 Rohini Khatri
Rohini Khatri Subbaya Subramanian
Subbaya Subramanian- 1Department of Surgery, Division of Basic and Translational Research, University of Minnesota, Minneapolis, MN, USA
- 2Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA
The study of regulatory non-coding RNAs has deepened our understanding of cancer on the molecular and clinical front. MicroRNAs, which encompass over 1000 short, endogenous nucleic acids, include important candidates that drive carcinogenesis. In this discussion, we review the literature on miR-135b, purporting its role as an oncogene in multiple cancers, and we highlight STAT3 inhibition as a potential therapeutic strategy mediated by miR-135b and demonstrate particular clinical relevance to colon cancer.
MicroRNA-135b (miR-135b) Overexpression and Oncogenic Behavior
miR-135b levels are elevated in a variety of cancers including breast, non-small cell lung cancer (NSCLC), prostate, and colon (1–3). Microarray analysis and quantitative reverse transcription-PCR (qRT-PCR) studies in NSCLC have demonstrated that miR-135b upregulation is far more robust in highly invasive lines compared to the less invasive. Overexpression of miR-135b conferred an increased tumorigenic ability to the relatively benign CL1-0 cells, resulting in more than a fourfold greater tumor burden in xenograft mouse models. In vivo, stable expression of a miR-135b antagonist decreased the number of metastatic tumor nodules in mice injected with highly invasive CL1-5-F4 cells shown to have high levels of miR-135b (4).
Clinically, high levels of miR-135b in lung cancer specimens significantly correlated with decreased overall survival (4). The investigators demonstrated that miR-135b suppresses key components of the Hippo pathway, a serine-threonine kinase pathway studied extensively in Drosophila melanogaster that plays a role in inhibition of overgrowth, regulation of cell division, and apoptosis. Mammalian orthologs within the Hippo pathways include LATS2 and LZTS1, which were identified as targets of miR-135b. Downregulation of LATS2 and LZTS1 and consequent nuclear localization of the transcriptional activator TAZ in patients with high miR-135b levels correlated with poor overall survival (4). Clinically, deregulation of this pathway also appears to be an important driver of hepatocellular and cholangiocarcinoma, although, to our knowledge, its regulation has not been linked to miR-135b in the setting of liver disease (5).
Microarray analysis and qRT-PCR also demonstrate that miR-135b levels are more than twofold higher in head and neck squamous cell carcinoma (HNSCC) in an orthotopic mouse model compared to controls (6). Parallel to the findings in lung cancer, in vitro studies with HNSCC also demonstrate that miR-135b overexpression confers an invasive phenotype – cells expressing miR-135b mimic displayed increased colony formation, cell migration, and proliferation. miR-135b mimics also induced increased tube formation of human umbilical vein endothelial cells, providing in vitro support for its role is promoting angiogenesis.
Investigators subsequently identified upregulation of hypoxia inducible factor 1 alpha (HIF-1α) and vascular endothelial growth factor A (VEGFA), which is a downstream target of HIF-1α, as a consequence of miR-135b overexpression. In vitro data suggests that miR-135b decreases available factor inhibiting HIF (FIH), which is also known as HIF-1α subunit inhibitor (HIFIAN). The Tgfbr1/Pten 2c knockout mice serving as the orthotopic HNSCC model in this study demonstrated decreased levels of Fih mRNA and protein in tongue squamous cell carcinoma compared to surrounding tongue epithelium and compared to control mice. These findings correlated with elevated levels of miR-135b and Hif-1α mRNA. When HNSCC cell lines were transfected with a miR-135b mimic, a corresponding increase in protein levels of HIF-1α and decrease in FIH was elicited.
Immunofluorescence and Western blot analysis demonstrated parallel correlation between elevated miR-135b levels and decreased FIH signal in the Tgfbr1/Pten 2cKO mice. Nuclear localization of HIF-1α and VEGFA signals were significantly increased in mice with elevated miR-135b levels. Finally, microvessel density of HNSCC in Tgfrb1 knockout mice was over fourfold greater compared to control Pten knockout HNSCC, providing in vivo data consistent with miR-135b promotion of angiogenesis.
