 Sander B. Frank
Sander B. Frank Cindy K. Miranti
Cindy K. Miranti- 1Laboratory of Integrin Signaling and Tumorigenesis, Van Andel Research Institute, Grand Rapids, MI, USA
- 2Genetics Graduate Program, Michigan State University, East Lansing, MI, USA
One of the foremost problems in the prostate cancer (PCa) field is the inability to distinguish aggressive from indolent disease, which leads to difficult prognoses and thousands of unnecessary surgeries. This limitation stems from the fact that the mechanisms of tumorigenesis in the prostate are poorly understood. Some genetic alterations are commonly reported in prostate tumors, including upregulation of Myc, fusion of Ets genes to androgen-regulated promoters, and loss of Pten. However, the specific roles of these aberrations in tumor initiation and progression are poorly understood. Likewise, the cell of origin for PCa remains controversial and may be linked to the aggressive potential of the tumor. One important clue is that prostate tumors co-express basal and luminal protein markers that are restricted to their distinct cell types in normal tissue. Prostate epithelium contains layer-specific stem cells as well as rare bipotent cells, which can differentiate into basal or luminal cells. We hypothesize that the primary oncogenic cell of origin is a transient-differentiating bipotent cell. Such a cell must maintain tight temporal and spatial control of differentiation pathways, thus increasing its susceptibility for oncogenic disruption. In support of this hypothesis, many of the pathways known to be involved in prostate differentiation can be linked to genes commonly altered in PCa. In this article, we review what is known about important differentiation pathways (Myc, p38MAPK, Notch, PI3K/Pten) in the prostate and how their misregulation could lead to oncogenesis. Better understanding of normal differentiation will offer new insights into tumor initiation and may help explain the functional significance of common genetic alterations seen in PCa. Additionally, this understanding could lead to new methods for classifying prostate tumors based on their differentiation status and may aid in identifying more aggressive tumors.
Introduction
Prostate cancer (PCa) is the most common non-skin cancer and second leading cause of cancer deaths in American men (1). Treatment for locally confined PCa is highly effective and typically involves radiation therapy or removal of the prostate gland. Such treatment, however, is not without considerable financial cost and a potentially negative impact on quality of life for the patient. Gleason grade, as determined by biopsy, is a moderate predictor of tumor aggressiveness, but even small, low grade tumors can become aggressive and metastasize (2). Researchers are currently working to understand and create better criteria to identify which primary tumors will be indolent and which will be aggressive (3). The ability to clearly identify which tumors are most likely to metastasize would potentially save thousands of men from unnecessary surgery when they are more likely to ultimately die of other causes (4). At the same time, the ability to accurately and reliably identify aggressive tumors would aid physicians and patients in deciding how to administer treatment. When detected too late or left untreated, PCa becomes metastatic and the primary therapeutic option becomes androgen deprivation. Despite initial effectiveness, androgen deprivation therapy invariably leads to the emergence of castration resistant disease which is incurable and accounts for the vast majority of the nearly 30,000 PCa deaths each year (1).
Whether contemplating cancer or a mechanical watch, it is impossible to fix something without understanding how it broke. Through years of work, PCa researchers have identified what is broken but are still working to understand how it broke. Although we know which oncogenes and tumor suppressors are most frequently altered in PCa, we do not understand how they drive tumor initiation (i.e., oncogenesis) (5). Without a thorough understanding of oncogenesis researchers will continue to struggle for a comprehensive understanding of PCa. The PCa field is currently stuck with a host of critical but unanswered questions: why are these particular genes mutated so frequently in PCa? How do the alterations drive oncogenesis? Does the timing and order of these mutations dictate tumor aggressiveness?
There are a few oncogenes and tumor suppressors that are mutated across a wide range of cancers, such as Ras and p53 (6, 7). However, most cancers contain a distinctive set of “driver mutations,” thus revealing multiple oncogenic routes that are highly dependent on not only the specific pathways disrupted but also the cell of origin (8). Many of the genes commonly altered in PCa are also involved in normal epithelial differentiation. With these observations in hand, we hypothesize that prostate oncogenesis arises as a defect in epithelial differentiation. Unfortunately, research is lacking in understanding the detailed signaling mechanisms of normal prostate differentiation. In this review we will describe key genes that are altered in PCa and what is known about their roles in epithelial differentiation. Additionally, we will propose some hypothetical models for how oncogenic alterations of these important pathways could potentially drive a normal trans-differentiating prostate epithelial cell to become tumorigenic.
Prostate Background
Prostate adenocarcinoma arises from the epithelia of the gland. Prostate epithelium is organized in a bilayer of basal and luminal cells along with a few rare embedded neuroendocrine cells (Figure 1). The epithelium is surrounded by a laminin (LM5, LM10) and collagen (COLIV, COLVII) matrix and fibromuscular stromal cells which transmit signals to regulate the epithelium (9). The prostate epithelium contains layer-specific markers, with the basal layer characterized by p63, basal keratins (K5, K14), and integrins (α6β4, α3β1) among others. The luminal layer contains markers such as TMPRSS2, luminal keratins (K8, K18), and androgen receptor (AR). Prostate tumors are characterized by a loss of basal cells and reduced matrix diversity (i.e., loss of LM5 and COLIV) (Figure 1). Moreover, tumor cells generally express a luminal phenotype driven by AR. However, tumors also express basal integrins, especially α6β1 which is an abnormal pairing that drives PCa growth and survival (10, 11). Similarly, tumor cells often co-express basal and luminal keratins, such as K5 and K8 (12–14). Other basal markers reportedly expressed in tumor cells include Bcl2, EGFR, and Met (15–20). The co-expression of a subset of luminal and basal markers supports the hypothesis that prostate tumors arise from the disruption of differentiation pathways which normally restrict basal and luminal marker expression to their respective cell types. However, the cell of origin, i.e., the cell that is the oncogenic target that gives rise to the tumor, has not been clearly identified in PCa.
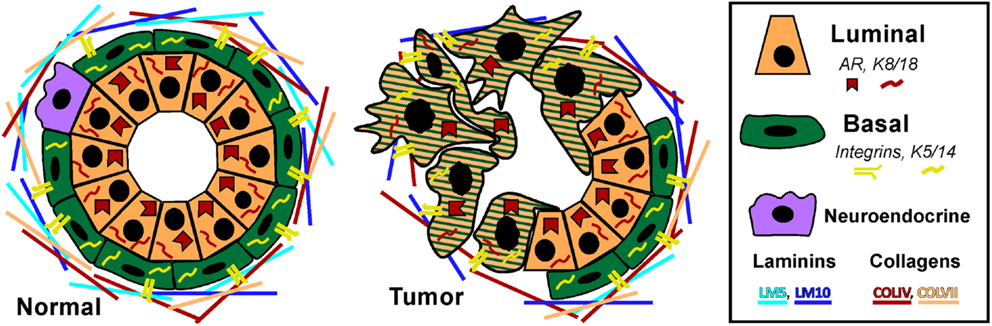
Figure 1. Prostate epithelial gland structure. The normal prostate epithelium is composed of a bilayer of basal and luminal cells and a few rare neuroendocrine cells. The epithelium is separated from the underlying stroma by a basement membrane containing laminins (LM5, LM10) and collagens (COLIV, COLVII). Basal cells express integrins that specifically interact with the basement membrane, namely α6β4, α3β1, and α2β1 as well as basal keratins K5 and K14. Luminal cells do not express integrins, but do express AR and keratins K8 and K18. A prominent characteristic of prostate tumors is the loss of basal cells and a loss of LM5 and COLIV. Correspondingly, the integrins that interact with these matrices, α3β1 and α6β4 are lost via loss of the α3 and β4 subunits, leaving integrin α6β1 which prefers the LM10 matrix. Similarly, tumor cells often co-express basal and luminal keratins, such as K5 and K8.
The struggle to define a clear cell of origin is complicated by the fact that the mechanism of prostate epithelial differentiation is not well understood. In the adult prostate, luminal cells are regularly shed and replaced by cells from the basal layer through differentiation (21). A simplistic view of this observation is that a basal progenitor or stem cell gives rise to the both basal and luminal populations through a transient-differentiation or amplification process (12, 22–24). However, findings from mouse models paint a more complicated picture of basal, luminal, and bipotent progenitors. Ousset et al. utilized cell lineage tracing to clearly demonstrate the existence of layer-specific epithelial progenitor cells in the developing mouse prostate (25). Wang et al. demonstrated that a luminal progenitor, marked by expression of Nkx3.1, resists luminal regression induced by castration and repopulates the majority of the mouse prostate during regeneration with androgen (26). On the other hand, using tissue recombination and renal capsule implants, Leong et al. showed that a single prostate stem cell is able to produce both epithelial layers (27). The Witte group also identified basal stem-like cells in the mouse prostate that produce both basal and luminal cells (28–31). Other researchers also identified a small population of bipotent progenitor cells that give rise to both basal and luminal cells (25, 26). These rare bipotent cells are marked by their co-expression of basal and luminal keratins (K5/K8) and are also found in the developing human prostate (25, 32). Thus, the mouse studies support the idea there are at least three different prostate epithelial progenitor populations, but which ones initiate PCa still remains unresolved.
Several studies demonstrate either basal or luminal progenitors can be the initiating cancer cell. The Witte group demonstrated that oncogenic disruption in the basal cell population drives tumor formation in mice (33–35). On the other hand, two groups reported that both basal and luminal epithelial cells can give rise to tumors upon knockout of PTEN (14, 36). Thus, mouse studies suggest distinct stem cell populations may be responsible for tumor initiation and seemingly disfavor the transient amplification theory. However, introduction of genetic mutations early in development and puberty in mice does not reflect the normal situation in humans were oncogenesis occurs in a fully differentiated gland. Moreover, the signals and cell types that regulate gland maintenance vs. development may be different. Transgenic mouse models rely on Cre recombination via commonly used “layer-specific” promoters, such as K5 or Nkx3.1. Thus, these studies still leave open the possibility that it is not a pure basal or luminal cell that becomes oncogenic, but rather a bipotent or transient-differentiating cell expressing multiple layer markers. Better understanding of prostate differentiation will be required to validate the transient-differentiating hypothesis of oncogenesis.
While studies in the mouse are highly informative, translation of these findings to understanding the human organ is complicated due to a lack of models for studying human oncogenesis. The mouse model is useful for genetic manipulations, but it is not without limitations (37, 38). For example, mice are not prone to develop spontaneous PCa like humans. Secondly, although the mouse and human prostate have similar cell types, the structure is different; the mouse prostate is lobular while the human is compact and consists of zones (39, 40). Additionally, there are far fewer basal cells in the mouse prostate and some luminal cells directly contact the basement membrane, unlike in humans where there is a continuous layer of basal cells. Based on these important differences, there is reason to consider that signaling mechanisms for differentiation in human and mouse epithelial cells may be different. As an alternative to transgenic mouse models, some researchers are using human prostate cells and xenografts in mice to study prostate development and differentiation. The Cunha group found that human basal cells can be induced to form a basal and luminal bilayer when combined with rat urogenital sinus mesenchyme and implanted in the mouse renal capsule (41). The Witte group developed a similar method to isolate and genetically modify epithelial progenitor cells from human prostates (42). The isolated progenitor cells were infected with virus to allow manipulation of desired oncogenes/tumor suppressors, and then the cells were implanted into mice along with stroma. Using this approach, they found that the induction of Akt and Erg in human basal progenitors is sufficient to induce prostate intraepithelial neoplasia (PIN), a PCa precursor lesion, when xenografted into mice (33). Other groups are inducing the differentiation of primary basal cells in vitro, including our group which has developed a reliable in vitro differentiation model that recapitulates many aspects seen in vivo (17, 43–45). These reports demonstrate that human basal cells can be induced to differentiate into luminal cells in vitro, thus providing a model to study epithelial differentiation in a controlled setting using human cells. The ability to manipulate cells in vitro during differentiation and then implant them into mice provides a useful approach to study how manipulation of trans-differentiating human prostate epithelial cells can become tumorigenic.
Based on the slowly building knowledge of normal prostate differentiation, as well as findings from other epithelial tissues, it is becoming apparent that many of the pathways involved in normal epithelial differentiation are misregulated in PCa. In this review, we will describe some of these key pathways that are involved in both differentiation and cancer, with the goal of illuminating how prostate oncogenesis in humans may arise from a disruption of normal differentiation. Furthermore, aggressive tumors are pathologically characterized by a less differentiated phenotype, and the aggressiveness of the tumor may be tied to its cell of origin (14, 36). Better understanding of prostate differentiation pathways will help us understand how the normal cellular process goes awry in cancer. This understanding may one day lead to classification of prostate tumors based on the state of their altered differentiation pathways.
Common Genetic Alterations in Prostate Cancer
While there are still no widely accepted subcategories of PCa, there are well established genetic alterations associated with the disease. Fundamentally, prostate tumors rely on AR signaling. While AR mutations are rare in primary tumors, castration resistant tumors utilize a variety of genetic alterations to upregulate AR signaling (46–48). Beyond the AR alterations in advanced tumors, three of the most common genetic alterations in PCa are: overexpression of Myc, loss of the tumor suppressor Pten, and fusion of Ets genes with upstream AR regulated promoter sequences (e.g., TMPRSS2-Erg) (49, 50).
The Myc gene is commonly amplified in PCa (Table 1) and protein levels correlate with poor prognosis (51). Myc is a well-studied oncogene that drives the expression of thousands of targets, including genes required for cell growth and cell cycle progression. Myc overexpression in the mouse prostate is sufficient to drive adenocarcinoma but not metastasis (52). The importance of Myc in PCa is well established, though not entirely understood, and will be discussed in further detail in the next section.
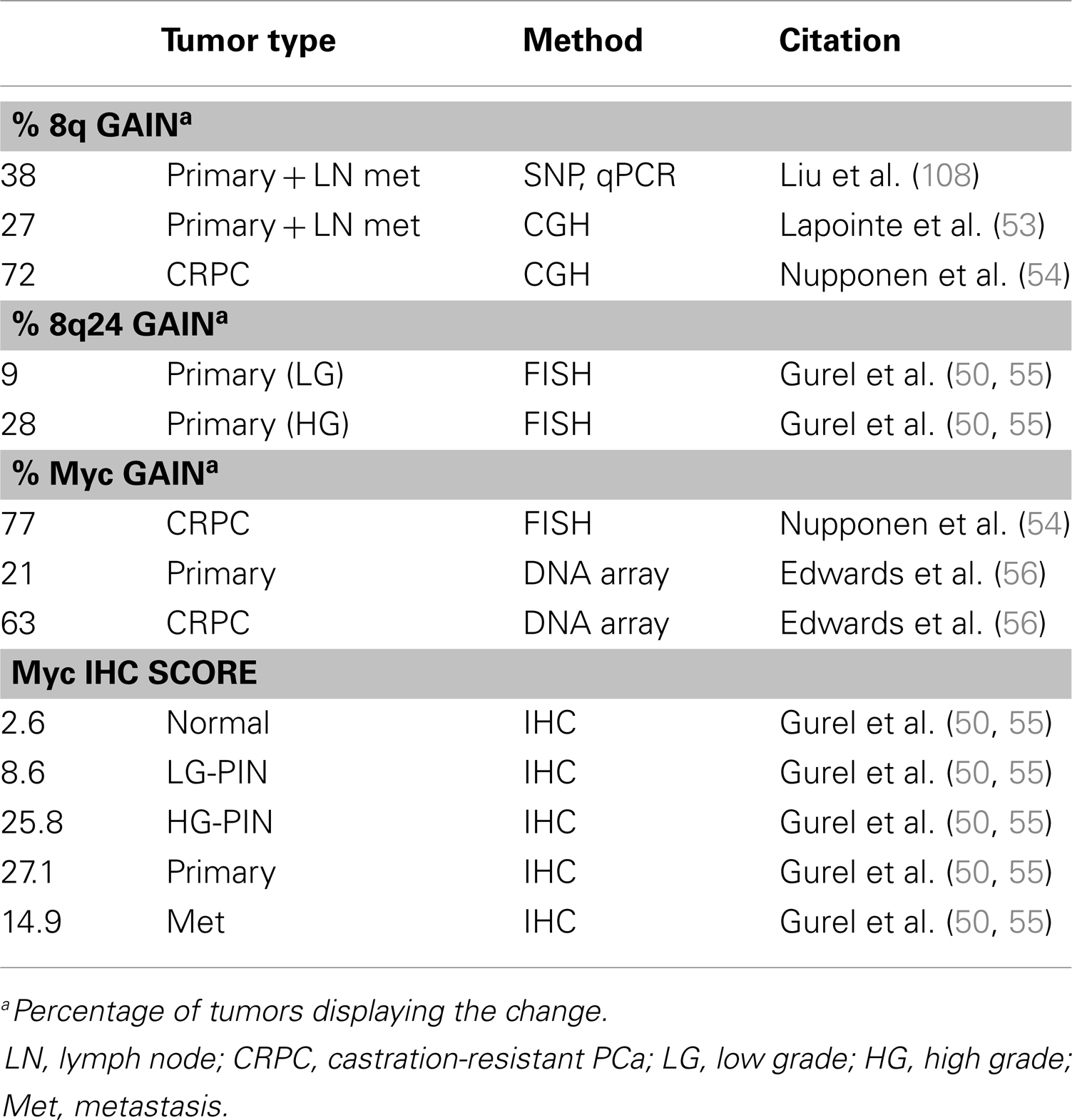
Table 1. Myc overexpression in PCa.
Another prevalent aberration in PCa is loss of the tumor suppressor Pten (Table 2), a negative regulator of the PI3K pathway. At least one copy of the Pten locus is lost in up to 65% of prostate tumors and complete loss of Pten protein is seen in ∼60% of late stage tumors (57–62) Loss of one copy of Pten greatly increases PCa progression in the TRAMP mouse model and Pten dosage has a marked impact on tumor latency and progression (63, 64). Moreover, complete loss of PTEN in the mouse prostate is sufficient to drive adenocarcinoma (65, 66). The role of Pten in PCa and differentiation will be discussed in further detail in a later section.
