion ex vivo (50, 55). Trio has been shown to confer guanine nucleotide exchange for Rac1 downstream of TWEAK-Fn14-induced Cdc42 activity, with subsequent promotion of cell migration (55). The role of Trio exchange for RhoA has been unexplored in GB, however given that Rac1 and RhoA may regulate divergent modes of invasion, it remains possible that Trio plays a role in the regulation of mesenchymal-amoeboid shifts in the migration of GB cells.
Dock
Within the Dock180 superfamily of proteins, Dock180 and Dock9 have been characterized in glioma. The Dock180 GEF signals as a bipartite GEF for Rac1 in connection with the PH domain of ELMO1, and expression of ELMO1 is elevated in a subset of GB (47, 114). Furthermore, the exogenous expression of ELMO1 and Dock180 in glioma cells enhances their migratory and invasive capacities in vitro and in brain tissue (76). In addition, the phosphorylation of Dock180 at tyrosine 1811 has been demonstrated to occur downstream of PDGFRα in glioma, promoting Rac1 activation and cell invasion, migration, and survival (72).
Dock9, also known as Zizimin1, is a Cdc42-specific Dock family GEF. Dock9 interacts with the plasma membrane via its N-terminal PH domain; the N-terminal region also serves to auto-inhibit the catalytic function of Dock9 (115, 116). Dock9 was initially characterized for its role in dendrite formation in rat hippocampal neurons (117), but has recently been shown to be important for GB cell invasion in the rat. GB cells with Zizimin1 knockdown displayed lower Rac1 and Cdc42 activity (79).
Roles for additional GEFs have yet to be defined in glioma, but further characterization of GEF localization and activation mechanisms may prove useful in understanding GB invasion.
Glioma Pro-Invasive Pathways Promote Tumor Survival
Cancer invasion and drug-resistance are increasingly being recognized as interconnected processes sharing overlapping pathways that together promote disease progression and therapy failure (7). In GB, the tumor response to external stresses, including from the tumor microenvironment or from chemotherapeutic or radiation treatment, involves coordinated pro-survival and pro-invasion signaling. The GB microenvironment is comprised of areas of hypoxia notably including pseudopalisading necrosis, one of the histological hallmarks of GB, which has been defined to consist of tumor cells actively invading away from a hypoxic core of tissue following a vaso-occlusive event. These hypoxia-induced migratory cells have increased tumor cell glycolysis and secrete pro-angiogenic factors (118–120), and increased microvessel density in astroglial brain tumors is a prognostic indicator of poor patient survival (121). Thus, tumor hypoxia in GB promotes both cell invasion into normal brain tissue and supports angiogenesis for tumor survival and growth. Glioma cells with the increased capacity for migration have a decreased expression of pro-apoptotic genes and are less sensitive to cytotoxic therapy-induced apoptosis (51, 53, 122–124). Several of the Rho GTPases and Rho GEFs shown to promote GB cell invasion have also been correlated to poor patient prognosis among GB tissue specimens (50, 56, 72, 101), and signaling through Rho GTPases is part of the coordinated survival response following treatment in GB.
For example, chemotherapeutic resistance in glioma cells has been shown to be promoted through Rac1-dependent Akt2 activity working upstream of the BCL2 family to promote cell survival (54). Activation of Akt2 led to increased MMP-9 expression and increased glioma cell migration and invasion (125). The inhibition of NF-κB has been shown to promote increased glioma cell death, which was synergistic under the combined treatment with TMZ, and led to decreased migration and invasion with decreased expression of invasion-related genes (126). Furthermore, overexpression of the pro-invasive TNFRSF member TROY increased glioma cell resistance to irradiation or TMZ treatment dependent upon Akt and NF-κB activity, and depletion of TROY in orthotopically implanted primary GB xenografts led to increased survival of mice (127). In addition, sub-optimal dosing of TMZ cooperatively reduces cell growth in EGFR inhibited glioma cells with concomitant Rho/ROCK-dependent inhibition of glioma cell invasion (96). Furthermore, glioma stem cells are increasingly being recognized as TMZ resistant, both through intrinsic resistance mechanisms and via interaction with the brain parenchyma toward the promotion of extrinsic resistance (128). Rac1 has been shown to be important for the maintenance of stemness and tumorigenicity in human glioma, whereby depletion of Rac1 suppressed glioma stem-like cell migration, invasion, and malignant transformation, while conferring enhanced radiation sensitivity to these glioma stem-like cells (129).
The irradiation of primary GB cells has been demonstrated to enhance cell invasion (10). TRAF2, which has been shown to be recruited upstream of SGEF-RhoG-Rac1 pro-invasive signaling in the TWEAK-Fn14 axis (56), when depleted in GB has also been shown to inhibit growth and confer radio-sensitization to tumor cells (130). Additional reports have characterized that signaling through TRAF2 promotes not only NF-κB activity, but also JNK/SAPK activity, inflammation, and cell migration and chemo- and radio-resistance of cancer cells (131–137). In GB, JNK is activated downstream of Rac1, whereby activated JNK translocated to paxillin-containing focal complexes leading to paxillin phosphorylation at a site known to regulate cell migration (138). Moreover, it has been suggested that Rac may promote radiation therapy-induced increased glioma cell invasion in concert with activated p38 and JNK signaling (139). In addition, the pharmacologic inhibition of auto-phosphorylation of focal adhesion kinase in GB was shown to inhibit cell invasion and increase cell apoptosis, an effect which was synergistic when treated with TMZ in vivo (140). Taken together, Rho GTPases and Rho GEFs are implicated in the regulation of invasion and survival signaling in GB (Figure 2).
