 Karl Heinimann1,2*
Karl Heinimann1,2*- 1Department of Biomedicine, Research Group Human Genomics, Basel, Switzerland
- 2Medical Genetics, University Hospital Basel, Basel, Switzerland
Microsatellite instability (MSI) is the molecular hallmark of DNA mismatch repair deficiency. Since its initial description in colorectal cancer (CRC) in 1993 and its association with Lynch syndrome, the most common inherited cancer predisposition world-wide, accumulating evidence suggests that MSI status may also be of concrete prognostic and predictive value in the management of sporadic CRC. This mini review aims at providing a concise survey of the molecular basis and the multifaceted role(s) of MSI status in today’s clinical practice.
Introduction
Twenty years ago, in 1993, in search for molecular clues to the pathogenesis of colorectal cancer (CRC) several research groups had made an intriguing observation: widespread somatic alterations at short, repetitive DNA sequences, referred to as replication error (RER+) phenotype or, more specifically, microsatellite instability (MSI) (1–3). They detected this novel form of genomic instability in about 10–15% of sporadic, predominantly proximally located, colorectal carcinomas as well as in most (>90%) of those from patients with the most common inherited cancer predisposition, Lynch syndrome (LS), also referred to as Hereditary Non-Polyposis Colon Cancer (HNPCC). The discovery allowed to link the hereditary disorder to a defect in a key DNA metabolic pathway, the mismatch repair (MMR) system, which had previously been known to cause MSI in Saccharomyces cerevisiae, and eventually opened up a new field in cancer research (4).
The Molecular Basis of MSI
Microsatellites, also referred to as short tandem repeats (STRs), consist of a few to several thousands of tandemly repeated motifs made up of one (mono) up to six (hexa) nucleotides and are thought to account for approximately 3% of the human genome (5). Given their number and the fact that they are scattered throughout the genome, their (hyper)variability in length coupled with a high degree of heterozygosity made them ideal polymorphic markers for genome mapping, population genetics, and genetic linkage studies as well as indispensable tools in forensics and transplantation medicine (“DNA fingerprint”).
Due to their repetitive sequence structure, it is not surprising that microsatellites exhibit a particularly high mutation rate compared to non-repetitive, unique DNA stretches. During replication of the nuclear genome DNA polymerases, due to slippage, often fail to correctly duplicate such microsatellite tracts. The resulting insertion/deletion loops can consequently lead to insertions or deletions of repeats, thereby altering the length of the respective microsatellite (replication error, RER). In eukaryotes these errors are corrected by the DNA MMR system: its heterodimers MSH2/MSH6 and MSH2/MSH3 detect the replication error (licensing step) and recruit the MLH1/PMS2 complex through which degradation of the mutated stretch and resynthesis are initiated (4). As originally observed in yeast, defects in MMR consequently result in the genome-wide accumulation of mutations at microsatellite loci, MSI, which has also been referred to as a “mutator phenotype” (6).
Assessment of MSI
Since MSI analysis of colorectal tumors provided a straight-forward, though indirect, means to identify LS patients great attention was given to the development of selection criteria and microsatellite markers to be used for testing. In 1996 and 2002 the National Cancer Institute in Bethesda, MD, held workshops in which the Bethesda guidelines for the identification of LS patients were defined (7, 8). MSI testing is recommended in patients who meet one of the following criteria: (a) diagnosed with CRC before age 50 years, (b) synchronous or metachronous CRC or other LS-related tumors, (c) CRC with typical MSI-high morphology and diagnosed before age 60 years, (d) CRC in one or more first-degree relatives with CRC or other LS-related tumors, one being diagnosed before age 50 years, and (e) CRC with two or more relatives with CRC or other LS-related tumors, regardless of age. The revised Bethesda guidelines thus incorporate personal as well as family history and pay attention to the fact that LS actually comprises a spectrum of different tumor types (endometrial, gastric, etc.).