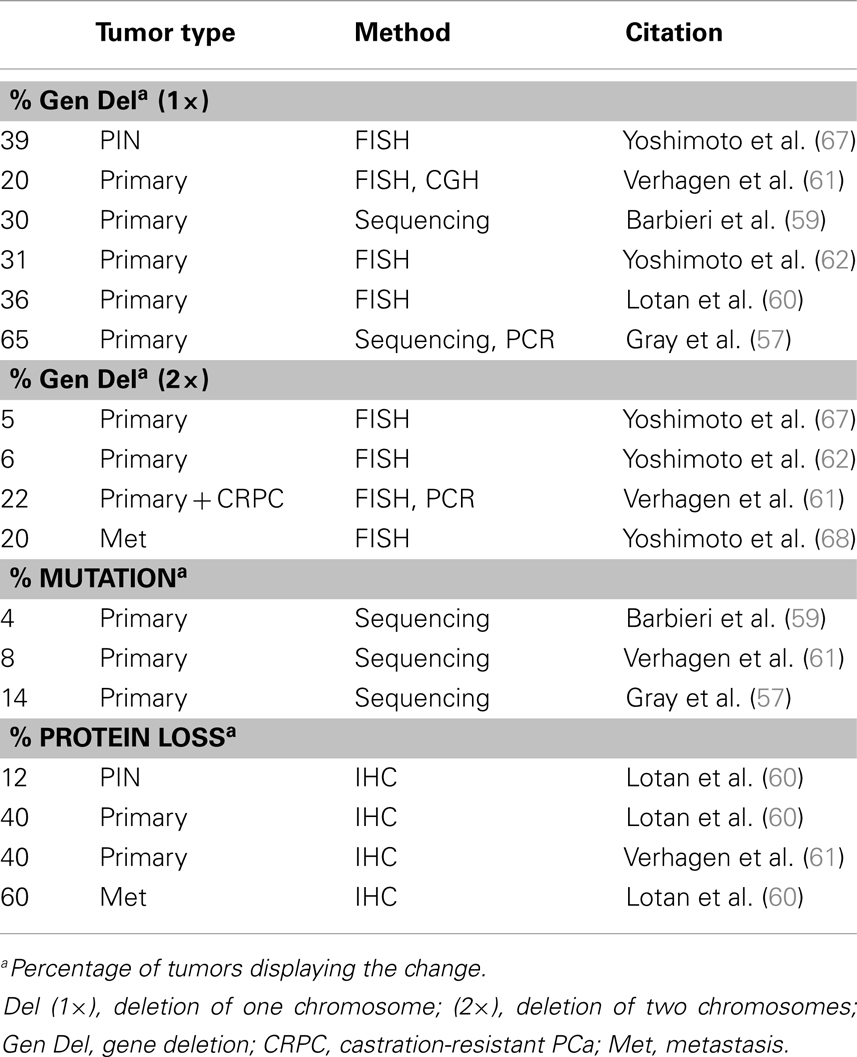
Table 2. Pten loss in PCa.
Activation of the Ets pathway is also a common occurrence in PCa (Table 3), most frequently through the fusion of the oncogene Erg downstream of the androgen-regulated promoter of TMPRSS2 (69, 70). Specific genetic rearrangements that drive tumor progression are relatively rare in solid cancers, but the TMPRSS2-ERG fusion is a notable exception and is observed in about 50% of prostate tumors (53, 62, 71–74). The identification of additional fusions of AR-driven promoters to other Ets members (as well as other targets) strongly suggests this type of rearrangement is a major driver of PCa (75–80). This has important implications about the role of AR in PCa development and may explain the dependency on AR for tumorigenesis. In the normal secretory epithelium, AR is primarily required for maintaining secretory functions and is not intrinsically required for survival or proliferation of the secretory epithelium (81, 82). In fact, AR is inhibitory to cell proliferation in normal cells (83, 84). But an opposite response is triggered in tumor cells, where both proliferation and survival depends on AR. The trigger is unknown, but prostate-specific oncogenes driven by AR are likely to be part of the answer.
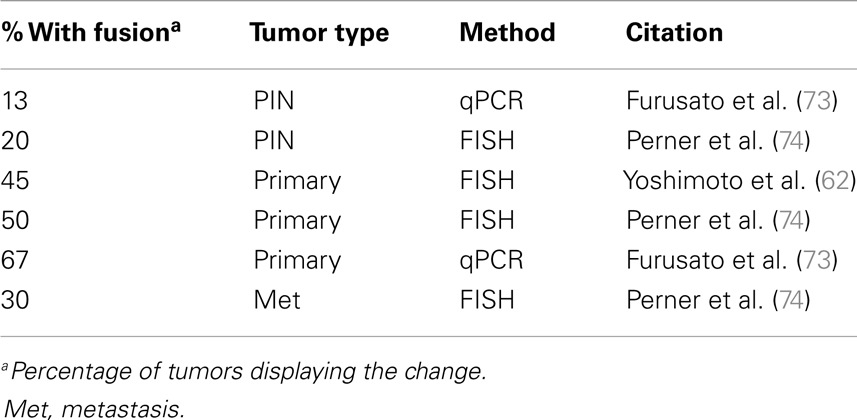
Table 3. TMPRSS2/Erg fusions in PCa.
Thus, the contribution of AR-driven Ets activation and the mechanisms that drive tumor initiation and tumor progression are in need of much further investigation. The Ets family of transcription factors can potentially regulate a wide range of cellular processes, including development, differentiation, invasion, and proliferation (85). Sun et al. reported that the TMPRSS2-Erg fusion activates Myc and prevents terminal prostate epithelial differentiation in the VCaP PCa line (77). Additionally, Yu et al. reported that Erg and AR-binding sites have considerable overlap and Erg functions in part by disrupting AR binding to its target genes in VCaP cells (76). Moreover, Yu et al. found that Erg activates EZH2, which is part of the polycomb repression complex and in turn down regulates an AR-driven differentiation program. The authors propose that TMPRSS2-ERG is likely to be an early mutational event that drives selection of cells with hyper activated or mutated AR to overcome the antagonistic effects of Erg activation on AR (76). Conversely, Chen et al. using transgenic mice and reported that Erg activation aids AR signaling by increasing AR binding to target genes, though only in the context of Pten loss (86). The Chen et al. group suggest that an explanation for the difference in their findings from those of Yu et al. is that the latter did their studies in VCaP cells, which retain Pten expression. If the Chen et al. finding is to be believed, then Ets activation may be a later event that must follow Pten loss. There are multiple mouse models of Erg overexpression, but only some of them produce PIN and none develop adenocarcinoma (75, 87–89). In the most aggressive model, overexpression of the N-terminal truncated Erg fusion product in luminal cells (via a modified probasin promoter) produces PIN in about 40% of mice but still fails to drive adenocarcinoma (75). The combination of Erg overexpression with single-copy loss of Pten drives progression to adenocarcinoma but does not result in metastasis (87, 88). These findings from mouse models further support the idea that Ets activation is a later event in PCa progression and must follow Pten loss. More research on Ets and its specific function in PCa tumorigenesis and/or progression are required to fully understand the significance of this common mutation.
As will be discussed in this review, pathways that are frequently altered in PCa (Myc, Pten, Erg) can be tied to normal prostate differentiation. Likewise, key epithelial differentiation pathways (p38MAPK, Notch) are also misregulated in human and mouse models of PCa. We propose the hypothesis that the ties between oncogenesis and differentiation are evidence that PCa arises from a transiently differentiating prostate epithelial cell. Moreover, while these differentiation pathways may not be direct drivers of PCa, we believe they are critical for oncogenesis via potential misregulation of Myc and PTEN during aberrant differentiation. In this review, we will discuss what is known about Myc, p38MAPK, Notch, and Pten and their roles in both cancer and differentiation with the goal of providing new insight into understanding PCa oncogenesis.
Myc
Myc Background
The general importance of Myc in PCa is well established, but it is less clear precisely how Myc drives tumor initiation and progression (51). In addition to its oncogenic role, Myc is also crucial for promoting epithelial differentiation (90, 91). Knowledge about normal prostate differentiation is limited, and much of it is based on mouse studies. More detailed investigations into the role of Myc in prostate differentiation may help us understand how its misregulation leads to PCa.
There are three genes in the Myc family: c-Myc, N-Myc, and L-Myc. c-Myc (Myc) is the best studied and most relevant in PCa. Myc is a basic helix-loop-helix transcription factor that typically functions as a heterodimer with a cofactor from the Max or Miz families (92). Transcriptional regulation by Myc is mediated through recruitment or activation of basal transcription machinery, promoting RNA Polymerase II elongation, or through recruitment of chromatin modifying enzymes (93, 94). The Myc/Max heterodimer is usually a transcriptional activator complex that competes with Mad/Max dimers for binding at E-box sites, the classic regulatory element recognized by Myc complexes. Myc also represses genes by binding with Sp1 or Miz1, which together repress transcription by blocking p300 (95, 96). Alternately, Myc can repress targets post-transcriptionally via activation of miRNAs (97, 98).
Myc is downstream of many pathways and is tightly regulated at the mRNA and protein levels (95, 99). Myc mRNA and protein have short half-lives and higher activity is usually associated with lower stability (100). Myc potentially regulates thousands of genes, with one estimate predicting as much as 15% of the genome (101, 102). While there are thousands of potential targets for Myc, its functional role in cellular processes is highly dependent on the level of expression, duration of activation, and expression of its cofactors.
Myc in Prostate Cancer
The vast majority of prostate tumors overexpress Myc (Table 1), which correlates with poor prognosis (51, 103, 104). While Myc mRNA is elevated in as many as 80% of prostate tumors, there is less certainty about Myc protein levels (51). The De Marzo group published a study showing that Myc protein expression is very low in normal prostate epithelium but higher and more nuclear localized in PIN and prostate tumors (55). The most common mechanism of Myc overexpression is through amplification of the gene locus, usually through gain of 8q. The narrower Myc region 8q24 is more selectively amplified in late metastatic tumors (50, 53, 105–108). However, early prostate tumors also overexpress Myc but rarely have Myc amplifications, suggesting other mechanisms driving Myc overexpression which are less well understood (51). Myc amplification is specifically observed in castration resistant tumors (54, 56). Bernard et al. demonstrated that Myc overexpression in the hormone-sensitive LNCaP line confers resistance to androgen deprivation or AR knockdown (109). Conversely, AR knockdown decreases Myc expression, indicating Myc is downstream of AR. In another study, the ability of AR to upregulate Myc was ligand independent (110). Alternatively, Myc reportedly upregulates AR, suggesting there may be feedback mechanisms between the two proteins (111, 112).
Another potential mechanism for Myc upregulation is via β-catenin, the downstream target of Wnt signaling. Constitutive β-catenin is sufficient to upregulate Myc and induce prostate tumor formation in a mouse model (113). Furthermore, the APC gene (an antagonist of β-catenin) is often silenced by hypermethylation in at least 50% of human prostate tumors (114, 115). However, the specific role of APC and β-catenin in human PCa is still unclear and it is unknown if the potential oncogenic activity is due to Myc upregulation. As will be discussed later, some groups report increased Notch signaling in prostate tumors, which may also drive transcription of Myc (116, 117).
Myc overexpression in the mouse prostate with a weak promoter drives low grade PIN but not adenocarcinoma (118). Using stronger variants of the probasin promoter to regulate Myc overexpression in luminal cells, researchers were able to drive progression to adenocarcinoma though not metastasis (52). In this model, when Myc is driven by the endogenous probasin promoter (Lo-Myc) mice take much longer to develop tumors than those with a stronger promoter (Hi-Myc) (52). Finally, mice with knockout of Mxi1 (a Myc antagonist) show prostate dysplasia but do not develop adenocarcinoma (119). All together, these models demonstrate that Myc can drive PCa in the mouse, and the level of Myc expression is related to the aggressiveness of carcinoma that develops.
Myc has many potential oncogenic and tumor-promoting targets. One group of genes known to be regulated by Myc is cell cycle regulators, such as E2F members, cyclins, and cyclin-dependent kinases (120). Additionally, Myc can regulate cell growth by upregulating tRNAs and rRNAs (120). Other important targets of Myc include stem cell genes, such as hTert and EZH2 (51, 76, 120). Myc is one of the four original genes whose overexpression was initially used to create pluripotent stem cells, along with Oct4, Sox2, and Klf4 (121). Although overexpression of Myc was later found not to be necessary for stem cell induction, Myc activity is required for embryonic stem cell self-renewal (122–125). Another key point regarding Myc is that the level and timing of its expression is critical for deciding what function the protein will play, for example deciding whether Myc drives proliferation or stem cell maintenance (120). However, which of these targets is critical for PCa development and progression is not clear. In summary, Myc amplification is common in late metastatic tumors and can act as a driver in mouse models but specific mechanisms of Myc regulation and downstream targets are poorly understood.
Myc in Prostate Differentiation
Beyond its multifaceted role in cancer, Myc is also important for differentiation. A shift from Myc/Max to Mad/Max binding is associated with terminal differentiation (126, 127). Transient expression of Myc aids induced pluripotent stem cell transformation while sustained expression stimulates down regulation of integrin α6 and drives differentiation of embryonic stem cells (128). In keratinocyte differentiation, Myc protein is expressed in the basal layer and decreases during differentiation of the suprabasal layers (129, 130). On the other hand, knockdown of Myc prevents in vitro keratinocyte proliferation while transient overexpression induces premature terminal differentiation (131–133). Overall a short, high spike in Myc appears to be required for proliferation, while a more moderate and extended increase in Myc is characteristic for differentiation (91). The role of Myc specifically in prostate differentiation has not been well investigated. Preliminary data from our lab indicate Myc follows a pattern of moderate increase over a brief period that is required during prostate epithelial differentiation in our in vitro model using primary human basal cells (unpublished data).
One mechanism by which Myc triggers differentiation is through its control of a cell adhesion program. About 40% of the genes downregulated upon Myc activation in mouse skin are involved in cell adhesion and cytoskeleton, including integrins α6, β1, and β4 (132). Integrin loss as the cells from the basal layer rise into the upper layers triggers keratinocyte differentiation (134). This adhesion profile is largely regulated via Miz1, given that a Myc mutant unable to bind Miz1 loses the ability to suppress integrin α6 and β1 transcription (132).
Another mechanism by which Myc may regulate differentiation is via interactions with chromatin remodeling proteins (94, 135, 136). Chromatin modifications are often associated with cell programing, such as patterns for stem or terminally differentiated cells (136, 137). Pellakuru et al. published a study looking at Myc and H3K27me3 in prostate differentiation and cancer (136). H3K27me3 is a marker of polycomb activity, which induces heterochromatin and gene repression. The group reported that basal prostate cells have lower levels of H3K27me3 than luminal cells as determined by immunostaining with human tissue sections (136). Furthermore, using a tissue micro array they also found that cases of human PIN show decreased H3K27me3 compared to normal luminal cells. Levels of H3K27me3 are also decreased in prostate tumors from Hi-Myc mice. Additionally, they showed that Myc knockdown in the PC3 and LNCaP PCa lines leads to an increase in H3K27me3 (136). The authors were unable to provide a mechanism for how Myc controls H3K27me3, but they previously reported that Myc upregulates EZH2, which is the catalytic member of the polycomb complex and is often overexpressed in PCa (136, 138). However, EZH2 overexpression does not correlate with higher H3K27me3 levels in this study, which led the authors to propose that regulation of EZH2 activation may be a separate event (136). Seemingly answering the idea proposed by Pellakuru et al. a later study reported that EZH2, upon phosphorylation at Ser21, plays a non-polycomb role in castration resistant PCa acting as an AR coactivator (139).
Myc Conclusion
Myc amplifications are very common in advanced prostate tumors but Myc is also upregulated in early tumors through currently unknown mechanisms (51). Normal upregulation of Myc is required for proliferation and differentiation, and it is the level and timing of Myc expression that largely determines which of those decisions the cell will make (91). The upregulation of Myc seen in PCa may explain how tumors arise from a transient amplifying or differentiating prostate cell which requires a temporary upregulation of Myc expression. However, additional oncogenic events are required to prevent terminal differentiation and death due to the oncogenic stress of sustained Myc activation (95). As typically happens with other cancers, loss of p53 can relieve apoptotic stress; however, p53 loss is a rare event in primary prostate tumors and is usually only seen in a small subset of metastatic tumors (140). Thus, other yet to be identified mechanisms must be involved in PCa development. Abnormal Myc expression and its role in regulating a cell adhesion program may also help explain why prostate tumors show a large general loss in integrin and matrix expression, except for the retention of the tumor-promoting integrin α6β1 pairing (11, 132, 141). Additionally, prolonged Myc activation in a transient-differentiating cell may drive changes in chromatin structure, as evidenced by the fact that basal and intermediate prostate cells show low levels of heterochromatin markings compared to tumors (136). There is accumulating evidence to suggest that Myc contributes to an altered differentiation program in PCa, but more studies are required to work out particular mechanisms.
p38MAPK
p38MAPK Background
The three classic branches of mitogen-activated protein kinase (MAPK) signaling are p38MAPK (p38), Erk, and Jnk. MAPK signaling involves kinase cascades that control a wide range of functions in the cell including proliferation, stress response, and differentiation (142). The MAPK pathways can regulate gene expression through a variety of mechanisms at the RNA and protein levels. Erk signaling is most classically associated with growth factor signaling, while Jnk and p38 are commonly associated with stress responses to insults such as inflammation and radiation (143). p38 and Jnk have specific direct upstream kinases: MKK3/6 activate p38 and MKK4/7 activate Jnk (though MKK4 can potentially activate p38 in some cases) (142). However, p38 and Jnk share some common activating kinases further upstream, such as Ask1 and Tak1 (143). This upstream convergence makes identifying the contribution of each pathway difficult. In epithelial differentiation, upstream p38 activation is via the receptor tyrosine kinase FGFR2, specifically the FGFR2b (FGFR2IIIb) isoforms, by KGF (FGF7) or FGF10 ligands (43, 144, 145). MAPKs are also negatively regulated by a host of MAPK phosphatases, which are dual-specificity protein phosphatases that inactivate MAPK members (146). While the MAPK pathways share some overlapping features, p38 has a distinctive role in epithelial differentiation (142).
There are four isoforms of p38: MAPK14 (p38α), MAPK11 (p38β), MAPK12 (p38γ), and MAPK13 (p38δ). They share about 60% gene homology and have some compensatory ability, though they also have differential target preferences (142). p38α is the most prevalent and ubiquitously expressed, while p38β is moderately expressed in many tissues and p38γ/δ are more tissue specific. The p38 kinases can signal through many different effectors, including other kinases, phosphatases, transcription factors, and mRNA binding proteins (142). Due to this range of potential targets, p38 can regulate gene expression at the transcriptional, post-transcriptional, and post-translational levels.
p38MAPK in Prostate Cancer
There are a handful of studies which investigated p38 signaling in PCa (Table 4). Unfortunately, most studies only interrogate p38α (generally referred to as p38). In the TRAMP mouse model of PCa, Uzgare et al. reported that p38 is highly activated in PIN lesions and more well-differentiated tumors but is absent in late stage and metastatic tumors (147). However, most other studies report that p38 activation correlates with PCa progression and treatment with a p38 inhibitor in a rat PCa model led to decreased angiogenesis and reduced tumor formation (148). Utilizing 25 primary prostate tumors and a combination of immunoblotting, ELISA, and IHC, Royuela et al. reported that phospho-p38 (p-p38) is upregulated in prostate tumors (149). Based on immunoblot analysis, tumors showed ∼50% higher expression of p-p38 than normal prostate. Furthermore, about 17% of normal prostate epithelium stains positive for p-p38 while nearly 90% of the tumor samples were positive (149). Additionally, a report by Lotan et al. demonstrated that MKK4 and MKK6 proteins are minimally expressed in normal prostate luminal cells, moderately expressed in basal cells, and highly upregulated in PIN lesions from human and mouse (TRAMP model) (150). However, this study did not look specifically at the active (phosphorylated) MKK proteins and also found that total MKK4/6 levels are not statistically different in low vs. high grade tumors (150). Ricote et al. looked at upstream (MKK6) and downstream (ATF-2, Elk-1) p38 targets in PCa progression (151). They reported that MKK6 is not detected in normal prostate samples, but it appears upregulated in PCa. Also, they detect p-ATF-2 and p-Elk-1 protein in normal basal cells but expression of both is higher in PCa (∼2.5- and ∼3-fold, respectively). ATF-2 and Elk-1 are also potential Jnk targets, but the authors did not detect any Jnk in the PCa samples so they attributed all of the ATF and Elk activation to p38 (151). Together, these reports suggest upregulated p38 activity in PCa progression, at least in part due to upregulation of the upstream activating kinases, such as MKK6.