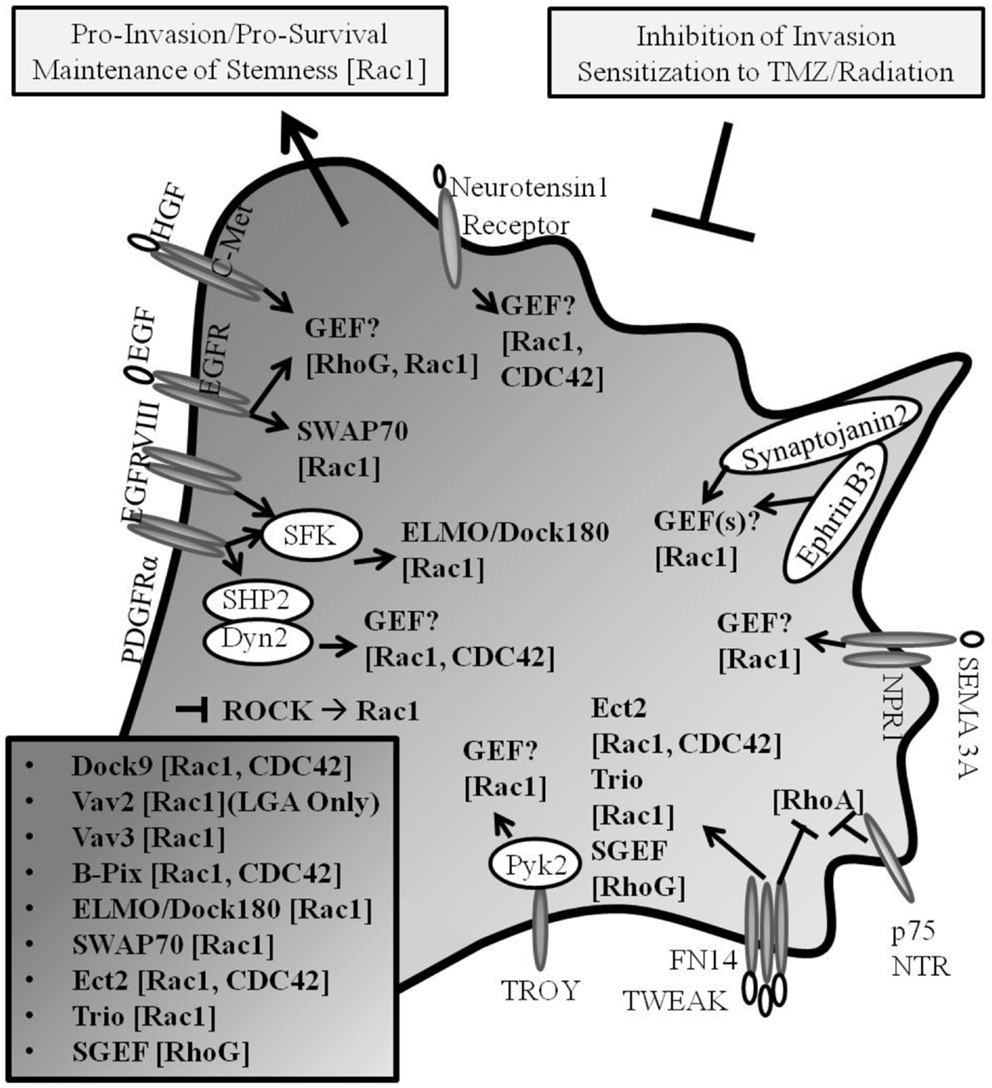
Figure 2. Rho GEF and Rho GTPase signaling in GB. Pro-invasive Rho GEF family members Dock9, Vav2, Vav3, β-Pix, ELMO/Dock180, SWAP-70, Ect2, Trio, and SGEF confer guanine nucleotide exchange for Rho GTPase family members Rac1, Cdc42, and RhoG in glioma progression (Known GEFs in glioma are summarized in insert with corresponding GTPases shown in brackets; LGA, lower grade astrocytoma only). Cancer invasion and drug-resistance share overlapping signaling pathways, including the signaling through Rho GTPases in glioma. The inhibition of mediators of invasion confers increased susceptibility to chemotherapeutic- and radiation-induced cell death.
Conclusion
Deregulated pathway signaling in GB tumors occurs within multiple processes including proliferation, metabolism, gliomagenesis, angiogenesis, survival, and invasion. Patient mortality is ultimately due to tumor spread and growth burden, thus the identification of key drivers of cell invasion can inform future targeted therapy development for use in clinical trials, with the intent to sensitize cells to combinatorial therapy with chemotherapeutic and radiologic interventions.
