 Alexandra Thiel1,2
Alexandra Thiel1,2 Ari Ristimäki1,2*
Ari Ristimäki1,2*
- 1Division of Pathology, HUSLAB and Haartman Institute, Helsinki University Central Hospital, University of Helsinki, Helsinki, Finland
- 2Genome-Scale Biology, Research Programs Unit, University of Helsinki, Helsinki, Finland
Different genetic aberrations of BRAF have been reported in various malignancies. BRAF is member of the RAS/RAF/MEK/ERK pathway and constitutive activity of this pathway can lead to increased cellular growth, invasion, and metastasis. The most common activating BRAF mutation in colorectal cancer is the V600E mutation, which is present in 5–15% of all tumors, and up to 80% of tumors with high microsatellite instability (MSI) harbor this mutation. BRAF mutation is associated with proximal location, higher age, female gender, MSI-H, high grade, and mucinous histology, and is a marker of poor prognosis in colorectal cancer. The role of BRAF mutation as a predictive marker in respect of EGFR targeted treatments is controversial. BRAF V600 selective inhibitors have been approved for the treatment of V600 mutation positive metastatic melanoma, but the response rates in colorectal cancer are poor. This might be due to innate resistance mechanisms of colorectal cancers against the treatment solely targeting BRAF. To overcome resistance the combination of treatments, simultaneous inhibition of BRAF and MEK or PI3K/mTOR, might emerge as a successful therapeutic concept.
Introduction
BRAF (v-raf murine sarcoma viral oncogene homolog B1) is a serine/threonine protein kinase of the RAF family. RAF proteins are kinases in RAS/RAF/MEK/ERK pathway. ARAF and CRAF are other family members of the RAF family, however, BRAF displays the best binding to RAS and has the highest phosphorylating activity (1). The RAS/RAF/MEK/ERK pathway usually responds to growth factors and cytokines. However, aberrant signaling of this pathway, for example by constantly active kinases can result in abnormal cellular growth, invasion, and metastasis (2).
BRAF is mutated at a high frequency in several cancers, although also amplification of the protein and aberrant splicing variants have been reported as well (1). The BRAF V600E mutation, deriving from a point mutation of the DNA (1799T → A) is the most common BRAF mutation and accounts for around 90% (3). BRAF V600E mutation is most prominent in melanoma (40–60%), papillary thyroid carcinoma (45%), low grade serous ovarian carcinoma (35%), and in colorectal adenocarcinoma (5–15%) (4). Other BRAF mutations include V600K and V600D/R, accounting for 16–29% and 3% of all BRAF mutations in melanoma, respectively (5, 6). Another activating BRAF mutation that is almost exclusively found in pilocytic astrocytomas is the KIAA1549-BRAF fusion, found in 66–100% of these tumors (7, 8).
Colorectal cancer development and progression can be divided into two separate pathways: chromosomal instability pathway and microsatellite instability (MSI) pathway. In roughly 75% of the cases, colorectal cancer develops through chromosomal instability pathway, and these tumors can harbor APC mutations (>90%), KRAS mutations (50%), TP53 mutations (70%), and allelic loss of 18q (80%) (9). MSI pathway covers approximately 15% of sporadic colorectal cancers and almost all Lynch syndrome (LS) cases. In cancers developing through the MSI pathway the DNA mismatch repair (MMR) function is dysfunctional, which leads to insertions and/or deletions of nucleotide repeats in the DNA (9). Remaining tumors belong to CpG island methylator pathway (CIMP) and Serrated Adenoma Pathway, and approximately one third of CIMP tumors are MSI-H while most of the serrated tumors have a deficient MLH1 gene due to promoter methylation.
Detection of BRAF Mutation in Colorectal Cancer
Until recently the detection of BRAF mutations was performed with Sanger sequencing or PCR-based assays. These methods require representative amount of malignant cells and extraction of the DNA. For specimens with a low content of tumor tissue, the DNA based protocols thus might not be sensitive enough to detect the BRAF mutations. A recent report compared the detection of BRAF mutations between two next generation sequencing (NGS) technologies and Sanger sequencing/q-PCR and found NGS to be reliable in detecting BRAF mutations and other standard-of-care mutations (10). Immunohistochemical (IHC) detection of BRAF V600E with a mutation specific antibody (clone VE1) was first described in metastatic melanoma and papillary thyroid carcinoma (11), and the antibody is currently commercially available (Figures 1A,B). The advantage of IHC lies in the minimal amount of the needed tissue and the availability of this technique in most pathological laboratories. Colorectal cancer has been analyzed with the BRAF V600E mutation specific antibody and most studies find high sensitivities and specificities (98.8–100%) in comparison with PCR-based methods or sequencing (12–16). In one study however, the sensitivity and specificity were only 71 and 74%, respectively (17). The choice of the positive control tissue and the amplification protocol seem to be crucial in successful detection of BRAF V600E mutation by IHC (16).