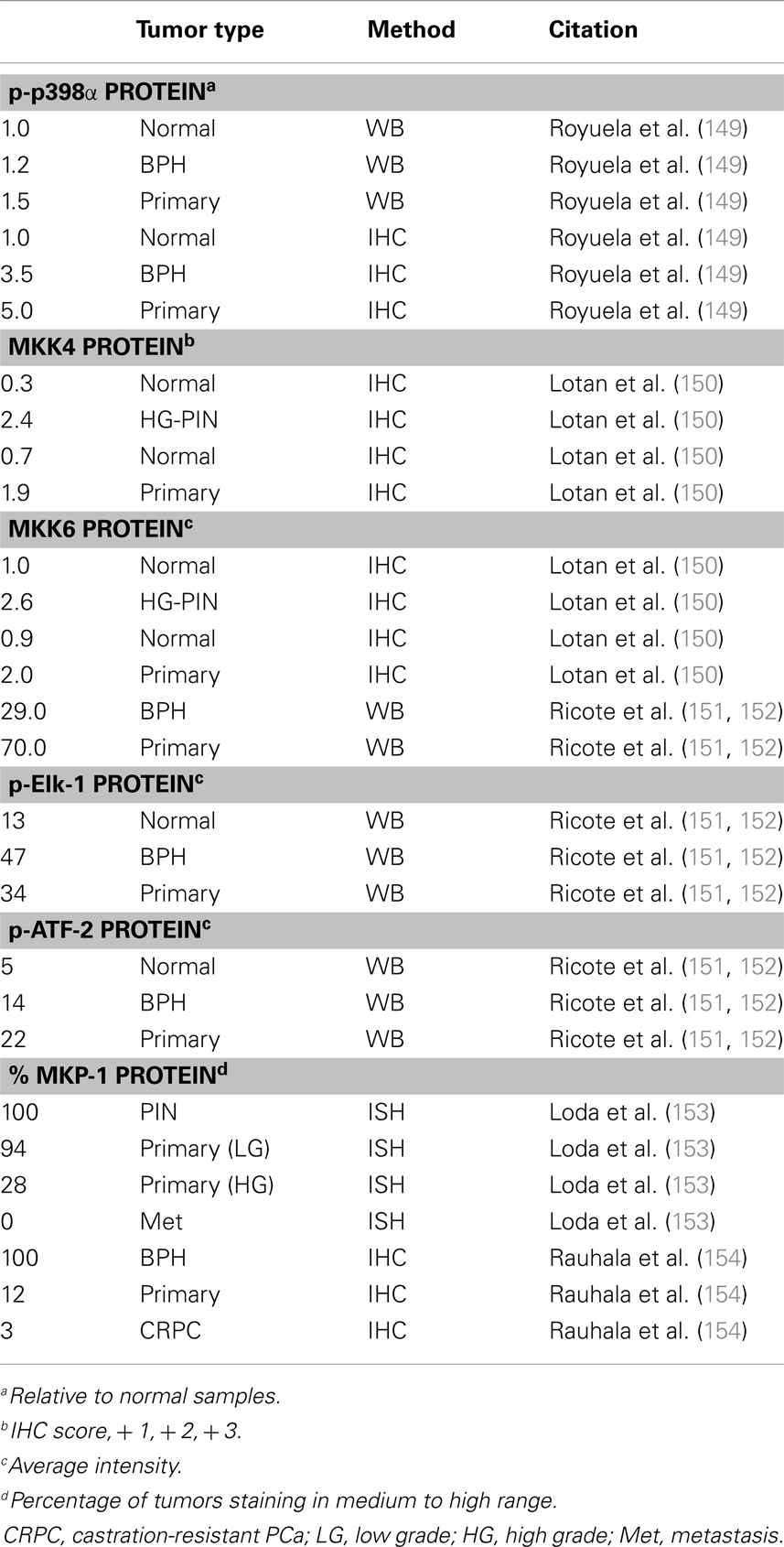
Table 4. p38 Signaling pathway in PCa.
MKP-1 (DUSP1) is a nuclear MAPK phosphatase that antagonizes Jnk and p38α/β activation (146). Several reports indicate MKP-1 is overexpressed in early prostate tumors but is downregulated in high grade and castration resistant tumors, as well as a portion of PIN lesions (153–157). MKP-1 can be activated by p38 in a negative feedback mechanism, so it is possible that down regulation of MKP-1 may be a necessary precursor to p38 upregulation in more advanced PCa tumors (158).
IL-6, a key regulator of inflammation, is also linked with PCa and p38 signaling (159–162). Ueda et al. reported that IL-6 activates transcription of AR targets in a p38-dependant manner in LNCaP cells (159). Ricote et al. reported that TNFα, a cytokine and known activator of MAPK stress response, induces apoptosis in LNCaP cells but not PC3. Moreover, TNFα activates p38 in LNCaP cells, and p38 inhibition increases apoptosis (152). Building on that finding, Gan et al. reported that LNCaP cells can be sensitized to docetaxel by blocking p38, which prevents p53 activation and apoptosis (163). Moreover, this was not observed with PC3 or DU145 cells, which do not have functional p53. These findings were supported by a second group which further investigated the role of p53 in docetaxel resistance in the same cell lines (164). Thus, over-activation of p38 is likely to trigger an apoptotic response without additional pathway alterations to compensate, which may include p53 loss in a subset of late PCa tumors but also likely involves other unknown mechanisms.
Though the FGFR2b receptor is crucial for differentiation, there are reports suggesting that growth factors such as EGFR and IGF1R can also activate p38 (165–167). Prostate tumors often show downregulation of FGFR2b and KGF (FGF7) and upregulation of other FGFs and FGFRs which drive proliferative (168). Overexpression of FGF10 in mouse prostate stromal cells causes adenocarcinoma when combined with normal mouse prostate epithelia and implanted in the mouse renal capsule (169). Furthermore, the degree of tumor progression correlated with the amount of FGF10-expressing stroma implanted, suggesting a dose-dependent function of FGF10 (169). Additionally, the FGF10 driven tumors are more resistant to androgen deprivation. This group also found that blocking FGFR1 activation in the epithelium with a dominant-negative mutant rescued oncogenic transformation, while dominant-negative FGFR2 only moderately reduced invasion (169). Moreover, another study reported that FGFR1 activation in prostate epithelium could drive PCa in the mouse (170). Whether the ability of FGF10 or FGFR1 to drive tumorigenesis is dependent on p38 was not determined. Thus, it is likely that the oncogenic potential of FGF10 is not through FGFR2b. Together, these findings support the idea that FGFR2b, which is a potential tumor suppressor in the prostate, inhibits tumor formation by driving differentiation (via p38) instead of proliferation (via other MAPKs or PI3K) (171). Moreover, alternate mechanisms of upstream p38 activation may contribute to PCa progression.
p38MAPK in Prostate Differentiation
p38 Promotes differentiation in a range of tissues including intestine, lung, bone, and cornea (172–175). Most research has focused on p38α and much less is known about the expression of specific p38 isoforms in the prostate. Our lab detected mRNA for all four isoforms in human prostate epithelia, but protein only for p38α and p38δ (unpublished data); the latter is often associated with endocrine glands but is also expressed in other epithelial cells such as keratinocytes (176). p38α knockout in mice is embryonic lethal, while p38γ or p38δ knockout results in apparently normal mice (177, 178). Despite the lack of an obvious phenotype, Schindler et al. that found p38δ−/− mice have normal skin but are resistant to skin tumor formation (179). While p38δ may have overlapping functions with p38α, there is evidence that it also has some unique functions that are not well defined (179, 180).
How p38 regulates epithelial differentiation is not well understood. In muscle differentiation, p38α/β (and possibly p38γ) activate MyoD and Mef2 transcription factors and the SWI-SNF chromatin remodeling complex, both of which are required for muscle differentiation (181). Other roles for p38 include inhibiting proliferation, which is a necessary prerequisite for differentiation (182–184). More specifically, p38 activity represses Erk and Jnk, which is reported to be a cellular switch from proliferation to differentiation (180, 183, 185). While p38 is essential in a range of differentiation models, investigation of its role in prostate differentiation is lacking, as is an understanding of the contribution of specific isoforms. However, p38 and its role in other differentiation models may serve as a good starting point for further investigation within the prostate.
Unlike other growth factors, KGF (FGF7) is an epithelial-specific differentiation factor that is typically secreted by surrounding stroma (186–188). KGF and FGF10 bind the same receptor, FGFR2b, and share many overlapping functions including upstream activation of p38 signaling (43, 144, 145). KGF or FGF10 is sufficient to drive prostate differentiation in vitro (17, 43). In mouse knockout models, FGF10 and FGFR2 are both required for proper development of the prostate (189, 190). Additionally, FGF10 overexpression can drive tumor formation as previously discussed, suggesting that the dosage of FGF10 is very important for proper prostate homeostasis (169). Thus, FGFR2b signaling through p38 is likely a critical step for prostate differentiation and aberrant expression of FGF ligands and receptors promotes PCa.
p38MAPK Conclusion
p38 activation correlates with PCa progression in many reports (149) (Table 4). Activation of p38 in PCa may be due to a combination of upregulated upstream kinases (MKK3/6) and downregulated MAPK phosphatases (150, 155–157). MKP-1 (DUSP1), which targets p38α/β, is often downregulated in late stage PCa tumors, which suggests p38δ may act as an early oncogenic activator while p38α is a late contributor. However, p38δ appears to play a tumor suppressive role in mouse skin and it would be useful to investigate p38δ in prostate tumors to see if its loss correlates with p38α overexpression (179). Alternatively, the role of p38 in PCa may be dictated by its activating receptor. This idea is supported by observations in PCa showing that more aggressive prostate tumors shift from expression of FGFR2b to FGFR2c, which would prevent differentiation and induce proliferative signals (171). Before the loss of FGFR2b, over activated p38 may drive basal cells to differentiate prematurely, which may partially explain the lack of basal cells in PCa tumors and the mixture of basal and luminal markers in cancer cells.
Notch
Notch Background
Notch is well known for its role in cell fate decisions, such as stem cell renewal, development, and differentiation (191). There are four Notch transmembrane receptors in rodents and mammals, Notch1–4, that are activated by transmembrane ligands on adjacent cells (192). In mammals, there are five classic ligands from two families: Jagged (Jag1/2) and Delta-like (Dll1/3/4). Recent work demonstrates that Notch signaling can also be activated by a variety of non-canonical proteins, such as Dlk1/2, LRP1, and TPS2 (193). The Notch receptor protein undergoes an initial cleavage event upon emergence from the ER and is then transported to the cell membrane. Upon ligand binding a second cleavage is initiated by ADAM10 and a final cleavage by the γ-secretase complex. The cleaved C-terminal receptor fragment, known as the Notch Intracellular Domain (NICD), translocates to the nucleus where it binds the repressive CSL protein [also known as RBPJκ, CBF1, Su(H), or Lag1] (192). The NICD/CSL complex recruits co-activators such as Mastermind-like (MAML1/2/3) and p300 which trigger a switch from repression to activation of the classic Notch target genes of the hairy and enhancer of split (Hes) family: Hes1-7 and Hey1/2/L (194). After activating transcription, the NICD fragment is quickly degraded and the Hes/Hey factors typically function in negative feedback loops by repressing their own transcription, thus critically controlling the temporal regulation of Notch. Notch also directly activates transcription of other targets, including p21/CDKN1A and Myc (195). While there is some overlap, the four different NICDs have some differential preferences for ligands and downstream targets, though these details are not thoroughly resolved (192, 196, 197). Adding to the complexity, Notch and CSL are reported to have some independent functions and do not always require each other for signaling (198–201). There are additional mechanisms of Notch regulation, such as endosomal and proteosomal turnover of the receptor as well as post-translational modifications of the ligands and receptors; these regulatory mechanisms will not be discussed in depth here but can be found in a variety of reviews (192, 194, 202).
Notch in Prostate Cancer
The Notch pathway is misregulated in many cancers, though the type of misregulation is tumor and cell-type specific (203, 204). The most studied model is T-cell Acute Lymphoblastic Leukemia, where Notch signaling is over activated in the majority of tumors (205–207). In other cancers, such as cutaneous and lung squamous cell carcinoma, Notch is understood to be a tumor suppressor (208). Notch1 loss drives skin cancer progression in mice in a non-cell autonomous matter due to loss of barrier cell function, which triggers an immune and growth cytokine response within the tumor microenvironment (209).
Within the PCa field, there are conflicting reports about whether the Notch pathway is tumor suppressive or oncogenic (191, 210, 211). Supporting the case for Notch as a tumor suppressor, Belandia and colleagues reported that Hey1 and HeyL are excluded from the nucleus upon the transition from benign to carcinoma in human prostate samples (212, 213) (Table 5). Furthermore, the same group showed that Hey1 and HeyL bind to AR and potentially function as AR co-repressors in the LNCaP line (213). Other studies similarly found a decrease in Notch1 and Hey1 protein in human PCa tumors compared to normal tissue (214, 215).
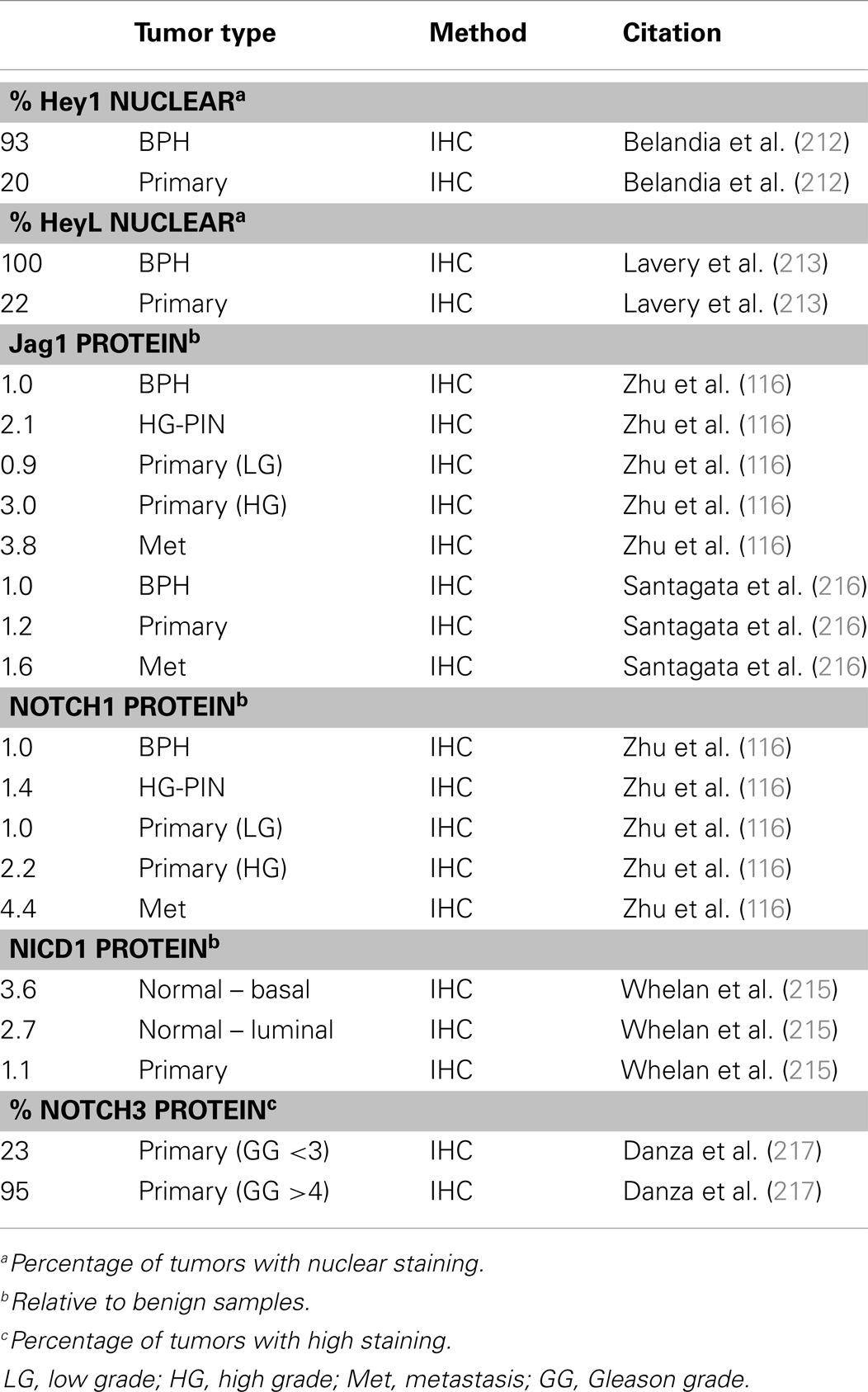
Table 5. Notch signaling in PCa.
Conversely, several reports demonstrate increased levels of Jag1 and Notch1 protein in high grade PCa tumors, implicating Notch as an oncogene (116, 117, 216, 218) (Table 5). Bin Hafeez et al. observe higher Notch1 protein staining in more aggressive prostate tumors: 64% positive staining for Gleason grade 4 tumors, 30% for high grade PIN, and 5% for normal tissue. Moreover, knockdown of Notch1 in PC3 cells decreases metastatic gene expression and decreases invasion in vitro (117). Furthermore, knockdown of CSL, which ablates downstream Notch activity, leads to decreased proliferation in PC3 PCa cells (219). Other groups reported that siRNA knockdown of Notch1 or Jag1 in PC3 cells decreases PC3 growth and colony formation, in part due to an increase in cell death (220, 221).