The acquisition of cell motility is complex and influenced by various intracellular and extracellular signaling events. The Rho family of GTPases are well-defined regulators of actin cytoskeletal dynamics (17, 24, 37), and have been characterized to contribute to most steps of cancer initiation and progression (141). Suppression of Rac activity selectively induces apoptosis in glioma cells but not in normal human astrocytes (142), thus proffering a rationale for the therapeutic inhibition of pro-migratory signaling pathways including those promoting Rac activation as an effective clinical option for GB. Increased support for the utility of Rho GTPase pathway inhibitors in treating cancer has led to the recent development of several strategies to identify drugs that will act against Rho GTPases, or their regulation by GEFs (143–151). These studies highlight the potential use of GEF inhibition and support both the rationale for and the feasibility of future GEF inhibitors for clinical use.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
GAP, GTPase-activating protein; GB, glioblastoma; GDI, guanine nucleotide dissociation inhibitor; GEF, guanine nucleotide exchange factor.
References
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114:97–109. doi: 10.1007/s00401-007-0278-6
2. Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS. The central nervous system. In: Frosch MP, Anthony DC, De Girolami U, editors. Robbins and Cotran Pathologic Basis of Disease, 8th ed. Philadelphia: Elsevier Saunders (2010). p. 1279–344.
3. Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol (2006) 1:97–117. doi:10.1146/annurev.pathol.1.110304.100043
4. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi:10.1016/S1470-2045(09)70025-7
5. Munson JM, Fried L, Rowson SA, Bonner MY, Karumbaiah L, Diaz B, et al. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci Transl Med (2012) 4:127ra136. doi:10.1126/scitranslmed.3003016
6. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell (2012) 150:165–78. doi:10.1016/j.cell.2012.04.042
7. Alexander S, Friedl P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med (2012) 18:13–26. doi:10.1016/j.molmed.2011.11.003
8. Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME. Molecular targets of glioma invasion. Cell Mol Life Sci (2007) 64:458–78. doi:10.1007/s00018-007-6342-5
9. Kwiatkowska A, Symons M. Signaling determinants of glioma cell invasion. Adv Exp Med Biol (2013) 986:121–41. doi:10.1007/978-94-007-4719-7_7
10. Zhai GG, Malhotra R, Delaney M, Latham D, Nestler U, Zhang M, et al. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the Rho signaling pathway. J Neurooncol (2006) 76:227–37. doi:10.1007/s11060-005-6499-4
11. Boureux A, Vignal E, Faure S, Fort P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol Biol Evol (2007) 24:203–16. doi:10.1093/molbev/msl145
12. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol (2005) 6:167–80. doi:10.1038/nrm1587
13. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature (2002) 420:629–35. doi:10.1038/nature01148
14. Vega FM, Ridley AJ. SnapShot: Rho family GTPases. Cell (2007) 129:1430. doi:10.1016/j.cell.2007.06.021
15. Riou P, Villalonga P, Ridley AJ. Rnd proteins: multifunctional regulators of the cytoskeleton and cell cycle progression. Bioessays (2010) 32:986–92. doi:10.1002/bies.201000060
16. Chardin P. Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol (2006) 7:54–62. doi:10.1038/nrm1788
17. Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell (1995) 81:53–62. doi:10.1016/0092-8674(95)90370-4
18. Ando S, Kaibuchi K, Sasaki T, Hiraoka K, Nishiyama T, Mizuno T, et al. Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J Biol Chem (1992) 267:25709–13.
19. Seabra MC. Membrane association and targeting of prenylated Ras-like GTPases. Cell Signal (1998) 10:167–72. doi:10.1016/S0898-6568(97)00120-4
20. Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal (1999) 11:545–54. doi:10.1016/S0898-6568(98)00063-1
21. Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci (2004) 117:1301–12. doi:10.1242/jcs.01118
22. Ding F, Yin Z, Wang HR. Ubiquitination in Rho signaling. Curr Top Med Chem (2011) 11:2879–87. doi:10.2174/156802611798281357
23. Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol (2010) 12:477–83. doi:10.1038/ncb2049
24. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell (1992) 70:401–10.
25. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell (1992) 70:389–99. doi:10.1016/0092-8674(92)90164-8
26. Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol (1995) 15:1942–52.
27. Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol (2011) 21:718–26. doi:10.1016/j.tcb.2011.08.002
28. Katoh H, Negishi M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature (2003) 424:461–4. doi:10.1038/nature01817
29. Katoh H, Hiramoto K, Negishi M. Activation of Rac1 by RhoG regulates cell migration. J Cell Sci (2006) 119:56–65. doi:10.1242/jcs.02720
30. Hiramoto K, Negishi M, Katoh H. Dock4 is regulated by RhoG and promotes Rac-dependent cell migration. Exp Cell Res (2006) 312:4205–16. doi:10.1016/j.yexcr.2006.09.006
31. Wennerberg K, Ellerbroek SM, Liu RY, Karnoub AE, Burridge K, Der CJ. RhoG signals in parallel with Rac1 and Cdc42. J Biol Chem (2002) 277:47810–7. doi:10.1074/jbc.M203816200
32. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol (2004) 265:23–32. doi:10.1016/j.ydbio.2003.06.003
33. Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell (2001) 106:489–98. doi:10.1016/S0092-8674(01)00471-8
34. Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, et al. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol (2003) 160:375–85. doi:10.1083/jcb.200208179
35. Cotteret S, Chernoff J. The evolutionary history of effectors downstream of Cdc42 and Rac. Genome Biol (2002) 3:REVIEWS0002. doi:10.1186/gb-2002-3-2-reviews0002
36. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science (1996) 273:245–8. doi:10.1126/science.273.5272.245
37. Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol (1996) 133:1403–15. doi:10.1083/jcb.133.6.1403
38. Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol (2003) 4:446–56. doi:10.1038/nrm1128
39. Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, et al. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science (1997) 275:1308–11. doi:10.1126/science.275.5304.1308
40. Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol (1999) 1:136–43. doi:10.1038/11056
41. Pankova K, Rosel D, Novotny M, Brabek J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci (2010) 67:63–71. doi:10.1007/s00018-009-0132-1
42. Sanz-Moreno V, Marshall CJ. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol (2010) 22:690–6. doi:10.1016/j.ceb.2010.08.020
43. Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, et al. Coordination of Rho GTPase activities during cell protrusion. Nature (2009) 461:99–103. doi:10.1038/nature08242
44. Shimizu T, Ihara K, Maesaki R, Kuroda S, Kaibuchi K, Hakoshima T. An open conformation of switch I revealed by the crystal structure of a Mg2+-free form of RHOA complexed with GDP. Implications for the GDP/GTP exchange mechanism. J Biol Chem (2000) 275:18311–7. doi:10.1074/jbc.M910274199
45. Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev (2002) 16:1587–609.
46. Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci (2002) 115:4901–13. doi:10.1242/jcs.00219
47. Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol (2002) 4:574–82.
48. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet (2012) 44:1006–14. doi:10.1038/ng.2359
49. Davis MJ, Ha BH, Holman EC, Halaban R, Schlessinger J, Boggon TJ. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc Natl Acad Sci USA (2013) 110:912–7. doi:10.1073/pnas.1220895110
50. Salhia B, Tran NL, Chan A, Wolf A, Nakada M, Rutka F, et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol (2008) 173:1828–38. doi:10.2353/ajpath.2008.080043
51. Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-kappaB and correlate with poor patient outcome. Cancer Res (2006) 66:9535–42. doi:10.1158/0008-5472.CAN-06-0418
52. Chan AY, Coniglio SJ, Chuang YY, Michaelson D, Knaus UG, Philips MR, et al. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene (2005) 24:7821–9. doi:10.1038/sj.onc.1208909
53. Tran NL, McDonough WS, Savitch BA, Sawyer TF, Winkles JA, Berens ME. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J Biol Chem (2005) 280:3483–92. doi:10.1074/jbc.M409906200
54. Fortin SP, Ennis MJ, Savitch BA, Carpentieri D, McDonough WS, Winkles JA, et al. Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol Cancer Res (2009) 7:1871–81. doi:10.1158/1541-7786.MCR-09-0194
55. Fortin SP, Ennis MJ, Schumacher CA, Zylstra-Diegel CR, Williams BO, Ross JT, et al. Cdc42 and the guanine nucleotide exchange factors Ect2 and Trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol Cancer Res (2012) 10(7):958–68. doi:10.1158/1541-7786.MCR-11-0616
56. Fortin Ensign SP, Mathews IT, Eschbacher JM, Loftus JC, Symons MH, Tran NL. The Src homology 3 domain-containing guanine nucleotide exchange factor is overexpressed in high-grade gliomas and promotes tumor necrosis factor-like weak inducer of apoptosis-fibroblast growth factor-inducible 14-induced cell migration and invasion via tumor necrosis factor receptor-associated factor 2. J Biol Chem (2013) 288(30):21887–97. doi:10.1074/jbc.M113.468686