To assess the presence of MSI in a given tumor the NCI workshop recommended to analyze a panel of five microsatellites including two mono- (BAT25, BAT26) and three di-nucleotide markers (D2S123, D5S346, and D17S250); depending on the mutational pattern, a secondary panel including additional mononucleotide (e.g., BAT40) and/or complex microsatellites (e.g., MYCL) should be tested (8). Alternatively, a panel of five quasimonomorphic mononucleotide repeats can be used which display even better sensitivity and which may obviate the need for normal tissue for comparison (9). Depending on the number of microsatellite markers displaying novel alleles, MSI can subsequently be rated as MSI-high (MSI-H, >2 out of 5 markers), MSI-low (MSI-L, 1 out of 5), or microsatellite stable (MSS, 0 out of 5).
The molecular dissection of CRCs into MSI-H and MSS tumors allowed to delineate two major, virtually exclusive pathways of genetic instability: chromosomal instability, which results in aneuploidy and is present in about 85% of CRC, and MSI. Whether MSI-L CRC constitute a pathogenetic class of their own is still a matter of debate: with regard to clinical, biological, and morphological parameters they closely resemble those of MSS CRC. Since the analytical sensitivity to detect MSI-L is dependent on the number of microsatellite markers analyzed, result interpretation, and comparison between different studies investigating MSI-L and MSS tumors are heavily compromised; furthermore, extensive genotyping efforts have failed to demonstrate fundamental differences (10, 11). Given these unresolved issues, a molecular subdivision into MSI-L and MSS CRC does currently not seem appropriate.
Role of MSI in Lynch Syndrome
The discovery of MSI in the majority of LS-related CRC led by analogy to a similar biochemical defect in yeast to the identification of the underlying MMR germ line mutations in MLH1, MSH2, MSH6, and PMS2. Heterozygous carriers of a MMR gene alteration are at a greatly increased lifetime risk to develop cancers of the LS tumor spectrum, mainly CRC (25–70%) and endometrial cancer (30–70%) (12). Knowledge of the underlying germ line mutation not only allows life-saving intensive-cancer surveillance but also gives asymptomatic family members the opportunity to clarify their carrier status; due to autosomal dominant inheritance offspring of a LS patient has an a priori chance of 50% of having inherited the pathogenic MMR mutation.
In contrast to their sporadic MSS CRC LS patients’ tumors typically have a comparatively favorable prognosis and absence of distant organ metastasis; together with the observation that LS cancers are accompanied by an intense immune response with dense lymphocytic infiltrates points to a possible protective effect by the immune system (13).
The revised Bethesda guidelines are probably the most commonly applied criteria to identify individuals with LS, yet many physicians dealing with familial CRC have the impression that LS per se remains underdiagnosed (14). An expert group therefore recently suggested to screen all individuals with CRC or endometrial cancer below age 70 by immunohistochemistry or MSI, both of which having similar clinical sensitivity and specificity. In order to discriminate between a hereditary (about 2–3% of all CRC) or a sporadic event (about 15%), tumors with immunohistochemical loss of MLH1 should then be further investigated for MLH1 promoter hypermethylation and targeted BRAF (V600E hotspot mutation) testing to decide on further (germ line) genetic testing. With high-throughput sequencing techniques entering routine genetic testing it is likely that these diagnostic screening algorithms will considerably change in the foreseeable future (15).
Role of MSI in Sporadic Cancer
Since their initial description it became evident that the 15% of sporadic MSI-H CRCs exhibit a distinct clinico-pathological profile, which they largely share with LS-related CRC and which distinguishes them from their MSS counterparts (Figure 1). Already in the seminal work by Thibodeau et al. (2) MSI-H CRC were predominantly located in the proximal colon and associated with increased patient survival and prognosis (2). Most of them were later found to exhibit loss of MLH1 protein expression which could be attributed to epigenetic silencing of the respective promoter, later also referred to as “CpG island methylator phenotype” since it often occurs in the context of global hypermethylation (16, 17). Regarding their molecular-histopathological profile MSI-H CRC display a diploid state, tend to be poorly differentiated, mucinous, and show prominent lymphocytic infiltration (15).