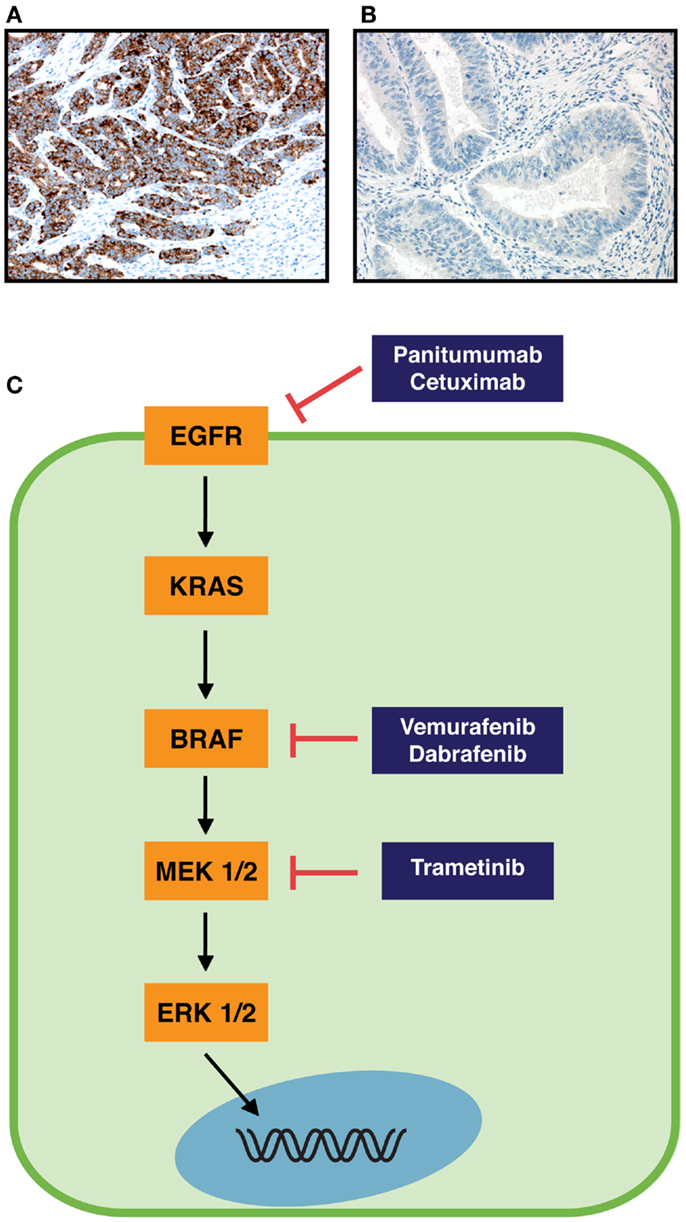
Figure 1. RAS-RAF pathway and immunohistochemical staining of colorectal cancer specimens with BRAF V600E mutation specific monoclonal antibody. (A) Strong immunopositivity in cancer cells with a BRAF V600E mutation. (B) No staining of cancer cells in a specimen without BRAF V600E mutation. Original magnifications are 200×. (C) Schematic RAS-RAF pathway (orange boxes) and inhibitors of components of this pathway (blue boxes). Arrows indicate an activation process, and blocked arrows an inhibition process.
Occurrence of BRAF Mutation in Colorectal Cancer
The frequency of BRAF V600E mutation differs in tumors with high MSI (MSI-H) compared to tumors that are microsatellite-stable (MSS). Whereas BRAF V600E mutation frequencies below 10% are reported for MSS tumors (3, 15, 16, 18), they range from 13 to 78% in MSI-H tumors, including cases with germ line mutation for one of the MMR genes (12, 15, 16, 18). In our consecutive colorectal cancer material BRAF V600E mutation was found in 78% of MSI-H and 8% of MSS tumors (p < 0.0001) (16). A recent study reported BRAF V600E mutation in 100% of sessile serrated adenomas/polyps, 94% of traditional serrated adenoma, and in 62% of micro vesicular hyperplastic polyps (19). BRAF V600E mutation in microvesicular hyperplastic polyps might indicate the polyps that have a higher risk for progression to adenomas/adenocarcinomas (19). The BRAF V600K mutation seems to be a rare event in colorectal cancer, at least in MSI-H tumors (16).