Studies in colon cancer also demonstrate a distinct increase in expression of miR-135b in both adenomas and carcinomas compared to normal epithelium (1, 7, 8). Its role in driving adenoma to carcinoma progression, however, has not been established. Nonetheless, the progressive increase in miR-135b expression from normal tissue to polyp to carcinoma, suggests that miR-135b deregulation is an early event that is amplified with increasing dysplasia. Nagel et al. support this hypothesis by demonstrating adenomatous polyposis coli (APC) to be an important target of miR-135b (7). On a genomic level, it is known that biallelic mutations in the APC gene are the primary initiating events in the adenoma to carcinoma sequence through deregulation of the β-catenin pathway. These mutations typically result from premature stop codons in the APC transcript and are detected in >70% of colon cancers. In cases where hypomorphic mutations are present, miR-135b activation may function as “third hit” to indeed suppress any residual tumor suppression (β-catenin regulating) activity. miR-135b expression is shown to be over 10-fold elevated in carcinoma compared to normal colon epithelium and clearly associated with decreased APC expression irrespective of chromosomal mutations. This tight association is supported by confirmation of a miR-135b binding site at the APC 3′ untranslated region and suggests that miR-135b upregulation leads to a more penetrant effect of APC derangement (7).
Regulation of miR-135b Expression
miR-135b is encoded in the first intron of LEMD1 gene, a cancer/testis antigen shown to be aberrantly expressed in colon cancers (9). miR-135b is located on 1q32.1 which shows DNA copy number gain in CRC progression (7). Lung cancer lines demonstrate alteration of LEMD1 and miR-135b expression when treated with a demethylating agent, suggesting a mode of epigenetic control in that cell population. Furthermore, investigators predicted that the transcription factor NF-κB has a potential binding site on the miR-135b promoter region (Figure 1). After demethylation, TNFα, a known potentiator of NF-κB transcription, indeed increased expression of miR-135b, suggesting a potential link between inflammatory signals and miR-135b transcription (4).
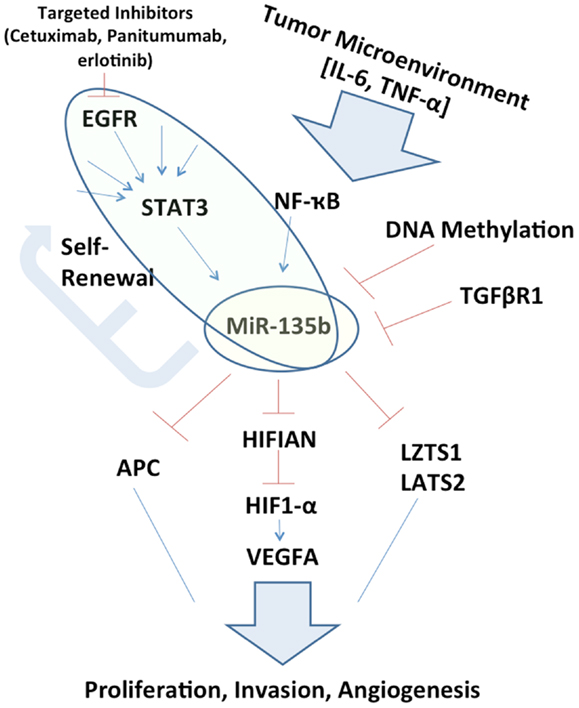
Figure 1. Schematic representation of miR-135b circuitry network. Studying the network surrounding miR-135b may elucidate links between otherwise far reaching pathways, such as EGFR/STAT3 signaling to Hippo (LZTS1, LATS2) and inhibition of growth. miR-135b silencing in vivo may itself be therapeutic and understanding its function may uncover additional therapeutic targets.
Interestingly, in the HNSCC study, miR-135b overexpression appeared to be induced from a knockdown of transforming growth factor beta receptor 1 (TGFBR1) (6). Clinically, mutations in TGFBR1 are associated with an increased risk of developing colon cancer (10).