While most research has focused on Notch1 or overall Notch activity, there are also a few papers reporting a specific role for Notch3 in PCa. Using prostate tumors with known patient outcome, Long et al. found Notch3 mRNA levels positively correlate with PCa recurrence (222). Moreover, of a 12-gene mRNA panel, Notch3 has the second highest prognostic ability for recurrence (222). Ross et al. reported that Notch3, Jag2, and Presenilin1 (a catalytic subunit of the γ-secretase complex) mRNA transcripts are upregulated in high grade prostate tumors (157). Notch is also implicated in PCa via a role in hypoxia. Exposure of LNCaP, PC3, and DU145 cell lines to prolonged hypoxia leads to down regulation of Notch1/2 mRNA and protein but has no effect on Notch3 (223). A follow up report found that hypoxia also induces changes in cholesterol and lipid rafts in the cell membrane, which increases colocalization of Notch3 and γ-secretase, leading to increased NICD3 expression (217). The study also measured Notch3 protein levels in PCa tumor sections and found Notch3 protein levels correlate positively with Gleason grade, thus supporting the Notch3 mRNA correlation reported by Long et al. (217, 222).
Notch in Prostate Differentiation
When it comes to cell fate decisions, Notch signaling is critical across most cell types. The Notch pathway has been studied in the prostate to some extent, but knowledge about specific mechanisms and signaling pathways is lacking. Many studies have been conducted in the mouse which, as discussed earlier, has some significant structural differences from the human. Treatment of rat prostates ex vivo with a γ-secretase inhibitor prevents lumen formation and treatment with the inhibitor in vivo prevents prostate regeneration following castration (220). A similar finding was reported for mouse prostates treated with γ-secretase inhibitors (224). As for receptor-specific studies, Notch1 is the most studied. Wang et al. used an interesting model where they made a transgenic mouse with a lethality gene (bacterial nitroreductase) under control of the Notch1 promoter, which would only be lethal in the presence of an inducing chemical (220). They took early developing mouse prostates and grew them ex vivo with or without the inducer and found that ablation of Notch1-expressing cells prevents proper organoid development and differentiation (220). In a follow up study, they utilized γ-secretase inhibitors and an interferon-inducible Notch1 mouse (Mx-Cre/Notch1flox) to study the effect of Notch1 loss on prostate development (214). They found that induced Notch1 knockout (in all cells of the prostate including the stroma) leads to increased proliferation and prostatic hyperplasia as well as co-expression of basal and luminal keratins (214). Moreover, Wu et al. utilized transgenic mice to investigate Notch in prostate development, reporting that Nkx3.1-Cre driven CSL knockout leads to decreased proliferation and differentiation defects in the prostate (225). Conversely, Notch1 constitutive activation (via PB-Cre or Nkx3.1-Cre driven NICD1) in the mouse prostate causes increased proliferation and hyperplasia (225). Both of these studies suggest that Notch signaling is required for proper differentiation, while Notch1 specifically appears to be crucial for maintenance of a proper and distinct basal layer. Notch1 also regulates p63, which is a classic basal cell marker in the prostate and a regulator of cell adhesion, including integrins (226–228). Therefore it is intriguing to consider that Notch signaling during prostate differentiation may need to strike a balance between downregulating adhesion through p63 while Notch1 must also maintain homeostatic basal cells. The balance between multiple Notch receptors and downstream targets may in fact be crucial for regulating the decision to stay basal or differentiate.
Studies on other Notch receptors in the mouse prostate are limited; though there are some studies in other tissues. For instance, Notch3 knockout mice develop normally (229). The NICD3 appears to be a weaker activator of downstream signaling than NICD1 and may actually antagonize NICD1 by competing for CSL (230). Dang et al. reported that constitutive Notch3 expression (via NICD3) in mouse lung epithelium prevents terminal differentiation and causes metaplasia and reduced epithelial branching (231). In esophageal differentiation, Notch1 activates Notch3, which in turn activates Hes5 and drives differentiation (232). In skin differentiation, Notch1/2/3 have all been detected in the interfollicular epidermis, but there is a shift in ligands from Jag2 in the basal layer to Jag1 in the upper layers (233). Moreover, in the hair follicle Notch1 is expressed primarily in the bottom of the niche, while the upper regions mainly express Notch2 or Notch3 (233). Such a mechanism where there is a shift in Notch receptor, ligand, and/or downstream targets during prostate differentiation is conceivable but has not yet been investigated.
Due to the structural differences in the mouse vs. the human prostate and the highly context-specific nature of the Notch pathway, further studies are needed to understand the role of Notch in human prostate tissue. For example, in mouse skin Notch1 and Notch2 are mainly expressed in the upper layers; however, in human skin Notch1 is expressed in all layers and Notch2 is mainly restricted to the basal layer (234). There have not been any reports clearly and uniformly demonstrating which components of the Notch pathway are expressed at the protein level in the normal human prostate (191). However, Wang et al. reported mRNA for all four receptors and most Hes/Hey members are expressed in human prostate samples, but only Notch1 and Hey1 levels are altered in PCa tumors (214). Notch1 is the most well-studied receptor in the prostate and it is found predominantly in basal cells of both mouse and human prostates (218, 235). Research on the other receptors is much less abundant. Recently, one study investigated Notch2 and Notch3 expression in PCa progression. They found low levels of Notch3 staining in normal prostate sections and decreased Notch2 expression with increasing tumor grade but did not report whether they detected Notch2 in normal prostate tissue (217). In a mouse model, Notch2 and Dlk1 protein were detected in developing mouse prostate stroma but not the epithelium (224). Understanding the specific role for each Notch receptor in prostate will require further investigation.
Notch Conclusion
While it is established that the Notch pathway is important for normal prostate differentiation, and it appears to be deregulated in PCa, specific details are still ambiguous. The Notch field is still working to define clear roles for the various ligands, receptors, and downstream targets. Moreover, any resolved mechanism is likely to be tissue and cell specific, as is common with this pathway (234). The lack of an NICD3-specific antibody makes identifying its contribution difficult to assess. Additionally, the transient nature of NICD activation and turnover makes it difficult to detect endogenous NICD via histological techniques or immunoprecipitation. Additionally, many studies rely on γ-secretase inhibitors, which fail to distinguish the function of specific Notch receptors. The same applies to studies that use knockdown of CSL to ablate Notch signaling. The limitations of γ-secretase inhibitors or CSL knockdown are apparent in a study from Yong et al. which saw differential effects depending on which technique they used (219). The explanations for such discrepancy are likely due to γ-secretase involvement in other Notch-independent functions and CSL-independent Notch signaling (200, 201). Specific knockdown of individual Notch components is more arduous but allows for a better understanding of the pathway.
There is conflicting data about whether Notch is acting as a tumor suppressor or an oncogene in PCa. While the role of Notch1 in normal prostate differentiation has been investigated, specific mechanisms remain elusive. One interesting idea is that Notch1 is known to transcriptionally upregulate Myc, which may explain how Notch signaling can function as an oncogene (236). The link between Notch-mediated repression of p63 could also explain why prostate tumors show loss of p63 (227). Notch as a tumor suppressor may be mediated through Hey2/L expression, which then acts as an AR co-repressor. Based on the complexity of Notch signaling, it is feasible that its role as an oncogene or tumor suppressor is dependent on which receptors and downstream transcription factors are being activated. For example, if Myc is the primary target, the pathway may be oncogenic; however, if HeyL is the primary target, it may be tumor suppressive. Due to the complexity of the Notch pathway, merely looking at a small selection of the ligands, receptors, or downstream factors may only be providing a small piece of the overall puzzle. As is seen with skin, temporal changes in Notch ligand expression are characteristic of differentiation (233). Perhaps the status of the Notch pathway in prostate tumors can be indicative of the cell of origin or be used to grade relative differentiation status. More thorough investigations of Notch signaling may help clarify its function in differentiation and resolve some of the conflicting findings about its role in oncogenesis and tumor progression.
PI3K/Pten
PI3K/Pten Background
The PI3K pathway regulates multiple processes in the cell including metabolism, proliferation, and survival (237). Multiple aspects of the PI3K pathway are frequently misregulated in many cancers (238, 239). PI3K is a lipid kinase that targets phosphoinositides, catalyzing the conversion of phosphatidylinositol 4,5-biphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). There are three classes of PI3K genes that all target PIP2 but each class varies by structure, regulation, and tissue specificity. PI3K class I members are implicated in cancer and are the most ubiquitously expressed. The class I PI3K family contains four members (PIK3CA, PIK3CB, PIK3CG, and PIK3CD) that code for the catalytic p110 subunit, while three other genes (PI3KR1-3) code for at least five isoforms of the p85 regulatory subunit (238). The p110α and p85α subunits are implicated most often in cancer (238, 239). Activation of a variety of receptors induces recruitment of the regulatory subunit, p85α, which brings the catalytic subunit, p110α, to the cell membrane to catalyze the conversion of PIP2 to PIP3. PIP3 recruits Akt which is then phosphorylated by Pdk1 and the mTORC2 complex. Activated Akt phosphorylates and activates the mTORC1 complex, which in turn regulates a range of targets such as S6K and 4EBP1, both of which are required for protein synthesis (240). Akt also phosphorylates other proteins commonly implicated in cancer, including GSK3β, BAD, MDM2, and p27 (CDKN1B) (238).
While mTORC1 activation by Akt can stimulate a negative feedback mechanism, the primary negative regulator of the PI3K pathway is Pten, a lipid phosphatase that converts PIP3 back to PIP2 and thus antagonizes Akt activity (58). However, Pten has functions beyond its role in regulating PI3K/Akt activity. First, the phosphatase region of Pten is related to other tyrosine phosphatase proteins and dual-specificity protein dephosphatases (58, 241) and as such can also dephosphorylate proteins like focal adhesion kinase (FAK) and cAMP response element binding protein (242, 243). Secondly, Pten has lipid-independent functions within the nucleus such as DNA repair via activation of RAD51 (58, 244, 245). Pten also interacts with p53 to arrest cell growth upon oxidative damage (246–248). Complete loss of Pten leads to major defects in DNA repair (245, 249). Finally, Pten also regulates cell polarity (250, 251).
Pten loss (via mutation, transcriptional repression, or deletion) is common in many cancers, including PCa (58, 252). Because of the ability for PTEN to regulate PI3K/Akt-dependent and independent pathways, it has the capacity to impact many aspects of cellular function, including DNA damage response, FAK activation, and cell polarity. Identifying the most important targets for the establishment, maintenance, or progression of prostate tumors is essential for a full understanding of PCa.
PI3K/Pten in Prostate Cancer
Loss of Pten expression in PCa was first recognized in the late 1990s and genetic loss of at least one allele occurs in as many as 65% of metastatic prostate tumors (57, 253, 254) (Table 2). Homozygous deletion of Pten occurs in <10% of primary tumors but occurs in about 20–50% of metastatic, castration resistant tumors (60, 61, 68). Genomic loss of Pten correlates with late stage PCa tumors and poor prognosis (60, 67, 255–257). Alterations in other members of the PI3K pathway are less common than Pten loss. Akt mutations are very rare and <5% of primary PCa tumors have mutations in p110α (PIK3CA), though 1–15% have amplification of the gene locus (5, 59, 258). Mutations in p85α (PIK3R1) are also very rare, though genomic deletions of its locus on 5q are more frequent and expression is downregulated in about 20% in primary tumors and 60% of metastases (5). Multiple studies looking at Pten protein expression by IHC found that while Pten deletions are readily apparent by protein loss, about 30–40% of tumors with loss of Pten protein do not show genomic loss of the Pten gene (60, 61, 259). While Pten deletions are quite common, mutations in the gene itself are rare in primary prostate tumors (5, 260, 261). Pten silencing by methylation is fairly common in other cancers but is not widely reported in PCa (262). Inactivation of Pten via alternate mechanisms, such as miRNA or competing-endogenous RNA, was recently reported, indicating this mechanism might be active in PCa (263–267).
Constitutive Akt expression in the mouse prostate, driven by the probasin promoter, leads to PIN but not tumor invasion, as is seen with total Pten knockout (65, 66, 268). These findings, in addition to the fact that Pten deletion is more common than other PI3K/Akt alterations, suggests that Pten contributes to prostate oncogenesis through multiple mechanisms beyond activation of PI3K/Akt signaling. Pten loss correlates with the TMPRSS2-Erg fusion in human tumor samples (5, 259, 269, 270). Furthermore, Erg activation combined with single-copy Pten loss accelerates PCa progression from PIN to adenocarcinoma in mice (87, 88, 271). In the TRAMP model, Pten haploinsufficiency in the prostate gives rise to larger tumors and decreased survival rates compared to the normal TRAMP mice (63). In humans, it is still not clear if the prognostic association of single-copy Pten loss is due to Pten haploinsufficiency or loss of heterozygosity, i.e., inactivation of the second allele via an alternate mechanism (60).
PI3K/Pten in Prostate Differentiation
While loss of Pten in PCa is well established, knowledge about how its loss contributes to tumor development and its role in normal prostate differentiation is very limited (14, 17, 272, 273). Nonetheless, PI3K and Pten are known regulators of epithelial differentiation in a variety of other models (274–278). The Rivard group reported that PI3K is required for E-cadherin assembly at tight junctions, which in turn activates Akt, downregulates Erk, and drives differentiation and survival (279–281). Calautti et al. utilized mouse primary keratinocytes to demonstrate that PI3K signaling is required for the initiation of differentiation and survival of differentiated suprabasal cells (282). Moreover, they found that PI3K signaling and differentiation require EGFR and E-cadherin engagement (282). Supporting these findings, luminal prostate cells also require E-cadherin and PI3K signaling for survival (17). However, the differentiated luminal cells, unlike the basal cells, do not express or require EGFR (17, 283). Thus, PI3K/Akt signaling is crucial for the emergence and survival of differentiated epithelia.
Most notable is the observation that Pten’s role in differentiation extends beyond regulating PI3K/Akt signaling. In a mammary ex vivo organoid model, Pten knockout causes disorganization of the epithelial bilayer (284). Moreover, Akt/mTORC1 signaling is necessary but not sufficient for proper differentiation in this model (284). Groszer et al. found that loss of Pten drives premature hematopoietic stem cell differentiation and increases the number of leukemia initiating cells (285–287). Using a kidney epithelium 3D culture model, the Mostov group found that Pten and the balance between PIP2 vs. PIP3 levels is necessary for establishing basal/apical polarity and lumen formation (250, 288). Upon siRNA knock down of Pten, the cells lost actin and PIP2 enrichment at the apical membrane which is required for recruitment of Annexin2 and subsequently Cdc42. Cdc42 then recruits Par6, which together function in a complex with Par3 and aPKC, which is known to regulate cell polarity (251, 289, 290). Pten may be targeted to the apical membrane via its association with tight junctions, though the precise mechanism is unknown. It was also reported that integrin β1 and laminin were required for establishment of basal polarity in the same kidney epithelium model, thus demonstrating that Pten and integrins likely cooperate to establish cell polarity (291). The Rivard group, using an intestine epithelium model, reported that Pten knockdown with shRNA leads to a loss of cell polarity and disruption of adherence junction formation (292). They followed up that study by creating an intestine-specific Pten−/− mouse model and found defects in differentiation in vivo (293). Thus, PI3K signaling and Pten both appear to be required for epithelial differentiation. PI3K may be primarily involved in survival of the differentiating cells while Pten appears to be important for establishing cell polarity which is required for organization of epithelial structure. Therefore, epithelial differentiation likely requires a delicate balance between Pten and PI3K activity; for example, Pten may be higher in early differentiation to set up polarity and lower in later differentiation to permit PI3K signaling for survival. Alternately, Pten levels may remain constant throughout differentiation with upregulation of PI3K signaling by other means in the new luminal cell.
PI3K/Pten Conclusion
Downregulation of Pten aids tumor growth (238). However, complete loss of Pten in normal cells can lead to cell arrest or apoptosis (245). In PCa, the initial loss of one copy of Pten may aid growth of the tumor, but other oncogenic events may be required for survival upon loss of heterozygosity. These additional events could include upregulation of other survival pathways, or in later stage tumors, loss of p53. Our lab previously reported that PCa cells utilize the combination of AR and α6β1 to activate a survival pathway independent of PI3K (11). Basal prostate cells do not require PI3K for survival, but luminal cells do (17, 283). Thus, it is conceivable that a transiently differentiating cell may not yet depend on PI3K for survival but already be activating AR and beginning to display the luminal phenotype. This hypothetical cell would combine the tumorigenic benefit of PI3K independent survival with AR activation, which in this context now becomes oncogenic. Additionally, loss of Pten likely disrupts polarity leading to disorganization of the bilayer, and altered homeostatic signaling between the stroma and epithelium, ultimately disrupting differentiation, which may be a prerequisite for tumorigenesis.
Crosstalk and Oncogenesis Models
Overview
As covered in this review, Myc, p38, Notch, and PI3K/Pten all play key roles in regulating differentiation and are all misregulated during oncogenesis. Myc upregulation and Pten loss are well established drivers of prostate oncogenesis. However, p38 and Notch have not yet been widely labeled as oncogenic drivers, though we believe that their misregulation in a transiently differentiating bipotent cell can contribute to misregulation of Myc and Pten and help drive oncogenesis. All these pathways are capable of crosstalking with each other, which may help explain their interconnected functions within the prostate. One of the great challenges facing PCa researchers is to understand prostate oncogenesis, which will allow better understanding of how to diagnose and treat PCa. Though there is much more investigation required, there is enough evidence in the literature to begin to formulate some ideas about how PCa may arise from defective differentiation. In this section we will review our hypothesized model for how these pathways drive normal differentiation and summarize potential mechanisms for how their misregulation and crosstalk could alter Myc and Pten to drive oncogenesis.
Model of Normal Differentiation
We favor the model of a transient-differentiating bipotent epithelial cell as the cell of origin for PCa. Based on the information presented in the preceding sections, we present a model of prostate epithelial differentiation (Figure 2). We hypothesize that the decision of a bipotent cell to differentiate toward a secretory phenotype is determined by stromal KGF (FGF7) activation of p38 through FGFR2b and MKK signaling. p38 affects a wide range of targets, including transcription factors, mRNA stability proteins, and other kinases/phosphatases. One action of p38 is to limit proliferation through Erk, while another is to upregulate Notch1 and Myc to drive differentiation. Upregulation of Pten, via a mechanism that may include Notch1 or other p38 target, is initially required to maintain chromosome stability and generate the PIP2 gradient at the apical membrane to establish cell polarity (Figure 2). The establishment of polarity is critical for initiating a differential gradient of Notch1 signaling basally to maintain stem-ness and Notch2/3/4 signaling apically toward the emerging luminal cell. Notch2/3/4 signaling in the emerging luminal cell downregulates p63, which along with Myc, downregulates integrin expression. Furthermore, as integrin expression diminishes in the transient-differentiating cell, there is a switch from Erk-dependent survival to a reliance on E-cadherin and PI3K survival pathways (17). Notch signaling through Hes1 limits Pten expression to allow effective PI3K/Akt signaling and survival of the luminal cell. Beyond promoting survival, PI3K negatively regulates Ask1, the upstream kinase required for p38 activation (272, 277, 294). Thus, PI3K upregulation in luminal cells may be required to terminate the p38 differentiation program (Figure 2).