Significance of BRAF Mutation in Colorectal Cancer
Connection to Clinicopathological Parameters
BRAF V600E mutations are associated with several clinicopathological parameters and the ones most often reported are: proximal location, higher age, female gender, MSI-H, high grade, and mucinous histology (16, 20–26). Whereas in most studies colorectal cancers are classified into proximal and distal location, Yamauchi et al. described a gradual linear increase of BRAF mutation, MSI-H, and high CpG island methylator phenotype frequency from rectum to ascending colon (27). The frequencies of all three factors were lower in cecum than in ascending colon, indicating that cecal cancers are a unique subtype (27).
High microsatellite instability is associated with a higher number of harvested lymph nodes (28, 29), and a recent study reported that BRAF V600E mutation was associated with a lower node harvest in the MSI-H group in colon cancer (30). The lymph node count is a predictor of long-term survival in colorectal cancer. Rather than just reflecting the quality of care, the lymph node count might be associated with several factors such as tumor location, tumor and host genetics, and immune interaction (30).
Prognostic Role
BRAF V600E mutation is associated with reduced survival (overall survival, disease-free survival, or cancer-specific survival) especially in MSS tumors (Table 1) (18, 21, 23, 24, 26, 31, 32). Its role in MSI-H tumors is not so clearly defined; while some studies attribute MSI-H tumors with excellent survival regardless of BRAF status (18), BRAF V600E mutation decreased overall survival independent of MSI status in another report (25). In addition, BRAF V600E mutation was associated with poor prognosis in all groups of advanced colorectal cancer (33) and was an independent prognostic factor for overall survival and cancer-specific survival in a pooled stage II/III cohort (22). In a couple of studies, no prognostic role was found to be associated with BRAF mutation (Table 1) (34, 35). Finally, in a meta-analysis that included 26 colorectal cancer studies, BRAF mutation was found to increase the risk of mortality (HR = 2.25, 95% CI: 1.82–2.83) (36).
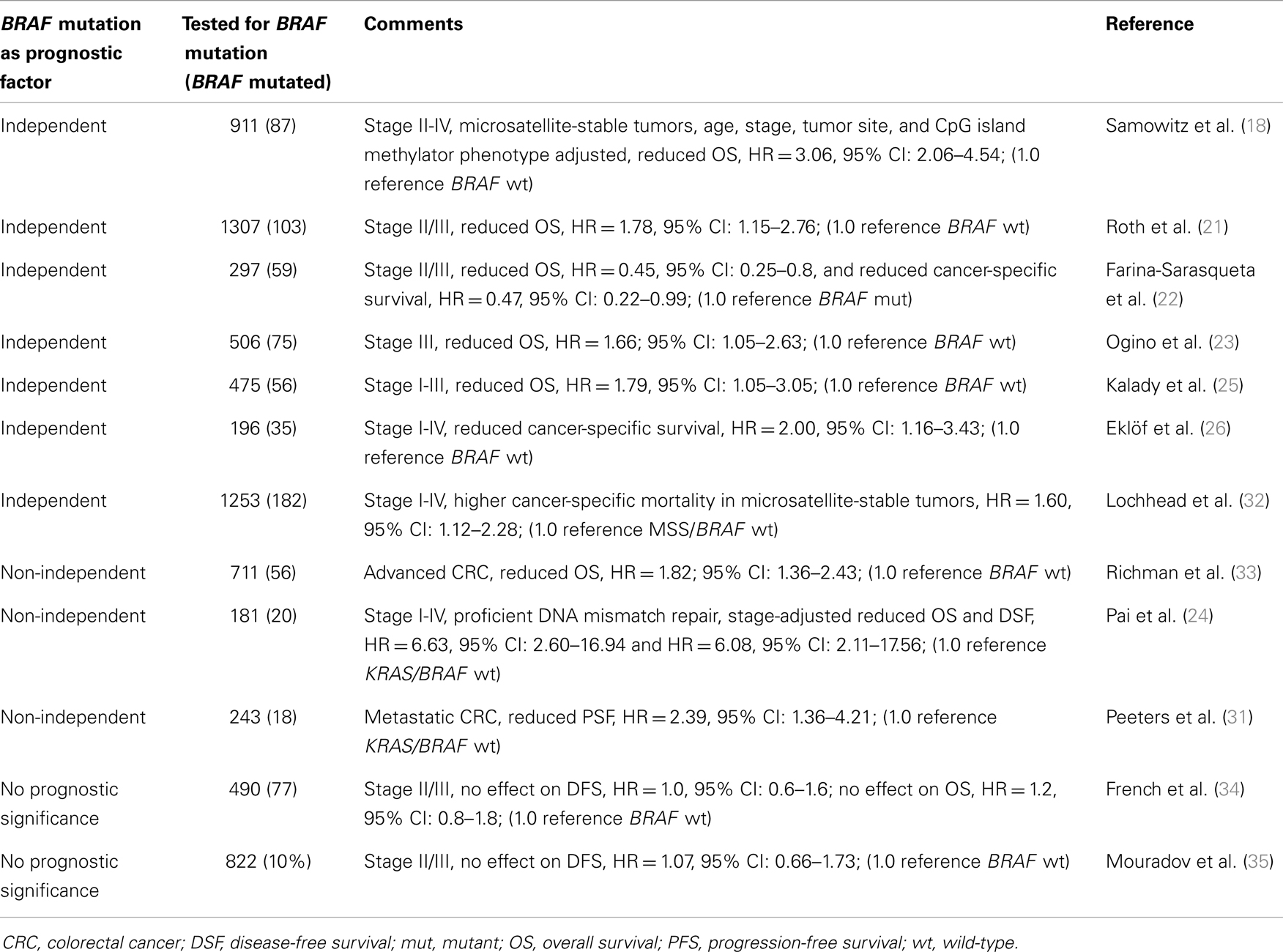
Table 1. BRAF mutation as prognostic factor in colorectal cancer.
