 Sonja Maria Wörmann
Sonja Maria Wörmann Hana Algül*
Hana Algül*- II. Medizinische Klinik, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
Pancreatic cancer (PC) is one of the most challenging tumor entities worldwide, characterized as a highly aggressive disease with dismal overall prognosis and an incidence rate equalling mortality rate. Over the last decade, substantial progress has been made to define the morphological changes and key genetic events in pancreatic carcinogenesis. And yet, it is still unclear what factors trigger PC. Some risk factors appear to be associated with sex, age, race/ethnicity, or other rare genetic conditions. Additionally, modifying factors such as smoking, obesity, diabetes, occupational risk factors, etc., increase the potential for acquiring genetic mutations that may result in PC. Another hallmark of PC is its poor response to radio- and chemo-therapy. Current chemotherapeutic regimens could not provide substantial survival benefit with a clear increase in overall survival. Recently, several new approaches to significantly improve the clinical outcome of PC have been described involving downstream signaling cascades desmoplasia and stromal response as well as tumor microenvironment, immune response, vasculature, and angiogenesis. This review summarizes major risk factors for PC and tries to illuminate relevant targets considerable for new therapeutic approaches.
Introduction and Facts
Pancreatic cancer (PC) is one of the most challenging tumor entities worldwide, characterized as a highly aggressive disease with dismal overall prognosis and an incidence rate (IR) equaling mortality rate (MR). Although over the past decade a downward trend in rates for most other major cancer sites could be observed, PC shows raising incidence and unfavorable MRs among men and women overall (1, 2). PC represents the eight leading cause of cancer-related death worldwide accounting for 4% of all cancers with approximately 266,669 deaths, out of 278,684 new cases in 2008 for both sexes (3–6), and displays the fourth-leading cause of cancer-related death in the U.S. (7), and the E.U. (1) among both men and women. Importantly, MRs are highest in more high-income areas of the world, intermediate in South and Central America and eastern Asia, and lowest in low-income areas (6, 8). Despite advances in surgery, chemotherapy, and radiation therapy, the prognosis of PC remains extremely poor with a 1- to 5-year overall survival rate about 25–6%, which is the lowest 5-year overall survival rate of any cancer in the U.S. (9), and a 5-year survival rate <10% in the E.U. (8). Prognosis of PC is largely determined by the stage (TNM classification) of disease at diagnosis. Between 2002 and 2008, around 8–9% of all U.S. PC patients were diagnosed with local disease, 25–27% with regional disease, and 52–56% with distant disease (10, 11). The median survival ranges from 4.5 months for the most advanced stage to 24.1 months for the earliest stage (9, 12). Due to the fact, that PC has a poor response to radio- and chemo-therapy (13), surgery provides at present the only potentially curative treatment prolonging survival. However, <15% (14) of all PC patients are candidates for surgical resection after which 5-year survival rarely exceeds 20–25% (15). The 5-year survival time for local disease is 24.1%, 9% for regional, 2% for distant disease, and the overall survival for the average patient reaches only 6% (10). The exact causes of PC are not known but some factors such as sex, age, race/ethnicity appear to be associated with PC. Additionally, modifying factors such as smoking, obesity, diabetes, occupational risk factors, etc., increase the potential for acquiring genetic mutations that may result in PC (Table 1).