57. Paulino VM, Yang Z, Kloss J, Ennis MJ, Armstrong BA, Loftus JC, et al. TROY (TNFRSF19) is overexpressed in advanced glial tumors and promotes glioblastoma cell invasion via Pyk2-Rac1 signaling. Mol Cancer Res (2010) 8:1558–67. doi:10.1158/1541-7786.MCR-10-0334
58. Servotte S, Camby I, Debeir O, Deroanne C, Lambert CA, Lapiere CM, et al. The in vitro influences of neurotensin on the motility characteristics of human U373 glioblastoma cells. Neuropathol Appl Neurobiol (2006) 32:575–84. doi:10.1111/j.1365-2990.2006.00760.x
59. Bagci T, Wu JK, Pfannl R, Ilag LL, Jay DG. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene (2009) 28:3537–50. doi:10.1038/onc.2009.204
60. Li X, Law JW, Lee AY. Semaphorin 5A and plexin-B3 regulate human glioma cell motility and morphology through Rac1 and the actin cytoskeleton. Oncogene (2012) 31:595–610. doi:10.1038/onc.2011.256
61. Li X, Lee AY. Semaphorin 5A and plexin-B3 inhibit human glioma cell motility through RhoGDIalpha-mediated inactivation of Rac1 GTPase. J Biol Chem (2010) 285:32436–45. doi:10.1074/jbc.M110.120451
62. Nakada M, Drake KL, Nakada S, Niska JA, Berens ME. Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res (2006) 66:8492–500. doi:10.1158/0008-5472.CAN-05-4211
63. Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol (2008) 20:235–41. doi:10.1016/j.ceb.2008.01.005
64. Chuang YY, Tran NL, Rusk N, Nakada M, Berens ME, Symons M. Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res (2004) 64:8271–5. doi:10.1158/0008-5472.CAN-04-2097
65. Wang Q, Wang JY, Zhang XP, Lv ZW, Fu D, Lu YC, et al. RLIP76 is overexpressed in human glioblastomas and is required for proliferation, tumorigenesis and suppression of apoptosis. Carcinogenesis (2013) 34:916–26. doi:10.1093/carcin/bgs401
66. Zhou X, Qian J, Hua L, Shi Q, Liu Z, Xu Y, et al. Geranylgeranyltransferase I promotes human glioma cell growth through Rac1 membrane association and activation. J Mol Neurosci (2013) 49:130–9. doi:10.1007/s12031-012-9905-3
67. Feng H, Hu B, Jarzynka MJ, Li Y, Keezer S, Johns TG, et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci USA (2012) 109:3018–23. doi:10.1073/pnas.1121457109
68. Feng H, Hu B, Vuori K, Sarkaria JN, Furnari FB, Cavenee WK, et al. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene (2013). doi:10.1038/onc.2013.198
69. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi:10.1016/j.ccr.2009.12.020
70. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA (1994) 91:7727–31. doi:10.1073/pnas.91.16.7727
71. The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. doi:10.1038/nature07385
72. Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T, et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest (2011) 121:4670–84. doi:10.1172/JCI58559
73. Hu B, Shi B, Jarzynka MJ, Yiin JJ, D’Souza-Schorey C, Cheng SY. ADP-ribosylation factor 6 regulates glioma cell invasion through the IQ-domain GTPase-activating protein 1-Rac1-mediated pathway. Cancer Res (2009) 69:794–801. doi:10.1158/0008-5472.CAN-08-2110
74. Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, et al. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol (2005) 7:270–7. doi:10.1038/ncb1227
75. Wang Z, Oh E, Thurmond DC. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem (2007) 282:9536–46. doi:10.1074/jbc.M610553200
76. Jarzynka MJ, Hu B, Hui KM, Bar-Joseph I, Gu W, Hirose T, et al. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res (2007) 67:7203–11. doi:10.1158/0008-5472.CAN-07-0473
77. Kwiatkowska A, Didier S, Fortin S, Chuang Y, White T, Berens ME, et al. The small GTPase RhoG mediates glioblastoma cell invasion. Mol Cancer (2012) 11:65. doi:10.1186/1476-4598-11-65
78. Feng H, Liu KW, Guo P, Zhang P, Cheng T, McNiven MA, et al. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene (2012) 31:2691–702. doi:10.1038/onc.2011.436
79. Hirata E, Yukinaga H, Kamioka Y, Arakawa Y, Miyamoto S, Okada T, et al. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J Cell Sci (2012) 125:858–68. doi:10.1242/jcs.089995
80. Kim JY, Oh MH, Bernard LP, Macara IG, Zhang H. The RhoG/ELMO1/Dock180 signaling module is required for spine morphogenesis in hippocampal neurons. J Biol Chem (2011) 286:37615–24. doi:10.1074/jbc.M111.268029
81. Franke K, Otto W, Johannes S, Baumgart J, Nitsch R, Schumacher S. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J (2012) 31:2908–21. doi:10.1038/emboj.2012.130
82. Samson T, Welch C, Monaghan-Benson E, Hahn KM, Burridge K. Endogenous RhoG is rapidly activated after epidermal growth factor stimulation through multiple guanine-nucleotide exchange factors. Mol Biol Cell (2010) 21:1629–42. doi:10.1091/mbc.E09-09-0809
83. Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, et al. A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell (2011) 21:681–93. doi:10.1016/j.devcel.2011.08.007
84. Iden S, Collard JG. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol (2008) 9:846–59. doi:10.1038/nrm2521
85. Berzat A, Hall A. Cellular responses to extracellular guidance cues. EMBO J (2010) 29:2734–45. doi:10.1038/emboj.2010.170