Predictive Role
It has been suggested that in order for metastatic colorectal cancer patients to receive a response for treatment with monoclonal antibodies targeting EGFR (panitumumab and cetuximab, Figure 1C), the BRAF gene needs to be present as wild-type (37, 38). Yuan et al. recently concluded in a meta-analysis that BRAF mutation is a predictive biomarker and indicates poor prognosis when metastatic colorectal cancer patients are treated with monoclonal antibodies against EGFR (39). In contrast to these results, a recent guideline does not recommend testing for BRAF mutations in colorectal cancer patients before anti-EGFR treatment (40). Garcia-Alfonso et al. (40) conclude that BRAF mutation is not predictive for anti-EGFR treatment in randomized trials. For patients (KRAS wild-type metastatic colorectal tumors) treated with chemotherapy/bevacizumab with or without cetuximab in the phase III CAIRO2 study, BRAF mutation was correlated to a shorter progression-free survival and overall survival, in both treatment arms (41). Similarly, BRAF mutation was not predictive for treatment with cetuximab, but was a marker of poor prognosis in metastatic colorectal cancer patients (KRAS wild-type) that were randomly assigned to treatment with FOLFIRI (irinotecan, fluorouracil, leucovorin) with or without cetuximab in the CRYSTAL study (42). The pooled analysis of the CRYSTAL and OPUS trials on metastatic colorectal cancer showed that BRAF mutation was not predictive for treatment with cetuximab in KRAS wild-type patients, but indicated poor prognosis (43). Finally, in a retrospective analysis of the PRIME study, BRAF mutation was not predictive for overall or progression-free survival in KRAS wild-type patients treated with FOLFOX4 (oxaliplatin, fluorouracil, leucovorin) with or without panitumumab (44).
As for treatment with standard chemotherapy agents (fluorouracil with irinotecan or oxaliplatin), BRAF V600E mutation was not predictive (33). Similarly, BRAF mutation was not predictive for fluorouracil-based therapy in mostly stage II colorectal cancer (45). A non-significant trend for better survival with fluorouracil/leucovorin + irinotecan (vs. fluorouracil/leucovorin alone) was detected in colorectal cancer stage III patients with BRAF V600E mutation (23).
Role in Identifying LS Patients
Lynch syndrome is a hereditary form of colorectal cancer that accounts for 1–3% of all CRC cases. It is the most common form of hereditary CRC and is caused by a germ line mutation of one of the MMR genes (46). As not all LS patients fulfill the Amsterdam II criteria or revised Bethesda guidelines, not all of them are detected in the routine clinical setting (47, 48). BRAF is usually present as wild-type in LS patients, and only 1.4% of the LS patients carry a BRAF V600E mutation (49). In sporadic colorectal cancer the BRAF V600E mutation rate ranges from 5 to 15% (4), and in the MSI-H group of consecutive primary colorectal cancers the BRAF V600E mutation rate reached 78% (16). This has led to the suggestion that the detection of BRAF V600E mutation might be a useful additional tool in finding LS patients, and several recent studies have used BRAF V600E IHC to implement this step (12, 15, 16).