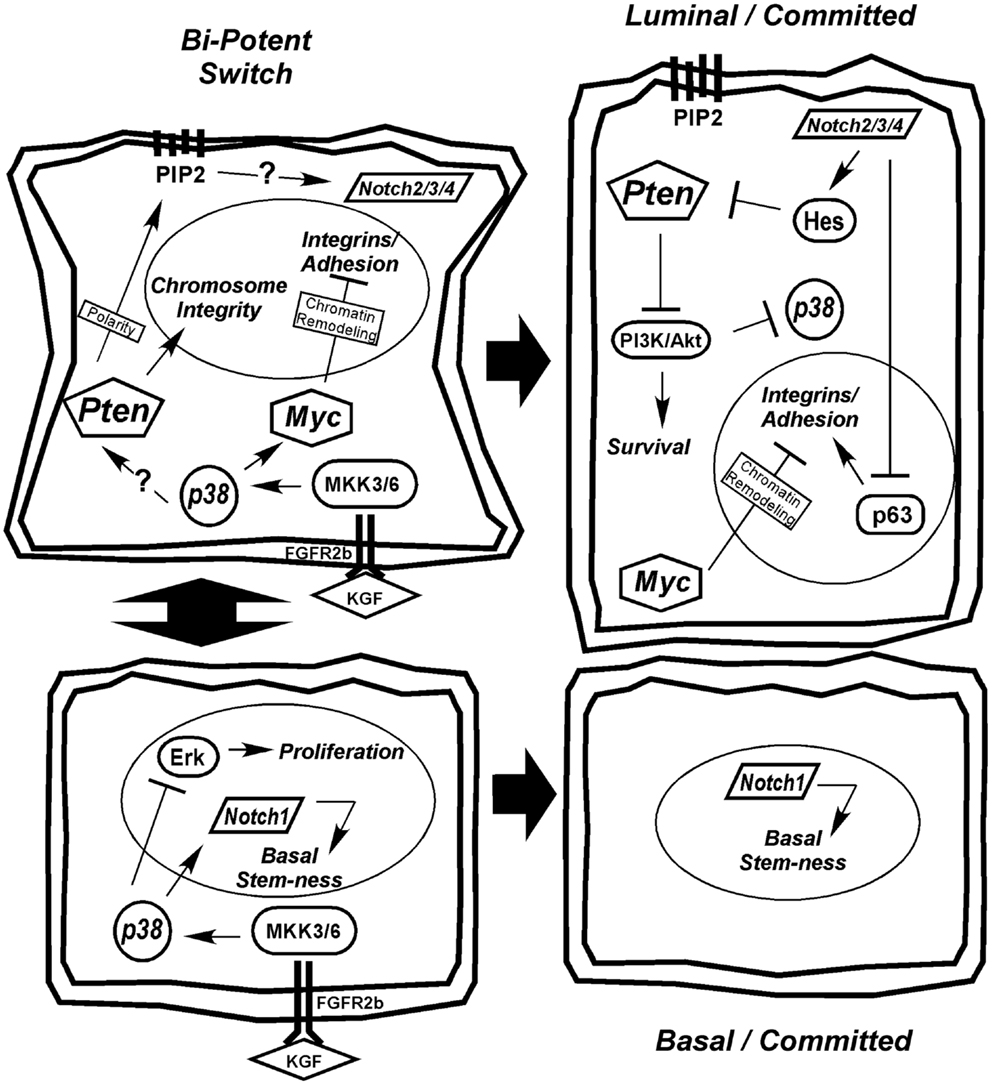
Figure 2. Hypothetical model for prostate epithelial differentiation. Upstream KGF(FGF7) from the stroma activates p38 through FGFR2b and MKK3/6, which plays multiple roles in differentiation, including inhibiting proliferation through suppression of Erk and upregulating differentiation determinants Notch1 and Myc. The transient-differentiating bipotent cell must make a decision between sustaining Notch1 signaling to commit to a basal phenotype or switching to Notch2/3/4 signaling to become luminal. The switch may be mediated by elevated Pten, via an unknown mechanism, to generate a PIP2 gradient at the apical membrane to establish cell polarity. Notch2/3/4 signaling in combination with Myc suppresses integrin expression allowing cells to detach. Luminal cells become dependent on PI3K signaling for survival as they lose matrix adhesion and integrin expression. Ultimately, Pten is partially suppressed to allow PI3K/Akt-dependent survival of the terminally differentiated luminal cell and the p38 differentiation program is terminated.
A critical question yet to be addressed in prostate secretory epithelial differentiation is how AR expression is upregulated. In Figure 3, we outline the few known mechanisms for controlling AR expression. Although, AR transcription is enhanced by androgen through AR and Myc binding sites in its promoter (Figure 3ii), this is not sufficient to induce AR expression in basal cells (17, 111). Low levels of AR mRNA or AR protein is detectable in AR-negative tumor cell lines (295, 296), suggesting AR expression is sensitive to both mRNA and protein stability factors. The mRNA PolyC-binding protein, HuR, binds polyC tracts in the 3′ UTR of AR mRNA and HuR (Figure 3i) is a known direct target of p38 (297–299). Thus, early signaling by p38 during differentiation may directly stabilize AR mRNA (Figure 3i). There are no reports linking Notch directly with AR expression, but stable AR protein expression requires loss of cell-matrix adhesion (17). Thus, the extent to which Notch is involved in reducing cell adhesion (300), would aid in enhancing AR expression.
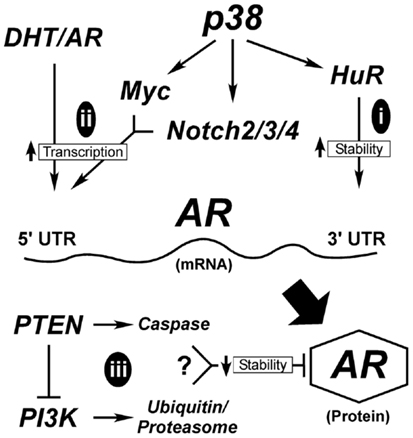
Figure 3. Hypothetical mechanisms for AR upregulation during differentiation. AR expression is tightly controlled both transcriptionally and post-transcriptionally. Early in differentiation prior to stable AR expression, p38 stimulates AR expression most likely by regulating mRNA stability (i). Once low levels of AR are made, androgen (DHT) binding to AR stimulates its binding to its own promoter through AR binding elements (ARE). This requires the cooperation of signals from p38 that stimulate Myc and Notch2/3/4 to suppress cell adhesion, ultimately elevating AR mRNA transcription (ii). AR protein is degraded through two different mechanisms (iii), the classical ubiquitinylation system and through caspase 3. Both Pten and PI3K are reported to influence these degradation pathways; although how these two contradictory signals are balanced is not clear.
Alternatively, AR protein stability may be enhanced during differentiation (Figure 3iii). Two mechanisms known to control AR protein stability are proteasomal degradation via ubiquitinylation and caspase 3-mediated degradation (301). Numerous AR-binding E3 ligases are known, but the upstream signals that suppress or activate them are less clear. One pathway involves Akt-dependent phosphorylation of Mdm2, which then targets AR for degradation (Figure 3). More intriguing is the role of Pten in promoting AR degradation by caspases (302). What is not clear is when during differentiation these pathways would be active, since they are antagonistic to each other and both result in AR loss. In Pten-negative tumors, active Akt would degrade AR resulting in AR loss. The reduced expression of AR in the prostate tumors of Pten-negative mice supports this idea (35). However, there must be a counteractive mechanism that keeps AR active in both tumors and secretory cells. Thus, even tumor cells are forced to balance signals to keep AR active.
Mechanisms for Oncogenic Myc Upregulation
Genomic amplification of Myc is common in advanced tumors, but early tumors must have alternate mechanisms of Myc upregulation which are not well defined (51). A moderate but transiently sustained increase in Myc expression is required for differentiation, but something must happen to prolong Myc activation in oncogenesis (91). Both p38 and Notch are essential for differentiation and each can drive Myc expression. Thus, it is possible that misregulation of either could sustain Myc activation during tumorigenesis as depicted in Figure 4.
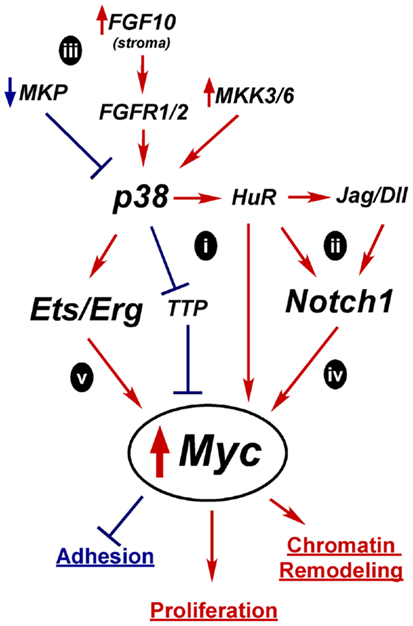
Figure 4. Hypothetical mechanisms for Myc upregulation in prostate oncogenesis. A tightly controlled increase in Myc expression is required for differentiation, but prolonged and excessive Myc activity is oncogenic and leads to increased proliferation, altered adhesion, and chromatin remodeling. Constitutive activation of p38 and Notch1 are two possible alternative mechanisms for upregulating Myc expression early in tumorigenesis. Active p38 can regulate mRNA stability proteins, such as TTP and HuR which affect Myc, Notch1, or Notch ligand mRNA half-life (i,ii). Overactive p38 signaling could result from loss of MAPK phosphatases (MKP), alternate growth factor receptor expression, or activation of MKKs (iii). Independently, overexpressed Notch ligands could stimulate Notch1 leading to increased Myc transcription (iv). Activation of p38 may enhance Erg/Ets fusion gene activity to drive Myc overexpression(v).
In the case of p38, one potential mechanism for increasing Myc activity is through mRNA stability (Figure 4i). Myc mRNA turnover is tightly controlled and p38 directly phosphorylates stability proteins that bind mRNAs like Myc that have stability control elements such as AU-rich or PolyC tracts (51, 303). p38 inactivates mRNA destabilizing proteins such as TTP, which upregulates Myc mRNA stability in rapamycin-treated cancer cells (304). Conversely, p38 activates HuR (297). HuR reportedly binds Myc mRNA, although there are conflicting reports as to whether this increases or decreases Myc stability (305–310). p38 may indirectly enhance Myc expression by increasing Notch ligand or receptor mRNA stability in an analogous fashion (Figure 4ii) (236). Notch1 has an AU-rich element and is stabilized via p38 activity in a leech zygote model (311). Additionally, the Notch ligand Dll4 was reported to be upregulated by HuR in mouse neuroepithelial cell (312). Whether Myc, Notch1, or Dll1 is regulated by p38 through mRNA stability in the prostate is unknown.
Several mechanisms could lead to aberrant p38 activation in tumors (Figure 4iii). Alterations that reportedly upregulate p38 signaling in PCa include loss of MAPK phosphatases, upregulation of upstream kinases MKK3/6, increased FGF10 expression in the stroma, and increased expression of proliferation-driving FGFRs (168). Prolonged p38 activation in a transiently differentiating bipotent cell would derail differentiation by preventing the required downregulation of Myc. Mutations that upregulate Notch1 signaling, such as increased ligand expression or increased receptor mRNA stability, could also lead to sustained Myc expression or activity (Figure 4iv).
The Ets fusion genes that are so prevalent in PCa provide another mechanism by which Myc could be upregulated (Figure 4v). TMPRSS2-Erg upregulates Myc in VCaP cells (77) and Etv4 binds Myc enhancers in PC3 cells (313). Etv4 knockdown decreases Myc expression. However, Etv4 overexpression in normal prostate epithelial cells did not alter Myc levels, suggesting other oncogenic alterations are required for this crosstalk (313). One of the downstream targets of p38 signaling is Elk-1, a member of the Ets family, which is upregulated in PCa (151). Thus, increased p38 signaling may be complementary or necessary for full Ets activation in PCa.
In summation, due to the normal upregulation of Myc as a part of differentiation, we hypothesize that alterations in one or more of these pathways would be enough to push Myc expression to initially stimulate differentiation. But its failure to subsequently be downregulated generates an oncogenic event resulting in only a partially committed, yet proliferative, cell with an altered chromatin program and retained matrix adherence. This model will need further testing to determine its relevance in human prostate epithelial differentiation and oncogenesis.
Mechanisms for Oncogenic Pten Downregulation
Another key oncogenic event in PCa is downregulation of Pten. There are several potential mechanisms whereby misregulation of normal differentiation signals could lead to Pten loss as outlined in Figure 5. How Pten expression is controlled during differentiation is not clear, but it is required to maintain epithelial integrity and structure. In fact, downregulation of Pten may be a prerequisite to Myc-driven oncogenesis, since elevated and prolonged Myc expression can induce apoptosis, potentially via Pten activation of p53 and Rad51 (Figure 5i) (58, 95, 314).
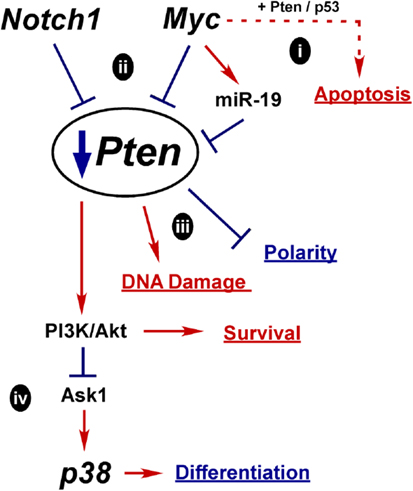
Figure 5. Hypothetical mechanisms for Pten down regulation in prostate oncogenesis. Myc or Notch1 could each act to suppress Pten expression early in oncogenesis. Loss of Pten may be a prerequisite for Myc upregulation to relieve Myc induced apoptosis (i). However, moderate activation of Myc could also contribute to Pten downregulation, via either transcriptional repression or induction of miR-19. Increased Notch1 signaling may also transcriptionally downregulate Pten (ii). Ultimely downregulation of Pten can lead to more DNA damage, loss of polarity, and increased PI3K/Akt survival signaling (iii). Loss of Pten can lead to stimulation of p38 via activation of upstream MAPKs, such as Ask1, further contributing to oncogenesis(iv).
About 30–40% of tumors with loss of Pten protein do not show genomic loss of the gene, indicating alternate mechanisms of inactivation (60). Crosstalk between Pten, Notch, and Myc is reported in the literature and we hypothesize that oncogenic action of Notch or Myc is sufficient to limit Pten expression early in oncogenesis (Figure 5ii). One mechanism may be similar to what is observed in Notch-driven T-ALL, where Hes1 and Myc both transcriptionally repress Pten (315, 316). In T-ALL, Notch inhibition kills cells by blocking Pten downregulation; however, resistant cells develop alternate mechanisms to upregulate Akt (316). In the prostate, NICD1 expression in mouse luminal cells (via the probasin promoter) leads to hyperplasia and decreases Pten expression in both epithelial and stromal cells (225). Likewise, CSL knockout in luminal cells (via the Nkx3.1 promoter) leads to increased Pten. Additionally, Myc activation of miR-19 targets and downregulates Pten in a mouse Myc-driven B-cell lymphoma model (317). Thus, under oncogenic conditions, upregulated Notch or Myc suppresses Pten expression and potentially drives tumor initiation (Figure 5ii) (223, 317).
We further hypothesize that Pten aids in temporally or spatially controlling the Notch switch that commits the cell to either the basal (Notch1) or luminal (Notch2/3/4) lineage (Figure 2). The necessary oncogenic consequence of Pten inactivation would be disruption of cell polarity, failure to maintain epithelial structure, and impaired differentiation (Figure 5iii). Because Pten is also critical for controlling DNA damage, another consequence of Pten loss would be increased susceptibility of the cell to mutation.
The best characterized result of Pten loss is increased survival signaling through PI3K/Akt, which is consistent with the switch to dependence on PI3K/Akt for normal secretory cell survival during differentiation (17). Another consequence of PI3K/Akt activation is suppression Ask1 (Figure 5iv), the upstream kinase required for p38 activation (272, 277, 294). If suppression of p38 signaling due to Pten loss occurs too early in the differentiation program, cells will not complete the process and become arrested. However, PI3K activation in tumor cells was shown to activate p38, which then stimulates EGFR and creates a positive feedback loop further activating PI3K (318). The mechanism of how PI3K activates p38 is not clear, but other oncogenic mutations may reverse the normal regulation by Ask1 and activate p38. Whether any of these events are major mechanisms for Pten loss or whether the downstream consequences are critical for prostate oncogenesis has yet to be demonstrated, but this model provides a sampling of potential mechanisms that should be further investigated.
Conclusion
In conclusion, pathways known to be involved in normal prostate differentiation (Myc, p38, Notch, and PI3K/Pten) can be linked to PCa. These pathways have complicated interactions; multiple functions, multiple targets, many levels of regulation, and significant crosstalk. To better understand prostate oncogenesis, these pathways need to be more thoroughly investigated in human prostate models. Differentiation is a very tightly regulated process where cells undergo major reprograming. We have provided a few, though certainly not exhaustive, potential models for how oncogenic disruption of specific differentiation pathways can drive tumor initiation. We hypothesize that the oncogenic cell of origin for PCa is not a committed basal or luminal stem cell, but rather a transient-differentiating bipotent cell. The bipotent K5/K8 cells found in both human and mouse prostates are the most likely candidates for prostate oncogenic transformation (25, 32). These cells must maintain very tight temporal and spatial control of the differentiation pathways and thus may be the easiest targets for oncogenic disruption. Only through better understanding of oncogenesis will we find new ways to classify prostate tumors and better predict tumor aggressiveness. Such discoveries will ultimately provide physicians and patients with more information when deciding how to treat this widespread disease.
Authors Contribution
Sander B. Frank was responsible for researching and writing the review. Cindy K. Miranti was responsible for editing, validating citations, and communicating the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding was provided by the Association for Internal Cancer Research (Sander B. Frank) and the Van Andel Research Institute (Cindy K. Miranti).