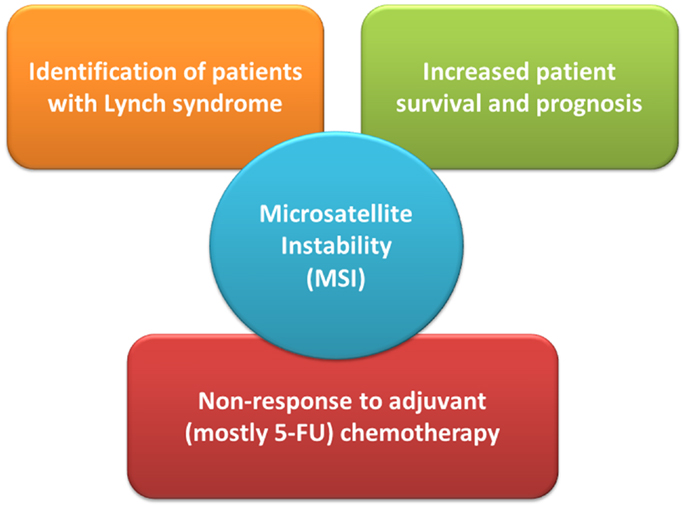
Figure 1. The role of microsatellite instability (MSI) status in clinical management of colorectal cancer patients. 5-FU denotes 5-fluoro-uracil.
A recent meta-analysis by Guastadisegni et al. who pooled data from 31 studies reporting survival in 12782 patients confirmed the initial observations between MSI-H status and a more favorable prognosis: patients with stages I-IV MSI-H CRC appeared to have a statistically significantly better outcome in terms of overall survival, disease-specific as well as disease-free survival (18). Moreover, results from a recent Norwegian study have shown that MSI status had an independent positive prognostic impact on stage II CRC patients after complete resection (19). How these findings and the inclusion of additional molecular markers may eventually impact on routine clinical and surgical practice, however, remains to be seen.
Guastadisegni et al. also investigated the effect of standard, 5-fluoro-uracil (5-FU) based chemotherapy. In the context of MSI and an underlying (hereditary or sporadic) defect in MMR the use of 5-FU in MSI-H CRC merits particular attention: as demonstrated in numerous in vitro studies, inactivation of the MMR system can result in resistance, or rather tolerance (i.e., failure to induce cell-cycle arrest), to 5-FU treatment (4, 20). In line with these observations the meta-analysis found a clear significant beneficial effect of 5-FU therapy in patients with MSS CRC only. Further studies by Des Guetz et al. (meta-analysis) and Sargent et al. provided comparable findings in that relapse-free survival was similar for treated and untreated MSI-H patients demonstrating MSI-H status as a predictive factor of non-response to adjuvant, mostly 5-FU-based chemotherapy (21, 22). The high variability in treatment response observed in the MSI-H CRC group may actually reflect the (in) efficiency of other DNA repair systems, like base-excision repair, to repair/tolerate chemotherapy-induced DNA lesions. Overall, current data advocate CRC patient stratification by determining the tumor’s MMR status, either by testing for MSI or immunohistochemical analysis of MMR proteins, in order to decide on adjuvant chemotherapy on an individual basis.
Concluding Remarks
As exemplified by the rapidly growing list of cancer genomes analyzed by means of ever more complete as well as cost-effective high-throughput (“next generation”) sequencing methods, a novel era of truly personalized medicine seems to be at hand (23). It is the hope of the author that, despite the inherent difficulties and (data) challenges when trying to get a deeper understanding of biological systems as complex as cancer, the novel “omics” and systems biology approaches will not only allow more comprehensive insights into tumor biology but eventually result in individual patient (tumor)-tailored treatment and, last not least, enable true cancer prevention.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author gratefully acknowledges the financial support of his research projects by the Krebsliga beider Basel, the Krebsliga Zentralschweiz, and Oncosuisse.
References
1. Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, et al. Clues to the pathogenesis of familial colorectal cancer. Science (1993) 260:812–6. doi: 10.1126/science.8484121
2. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science (1993) 260:816–9. doi:10.1126/science.8484122
3. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature (1993) 363:558–61. doi:10.1038/363558a0
4. Peña-Diaz J, Jiricny J. Mammalian mismatch repair: error-free or error-prone? Trends Biochem Sci (2012) 37:1–9. doi:10.1016/j.tibs.2012.03.001
5. Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet (2004) 5:435–45. doi:10.1038/nrg1348
6. Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell (1993) 75:1227–36. doi:10.1016/0092-8674(93)90331-J
7. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res (1998) 58:5248–57.
8. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst (2004) 96:261–8. doi:10.1093/jnci/djh281
9. Buhard O, Suraweera N, Lectard A, Duval A, Hamelin R. Quasimonomorphic mononucleotide repeats for high-level microsatellite instability analysis. Dis Markers (2004) 20:251–7. doi:10.1155/2004/159347
10. Laiho P, Launonen V, Lahermo P, Esteller M, Guo M, Herman JG, et al. Low-level microsatellite instability in most colorectal carcinomas. Cancer Res (2002) 62:1166–70.
11. Wright CM, Dent OF, Newland RC, Barker M, Chapuis PH, Bokey EL, et al. Low level microsatellite instability may be associated with reduced cancer specific survival in sporadic stage C colorectal carcinoma. Gut (2005) 54:103–8. doi:10.1136/gut.2003.034579
12. Vasen HFA, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut (2013) 62:812–23. doi:10.1136/gutjnl-2012-304356
13. von Knebel Doeberitz M, Kloor M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam Cancer (2013) 12:307–12. doi:10.1007/s10689-013-9662-7
14. Trano G, Wasmuth HH, Sjursen W, Hofsli E, Vatten LJ. Awareness of heredity in colorectal cancer patients is insufficient among clinicians: a Norwegian population-based study. Colorectal Dis (2009) 11:456–61. doi:10.1111/j.1463-1318.2009.01830.x
15. Geiersbach KB, Samowitz WS. Microsatellite instability and colorectal cancer. Arch Pathol Lab Med (2011) 135:1269–77. doi:10.5858/arpa.2011-0035-RA
16. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res (1997) 57:808–11.
17. Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, et al. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology (2005) 129:837–45. doi:10.1053/j.gastro.2005.06.020
18. Guastadisegni C, Colafranceschi M, Ottini L, Dogliotti E. Microsatellite instability as a marker of prognosis and response to therapy: a meta-analysis of colorectal cancer survival data. Eur J Cancer (2010) 46:2788–98. doi:10.1016/j.ejca.2010.05.009
19. Merok MA, Ahlquist T, Royrvik EC, Tufteland KF, Hektoen M, Sjo OH, et al. Microsatellite instability has a positive prognostic impact on stage II colorectal cancer after complete resection: results from a large, consecutive Norwegian series. Ann Oncol (2013) 24:1274–82. doi:10.1093/annonc/mds614
20. Jover R, Fau Zapater P, Zapater P, Fau Castells A, Castells A, Fau Llor X, et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur J Cancer (2009) 2(45):365–73. doi:10.1016/j.ejca.2008.07.016
21. Des Guetz G, Uzzan B, Nicolas P, Schischmanoff O, Perret GY, Morere JF. Microsatellite instability does not predict the efficacy of chemotherapy in metastatic colorectal cancer. A systematic review and meta-analysis. Anticancer Res (2009) 29:1615–20.
22. Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol (2010) 28:3219–26. doi:10.1200/JCO.2009.27.1825
Keywords: microsatellite instability, mismatch repair, Lynch syndrome, hereditary non-polyposis colon cancer, colorectal cancer, therapy, prognosis
Citation: Heinimann K (2013) Toward a molecular classification of colorectal cancer: the role of microsatellite instability status. Front. Oncol. 3:272. doi: 10.3389/fonc.2013.00272
Received: 10 September 2013; Paper pending published: 04 October 2013;
Accepted: 18 October 2013; Published online: 31 October 2013.
Edited by:
Alessandro Lugli, University of Bern, SwitzerlandReviewed by:
Gieri Cathomas, Kantonales Institut für Pathologie Liestal, SwitzerlandHiroaki Taniguchi, The University of Tokyo, Japan
Copyright: © 2013 Heinimann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karl Heinimann, Department of Biomedicine, Research Group Human Genomics, Mattenstrasse 28, 4058 Basel, Switzerland e-mail:a2FybC5oZWluaW1hbm5AdXNiLmNo