BRAF Inhibitors in Treatment of Cancer
The first RAF inhibitor, sorafenib, was not effective in clinical use for metastatic melanoma, as it did not improve median overall survival in randomized, double-blind, placebo-controlled phase III studies, when given in combination with paclitaxel and carboplatin as second-line treatment or to chemotherapy-naïve patients (50–52). The reason for the disappointing results with sorafenib in melanoma might be that this multi-targeted tyrosine kinase inhibitor has a higher affinity for isoforms other than BRAF and targets several other pathways as well (50, 53). However, in advanced hepatocellular carcinoma, the median survival time was increased by nearly 3 month in patients treated with sorafenib, in a phase III, double-blind, placebo-controlled trial (54). Vemurafenib (PLX4032) and dabrafenib (GSK2118436) are approved for treatment of unresectable or metastatic melanoma (Food and Drug Administration) and vemurafenib is also approved by the European commission/European Medicines Agency. Both selectively inhibit the BRAF V600 mutated form of BRAF, inhibit phosphorylation of ERK, and have high clinical response rates in melanoma patients (Figure 1C) (50, 53). Whereas patients with BRAF V600 mutated melanomas had a clear survival benefit when treated with BRAF inhibitors, the response rate in metastatic colorectal carcinoma (harboring BRAF V600E mutation) was poor, since only one patient (1/19) displayed a partial response and 4 out of 19 patients a minor response (55, 56). It has been noted already in xenografts from BRAF V600E mutant colorectal cancer cell lines that tumor growth inhibition was most efficient when vemurafenib was combined with EGRF or Akt inhibitors and/or chemotherapeutic agents (57).
Resistance to BRAF Inhibition in Melanoma and Colorectal Cancer
BRAF V600E mutant melanomas initially have a good response rate. However, most of them acquire a drug resistance after 6–7 months, and roughly 10% have tumor progression at earlier stages (53, 55). BRAF V600E mutated colorectal cancer on the contrary, seems to display an innate resistance to inhibition with BRAF inhibitors, which was also demonstrated in colorectal cancer cell lines (55, 58, 59). The mechanisms of resistance can be grouped according to their dependence on ERK signaling (60). ERK-dependent resistance mechanisms can occur via activating MEK1 mutations (61), activating NRAS mutations (62), COT overexpression (63), elevated CRAS activity (64), BRAF V600E alternative splicing or amplification (65). ERK-independent mechanisms include the PI3K pathway (66), overexpression of PDGFRβ (62), IGF1R activation (67), and hepatocyte growth factor (59). Importantly, Romano et al. report that different mechanisms of resistance can occur in the same patient at different metastatic locations (68).
In BRAF V600E mutant colorectal cancer cells the amplification of the BRAF gene was identified as mechanism of resistance to MEK and BRAF inhibition (69). Two studies detected the critical role of EGFR in BRAF V600E mutant colorectal cancer cells that did not respond to BRAF inhibition (58, 70). Corocan et al. reported that BRAF V600E mutant colorectal cancer cell lines harbored more phospho-EGRF than melanomas with the same mutation, and reactivated MAPK signaling via EGFR (58). Prahallad et al. described a rapid feedback activation of EGFR (via CDC25C inhibition) upon RAF inhibition, and EGFR was highly expressed BRAF V600E mutant colorectal cancer cells as compared to BRAF V600E mutant melanoma cells (70).
Overcoming of Resistance and Combination Treatments
To overcome resistances upon treatment with a BRAF inhibitor, targeting novel downstream kinases of the pathway or combination of therapies might be helpful. As for melanoma treatment, the combination of vemurafenib with the HDM2 inhibitor nutlin-3 (leading to p53 restoration), has shown synergistic effect on inducing apoptosis and suppressing tumor growth in melanoma cell lines and xenografts (71). Novel combinatorial treatment options include BRAF inhibition simultaneously with PI3K/mTOR as shown in colorectal cell lines and animal models (72–74). Coffee et al. used the Apc-Braf mouse model (mice bearing a BrafV600E allele) and showed that concomitant inhibition of PI3K/mTOR and BRAF resulted in tumor regression due to induction of apoptosis and decrease in proliferation (73). Also Rad et al. reported the potent growth inhibitory effect of combined BRAF/PI3K inhibition on xenografts of BRAF mutant mouse and human colorectal cancer cell lines (74). Furthermore, MEK inhibition alone caused regression of xenografted and orthotopically transplanted tumors, and reduced proliferation in tumors of BrafLSL-V637E/+ mice (orthologous to human BRAF V600E mutation) (74). A combined inhibition of BRAF (dabrafenib 150 mg) and MEK1/2 (trametinib, 1 or 2 mg) was performed in metastatic melanoma patients with BRAF V600E mutation, in a open-label phase II study with randomly assigned patients. Both median progression-free survival (9.4 vs. 5.8 months) and complete/partial response (76 vs. 54%) were significantly improved in the combination group (150 + 2 mg) vs. dabrafenib immunotherapy (75). Both dabrafenib and trametinib, were recently (May 2013) approved by the Food and Drug Administration for treatment of metastatic/inoperable melanoma (Figure 1C).
Conclusion
BRAF V600E mutation is a marker of poor prognosis in colorectal cancer. Detection of this mutation can also be used to identify LS patients. Targeted treatment of BRAF V600E mutation is in use in advanced melanoma. However, the response is short-lived in melanoma patients, due to the development of acquired resistance. In colorectal cancer targeted treatment of mutated BRAF is not feasible due to the innate resistance. New insights into possible resistance mechanisms were reported recently, and combinatorial treatment options might impact therapy of tumors carrying a BRAF mutation.