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi: 10.3322/caac.21166
2. Warlick C, Trock BJ, Landis P, Epstein JI, Carter HB. Delayed versus immediate surgical intervention and prostate cancer outcome. J Natl Cancer Inst (2006) 98(5):355–7. doi:10.1093/jnci/djj072
3. Williams SB, Salami S, Regan MM, Ankerst DP, Wei JT, Rubin MA, et al. Selective detection of histologically aggressive prostate cancer: an Early Detection Research Network prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer (2012) 118(10):2651–8. doi:10.1002/cncr.26396
4. Klotz L. Active surveillance with selective delayed intervention: using natural history to guide treatment in good risk prostate cancer. J Urol (2004) 172(5 Pt 2):S48–50. doi:10.1097/01.ju.0000141712.79986.77
5. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell (2010) 18(1):11–22. doi:10.1016/j.ccr.2010.05.026
6. Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene (2007) 26(15):2226–42. doi:10.1038/sj.onc.1210291
7. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer (2007) 7(4):295–308. doi:10.1038/nrc2175
8. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339(6127):1546–58. doi:10.1126/science.1235122
9. Cunha GR. Mesenchymal-epithelial interactions: past, present, and future. Differentiation (2008) 76(6):578–86. doi:10.1111/j.1432-0436.2008.00290.x
10. Cress AE, Rabinovitz I, Zhu W, Nagle RB. The α6β1 and α6β4 integrins in human prostate cancer progression. Cancer Metastasis Rev (1995) 14(3):219–28. doi:10.1007/BF00690293
11. Lamb LE, Zarif JC, Miranti CK. The androgen receptor induces integrin alpha6beta1 to promote prostate tumor cell survival via NF-kappaB and Bcl-xL Independently of PI3K signaling. Cancer Res (2011) 71(7):2739–49. doi:10.1158/0008-5472.CAN-10-2745
12. Schalken JA, van Leenders G. Cellular and molecular biology of the prostate: stem cell biology. Urology (2003) 62(5 Suppl 1):11–20. doi:10.1016/S0090-4295(03)00758-1
13. Verhagen AP, Ramaekers FC, Aalders TW, Schaafsma HE, Debruyne FM, Schalken JA. Colocalization of basal and luminal cell-type cytokeratins in human prostate cancer. Cancer Res (1992) 52(22):6182–7.
14. Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol (2013) 15(3):274–83. doi:10.1038/ncb2697
15. de Muga S, Hernandez S, Agell L, Salido M, Juanpere N, Lorenzo M, et al. Molecular alterations of EGFR and PTEN in prostate cancer: association with high-grade and advanced-stage carcinomas. Mod Pathol (2010) 23(5):703–12. doi:10.1038/modpathol.2010.45
16. Peraldo-Neia C, Migliardi G, Mello-Grand M, Montemurro F, Segir R, Pignochino Y, et al. Epidermal growth factor receptor (EGFR) mutation analysis, gene expression profiling and EGFR protein expression in primary prostate cancer. BMC Cancer (2011) 11:31. doi:10.1186/1471-2407-11-31
17. Lamb LE, Knudsen BS, Miranti CK. E-cadherin-mediated survival of androgen-receptor-expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. J Cell Sci (2010) 123(Pt 2):266–76. doi:10.1242/jcs.054502
18. van Leenders G, van Balken B, Aalders T, Hulsbergen-van de Kaa C, Ruiter D, Schalken J. Intermediate cells in normal and malignant prostate epithelium express c-MET: implications for prostate cancer invasion. Prostate (2002) 51(2):98–107. doi:10.1002/pros.10073
19. Carvalho JR, Filipe L, Costa VL, Ribeiro FR, Martins AT, Teixeira MR, et al. Detailed analysis of expression and promoter methylation status of apoptosis-related genes in prostate cancer. Apoptosis (2010) 15(8):956–65. doi:10.1007/s10495-010-0508-6
20. McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, et al. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res (1992) 52(24):6940–4.
21. English HF, Santen RJ, Isaacs JT. Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. Prostate (1987) 11(3):229–42. doi:10.1002/pros.2990110304
22. Isaacs JT, Coffey DS. Etiology and disease process of benign prostatic hyperplasia. Prostate Suppl (1989) 2:33–50. doi:10.1002/pros.2990150506
23. De Marzo AM, Meeker AK, Epstein JI, Coffey DS. Prostate stem cell compartments: expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am J Pathol (1998) 153(3):911–9. doi:10.1016/S0002-9440(10)65632-5
24. Tokar EJ, Ancrile BB, Cunha GR, Webber MM. Stem/progenitor and intermediate cell types and the origin of human prostate cancer. Differentiation (2005) 73(9–10):463–73. doi:10.1111/j.1432-0436.2005.00047.x
25. Ousset M, Van Keymeulen A, Bouvencourt G, Sharma N, Achouri Y, Simons BD, et al. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat Cell Biol (2012) 14(11):1131–8. doi:10.1038/ncb2600
26. Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature (2009) 461(7263):495–500. doi:10.1038/nature08361
27. Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature (2008) 456(7223):804–8. doi:10.1038/nature07427
28. Garraway IP, Sun W, Tran CP, Perner S, Zhang B, Goldstein AS, et al. Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate (2010) 70(5):491–501. doi:10.1002/pros.21083
29. Lawson DA, Xin L, Lukacs RU, Cheng D, Witte ON. Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci U S A (2007) 104(1):181–6. doi:10.1073/pnas.0609684104
30. Xin L, Ide H, Kim Y, Dubey P, Witte ON. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A (2003) 1:11896–903. doi:10.1073/pnas.1734139100
31. Xin L, Lawson DA, Witte ON. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc Natl Acad Sci U S A (2005) 102(19):6942–7. doi:10.1073/pnas.0502320102
32. Xue Y, Smedts F, Debruyne FM, de la Rosette JJ, Schalken JA. Identification of intermediate cell types by keratin expression in the developing human prostate. Prostate (1998) 34(4):292–301. doi:10.1002/(SICI)1097-0045(19980301)34:4<292::AID-PROS7>3.0.CO;2-J
33. Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science (2010) 329(5991):568–71. doi:10.1126/science.1189992
34. Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, Witte ON. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci U S A (2010) 107(6):2610–5. doi:10.1073/pnas.0913873107
35. Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A (2006) 103(5):1480–5. doi:10.1073/pnas.0510652103
36. Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell (2012) 21(2):253–65. doi:10.1016/j.ccr.2012.01.005
37. Pienta KJ, Abate-Shen C, Agus DB, Attar RM, Chung LW, Greenberg NM, et al. The current state of preclinical prostate cancer animal models. Prostate (2008) 68(6):629–39. doi:10.1002/pros.20726
38. Valkenburg KC, Williams BO. Mouse models of prostate cancer. Prostate Cancer (2011) 2011:895238. doi:10.1155/2011/895238
39. Abate-Shen C, Shen MM. Mouse models of prostate carcinogenesis. Trends Genet (2002) 18(5):S1–5. doi:10.1016/S0168-9525(02)02683-5
40. McNeal JE. The zonal anatomy of the prostate. Prostate (1981) 2(1):35–49. doi:10.1002/pros.2990020105
41. Hayward SW, Haughney PC, Rosen MA, Greulich KM, Weier HU, Dahiya R, et al. Interactions between adult human prostatic epithelium and rat urogenital sinus mesenchyme in a tissue recombination model. Differentiation (1998) 63(3):131–40. doi:10.1046/j.1432-0436.1998.6330131.x
42. Goldstein AS, Drake JM, Burnes DL, Finley DS, Zhang H, Reiter RE, et al. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat Protoc (2011) 6(5):656–67. doi:10.1038/nprot.2011.317
43. Heer R, Collins AT, Robson CN, Shenton BK, Leung HY. KGF suppresses α2β1 integrin function and promotes differentiation of the transient amplifying population in human prostatic epithelium. J Cell Sci (2006) 119(Pt 7):1416–24. doi:10.1242/jcs.02802
44. Peehl DM, Wong ST, Stamey TA. Vitamin A regulates proliferation and differentiation of human prostatic epithelial cells. Prostate (1993) 23(1):69–78. doi:10.1002/pros.2990230107
45. van Leenders GJ, Schalken JA. Epithelial cell differentiation in the human prostate epithelium: implications for the pathogenesis and therapy of prostate cancer. Crit Rev Oncol Hematol (2003) 27(46):S3–10. doi:10.1016/S1040-8428(03)00059-3
46. Bluemn EG, Nelson PS. The androgen/androgen receptor axis in prostate cancer. Curr Opin Oncol (2012) 24(3):251–7. doi:10.1097/CCO.0b013e32835105b3
47. Tsao CK, Small AC, Galsky MD, Oh WK. Overcoming castration resistance in prostate cancer. Curr Opin Urol (2012) 22(3):167–74. doi:10.1097/MOU.0b013e3283523b8b
48. Waltering KK, Urbanucci A, Visakorpi T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol (2012) 360(1–2):38–43. doi:10.1016/j.mce.2011.12.019
49. da Silva HB, Amaral EP, Nolasco EL, de Victo NC, Atique R, Jank CC, et al. Dissecting major signaling pathways throughout the development of prostate cancer. Prostate Cancer (2013) 920612(10):29. doi:10.1155/2013/920612
50. Gurel B, Iwata T, Koh CM, Yegnasubramanian S, Nelson WG, De Marzo AM. Molecular alterations in prostate cancer as diagnostic, prognostic, and therapeutic targets. Adv Anat Pathol (2008) 15(6):319–31. doi:10.1097/PAP.0b013e31818a5c19
51. Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and prostate cancer. Genes Cancer (2010) 1(6):617–28. doi:10.1177/1947601910379132
52. Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell (2003) 4(3):223–38. doi:10.1016/S1535-6108(03)00197-1
53. Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, et al. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res (2007) 67(18):8504–10. doi:10.1158/0008-5472.CAN-07-0673
54. Nupponen NN, Kakkola L, Koivisto P, Visakorpi T. Genetic alterations in hormone-refractory recurrent prostate carcinomas. Am J Pathol (1998) 153(1):141–8. doi:10.1016/S0002-9440(10)65554-X
55. Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol (2008) 21(9):1156–67. doi:10.1038/modpathol.2008.111
56. Edwards J, Krishna NS, Witton CJ, Bartlett JM. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin Cancer Res (2003) 9(14):5271–81.
57. Gray IC, Stewart LM, Phillips SM, Hamilton JA, Gray NE, Watson GJ, et al. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br J Cancer (1998) 78(10):1296–300. doi:10.1038/bjc.1998.674
58. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi:10.1038/nrm3330
59. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet (2012) 44(6):685–9. doi:10.1038/ng.2279
60. Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res (2011) 17(20):6563–73. doi:10.1158/1078-0432.CCR-11-1244
61. Verhagen PC, van Duijn PW, Hermans KG, Looijenga LH, van Gurp RJ, Stoop H, et al. The PTEN gene in locally progressive prostate cancer is preferentially inactivated by bi-allelic gene deletion. J Pathol (2006) 208(5):699–707. doi:10.1002/path.1929
62. Yoshimoto M, Ding K, Sweet JM, Ludkovski O, Trottier G, Song KS, et al. PTEN losses exhibit heterogeneity in multifocal prostatic adenocarcinoma and are associated with higher Gleason grade. Mod Pathol (2013) 26(3):435–47. doi:10.1038/modpathol.2012.162
63. Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, et al. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A (2001) 98(20):11563–8. doi:10.1073/pnas.201167798
64. Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol (2003) 1(3):27. doi:10.1371/journal.pbio.0000059
65. Ma X, Ziel-van der Made AC, Autar B, van der Korput HA, Vermeij M, van Duijn P, et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res (2005) 65(13):5730–9. doi:10.1158/0008-5472.CAN-04-4519
66. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell (2003) 4(3):209–21. doi:10.1016/S1535-6108(03)00215-0
67. Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, Soares FA, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer (2007) 97(5):678–85. doi:10.1038/sj.bjc.6603924
68. Yoshimoto M, Cutz JC, Nuin PA, Joshua AM, Bayani J, Evans AJ, et al. Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet Cytogenet (2006) 169(2):128–37. doi:10.1016/j.cancergencyto.2006.04.003
69. Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol (2009) 6(8):429–39. doi:10.1038/nrurol.2009.127
70. Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol (2009) 56(2):275–86. doi:10.1016/j.eururo.2009.04.036
71. Demichelis F, Fall K, Perner S, Andren O, Schmidt F, Setlur SR, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene (2007) 26(31):4596–9. doi:10.1038/sj.onc.1210237
72. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science (2005) 310(5748):644–8. doi:10.1126/science.1117679
73. Furusato B, Gao CL, Ravindranath L, Chen Y, Cullen J, McLeod DG, et al. Mapping of TMPRSS2-ERG fusions in the context of multi-focal prostate cancer. Mod Pathol (2008) 21(2):67–75. doi:10.1038/modpathol.3800981
74. Perner S, Mosquera JM, Demichelis F, Hofer MD, Paris PL, Simko J, et al. TMPRSS2-ERG fusion prostate cancer: an early molecular event associated with invasion. Am J Surg Pathol (2007) 31(6):882–8. doi:10.1097/01.pas.0000213424.38503.aa
75. Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia (2008) 10(2):177–88. doi:10.1593/neo.07822
76. Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell (2010) 17(5):443–54. doi:10.1016/j.ccr.2010.03.018
77. Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene (2008) 27(40):5348–53. doi:10.1038/onc.2008.183
78. Wang J, Cai Y, Yu W, Ren C, Spencer DM, Ittmann M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer Res (2008) 68(20):8516–24. doi:10.1158/0008-5472.CAN-08-1147
79. Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer (2008) 8(7):497–511. doi:10.1038/nrc2402
80. Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med (2010) 16(7):793–8. doi:10.1038/nm.2166
81. Cunha GR, Ricke W, Thomson A, Marker PC, Risbridger G, Hayward SW, et al. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J Steroid Biochem Mol Biol (2004) 92(4):221–36. doi:10.1016/j.jsbmb.2004.10.017
82. Wu CT, Altuwaijri S, Ricke WA, Huang SP, Yeh S, Zhang C, et al. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc Natl Acad Sci U S A (2007) 104(31):12679–84. doi:10.1073/pnas.0704940104
83. Berger R, Febbo PG, Majumder PK, Zhao JJ, Mukherjee S, Signoretti S, et al. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res (2004) 64(24):8867–75. doi:10.1158/0008-5472.CAN-04-2938
84. Whitacre DC, Chauhan S, Davis T, Gordon D, Cress AE, Miesfeld RL. Androgen induction of in vitro prostate cell differentiation. Cell Growth Differ (2002) 13(1):1–11.
85. Shaikhibrahim Z, Wernert N. ETS transcription factors and prostate cancer: the role of the family prototype ETS-1 (review). Int J Oncol (2012) 40(6):1748–54. doi:10.3892/ijo.2012.1380
86. Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med (2013) 19(8):1023–9. doi:10.1038/nm.3216
87. Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet (2009) 41(5):619–24. doi:10.1038/ng.370
88. King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet (2009) 41(5):524–6. doi:10.1038/ng.371
89. Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A (2008) 105(6):2105–10. doi:10.1073/pnas.0711711105
90. Gandarillas A. The mysterious human epidermal cell cycle, or an oncogene-induced differentiation checkpoint. Cell Cycle (2012) 11(24):4507–16. doi:10.4161/cc.22529
91. Watt FM, Frye M, Benitah SA. MYC in mammalian epidermis: how can an oncogene stimulate differentiation? Nat Rev Cancer (2008) 8(3):234–42. doi:10.1038/nrc2328
92. Cascon A, Robledo M. MAX and MYC: a heritable breakup. Cancer Res (2012) 72(13):3119–24. doi:10.1158/0008-5472.CAN-11-3891
93. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc regulates transcriptional pause release. Cell (2010) 141(3):432–45. doi:10.1016/j.cell.2010.03.030
94. Varlakhanova NV, Knoepfler PS. Acting locally and globally: Myc’s ever-expanding roles on chromatin. Cancer Res (2009) 69(19):7487–90. doi:10.1158/0008-5472.CAN-08-4832
95. Luscher B, Vervoorts J. Regulation of gene transcription by the oncoprotein MYC. Gene (2012) 494(2):145–60. doi:10.1016/j.gene.2011.12.027
96. Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M. An alternative pathway for gene regulation by Myc. EMBO J (1997) 16(18):5672–86. doi:10.1093/emboj/16.18.5672
97. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet (2008) 40(1):43–50. doi:10.1038/ng.2007.30
98. Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol (2010) 12(3):247–56. doi:10.1038/ncb2024
99. Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res (2011) 110:77–106. doi:10.1016/B978-0-12-386469-7.00004-9
100. Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem (2006) 281(46):34725–9. doi:10.1074/jbc.R600017200
101. Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell (2004) 116(4):499–509. doi:10.1016/S0092-8674(04)00127-8
102. Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A (2003) 100(14):8164–9. doi:10.1073/pnas.1332764100
103. Buttyan R, Sawczuk IS, Benson MC, Siegal JD, Olsson CA. Enhanced expression of the c-myc protooncogene in high-grade human prostate cancers. Prostate (1987) 11(4):327–37. doi:10.1002/pros.2990110405
104. Fleming WH, Hamel A, MacDonald R, Ramsey E, Pettigrew NM, Johnston B, et al. Expression of the c-myc protooncogene in human prostatic carcinoma and benign prostatic hyperplasia. Cancer Res (1986) 46(3):1535–8.
105. Al Olama AA, Kote-Jarai Z, Giles GG, Guy M, Morrison J, Severi G, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet (2009) 41(10):1058–60. doi:10.1038/ng.452
106. Porkka KP, Tammela TL, Vessella RL, Visakorpi T. RAD21 and KIAA0196 at 8q24 are amplified and overexpressed in prostate cancer. Genes Chromosomes Cancer (2004) 39(1):1–10. doi:10.1002/gcc.10289
107. Tsuchiya N, Kondo Y, Takahashi A, Pawar H, Qian J, Sato K, et al. Mapping and gene expression profile of the minimally overrepresented 8q24 region in prostate cancer. Am J Pathol (2002) 160(5):1799–806. doi:10.1016/S0002-9440(10)61126-1
108. Liu W, Xie CC, Zhu Y, Li T, Sun J, Cheng Y, et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia (2008) 10(8):897–907.
109. Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH. Myc confers androgen-independent prostate cancer cell growth. J Clin Invest (2003) 112(11):1724–31. doi:10.1172/JCI200319035
110. Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One (2013) 8(5):e63563. doi:10.1371/journal.pone.0063563
111. Grad JM, Dai JL, Wu S, Burnstein KL. Multiple androgen response elements and a Myc consensus site in the androgen receptor (AR) coding region are involved in androgen-mediated up-regulation of AR messenger RNA. Mol Endocrinol (1999) 13(11):1896–911. doi:10.1210/me.13.11.1896
112. Nadiminty N, Tummala R, Lou W, Zhu Y, Zhang J, Chen X, et al. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of Myc expression in prostate cancer cells. J Biol Chem (2012) 287(2):1527–37. doi:10.1074/jbc.M111.278705
113. Yu X, Wang Y, Jiang M, Bierie B, Roy-Burman P, Shen MM, et al. Activation of beta-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate (2009) 69(3):249–62. doi:10.1002/pros.20877
114. Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol (2004) 202(2):233–40. doi:10.1002/path.1503
115. Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res (2004) 64(6):1975–86. doi:10.1158/0008-5472.CAN-03-3972
116. Zhu H, Zhou X, Redfield S, Lewin J, Miele L. Elevated Jagged-1 and Notch-1 expression in high grade and metastatic prostate cancers. Am J Transl Res (2013) 5(3):368–78.