In lymphoma cells, LEMD1 and miR-135b expression were demonstrated to be under STAT3 regulation (11). Signal transducer and activator of transcription (STAT) proteins are a family of cytoplasmic transcription factors that mediate external cytokine signaling and growth factor stimulation to transcription of genes involved in proliferation, differentiation, cell survival, development, and inflammation (12, 13). While there are several isoforms, constitutive activation of STAT3 has been identified to activate anti-apoptotic pathways and proliferative behaviors in a variety of cancers (12, 14).
If miR-135b expression is pervasively regulated by STAT3 in a variety of cancer types, it may perform a key exchange between the tumor microenvironment to inhibition of tumor suppressors. This extracellular ligand stimulation may also explain differences that may be observed in the role of miR-135b between one organ system and another, owing to the variable tumor microenvironments (Figure 1). This potential signaling cascade also elucidates the mechanism behind therapeutic benefits of STAT3 inhibition (13).
STAT3 and anti-EGFR Therapy
miR-135b may also represent a mechanism by which a network of tumor suppressors is downregulated despite targeted therapy against the cancer cells. The most apparent example would be the upstream activation of STAT3 via epidermal growth factor receptor regulation (EGFR). Multiple anti-EGFR drugs are utilized for treatment of HNSCC, bladder cancer, NSCLC, and colon cancer. The patients that qualify for this class of medications represent a selected group, and some continue to experience disease progression during treatment.
There is evidence that amongst HNSCC and NSCLC cells that are resistant to anti-EGFR therapy, STAT3 remains persistently phosphorylated in its active state (15, 16). The presence of phosphorylated STAT3 suggests a mode of activation that bypasses the EGFR signaling cascade (15). Concurrently, aggressive efforts are geared toward developing clinically available STAT3 inhibitors to restore the desired therapeutic effect or even replace anti-EGFR therapy (13, 16).
In treatment of colon cancer, anti-EGFR therapy is generally reserved for cases without KRAS mutations (17). What remains to be understood is the role of phosphorylated STAT3 as a predictor of response to anti-EGFR therapy. In a recent retrospective study, investigators examined levels of phosphorylated STAT3 amongst patients receiving anti-EGFR therapy. Multivariate analysis demonstrated that absence of phosphorylated STAT3 was associated with improved overall survival and increase time to progression of the disease (18). This finding suggests that if phosphorylated STAT3 is truly a marker of poor outcome, mir-135b also has the potential to be explored as a biomarker and driver of resistance to anti-EGFR therapy.
Persistent STAT3 activation also has a specific association with cell proliferation and tumor growth in colon cancer. In a study of 32 tumor specimens, 29 demonstrated STAT3 binding to DNA via electrophoretic mobility shift assay (EMSA). Immunohistochemistry of colon cancer biopsies confirmed presence of phosphorylated STAT3 in the nuclei of dedifferentiated epithelium but not in surrounding normal colonic crypts (14). When colon cancer cell lines were transfected with constitutively active STAT3, cell proliferation was increased, whereas transformation of the cells with a dominant negative construct of STAT resulted in decreased proliferation.
This study also points to a potential limitation of in vitro models in studying the role of phosphorylated STAT3, and presumably, miR-135b. In multiple colon cancer lines, investigators confirmed the abundance of STAT3 expression by Western Blot, similar to analysis of tumor specimens. However, when STAT3-DNA binding was assessed via EMSA, none of the cell lines demonstrated constitutive binding. Upon treating the cells with interleukin-6 (IL-6), a known activator of the STAT3 pathway, constitutive DNA binding was successfully demonstrated. Similarly, when the cell lines were injected subcutaneously to produce xenograft tumors where they were presumably exposed to in vivo paracrine and autocrine signaling, EMSA experiments confirmed abundance of DNA-bound STAT3.