Authors’ Note
After acceptance of this review, a novel study reported the combined use of BRAF V600E and MMR immunohistochemistry as a prognostic tool in colorectal cancer (Toon CW, Chou A, DeSilva K, Chan J, Patterson J, Clarkson A, et al. BRAFV600E immunohistochemistry in conjunction with mismatch repair status predicts survival in patients with colorectal cancer. Modern Pathol (2013) Oct 25. doi: 10.1038/modpathol.2013.200). The authors restricted their analysis to only immunohistochemistry of BRAF V600E and MMR status on 1426 consecutive colorectal cancer cases, and found that MSS/BRAF V600E mutant tumor status was a marker for poor prognosis in univariate analysis when compared to MSS/BRAF wild type tumors (HR = 1.79, 95% CI: 1.24–2.60). Immunohistochemical screening for BRAF V600E mutation and MMR gene expression thus can facilitate the detection of Lynch syndrome patients and can also identify subgroups with a poor prognosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants from the Sigrid Julius Foundation (Ari Ristimäki), Finnish Cancer Organization (Ari Ristimäki), and Helsinki University Central Hospital Research Funds and HUSLAB R&D Funds (Ari Ristimäki).
References
1. Niault TS, Baccarini M. Targets of raf in tumorigenesis. Carcinogenesis (2010) 31:1165–74. doi:10.1093/carcin/bgp337
2. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget (2012) 3:954–87.
3. Roskoski R Jr. RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun (2010) 399:313–7. doi:10.1016/j.bbrc.2010.07.092
4. Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology (2013) 45:346–56. doi:10.1097/PAT.0b013e328360b61d
5. Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer (2011) 104:392–8. doi:10.1038/sj.bjc.6606030
6. Kudchadkar R, Paraiso KH, Smalley KS. Targeting mutant BRAF in melanoma: current status and future development of combination therapy strategies. Cancer J (2012) 18:124–31. doi:10.1097/PPO.0b013e31824b436e
7. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene (2009) 28:2119–23. doi:10.1038/onc.2009.73
8. Lawson AR, Tatevossian RG, Phipps KP, Picker SR, Michalski A, Sheer D, et al. RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol (2010) 120:271–3. doi:10.1007/s00401-010-0693-y
9. Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO: Classification of Tumours of the Digestive System, Fourth Edition. Lyon: IARC Press (2010).
10. McCourt CM, McArt DG, Mills K, Catherwood MA, Maxwell P, Waugh DJ, et al. Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS One (2013) 8:e69604. doi:10.1371/journal.pone.0069604
11. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol (2011) 122:11–9. doi:10.1007/s00401-011-0841-z
12. Capper D, Voigt A, Bozukova G, Ahadova A, Kickingereder P, von Deimling A, et al. BRAF V600E-specific immunohistochemistry for the exclusion of Lynch syndrome in MSI-H colorectal cancer. Int J Cancer (2013) 133:1624–30. doi:10.1002/ijc.28183
13. Affolter K, Samowitz W, Tripp S, Bronner MP. BRAF V600E mutation detection by immunohistochemistry in colorectal carcinoma. Genes Chromosomes Cancer (2013) 52:748–52. doi:10.1002/gcc.22070
14. Sinicrope FA, Smyrk TC, Tougeron D, Thibodeau SN, Singh S, Muranyi A, et al. Mutation-specific antibody detects mutant BRAF protein expression in human colon carcinomas. Cancer (2013) 119:2765–70. doi:10.1002/cncr.28133
15. Toon CW, Walsh MD, Chou A, Capper D, Clarkson A, Sioson L, et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for Lynch syndrome. Am J Surg Pathol (2013) 37:1592–602. doi:10.1097/PAS.0b013e31828f233d
16. Thiel A, Heinonen M, Kantonen J, Gylling A, Lahtinen L, Korhonen M, et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Arch (2013). doi:10.1007/s00428-013-1470-9
17. Adackapara CA, Sholl LM, Barletta JA, Hornick JL. Immunohistochemistry using the BRAF V600E mutation-specific monoclonal antibody VE1 is not a useful surrogate for genotyping in colorectal adenocarcinoma. Histopathology (2013) 63:187–93. doi:10.1111/his.12154
18. Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res (2005) 65:6063–9. doi:10.1158/0008-5472.CAN-05-0404
19. Mesteri I, Bayer G, Meyer J, Capper D, Schoppmann SF, von Deimling A, et al. Improved molecular classification of serrated lesions of the colon by immunohistochemical detection of BRAF V600E. Mod Pathol (2013). doi:10.1038/modpathol.2013.126
20. Zlobec I, Bihl MP, Schwarb H, Terracciano L, Lugli A. Clinicopathological and protein characterization of BRAF- and K-RAS-mutated colorectal cancer and implications for prognosis. Int JCancer (2010) 127:367–80. doi:10.1002/ijc.25042
21. Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol (2010) 28:466–74. doi:10.1200/JCO.2009.23.3452