86. Meller J, Vidali L, Schwartz MA. Endogenous RhoG is dispensable for integrin-mediated cell spreading but contributes to Rac-independent migration. J Cell Sci (2008) 121:1981–9. doi:10.1242/jcs.025130
87. Bigarella CL, Borges L, Costa FF, Saad ST. ARHGAP21 modulates FAK activity and impairs glioblastoma cell migration. Biochim Biophys Acta (2009) 1793:806–16. doi:10.1016/j.bbamcr.2009.02.010
88. Yiin JJ, Hu B, Jarzynka MJ, Feng H, Liu KW, Wu JY, et al. Slit2 inhibits glioma cell invasion in the brain by suppression of Cdc42 activity. Neuro Oncol (2009) 11:779–89. doi:10.1215/15228517-2008-017
89. Yan B, Chour HH, Peh BK, Lim C, Salto-Tellez M. RhoA protein expression correlates positively with degree of malignancy in astrocytomas. Neurosci Lett (2006) 407:124–6. doi:10.1016/j.neulet.2006.08.032
90. Malchinkhuu E, Sato K, Maehama T, Mogi C, Tomura H, Ishiuchi S, et al. S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem Biophys Res Commun (2008) 366:963–8. doi:10.1016/j.bbrc.2007.12.054
91. Johnston AL, Lun X, Rahn JJ, Liacini A, Wang L, Hamilton MG, et al. The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol (2007) 5:e212. doi:10.1371/journal.pbio.0050212
92. Salhia B, Rutten F, Nakada M, Beaudry C, Berens M, Kwan A, et al. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res (2005) 65:8792–800. doi:10.1158/0008-5472.CAN-05-0160
93. Manning TJ Jr, Parker JC, Sontheimer H. Role of lysophosphatidic acid and rho in glioma cell motility. Cell Motil Cytoskeleton (2000) 45:185–99. doi:10.1002/(SICI)1097-0169(200003)45:3<185::AID-CM2>3.0.CO;2-G
94. Seasholtz TM, Radeff-Huang J, Sagi SA, Matteo R, Weems JM, Cohen AS, et al. Rho-mediated cytoskeletal rearrangement in response to LPA is functionally antagonized by Rac1 and PIP2. J Neurochem (2004) 91:501–12. doi:10.1111/j.1471-4159.2004.02749.x
95. Goldberg L, Kloog Y. A Ras inhibitor tilts the balance between Rac and Rho and blocks phosphatidylinositol 3-kinase-dependent glioblastoma cell migration. Cancer Res (2006) 66:11709–17. doi:10.1158/0008-5472.CAN-06-1878
96. Ramis G, Thomas-Moya E, Fernandez de Mattos S, Rodriguez J, Villalonga P. EGFR inhibition in glioma cells modulates Rho signaling to inhibit cell motility and invasion and cooperates with temozolomide to reduce cell growth. PLoS ONE (2012) 7:e38770. doi:10.1371/journal.pone.0038770
97. Lefranc F, Sauvage S, Van Goietsenoven G, Megalizzi V, Lamoral-Theys D, Debeir O, et al. Narciclasine, a plant growth modulator, activates Rho and stress fibers in glioblastoma cells. Mol Cancer Ther (2009) 8:1739–50. doi:10.1158/1535-7163.MCT-08-0932
98. Danussi C, Akavia UD, Niola F, Jovic A, Lasorella A, Pe’er D, et al. RHPN2 drives mesenchymal transformation in malignant glioma by triggering RhoA activation. Cancer Res (2013) 73(16):5140–50. doi:10.1158/0008-5472.CAN-13-1168-T
99. Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene (2009) 28:1570–83. doi:10.1038/onc.2009.2
100. Mertsch S, Oellers P, Wendling M, Stracke W, Thanos S. Dissecting the inter-substrate navigation of migrating glioblastoma cells with the stripe assay reveals a causative role of ROCK. Mol Neurobiol (2013) 48(1):169–79. doi:10.1007/s12035-013-8429-3
101. Seol HJ, Smith CA, Salhia B, Rutka JT. The guanine nucleotide exchange factor SWAP-70 modulates the migration and invasiveness of human malignant glioma cells. Transl Oncol (2009) 2:300–9.
102. Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol (1999) 147:921–8. doi:10.1083/jcb.147.5.921
103. Matthews HK, Delabre U, Rohn JL, Guck J, Kunda P, Baum B. Changes in Ect2 localization couple actomyosin-dependent cell shape changes to mitotic progression. Dev Cell (2012) 23:371–83. doi:10.1016/j.devcel.2012.06.003
104. Weeks A, Okolowsky N, Golbourn B, Ivanchuk S, Smith C, Rutka JT. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am J Pathol (2012) 181:662–74. doi:10.1016/j.ajpath.2012.04.011
105. Frank SR, Hansen SH. The PIX-GIT complex: a G protein signaling cassette in control of cell shape. Semin Cell Dev Biol (2008) 19:234–44. doi:10.1016/j.semcdb.2008.01.002
106. Wang H, Han M, Whetsell W Jr, Wang J, Rich J, Hallahan D, et al. Tax-interacting protein 1 coordinates the spatiotemporal activation of Rho GTPases and regulates the infiltrative growth of human glioblastoma. Oncogene (2013). doi:10.1038/onc.2013.97
107. Borggrefe T, Wabl M, Akhmedov AT, Jessberger R. A B-cell-specific DNA recombination complex. J Biol Chem (1998) 273:17025–35. doi:10.1074/jbc.273.27.17025
108. Qi H, Fournier A, Grenier J, Fillion C, Labrie Y, Labrie C. Isolation of the novel human guanine nucleotide exchange factor Src homology 3 domain-containing guanine nucleotide exchange factor (SGEF) and of C-terminal SGEF, an N-terminally truncated form of SGEF, the expression of which is regulated by androgen in prostate cancer cells. Endocrinology (2003) 144:1742–52.
109. Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, DeMali KA, et al. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell (2004) 15:3309–19. doi:10.1091/mbc.E04-02-0146
110. Krishna Subbaiah V, Massimi P, Boon SS, Myers MP, Sharek L, Garcia-Mata R, et al. The invasive capacity of HPV transformed cells requires the hDlg-dependent enhancement of SGEF/RhoG activity. PLoS Pathog (2012) 8:e1002543. doi:10.1371/journal.ppat.1002543
111. Garcia JL, Couceiro J, Gomez-Moreta JA, Gonzalez Valero JM, Briz AS, Sauzeau V, et al. Expression of VAV1 in the tumour microenvironment of glioblastoma multiforme. J Neurooncol (2012) 110:69–77. doi:10.1007/s11060-012-0936-y
112. Bagchi S, Liao Z, Gonzalez FA, Chorna NE, Seye CI, Weisman GA, et al. The P2Y2 nucleotide receptor interacts with alphav integrins to activate Go and induce cell migration. J Biol Chem (2005) 280:39050–7. doi:10.1074/jbc.M504819200
113. Debant A, Serra-Pages C, Seipel K, O’Brien S, Tang M, Park SH, et al. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci USA (1996) 93:5466–71. doi:10.1073/pnas.93.11.5466
114. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell (2006) 9:157–73. doi:10.1016/j.ccr.2006.02.019
115. Meller N, Westbrook MJ, Shannon JD, Guda C, Schwartz MA. Function of the N-terminus of zizimin1: autoinhibition and membrane targeting. Biochem J (2008) 409:525–33. doi:10.1042/BJ20071263
116. Meller N, Irani-Tehrani M, Kiosses WB, Del Pozo MA, Schwartz MA. Zizimin1, a novel Cdc42 activator, reveals a new GEF domain for Rho proteins. Nat Cell Biol (2002) 4:639–47. doi:10.1038/ncb835
117. Kuramoto K, Negishi M, Katoh H. Regulation of dendrite growth by the Cdc42 activator Zizimin1/Dock9 in hippocampal neurons. J Neurosci Res (2009) 87:1794–805. doi:10.1002/jnr.21997
118. Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci USA (2011) 108:3749–54. doi:10.1073/pnas.1014480108
119. Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res (2004) 64:920–7. doi:10.1158/0008-5472.CAN-03-2073
120. Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol (2006) 65:529–39. doi:10.1097/00005072-200606000-00001
121. Leon SP, Folkerth RD, Black PM. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer (1996) 77:362–72. doi:10.1002/(SICI)1097-0142(19960115)77:2<362::AID-CNCR20>3.0.CO;2-Z
122. Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough WS, et al. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia (2005) 7:7–16. doi:10.1593/neo.04535
123. Mariani L, Beaudry C, McDonough WS, Hoelzinger DB, Demuth T, Ross KR, et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: a cDNA microarray analysis. J Neurooncol (2001) 53:161–76. doi:10.1023/A:1012253317934
124. Kislin KL, McDonough WS, Eschbacher JM, Armstrong BA, Berens ME. NHERF-1: modulator of glioblastoma cell migration and invasion. Neoplasia (2009) 11:377–87.
125. Zhang B, Gu F, She C, Guo H, Li W, Niu R, et al. Reduction of Akt2 inhibits migration and invasion of glioma cells. Int J Cancer (2009) 125:585–95. doi:10.1002/ijc.24314
126. Brassesco MS, Roberto GM, Morales AG, Oliveira JC, Delsin LE, Pezuk JA, et al. Inhibition of NF- kappa B by dehydroxymethylepoxyquinomicin suppresses invasion and synergistically potentiates temozolomide and gamma -radiation cytotoxicity in glioblastoma cells. Chemother Res Pract (2013) 2013:593020.