These findings suggest that the tumor microenvironment plays an important role in inducing oncogenic signaling pathways. IL-6 levels are elevated in serum and tumor samples of multiple cancers, and IL-6 and STAT3 activation constitute an important pathway in tumorigenesis in colitis associated cancers (19, 20). Moreover, miR-135b expression was elevated in tumors in mouse models of APC mutations as well as inflammatory bowel disease (21). If miR-135b expression is in fact tightly regulated by STAT3 in a variety of cancers, in vitro studies may be under-representing the degree of upregulation and oncogenicity that miR-135b contributes in vivo, particularly in colon cancer and the subpopulation of colitis associated cancers. miR-135b upregulation may be the common initiating pathway between malignancy derived from somatic mutations and those derived from inflammatory backgrounds where Wnt derangement is typically a later event. In vivo overexpression of miR-135b may help elucidate the role in tumorigenesis, particularly in the context of existing APC mutations and colitis models.
miR-135b in Cancer Stem Cells
The role of miR-135b in cancer stem cells (CSCs) is also emerging and supported by the potential role of STAT3 in the same context. Through a screen for microRNAs deregulated in CSC in pediatric solid tumor lines, investigators identified miR-135b as highly expressed in the stem-cell enriched cell population (22). A stemness assay confirmed that in vitro blockade of miR-135b demonstrated a markedly diminished ability of the neuroblastoma and medulloblastoma cells to form neurospheres, thereby alluding to the role of miR-135b in self-renewal and proliferation (22).
In colon cancer cell lines, investigators utilized CD133 and aldehyde dehydrogenase 1 (ALDH1) as selective markers for CSC and demonstrated increased ability of this selected cell population in producing subcutaneous tumors in xenograft models (23). This selected cell population demonstrated increased levels of phosphorylated STAT3 compared to the unsorted cells. Furthermore, immunohistochemistry/immunofluorescence analysis of over 100 colon cancer microarrays demonstrated that co-localization of phosphorylated STAT3 and ALDH1 or CD133 occurred in over 60% of the cases. An anchorage-independent assay showed that pharmacological inhibition of STAT3 in the CSCs markedly decreased tumorsphere formation. Furthermore, treatment of the cell lines with STAT3 inhibitors decreased the percentage population of ALDH1+/CD133+ cells in contrast to treatment with doxorubicin or 5-flurouracil which increased the ALDH1+/CD133+ subpopulation, suggesting that STAT3 plays a unique role in CSC propagation.
The parallel findings of miR-135b suppression in pediatric CSC and STAT3 blockade in colon CSC strongly allude to miR-135b as being an important driver in maintaining cell oncogenicity, particularly in the self-renewing behaviors of CSCs. Further experimentation to assess miR-135b levels with better defined markers of stemness in colon cancer would be needed to provide conclusive evidence, and would be supported by differential expression of miR-135b along the crypt-villus axis.
Conclusion
With the knowledge that we have about the regulation of miR-135b, its important downstream targets, and the oncogenic impact produced from its dysregulation, the data begs for more conclusive studies, particularly in vivo assessment of its role in colon cancer. miR-135b emerges as one example of a microRNA that may deepen our understanding of resistance to current cancer therapy, and possibly serve as an adjunct to guiding anti-EGFR treatment in colon cancer patients, particularly as its role is better explored in KRAS mutants. Direct blockade of miR-135b may itself serve as a therapeutic intervention against a network of events driving oncogenesis and even target the self-renewing CSCs. Understanding this valuable molecular target and placing it in our diagnostic and therapeutic armamentarium will enhance our options against colon and other cancers in an era of personalized medicine.
Acknowledgments
Rohini Khatri is supported by the Colon Cancer Surgical Oncology Training Program from the Department of Surgery, University of Minnesota.