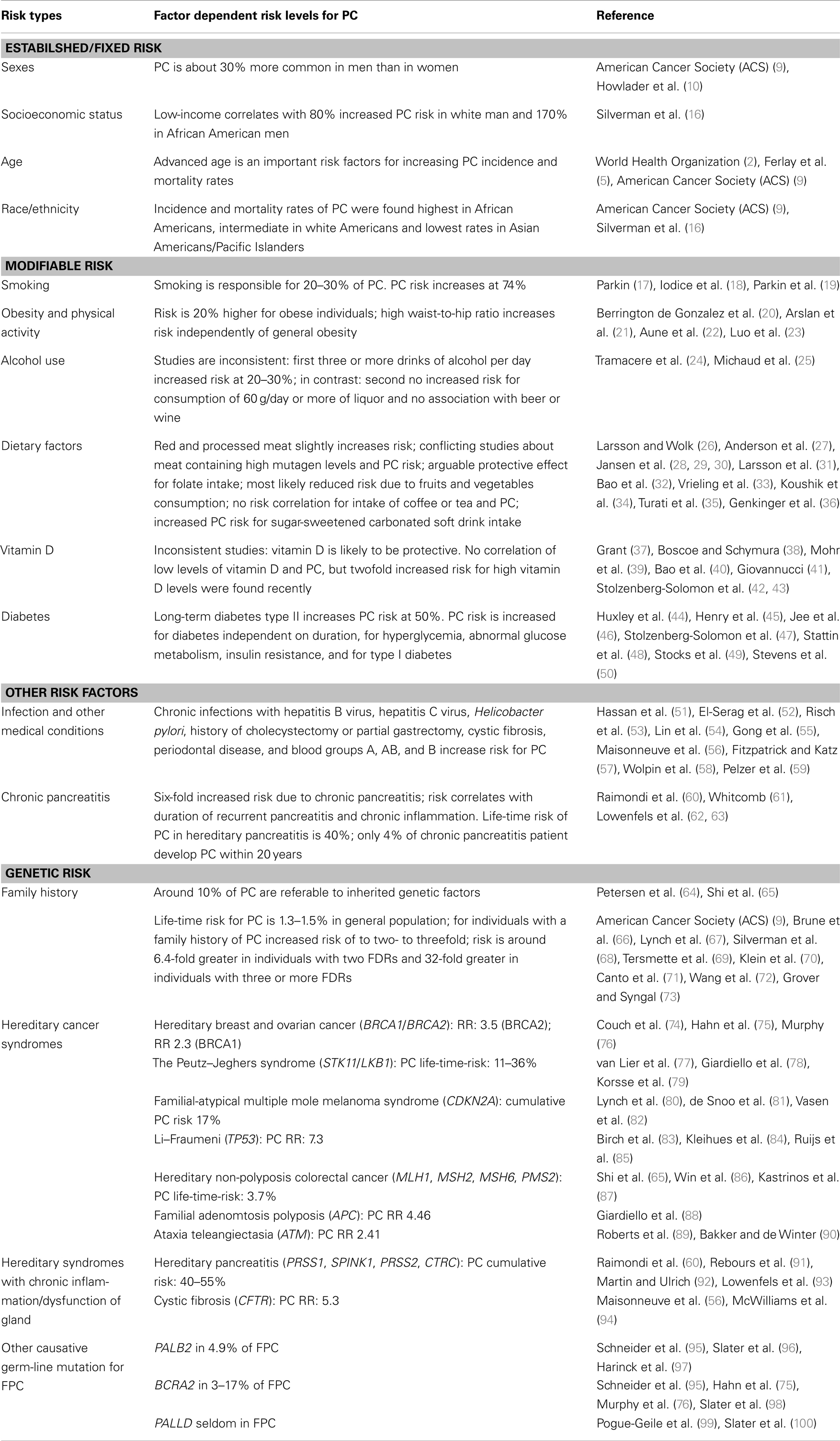
Table 1. Risk factors for PC.
Established Risks
Sexes
Pancreatic cancer is about 30% more common in men than in women. Data from U.S. (9, 10), E.U., and worldwide (4, 101) demonstrate higher incidence and MRs (ASR per 100,000 persons of 13.6/12.5) for men compared to women (ASR per 100,000 persons of 10.5/9.5). The observed gender specific disparity might be attributed to differential lifestyle habits such as use of tobacco, alcohol, etc., hormone dependence has not been confirmed so far (102).
Socioeconomic status
Interestingly, socioeconomic status measured as years of education negatively correlates with PC mortality (103). In addition, low income was associated with an 80% increased PC risk in white man along with an even higher increased risk of 170% in African American men after accounting for differences in heavy alcohol drinking, smoking, and dietary factors (16).
Age
Advanced age is one of the most important risk factors for increasing PC incidence and MRs. Notably, the risk is low until the age of 50, but steeply increases after that. Retrospectively, PC IRs in 35- to 39-year-old men increased from 1.2 up to 100.5 among subjects reaching 85 years and more. Similar findings could be observed in women, with increasing IRs from 1.0 (35–39 years of age) to 87.7 (over 85 years of age) (2, 5, 9). The median age of PC is diagnosed at 71 years (9) in the U.S. and 72 years in the U.K. (17), meaning that about half of all patients develop the disease at an age higher than 71/72 years. An even greater risk was found in patients with familial PC as the average age at diagnosis in these individuals is around 68 (66).