117. Bin Hafeez B, Adhami VM, Asim M, Siddiqui IA, Bhat KM, Zhong W, et al. Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin Cancer Res (2009) 15(2):452–9. doi:10.1158/1078-0432.CCR-08-1631
118. Zhang X, Lee C, Ng PY, Rubin M, Shabsigh A, Buttyan R. Prostatic neoplasia in transgenic mice with prostate-directed overexpression of the c-myc oncoprotein. Prostate (2000) 43(4):278–85. doi:10.1002/1097-0045(20000601)43:4<278::AID-PROS7>3.0.CO;2-4
119. Schreiber-Agus N, Meng Y, Hoang T, Hou H Jr, Chen K, Greenberg R, et al. Role of Mxi1 in ageing organ systems and the regulation of normal and neoplastic growth. Nature (1998) 393(6684):483–7. doi:10.1038/31008
120. Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev (2008) 22(20):2755–66. doi:10.1101/gad.1712408
121. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
122. Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A (2010) 107(20):9222–7. doi:10.1073/pnas.1004584107
123. Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol (2008) 26(1):101–6. doi:10.1038/nbt1374
124. Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development (2005) 132(5):885–96. doi:10.1242/dev.01670
125. Varlakhanova NV, Cotterman RF, deVries WN, Morgan J, Donahue LR, Murray S, et al. Myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation (2010) 80(1):9–19. doi:10.1016/j.diff.2010.05.001
126. Xu D, Popov N, Hou M, Wang Q, Bjorkholm M, Gruber A, et al. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci U S A (2001) 98(7):3826–31. doi:10.1073/pnas.071043198
127. Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, et al. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev (2001) 15(16):2042–7. doi:10.1101/gad.907901
128. Sumi T, Tsuneyoshi N, Nakatsuji N, Suemori H. Apoptosis and differentiation of human embryonic stem cells induced by sustained activation of c-Myc. Oncogene (2007) 26(38):5564–76. doi:10.1038/sj.onc.1210353
129. Bull JJ, Muller-Rover S, Patel SV, Chronnell CM, McKay IA, Philpott MP. Contrasting localization of c-Myc with other Myc superfamily transcription factors in the human hair follicle and during the hair growth cycle. J Invest Dermatol (2001) 116(4):617–22. doi:10.1046/j.1523-1747.2001.12771234.x
130. Gandarillas A, Watt FM. Changes in expression of members of the fos and jun families and myc network during terminal differentiation of human keratinocytes. Oncogene (1995) 11(7):1403–7.
131. Gandarillas A, Watt FM. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev (1997) 11(21):2869–82. doi:10.1101/gad.11.21.2869
132. Gebhardt A, Frye M, Herold S, Benitah SA, Braun K, Samans B, et al. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J Cell Biol (2006) 172(1):139–49. doi:10.1083/jcb.200506057
133. Hashiro M, Matsumoto K, Okumura H, Hashimoto K, Yoshikawa K. Growth inhibition of human keratinocytes by antisense c-myc oligomer is not coupled to induction of differentiation. Biochem Biophys Res Commun (1991) 174(1):287–92. doi:10.1016/0006-291X(91)90518-C
134. Watt FM. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J (2002) 21(15):3919–26. doi:10.1093/emboj/cdf399
135. McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol (2000) 20(2):556–62. doi:10.1128/MCB.20.2.556-562.2000
136. Pellakuru LG, Iwata T, Gurel B, Schultz D, Hicks J, Bethel C, et al. Global levels of H3K27me3 track with differentiation in vivo and are deregulated by MYC in prostate cancer. Am J Pathol (2012) 181(2):560–9. doi:10.1016/j.ajpath.2012.04.021
137. Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell (2008) 2(5):437–47. doi:10.1016/j.stem.2008.03.021
138. Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget (2011) 2(9):669–83.
139. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science (2012) 338(6113):1465–9. doi:10.1126/science.1227604
140. Dong JT. Prevalent mutations in prostate cancer. J Cell Biochem (2006) 97(3):433–47. doi:10.1002/jcb.20696
141. Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development (2003) 130(12):2793–808. doi:10.1242/dev.00462
142. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J (2010) 429(3):403–17. doi:10.1042/BJ20100323
143. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer (2009) 9(8):537–49. doi:10.1038/nrc2694
144. Belleudi F, Purpura V, Torrisi MR. The receptor tyrosine kinase FGFR2b/KGFR controls early differentiation of human keratinocytes. PLoS One (2011) 6(9):21. doi:10.1371/journal.pone.0024194
145. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer (2010) 10(2):116–29. doi:10.1038/nrc2780
146. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev (2008) 27(2):253–61. doi:10.1007/s10555-008-9123-1
147. Uzgare AR, Kaplan PJ, Greenberg NM. Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate (2003) 55(2):128–39. doi:10.1002/pros.10212
148. Rajashekhar G, Kamocka M, Marin A, Suckow MA, Wolter WR, Badve S, et al. Pro-inflammatory angiogenesis is mediated by p38 MAP kinase. J Cell Physiol (2011) 226(3):800–8. doi:10.1002/jcp.22404
149. Royuela M, Arenas MI, Bethencourt FR, Sanchez-Chapado M, Fraile B, Paniagua R. Regulation of proliferation/apoptosis equilibrium by mitogen-activated protein kinases in normal, hyperplastic, and carcinomatous human prostate. Hum Pathol (2002) 33(3):299–306. doi:10.1053/hupa.2002.32227
150. Lotan TL, Lyon M, Huo D, Taxy JB, Brendler C, Foster BA, et al. Up-regulation of MKK4, MKK6 and MKK7 during prostate cancer progression: an important role for SAPK signalling in prostatic neoplasia. J Pathol (2007) 212(4):386–94. doi:10.1002/path.2194
151. Ricote M, Garcia-Tunon I, Bethencourt F, Fraile B, Onsurbe P, Paniagua R, et al. The p38 transduction pathway in prostatic neoplasia. J Pathol (2006) 208(3):401–7. doi:10.1002/path.1910
152. Ricote M, Garcia-Tunon I, Fraile B, Fernandez C, Aller P, Paniagua R, et al. P38 MAPK protects against TNF-alpha-provoked apoptosis in LNCaP prostatic cancer cells. Apoptosis (2006) 11(11):1969–75. doi:10.1007/s10495-006-0086-9
153. Loda M, Capodieci P, Mishra R, Yao H, Corless C, Grigioni W, et al. Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol (1996) 149(5):1553–64.
154. Rauhala HE, Porkka KP, Tolonen TT, Martikainen PM, Tammela TL, Visakorpi T. Dual-specificity phosphatase 1 and serum/glucocorticoid-regulated kinase are downregulated in prostate cancer. Int J Cancer (2005) 117(5):738–45. doi:10.1002/ijc.21270
155. Magi-Galluzzi C, Mishra R, Fiorentino M, Montironi R, Yao H, Capodieci P, et al. Mitogen-activated protein kinase phosphatase 1 is overexpressed in prostate cancers and is inversely related to apoptosis. Lab Invest (1997) 76(1):37–51.
156. Magi-Galluzzi C, Montironi R, Cangi MG, Wishnow K, Loda M. Mitogen-activated protein kinases and apoptosis in PIN. Virchows Arch (1998) 432(5):407–13. doi:10.1007/s004280050184
157. Ross AE, Marchionni L, Vuica-Ross M, Cheadle C, Fan J, Berman DM, et al. Gene expression pathways of high grade localized prostate cancer. Prostate (2011) 25(10):21373. doi:10.1002/pros.21373
158. Manetsch M, Che W, Seidel P, Chen Y, Ammit AJ. MKP-1: a negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cell Signal (2012) 24(4):907–13. doi:10.1016/j.cellsig.2011.12.013
159. Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem (2002) 277(9):7076–85. doi:10.1074/jbc.M108255200
160. Zauberman A, Zipori D, Krupsky M, Ben-Levy R. Stress activated protein kinase p38 is involved in IL-6 induced transcriptional activation of STAT3. Oncogene (1999) 18(26):3886–93. doi:10.1038/sj.onc.1202738
161. Culig Z, Puhr M. Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol (2012) 360(1–2):52–8. doi:10.1016/j.mce.2011.05.033
162. Lin DL, Whitney MC, Yao Z, Keller ET. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin Cancer Res (2001) 7(6):1773–81.
163. Gan L, Wang J, Xu H, Yang X. Resistance to docetaxel-induced apoptosis in prostate cancer cells by p38/p53/p21 signaling. Prostate (2011) 71(11):1158–66. doi:10.1002/pros.21331
164. Liu C, Zhu Y, Lou W, Nadiminty N, Chen X, Zhou Q, et al. Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate (2013) 73(4):418–27. doi:10.1002/pros.22583
165. Frey MR, Golovin A, Polk DB. Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J Biol Chem (2004) 279(43):44513–21. doi:10.1074/jbc.M406253200
166. Lewis DA, Spandau DF. UVB-induced activation of NF-κB is regulated by the IGF-1R and dependent on p38 MAPK. J Invest Dermatol (2008) 128(4):1022–9. doi:10.1038/sj.jid.5701127
167. McCawley LJ, Li S, Wattenberg EV, Hudson LG. Sustained activation of the mitogen-activated protein kinase pathway. A mechanism underlying receptor tyrosine kinase specificity for matrix metalloproteinase-9 induction and cell migration. J Biol Chem (1999) 274(7):4347–53. doi:10.1074/jbc.274.7.4347
168. Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer (2004) 11(4):709–24. doi:10.1677/erc.1.00535
169. Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell (2007) 12(6):572–85. doi:10.1016/j.ccr.2007.11.002
170. Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell (2007) 12(6):559–71. doi:10.1016/j.ccr.2007.11.004
171. Kwabi-Addo B, Ropiquet F, Giri D, Ittmann M. Alternative splicing of fibroblast growth factor receptors in human prostate cancer. Prostate (2001) 46(2):163–72. doi:10.1002/1097-0045(20010201)46:2<163::AID-PROS1020>3.0.CO;2-T
172. Houde M, Laprise P, Jean D, Blais M, Asselin C, Rivard N. Intestinal epithelial cell differentiation involves activation of p38 mitogen-activated protein kinase that regulates the homeobox transcription factor CDX2. J Biol Chem (2001) 276(24):21885–94. doi:10.1074/jbc.M100236200
173. Sharma GD, He J, Bazan HE. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem (2003) 278(24):21989–97. doi:10.1074/jbc.M302650200
174. Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, et al. p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet (2007) 39(6):750–8. doi:10.1038/ng2037
175. Wang X, Goh CH, Li B. p38 Mitogen-activated protein kinase regulates osteoblast differentiation through osterix. Endocrinology (2007) 148(4):1629–37. doi:10.1210/en.2006-1000
176. Dashti SR, Efimova T, Eckert RL. MEK6 regulates human involucrin gene expression via a p38α – and p38δ -dependent mechanism. J Biol Chem (2001) 276(29):27214–20. doi:10.1074/jbc.M100465200
177. Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38α results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med (2000) 191(5):859–70. doi:10.1084/jem.191.5.859
178. Sabio G, Arthur JS, Kuma Y, Peggie M, Carr J, Murray-Tait V, et al. p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J (2005) 24(6):1134–45. doi:10.1038/sj.emboj.7600578
179. Schindler EM, Hindes A, Gribben EL, Burns CJ, Yin Y, Lin MH, et al. p38delta Mitogen-activated protein kinase is essential for skin tumor development in mice. Cancer Res (2009) 69(11):4648–55. doi:10.1158/0008-5472.CAN-08-4455
180. Efimova T, Broome AM, Eckert RL. A regulatory role for p38δ MAPK in keratinocyte differentiation. Evidence for p38δ-ERK1/2 complex formation. J Biol Chem (2003) 278(36):34277–85. doi:10.1074/jbc.M302759200
181. Keren A, Tamir Y, Bengal E. The p38 MAPK signaling pathway: a major regulator of skeletal muscle development. Mol Cell Endocrinol (2006) 252(1–2):224–30. doi:10.1016/j.mce.2006.03.017
182. Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev (2005) 19(10):1175–87. doi:10.1101/gad.1306705
183. Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, et al. p38alpha Suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet (2007) 39(6):741–9. doi:10.1038/ng2033
184. Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, et al. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev (2000) 14(5):574–84. doi:10.1101/gad.14.5.574
185. Hall JP, Davis RJ. Inhibition of the p38 pathway upregulates macrophage JNK and ERK activities, and the ERK, JNK, and p38 MAP kinase pathways are reprogrammed during differentiation of the murine myeloid M1 cell line. J Cell Biochem (2002) 86(1):1–11. doi:10.1002/jcb.10187
186. Danilenko DM. Preclinical and early clinical development of keratinocyte growth factor, an epithelial-specific tissue growth factor. Toxicol Pathol (1999) 27(1):64–71. doi:10.1177/019262339902700113
187. Mehta PB, Robson CN, Neal DE, Leung HY. Keratinocyte growth factor activates p38 MAPK to induce stress fibre formation in human prostate DU145 cells. Oncogene (2001) 20(38):5359–65. doi:10.1038/sj.onc.1204688
188. Miki T, Bottaro DP, Fleming TP, Smith CL, Burgess WH, Chan AM, et al. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci U S A (1992) 89(1):246–50. doi:10.1073/pnas.89.1.246
189. Donjacour AA, Thomson AA, Cunha GR. FGF-10 plays an essential role in the growth of the fetal prostate. Dev Biol (2003) 261(1):39–54. doi:10.1016/S0012-1606(03)00250-1
190. Lin Y, Liu G, Zhang Y, Hu YP, Yu K, Lin C, et al. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development (2007) 134(4):723–34. doi:10.1242/dev.02765
191. Leong KG, Gao WQ. The notch pathway in prostate development and cancer. Differentiation (2008) 76(6):699–716. doi:10.1111/j.1432-0436.2008.00288.x
192. Kopan R, Ilagan MX. The canonical notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137(2):216–33. doi:10.1016/j.cell.2009.03.045
193. Wang MM. Notch signaling and notch signaling modifiers. Int J Biochem Cell Biol (2011) 43(11):1550–62. doi:10.1016/j.biocel.2011.08.005
194. Fischer A, Gessler M. Delta-notch – and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res (2007) 35(14):4583–96. doi:10.1093/nar/gkm477
195. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev (2006) 20(15):2096–109. doi:10.1101/gad.1450406
196. Shimizu K, Chiba S, Saito T, Kumano K, Hamada Y, Hirai H. Functional diversity among Notch1, Notch2, and Notch3 receptors. Biochem Biophys Res Commun (2002) 291(4):775–9. doi:10.1006/bbrc.2002.6528
197. Yuan Z, Friedmann DR, VanderWielen BD, Collins KJ, Kovall RA. Characterization of CSL (CBF-1, Su(H), Lag-1) mutants reveals differences in signaling mediated by Notch1 and Notch2. J Biol Chem (2012) 287(42):34904–16. doi:10.1074/jbc.M112.403287
198. Barolo S, Walker RG, Polyanovsky AD, Freschi G, Keil T, Posakony JW. A notch-independent activity of suppressor of hairless is required for normal mechanoreceptor physiology. Cell (2000) 103(6):957–69. doi:10.1016/S0092-8674(00)00198-7
199. Martinez Arias A, Zecchini V, Brennan K. CSL-independent Notch signalling: a checkpoint in cell fate decisions during development? Curr Opin Genet Dev (2002) 12(5):524–33. doi:10.1016/S0959-437X(02)00336-2
200. Johnson JE, Macdonald RJ. Notch-independent functions of CSL. Curr Top Dev Biol (2011) 97:55–74. doi:10.1016/B978-0-12-385975-4.00009-7
201. Minter LM, Osborne BA. Canonical and non-canonical notch signaling in CD4(+) T cells. Curr Top Microbiol Immunol (2012) 360:99–114. doi:10.1007/82_2012_233
202. Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell (2009) 16(5):633–47. doi:10.1016/j.devcel.2009.03.010
203. Roy M, Pear WS, Aster JC. The multifaceted role of notch in cancer. Curr Opin Genet Dev (2007) 17(1):52–9. doi:10.1016/j.gde.2006.12.001
204. Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res (2007) 67(5):1879–82. doi:10.1158/0008-5472.CAN-06-3958
205. Tzoneva G, Ferrando AA. Recent advances on NOTCH signaling in T-ALL. Curr Top Microbiol Immunol (2012) 360:163–82. doi:10.1007/82_2012_232
206. Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest (2012) 122(10):3398–406. doi:10.1172/JCI61269
207. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306(5694):269–71. doi:10.1126/science.1102160
208. Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A (2011) 108(43):17761–6. doi:10.1073/pnas.1114669108
209. Demehri S, Turkoz A, Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell (2009) 16(1):55–66. doi:10.1016/j.ccr.2009.05.016
210. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer (2011) 11(5):338–51. doi:10.1038/nrc3035
211. South AP, Cho RJ, Aster JC. The double-edged sword of Notch signaling in cancer. Semin Cell Dev Biol (2012) 23(4):458–64. doi:10.1016/j.semcdb.2012.01.017
212. Belandia B, Powell SM, Garcia-Pedrero JM, Walker MM, Bevan CL, Parker MG. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol Cell Biol (2005) 25(4):1425–36. doi:10.1128/MCB.25.4.1425-1436.2005
213. Lavery DN, Villaronga MA, Walker MM, Patel A, Belandia B, Bevan CL. Repression of androgen receptor activity by HEYL, a third member of the Hairy/Enhancer-of-split-related family of Notch effectors. J Biol Chem (2011) 286(20):17796–808. doi:10.1074/jbc.M110.198655
214. Wang XD, Leow CC, Zha J, Tang Z, Modrusan Z, Radtke F, et al. Notch signaling is required for normal prostatic epithelial cell proliferation and differentiation. Dev Biol (2006) 290(1):66–80. doi:10.1016/j.ydbio.2005.11.009
215. Whelan JT, Kellogg A, Shewchuk BM, Hewan-Lowe K, Bertrand FE. Notch-1 signaling is lost in prostate adenocarcinoma and promotes PTEN gene expression. J Cell Biochem (2009) 107(5):992–1001. doi:10.1002/jcb.22199
216. Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res (2004) 64(19):6854–7. doi:10.1158/0008-5472.CAN-04-2500
217. Danza G, Di Serio C, Ambrosio MR, Sturli N, Lonetto G, Rosati F, et al. Notch3 is activated by chronic hypoxia and contributes to the progression of human prostate cancer. Int J Cancer (2013) 1(10):28293. doi:10.1002/ijc.28293
218. Shou J, Ross S, Koeppen H, de Sauvage FJ, Gao WQ. Dynamics of notch expression during murine prostate development and tumorigenesis. Cancer Res (2001) 61(19):7291–7.