References
1. Bandres E, Cubedo E, Agirre X, Malumbres R, Zarate R, Ramirez N, et al. Identification by real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer (2006) 5:29. doi: 10.1186/1476-4598-5-29
2. Lowery AJ, Miller N, Devaney A, Mcneill RE, Davoren PA, Lemetre C, et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res (2009) 11:R27. doi:10.1186/bcr2257
3. Tong AW, Fulgham P, Jay C, Chen P, Khalil I, Liu S, et al. MicroRNA profile analysis of human prostate cancers. Cancer Gene Ther (2009) 16:206–16. doi:10.1038/cgt.2008.77
4. Lin CW, Chang YL, Chang YC, Lin JC, Chen CC, Pan SH, et al. MicroRNA-135b promotes lung cancer metastasis by regulating multiple targets in the hippo pathway and LZTS1. Nat Commun (2013) 4:1877. doi:10.1038/ncomms2876
5. Avruch J, Zhou D, Fitamant J, Bardeesy N. Mst1/2 signalling to yap: gatekeeper for liver size and tumour development. Br J Cancer (2011) 104:24–32. doi:10.1038/sj.bjc.6606011
6. Zhang L, Sun ZJ, Bian Y, Kulkarni AB. MicroRNA-135b acts as a tumor promoter by targeting the hypoxia-inducible factor pathway in genetically defined mouse model of head and neck squamous cell carcinoma. Cancer Lett (2013) 331:230–8. doi:10.1016/j.canlet.2013.01.003
7. Nagel R, Le Sage C, Diosdado B, Van Der Waal M, Oude Vrielink JA, Bolijn A, et al. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res (2008) 68:5795–802. doi:10.1158/0008-5472.CAN-08-0951
8. Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, Silverstein KA, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer (2009) 9:401. doi:10.1186/1471-2407-9-401
9. Yuki D, Lin YM, Fujii Y, Nakamura Y, Furukawa Y. Isolation of LEM domain-containing 1, a novel testis-specific gene expressed in colorectal cancers. Oncol Rep (2004) 12:275–80.
10. Lampropoulos P, Zizi-Sermpetzoglou A, Rizos S, Kostakis A, Nikiteas N, Papavassiliou AG. TGF-beta signalling in colon carcinogenesis. Cancer Lett (2012) 314:1–7. doi:10.1016/j.canlet.2011.09.041
11. Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N, et al. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood (2011) 118:6881–92. doi:10.1182/blood-2011-05-354654
12. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi:10.1038/nrc2734
13. Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs (2009) 18:45–56. doi:10.1517/13543780802565791
14. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia (2005) 7:545–55. doi:10.1593/neo.04571
15. Baba Y, Fujii M, Tokumaru Y, Kato Y. Present and future of EGFR inhibitors for head and neck squamous cell cancer. J Oncol (2012) 2012:986725. doi:10.1155/2012/986725
16. Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, et al. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res (2012) 18:4986–96. doi:10.1158/1078-0432.CCR-12-0792
17. Heinemann V, Stintzing S, Kirchner T, Boeck S, Jung A. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat Rev (2009) 35:262–71. doi:10.1016/j.ctrv.2008.11.005
18. Dobi E, Monnien F, Kim S, Ivanaj A, N’guyen T, Demarchi M, et al. Impact of STAT3 phosphorylation on the clinical effectiveness of anti-EGFR-based therapy in patients with metastatic colorectal cancer. Clin Colorectal Cancer (2013) 12:28–36. doi:10.1016/j.clcc.2012.09.002
19. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell (2009) 15:103–13. doi:10.1016/j.ccr.2009.01.001
20. Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol (2012) 30:1005–14. doi:10.1200/JCO.2010.31.8907
21. Necela BM, Carr JM, Asmann YW, Thompson EA. Differential expression of microRNAs in tumors from chronically inflamed or genetic (APC(Min/+)) models of colon cancer. PLoS One (2011) 6:e18501. doi:10.1371/journal.pone.0018501
22. Sanchez-Diaz PC, Hsiao TH, Chang JC, Yue D, Tan MC, Chen HI, et al. De-regulated microRNAs in pediatric cancer stem cells target pathways involved in cell proliferation, cell cycle and development. PLoS One (2013) 8:e61622. doi:10.1371/journal.pone.0061622
Keywords: colon cancer, microRNAs, miR-135b, STAT3, EGFR
Citation: Khatri R and Subramanian S (2013) MicroRNA-135b and its circuitry networks as potential therapeutic targets in colon cancer. Front. Oncol. 3:268. doi: 10.3389/fonc.2013.00268
Received: 30 August 2013; Accepted: 08 October 2013;
Published online: 22 October 2013.
Edited by:
Alessandro Lugli, University of Bern, SwitzerlandReviewed by:
Riccardo Fodde, Erasmus MC, NetherlandsCopyright: © 2013 Khatri and Subramanian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:c3VicmVlQHVtbi5lZHU=