22. Farina-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VE, Rutten HJ, et al. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol (2010) 21:2396–402. doi:10.1093/annonc/mdq258
23. Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res (2012) 18:890–900. doi:10.1158/1078-0432.CCR-11-2246
24. Pai RK, Jayachandran P, Koong AC, Chang DT, Kwok S, Ma L, et al. BRAF-mutated, microsatellite-stable adenocarcinoma of the proximal colon: an aggressive adenocarcinoma with poor survival, mucinous differentiation, and adverse morphologic features. Am J Surg Pathol (2012) 36:744–52. doi:10.1097/PAS.0b013e31824430d7
25. Kalady MF, Dejulius KL, Sanchez JA, Jarrar A, Liu X, Manilich E, et al. BRAF mutations in colorectal cancer are associated with distinct clinical characteristics and worse prognosis. Dis Colon Rectum (2012) 55:128–33. doi:10.1097/DCR.0b013e31823c08b3
26. Eklöf V, Wikberg ML, Edin S, Dahlin AM, Jonsson BA, Oberg A, et al. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer (2013) 108:2153–63. doi:10.1038/bjc.2013.212
27. Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut (2012) 61:847–54. doi:10.1136/gutjnl-2011-300865
28. Soreide K, Nedrebo BS, Soreide JA, Slewa A, Korner H. Lymph node harvest in colon cancer: influence of microsatellite instability and proximal tumor location. World J Surg (2009) 33:2695–703. doi:10.1007/s00268-009-0255-4
29. Eveno C, Nemeth J, Soliman H, Praz F, de The H, Valleur P, et al. Association between a high number of isolated lymph nodes in T1 to T4 N0M0 colorectal cancer and the microsatellite instability phenotype. Arch Surg (2010) 145:12–7. doi:10.1001/archsurg.2009.224
30. Berg M, Guriby M, Nordgard O, Nedrebo BS, Ahlquist TC, Smaaland R, et al. Influence of microsatellite instability, KRAS and BRAF mutations on lymph node harvest in stage I-III colon cancers. Mol Med (2013). doi:10.2119/molmed.2013.00049
31. Peeters M, Oliner K, Parker A, Siena S, Van Cutsem E, Huang J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase 3 study of metastatic colorectal cancer. Clin Cancer Res (2013) 19:1902–12. doi:10.1158/1078-0432.CCR-12-1913
32. Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst (2013) 105:1151–6. doi:10.1093/jnci/djt173
33. Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol (2009) 27:5931–7. doi:10.1200/JCO.2009.22.4295
34. French AJ, Sargent DJ, Burgart LJ, Foster NR, Kabat BF, Goldberg R, et al. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res (2008) 14:3408–15. doi:10.1158/1078-0432.CCR-07-1489
35. Mouradov D, Domingo E, Gibbs P, Jorissen RN, Li S, Soo PY, et al. Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol (2013) 108:1785–93. doi:10.1038/ajg.2013.292
36. Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A, Li G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PLoS One (2012) 7:e47054. doi:10.1371/journal.pone.0047054
37. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol (2008) 26:5705–12. doi:10.1200/JCO.2008.18.0786
38. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol (2010) 11:753–62. doi:10.1016/S1470-2045(10)70130-3
39. Yuan ZX, Wang XY, Qin QY, Chen DF, Zhong QH, Wang L, et al. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: a meta-analysis. PLoS One (2013) 8:e65995. doi:10.1371/journal.pone.0065995
40. Garcia-Alfonso P, Salazar R, Garcia-Foncillas J, Musulen E, Garcia-Carbonero R, Paya A, et al. Guidelines for biomarker testing in colorectal carcinoma (CRC): a national consensus of the Spanish society of pathology (SEAP) and the Spanish society of medical oncology (SEOM). Clin Transl Oncol (2012) 14:726–39. doi:10.1007/s12094-012-0856-5
41. Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink-Borger ME, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer (2010) 46:1997–2009. doi:10.1016/j.ejca.2010.03.036
42. Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol (2011) 29:2011–9. doi:10.1200/JCO.2010.33.5091
43. Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer (2012) 48:1466–75. doi:10.1016/j.ejca.2012.02.057
44. Oliner K, Douillard J, Siena S, Tabernero J, Burkes R, Barugel M, et al. Evaluation of KRAS, NRAS, and BRAF mutations in PRIME: panitumumab with FOLFOX4 as first-line treatment in metastatic colorectal cancer (mCRC). Ann Oncol (2013) 24(Suppl 4):iv23–4. doi:10.1093/annonc/mdt201.31
45. Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol (2011) 29:1261–70. doi:10.1200/JCO.2010.30.1366
46. Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut (2013) 62:812–23. doi:10.1136/gutjnl-2012-304356