127. Loftus JC, Dhruv H, Tuncali S, Kloss J, Yang Z, Schumacher CA, et al. TROY (TNFRSF19) promotes glioblastoma cell survival signaling and therapeutic resistance. Mol Cancer Res (2013) 11(8):865–74. doi:10.1158/1541-7786.MCR-13-0008
128. Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells – much more complex than expected. Mol Cancer (2011) 10:128. doi:10.1186/1476-4598-10-128
129. Yoon CH, Hyun KH, Kim RK, Lee H, Lim EJ, Chung HY, et al. The small GTPase Rac1 is involved in the maintenance of stemness and malignancies in glioma stem-like cells. FEBS Lett (2011) 585:2331–8. doi:10.1016/j.febslet.2011.05.070
130. Zheng M, Morgan-Lappe SE, Yang J, Bockbrader KM, Pamarthy D, Thomas D, et al. Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res (2008) 68:7570–8. doi:10.1158/0008-5472.CAN-08-0632
131. Yang HJ, Youn H, Seong KM, Jin YW, Kim J, Youn B. Phosphorylation of ribosomal protein S3 and antiapoptotic TRAF2 protein mediates radioresistance in non-small cell lung cancer cells. J Biol Chem (2013) 288:2965–75. doi:10.1074/jbc.M112.385989
132. Lin WJ, Su YW, Lu YC, Hao Z, Chio II, Chen NJ, et al. Crucial role for TNF receptor-associated factor 2 (TRAF2) in regulating NFkappaB2 signaling that contributes to autoimmunity. Proc Natl Acad Sci USA (2011) 108:18354–9. doi:10.1073/pnas.1109427108
133. Jang KW, Lee KH, Kim SH, Jin T, Choi EY, Jeon HJ, et al. Ubiquitin ligase CHIP induces TRAF2 proteasomal degradation and NF-kappaB inactivation to regulate breast cancer cell invasion. J Cell Biochem (2011) 112:3612–20. doi:10.1002/jcb.23292
134. Natoli G, Costanzo A, Guido F, Moretti F, Bernardo A, Burgio VL, et al. Nuclear factor kB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J Biol Chem (1998) 273:31262–72. doi:10.1074/jbc.273.47.31262
135. Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science (1995) 269:1424–7. doi:10.1126/science.7544915
136. Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell (1996) 87:565–76. doi:10.1016/S0092-8674(00)81375-6
137. Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, et al. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science (1997) 275:200–3. doi:10.1126/science.275.5297.200
138. Nomura N, Nomura M, Mizuki N, Hamada J. Rac1 mediates phorbol 12-myristate 13-acetate-induced migration of glioblastoma cells via paxillin. Oncol Rep (2008) 20:705–11.
139. Hwang SY, Jung JW, Jeong JS, Kim YJ, Oh ES, Kim TH, et al. Dominant-negative Rac increases both inherent and ionizing radiation-induced cell migration in C6 rat glioma cells. Int J Cancer (2006) 118:2056–63. doi:10.1002/ijc.21574
140. Golubovskaya VM, Huang G, Ho B, Yemma M, Morrison CD, Lee J, et al. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol Cancer Ther (2013) 12:162–72. doi:10.1158/1535-7163.MCT-12-0701
141. Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett (2008) 582:2093–101. doi:10.1016/j.febslet.2008.04.039
142. Senger DL, Tudan C, Guiot MC, Mazzoni IE, Molenkamp G, LeBlanc R, et al. Suppression of Rac activity induces apoptosis of human glioma cells but not normal human astrocytes. Cancer Res (2002) 62:2131–40.
143. Surviladze Z, Young SM, Sklar LA. High-throughput flow cytometry bead-based multiplex assay for identification of Rho GTPase inhibitors. Methods Mol Biol (2012) 827:253–70. doi:10.1007/978-1-61779-442-1_17
144. Marchioni F, Zheng Y. Targeting rho GTPases by peptidic structures. Curr Pharm Des (2009) 15:2481–7. doi:10.2174/138161209788682334
145. Nassar N, Cancelas J, Zheng J, Williams DA, Zheng Y. Structure-function based design of small molecule inhibitors targeting Rho family GTPases. Curr Top Med Chem (2006) 6:1109–16. doi:10.2174/156802606777812095
146. Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, et al. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem (2012) 287:13228–38. doi:10.1074/jbc.M111.334524
147. Ortiz-Sanchez JM, Nichols SE, Sayyah J, Brown JH, McCammon JA, Grant BJ. Identification of potential small molecule binding pockets on Rho family GTPases. PLoS ONE (2012) 7:e40809. doi:10.1371/journal.pone.0040809
148. Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci USA (2010) 107:9446–51. doi:10.1073/pnas.0911863107
149. Blangy A, Fort P. Using a modified yeast two-hybrid system to screen for chemical GEF inhibitors. Methods Mol Biol (2012) 928:81–95. doi:10.1007/978-1-62703-008-3_7
150. Schmidt S, Diriong S, Mery J, Fabbrizio E, Debant A. Identification of the first Rho-GEF inhibitor, TRIPalpha, which targets the RhoA-specific GEF domain of Trio. FEBS Lett (2002) 523:35–42. doi:10.1016/S0014-5793(02)02928-9
Keywords: glioblastoma, invasion, survival, RhoGTPase, Rac1, Cdc42, RhoA, GEFs
Citation: Fortin Ensign SP, Mathews IT, Symons MH, Berens ME and Tran NL (2013) Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front. Oncol. 3:241. doi: 10.3389/fonc.2013.00241
Received: 12 July 2013; Paper pending published: 30 July 2013;
Accepted: 02 September 2013; Published online: 04 October 2013.
Edited by:
Giuseppe Merla, IRCCS Casa Sollievo della Sofferenza, ItalyReviewed by:
Lucia Micale, IRCCS, Casa Sollievo della Sofferenza Hospital, ItalyPriam Villalonga, Universitat de les Illes Balears, Spain
Copyright: © 2013 Fortin Ensign, Mathews, Symons, Berens and Tran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nhan L. Tran, Translational Genomics Research Institute, 445 N 5th Street, Suite 400, Phoenix, AZ 85004, USA e-mail:bnRyYW5AdGdlbi5vcmc=