219. Yong T, Sun A, Henry MD, Meyers S, Davis JN. Down regulation of CSL activity inhibits cell proliferation in prostate and breast cancer cells. J Cell Biochem (2011) 112(9):2340–51. doi:10.1002/jcb.23157
220. Wang XD, Shou J, Wong P, French DM, Gao WQ. Notch1-expressing cells are indispensable for prostatic branching morphogenesis during development and re-growth following castration and androgen replacement. J Biol Chem (2004) 279(23):24733–44. doi:10.1074/jbc.M401602200
221. Ye QF, Zhang YC, Peng XQ, Long Z, Ming YZ, He LY. Silencing Notch-1 induces apoptosis and increases the chemosensitivity of prostate cancer cells to docetaxel through Bcl-2 and Bax. Oncol Lett (2012) 3(4):879–84. doi:10.3892/ol.2012.572
222. Long Q, Johnson BA, Osunkoya AO, Lai YH, Zhou W, Abramovitz M, et al. Protein-coding and microRNA biomarkers of recurrence of prostate cancer following radical prostatectomy. Am J Pathol (2011) 179(1):46–54. doi:10.1016/j.ajpath.2011.03.008
223. Danza G, Di Serio C, Rosati F, Lonetto G, Sturli N, Kacer D, et al. Notch signaling modulates hypoxia-induced neuroendocrine differentiation of human prostate cancer cells. Mol Cancer Res (2012) 10(2):230–8. doi:10.1158/1541-7786.MCR-11-0296
224. Orr B, Grace OC, Vanpoucke G, Ashley GR, Thomson AA. A role for notch signaling in stromal survival and differentiation during prostate development. Endocrinology (2009) 150(1):463–72. doi:10.1210/en.2008-0383
225. Wu X, Xu K, Zhang L, Deng Y, Lee P, Shapiro E, et al. Differentiation of the ductal epithelium and smooth muscle in the prostate gland are regulated by the Notch/PTEN-dependent mechanism. Dev Biol (2011) 356(2):337–49. doi:10.1016/j.ydbio.2011.05.659
226. Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 Regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol (2006) 8(6):551–61. doi:10.1038/ncb1420
227. Nguyen BC, Lefort K, Mandinova A, Antonini D, Devgan V, Della Gatta G, et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev (2006) 20(8):1028–42. doi:10.1101/gad.1406006
228. Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development (2012) 139(4):772–82. doi:10.1242/dev.071191
229. Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, et al. Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis (2003) 37(3):139–43. doi:10.1002/gene.10241
230. Beatus P, Lundkvist J, Oberg C, Lendahl U. The notch 3 intracellular domain represses notch 1-mediated activation through Hairy/Enhancer of split (HES) promoters. Development (1999) 126(17):3925–35.
231. Dang TP, Eichenberger S, Gonzalez A, Olson S, Carbone DP. Constitutive activation of Notch3 inhibits terminal epithelial differentiation in lungs of transgenic mice. Oncogene (2003) 22(13):1988–97. doi:10.1038/sj.onc.1206230
232. Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L, et al. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology (2010) 139(6):2113–23. doi:10.1053/j.gastro.2010.08.040
233. Watt FM, Estrach S, Ambler CA. Epidermal notch signalling: differentiation, cancer and adhesion. Curr Opin Cell Biol (2008) 20(2):171–9. doi:10.1016/j.ceb.2008.01.010
234. Okuyama R, Tagami H, Aiba S. Notch signaling: its role in epidermal homeostasis and in the pathogenesis of skin diseases. J Dermatol Sci (2008) 49(3):187–94. doi:10.1016/j.jdermsci.2007.05.017
235. Brown MD, Gilmore PE, Hart CA, Samuel JD, Ramani VA, George NJ, et al. Characterization of benign and malignant prostate epithelial Hoechst 33342 side populations. Prostate (2007) 67(13):1384–96. doi:10.1002/pros.20620
236. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A (2006) 103(48):18261–6. doi:10.1073/pnas.0606108103
237. Franke TF. PI3K/Akt: getting it right matters. Oncogene (2008) 27(50):6473–88. doi:10.1038/onc.2008.313
238. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer (2009) 9(8):550–62. doi:10.1038/nrc2664
239. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev (2010) 20(1):87–90. doi:10.1016/j.gde.2009.11.002
240. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene (2006) 25(48):6416–22. doi:10.1038/sj.onc.1209888
241. Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J (2009) 418(3):475–89. doi:10.1042/BJ20082234
242. Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res (2011) 71(8):2821–5. doi:10.1158/0008-5472.CAN-10-3399
243. Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science (1998) 280(5369):1614–7. doi:10.1126/science.280.5369.1614
244. Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, et al. Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. J Cell Sci (2006) 119(Pt 24):5160–8. doi:10.1242/jcs.000133
245. Ming M, He YY. PTEN in DNA damage repair. Cancer Lett (2012) 319(2):125–9. doi:10.1016/j.canlet.2012.01.003
246. Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol (2008) 28(10):3281–9. doi:10.1128/MCB.00310-08
247. Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell (2003) 3(2):117–30. doi:10.1016/S1535-6108(03)00021-7
248. Zhang XP, Liu F, Wang W. Two-phase dynamics of p53 in the DNA damage response. Proc Natl Acad Sci U S A (2011) 108(22):8990–5. doi:10.1073/pnas.1100600108
249. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell (2007) 128(1):157–70. doi:10.1016/j.cell.2006.11.042
250. Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, et al. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell (2007) 128(2):383–97. doi:10.1016/j.cell.2006.11.051
251. Tepass U. The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu Rev Cell Dev Biol (2012) 28:655–85. doi:10.1146/annurev-cellbio-092910-154033
252. Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review). Int J Oncol (2012) 40(3):639–44. doi:10.3892/ijo.2011.1312
253. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275(5308):1943–7. doi:10.1126/science.275.5308.1943
254. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet (1997) 15(4):356–62. doi:10.1038/ng0497-356
255. McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res (1999) 59(17):4291–6.
256. Schmitz M, Grignard G, Margue C, Dippel W, Capesius C, Mossong J, et al. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int J Cancer (2007) 120(6):1284–92. doi:10.1002/ijc.22359
257. Cuzick J, Yang ZH, Fisher G, Tikishvili E, Stone S, Lanchbury JS, et al. Prognostic value of PTEN loss in men with conservatively managed localised prostate cancer. Br J Cancer (2013) 108(12):2582–9. doi:10.1038/bjc.2013.248
258. Sun X, Huang J, Homma T, Kita D, Klocker H, Schafer G, et al. Genetic alterations in the PI3K pathway in prostate cancer. Anticancer Res (2009) 29(5):1739–43.
259. Han B, Mehra R, Lonigro RJ, Wang L, Suleman K, Menon A, et al. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod Pathol (2009) 22(8):1083–93. doi:10.1038/modpathol.2009.69
260. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature (2011) 470(7333):214–20. doi:10.1038/nature09744
261. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi:10.1038/nature11125
262. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer (2011) 11(4):289–301. doi:10.1038/nrc3037
263. Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA (2010) 304(24):2724–31. doi:10.1001/jama.2010.1877
264. Muller I, Wischnewski F, Pantel K, Schwarzenbach H. Promoter- and cell-specific epigenetic regulation of CD44, Cyclin D2, GLIPR1 and PTEN by methyl-CpG binding proteins and histone modifications. BMC Cancer (2010) 10:297. doi:10.1186/1471-2407-10-297
265. Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b ∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal (2010) 3(117):2000594. doi:10.1126/scisignal.2000594
266. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature (2010) 465(7301):1033–8. doi:10.1038/nature09144
267. Wu Z, He B, He J, Mao X. Upregulation of miR-153 promotes cell proliferation via downregulation of the PTEN tumor suppressor gene in human prostate cancer. Prostate (2013) 73(6):596–604. doi:10.1002/pros.22600
268. Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A (2003) 100(13):7841–6. doi:10.1073/pnas.1232229100
269. Reid AH, Attard G, Brewer D, Miranda S, Riisnaes R, Clark J, et al. Novel, gross chromosomal alterations involving PTEN cooperate with allelic loss in prostate cancer. Mod Pathol (2012) 25(6):902–10. doi:10.1038/modpathol.2011.207
270. Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, Ludkovski O, et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol (2008) 21(12):1451–60. doi:10.1038/modpathol.2008.96
271. Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A (2009) 106(30):12465–70. doi:10.1073/pnas.0905931106
272. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol (2001) 21(3):893–901. doi:10.1128/MCB.21.3.893-901.2001
273. Lu TL, Huang YF, You LR, Chao NC, Su FY, Chang JL, et al. Conditionally ablated Pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice. Am J Pathol (2013) 182(3):975–91. doi:10.1016/j.ajpath.2012.11.025
274. Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci (2013) 1280:30–4. doi:10.1111/nyas.12059
275. Watanabe H, Saito H, Ueda J, Evers BM. Regulation of pancreatic duct cell differentiation by phosphatidylinositol-3 kinase. Biochem Biophys Res Commun (2008) 370(1):33–7. doi:10.1016/j.bbrc.2008.03.012
276. Oyama K, Okawa T, Nakagawa H, Takaoka M, Andl CD, Kim SH, et al. AKT induces senescence in primary esophageal epithelial cells but is permissive for differentiation as revealed in organotypic culture. Oncogene (2007) 26(16):2353–64. doi:10.1038/sj.onc.1210025
277. Sayama K, Yamasaki K, Hanakawa Y, Shirakata Y, Tokumaru S, Ijuin T, et al. Phosphatidylinositol 3-kinase is a key regulator of early phase differentiation in keratinocytes. J Biol Chem (2002) 277(43):40390–6. doi:10.1074/jbc.M112423200
278. Uzan B, Figeac F, Portha B, Movassat J. Mechanisms of KGF mediated signaling in pancreatic duct cell proliferation and differentiation. PLoS One (2009) 4(3):6. doi:10.1371/journal.pone.0004734
279. Laprise P, Chailler P, Houde M, Beaulieu JF, Boucher MJ, Rivard N. Phosphatidylinositol 3-kinase controls human intestinal epithelial cell differentiation by promoting adherens junction assembly and p38 MAPK activation. J Biol Chem (2002) 277(10):8226–34. doi:10.1074/jbc.M110235200
280. Laprise P, Langlois MJ, Boucher MJ, Jobin C, Rivard N. Down-regulation of MEK/ERK signaling by E-cadherin-dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J Cell Physiol (2004) 199(1):32–9. doi:10.1002/jcp.10432
281. Lemieux E, Boucher MJ, Mongrain S, Boudreau F, Asselin C, Rivard N. Constitutive activation of the MEK/ERK pathway inhibits intestinal epithelial cell differentiation. Am J Physiol Gastrointest Liver Physiol (2011) 301(4):7. doi:10.1152/ajpgi.00508.2010
282. Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PF. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J Biol Chem (2005) 280(38):32856–65. doi:10.1074/jbc.M506119200
283. Edick MJ, Tesfay L, Lamb LE, Knudsen BS, Miranti CK. Inhibition of integrin-mediated crosstalk with epidermal growth factor receptor/Erk or Src signaling pathways in autophagic prostate epithelial cells induces caspase-independent death. Mol Biol Cell (2007) 18(7):2481–90. doi:10.1091/mbc.E06-04-0261
284. Ghosh S, Varela L, Sood A, Park BH, Lotan T. mTOR signaling feedback modulates mammary epithelial differentiation and restrains invasion downstream of PTEN loss. Cancer Res (2013) 73(16):5218–31. doi:10.1158/0008-5472.CAN-13-0429
285. Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science (2001) 294(5549):2186–9. doi:10.1126/science.1065518
286. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature (2006) 441(7092):475–82. doi:10.1038/nature04703
287. Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature (2006) 441(7092):518–22. doi:10.1038/nature04747
288. Gassama-Diagne A, Yu W, ter Beest M, Martin-Belmonte F, Kierbel A, Engel J, et al. Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat Cell Biol (2006) 8(9):963–70. doi:10.1038/ncb1461
289. Horne-Badovinac S, Lin D, Waldron S, Schwarz M, Mbamalu G, Pawson T, et al. Positional cloning of heart and soul reveals multiple roles for PKCλ in zebrafish organogenesis. Curr Biol (2001) 11(19):1492–502. doi:10.1016/S0960-9822(01)00458-4
290. Munro EM. PAR proteins and the cytoskeleton: a marriage of equals. Curr Opin Cell Biol (2006) 18(1):86–94. doi:10.1016/j.ceb.2005.12.007
291. Yu W, Datta A, Leroy P, O’Brien LE, Mak G, Jou TS, et al. Beta1-integrin orients epithelial polarity via Rac1 and laminin. Mol Biol Cell (2005) 16(2):433–45. doi:10.1091/mbc.E04-05-0435
292. Langlois MJ, Bergeron S, Bernatchez G, Boudreau F, Saucier C, Perreault N, et al. The PTEN phosphatase controls intestinal epithelial cell polarity and barrier function: role in colorectal cancer progression. PLoS One (2010) 5(12):0015742. doi:10.1371/journal.pone.0015742
293. Roy SA, Langlois MJ, Carrier JC, Boudreau F, Rivard N, Perreault N. Dual regulatory role for phosphatase and tensin homolog in specification of intestinal endocrine cell subtypes. World J Gastroenterol (2012) 18(14):1579–89. doi:10.3748/wjg.v18.i14.1579
294. Vazquez de la Torre A, Junyent F, Folch J, Pelegri C, Vilaplana J, Auladell C, et al. PI3 k/akt inhibition induces apoptosis through p38 activation in neurons. Pharmacol Res (2013) 70(1):116–25. doi:10.1016/j.phrs.2013.01.007
295. Alimirah F, Chen J, Basrawala Z, Xin H, Choubey D. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: implications for the androgen receptor functions and regulation. FEBS Lett (2006) 580(9):2294–300. doi:10.1016/j.febslet.2006.03.041
296. Buchanan G, Craft PS, Yang M, Cheong A, Prescott J, Jia L, et al. PC-3 cells with enhanced androgen receptor signaling: a model for clonal selection in prostate cancer. Prostate (2004) 60(4):352–66. doi:10.1002/pros.20079
297. Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol (2009) 29(16):4341–51. doi:10.1128/MCB.00210-09
298. Yeap BB, Wilce JA, Leedman PJ. The androgen receptor mRNA. Bioessays (2004) 26(6):672–82. doi:10.1002/bies.20051
299. Barker A, Epis MR, Porter CJ, Hopkins BR, Wilce MC, Wilce JA, et al. Sequence requirements for RNA binding by HuR and AUF1. J Biochem (2012) 151(4):423–37. doi:10.1093/jb/mvs010
300. Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A (2010) 107(11):5012–7. doi:10.1073/pnas.1000896107
301. Jaworski T. Degradation and beyond: control of androgen receptor activity by the proteasome system. Cell Mol Biol Lett (2006) 11(1):109–31. doi:10.2478/s11658-006-0011-9
302. Lin HK, Hu YC, Lee DK, Chang C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol Endocrinol (2004) 18(10):2409–23. doi:10.1210/me.2004-0117
303. Dean JL, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal (2004) 16(10):1113–21. doi:10.1016/j.cellsig.2004.04.006
304. Marderosian M, Sharma A, Funk AP, Vartanian R, Masri J, Jo OD, et al. Tristetraprolin regulates Cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene (2006) 25(47):6277–90. doi:10.1038/sj.onc.1209645
305. Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev (2009) 23(15):1743–8. doi:10.1101/gad.1812509
306. Liu L, Rao JN, Zou T, Xiao L, Wang PY, Turner DJ, et al. Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol Biol Cell (2009) 20(23):4885–98. doi:10.1091/mbc.E09-07-0550
307. Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem (1996) 271(14):8144–51. doi:10.1074/jbc.271.14.8144
308. Cho SJ, Jung YS, Zhang J, Chen X. The RNA-binding protein RNPC1 stabilizes the mRNA encoding the RNA-binding protein HuR and cooperates with HuR to suppress cell proliferation. J Biol Chem (2012) 287(18):14535–44. doi:10.1074/jbc.M111.326827
309. Jiang P, Smith AD, Li R, Rao JN, Liu L, Donahue JM, et al. Sphingosine kinase 1 overexpression stimulates intestinal epithelial cell proliferation through increased c-Myc translation. Am J Physiol Cell Physiol (2013) 304(12):10. doi:10.1152/ajpcell.00271.2012
310. Talwar S, Jin J, Carroll B, Liu A, Gillespie MB, Palanisamy V. Caspase-mediated cleavage of RNA-binding protein HuR regulates c-Myc protein expression after hypoxic stress. J Biol Chem (2011) 286(37):32333–43. doi:10.1074/jbc.M111.255927
311. Gonsalves FC, Weisblat DA. MAPK regulation of maternal and zygotic Notch transcript stability in early development. Proc Natl Acad Sci U S A (2007) 104(2):531–6. doi:10.1073/pnas.0609851104
312. Garcia-Dominguez DJ, Morello D, Cisneros E, Kontoyiannis DL, Frade JM. Stabilization of Dll1 mRNA by Elavl1/HuR in neuroepithelial cells undergoing mitosis. Mol Biol Cell (2011) 22(8):1227–39. doi:10.1091/mbc.E10-10-0808
313. Hollenhorst PC, Paul L, Ferris MW, Graves BJ. The ETS gene ETV4 is required for anchorage-independent growth and a cell proliferation gene expression program in PC3 prostate cells. Genes Cancer (2011) 1(10):1044–52. doi:10.1177/1947601910395578
314. Shortt J, Martin BP, Newbold A, Hannan KM, Devlin JR, Baker AJ, et al. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood (2013) 121(15):2964–74. doi:10.1182/blood-2012-08-446096
315. Gutierrez A, Grebliunaite R, Feng H, Kozakewich E, Zhu S, Guo F, et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J Exp Med (2011) 208(8):1595–603. doi:10.1084/jem.20101691
316. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med (2007) 13(10):1203–10. doi:10.1038/nm1636
317. Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17 ∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev (2009) 23(24):2806–11. doi:10.1101/gad.1872909
Keywords: prostate cancer, differentiation, Myc, Pten, notch, p38MAPK
Citation: Frank SB and Miranti CK (2013) Disruption of prostate epithelial differentiation pathways and prostate cancer development. Front. Oncol. 3:273. doi: 10.3389/fonc.2013.00273
Received: 31 July 2013; Accepted: 18 October 2013;
Published online: 31 October 2013.
Edited by:
Andreas Birbach, Medical University of Vienna, AustriaReviewed by:
David Ian Quinn, University of Southern California, USABeatrice S. Knudsen, Cedars Sinai Hospital, USA
Copyright: © 2013 Frank and Miranti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cindy K. Miranti, Laboratory of Integrin Signaling and Tumorigenesis, Van Andel Research Institute, 333 Bostwick Avenue NE, Grand Rapids, MI 49503, USA e-mail:Y2luZHkubWlyYW50aUB2YWkub3Jn