47. Weissman SM, Burt R, Church J, Erdman S, Hampel H, Holter S, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National society of genetic counselors and the collaborative group of the Americans on inherited colorectal cancer joint practice guideline. J Genet Couns (2012) 21:484–93. doi:10.1007/s10897-011-9465-7
48. Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA (2012) 308:1555–65. doi:10.1001/jama.2012.13088
49. Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet (2012) 49:151–7. doi:10.1136/jmedgenet-2011-100714
50. Mandala M, Voit C. Targeting BRAF in melanoma: biological and clinical challenges. Crit Rev Oncol Hematol (2013) 87:239–55. doi:10.1016/j.critrevonc.2013.01.003
51. Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C, Hersey P, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol (2009) 27:2823–30. doi:10.1200/JCO.2007.15.7636
52. Flaherty KT, Lee SJ, Zhao F, Schuchter LM, Flaherty L, Kefford R, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol (2013) 31:373–9. doi:10.1200/JCO.2012.42.1529
53. Salama AK, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clin Cancer Res (2013) 19:4326–34. doi:10.1158/1078-0432.CCR-13-0779
54. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359:378–90. doi:10.1056/NEJMoa0708857
55. Lo RS. Receptor tyrosine kinases in cancer escape from BRAF inhibitors. Cell Res (2012) 22:945–7. doi:10.1038/cr.2012.78
56. Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors [abstract]. J Clin Oncol (2010) 28(Suppl):3534.
57. Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res (2012) 72:779–89. doi:10.1158/0008-5472.CAN-11-2941
58. Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov (2012) 2:227–35. doi:10.1158/2159-8290.CD-11-0341
59. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature (2012) 487:500–4. doi:10.1038/nature11183
60. Corcoran RB, Settleman J, Engelman JA. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget (2011) 2:336–46.
61. Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A (2009) 106:20411–6. doi:10.1073/pnas.0905833106
62. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature (2010) 468:973–7. doi:10.1038/nature09626
63. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature (2010) 468:968–72. doi:10.1038/nature09627
64. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell (2010) 140:209–21. doi:10.1016/j.cell.2009.12.040
65. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature (2011) 480:387–90. doi:10.1038/nature10662
66. Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res (2011) 71:2750–60. doi:10.1158/0008-5472.CAN-10-2954
67. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell (2010) 18:683–95. doi:10.1016/j.ccr.2010.11.023
68. Romano E, Pradervand S, Paillusson A, Weber J, Harshman K, Muehlethaler K, et al. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin Cancer Res (2013) 19:5749–57. doi:10.1158/1078-0432.CCR-13-0661
69. Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal (2010) 3:ra84. doi:10.1126/scisignal.2001148
70. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature (2012) 483:100–3. doi:10.1038/nature10868
71. Ji Z, Kumar R, Taylor M, Rajadurai A, Marzuka-Alcala A, Chen YE, et al. Vemurafenib synergizes with nutlin-3 to deplete survivin and suppress melanoma viability and tumor growth. Clin Cancer Res (2013) 19:4383–91. doi:10.1158/1078-0432.CCR-13-0074
72. Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R Jr, Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res (2013) 19:657–67. doi:10.1158/1078-0432.CCR-11-1446
73. Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res (2013) 19:2688–98. doi:10.1158/1078-0432.CCR-12-2556
74. Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, et al. A genetic progression model of braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell (2013) 24:15–29. doi:10.1016/j.ccr.2013.05.014
Keywords: BRAF, colorectal cancer, Lynch syndrome, microsatellite instability, V600E, V600K, vemurafenib, dabrafenib
Citation: Thiel A and Ristimäki A (2013) Toward a molecular classification of colorectal cancer: the role of BRAF. Front. Oncol. 3:281. doi: 10.3389/fonc.2013.00281
Received: 30 August 2013; Paper pending published: 16 September 2013;
Accepted: 31 October 2013; Published online: 15 November 2013.
Edited by:
Alessandro Lugli, University of Bern, SwitzerlandReviewed by:
Kjetil Søreide, Stavanger University Hospital, NorwayRupert Langer, University of Bern, Switzerland
Copyright: © 2013 Thiel and Ristimäki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ari Ristimäki, Division of Pathology and Genetics, Helsinki University Central Hospital, HUSLAB, P.O. Box 400, Helsinki FIN-00029 HUS, Finland e-mail:YXJpLnJpc3RpbWFraUBoZWxzaW5raS5maQ==