Race/ethnicity
The IRs and MRs of PC vary across different ethnic groups. In the U.S. the highest rates were found in African Americans (IR:15.3/MR: 13.8), followed by intermediate rates in white Americans (IR:11.6/MR:10.7), and lowest rates in Asian Americans/Pacific Islanders (IR: 8.8/MR: 7.5; data respectively for 2005–2009) (9). Commonly, IRs are higher in African Americans than in white Americans at every age. The racial disparity might be almost entirely explained by factors such as cigarette smoking and long-term diabetes mellitus for men and by moderate/heavy alcohol consumption and elevated body mass index for woman (16).
Modifiable Risks
Smoking
Smoking is the most important risk factor for PC and estimated to be responsible for approximately 20–30% of PC (17–19). A dose- and duration-related risk increase contributing to an earlier age of PC onset was found in several studies (104–106). Smokers face a 74% higher risk for PC compared to non-smokers (107). A European-wide study in 2012 provided evidence for a 27% risk increase for every five cigarettes smoked per day (108). Interestingly, several studies showed evidence that the deleterious effects of alcohol and tobacco in PC occurrence and mortality appear to resolve after 10 years of abstinence (18, 106, 109). For smokers, the risk can even be reduced to levels of non-smokers after 5 years of cessation as assessed by a European-wide prospective study in 2009 (109). Smokers with a family history of PC have an even greater risk of developing PC than non-smokers (66). For secondhand smoke exposure and PC the European (EPIC) study showed that passive smoking can increase the risk of PC by 50% (109) and children who are exposed daily to tobacco smoke have double the risk of developing PC later in life (110). In addition, cigar and pipe smoking (18, 111) as well as the use of smokeless tobacco (112) and moist snuff (113) have also been associated with increased PC risk (114).
Obesity and physical activity
Several studies have linked obesity and overweight to increased PC risk. The risk for developing PC was found to be around 20% higher for obese compared to normal weight individuals (20, 21). An estimated 12% of all PC cases in the UK are attributed to overweight and obesity (17, 19). In both men and women a BMI of 25 was associated with an increased risk for PC, but that risk was more pronounced in humans with a BMI of 35 or greater with an overall increase by 10% per five-point increase in BMI (22). Interestingly, having a high waist-to-hip ratio increases PC risk independently of general obesity (21, 22). Specifically, a 70% increase in PC risk for women having a high waist-to-hip ratio was assessed (23). Moreover, overweight and obesity during early adulthood where not only associated with a greater risk but also earlier disease onset of PC. Thereby, obesity at an older age was correlated to a lower overall survival in patients with PC (115). The mechanisms behind these observations are not well understood, however chronic inflammation, mediated by molecules such as TNFα and IL-6, feedback loops associated with the obese state, and disruptions in autophagy promoting ER stress and mitochondrial dysfunction are likely to be involved (116). Supporting this hypothesis, a study suggested that dietary and other lifestyle factors influencing insulin resistance are associated with PC risk (117). At present, there is no well-defined evidence correlating physical activity and PC risk (118, 119). More recently, recreational physical activity was proposed to exert a protective effect on PC risk (120), however this was not confirmed by a previous study (121).
Alcohol use
There is no clear evidence whether alcohol use causes PC or not. In several but not all previously performed studies alcohol use was correlated to increased risk for developing PC. A modest increase in PC risk was observed with consumption of 30 or more grams of alcohol per day (122). A meta-analysis found that consumption of three or more drinks of alcohol per day was associated with a 20–30% increased risk of PC (24). However, a recent large nested case-control study in 2010 showed no increased risk, even at consumption of 60 g/day or more of liquor, and found no association with beer or wine (25).
Dietary factors
A number of dietary factors have been assessed regarding their association with PC risk. There is some evidence that consumption of red and processed meat may slightly increase PC risk (26). However, studies are conflicting about meat containing high mutagen levels and increased risk of PC (27, 28). A protective effect has been reported previously for folate intake (29), although it could not be confirmed in a recent large analysis (30). At present, opposing evidence exists regarding the effect of fruit and vegetable consumption on PC risk (31–33). A recent study suggests, that most nutrients obtained through consumption of fruits and vegetables may reduce the risk of developing PC (34). Overall, no association was observed for intake of coffee or tea and PC (35, 36). Interestingly, a positive association for risk of PC was found in the case of sugar-sweetened carbonated soft drink intake raising insulin and glucose levels, and thus promoting obesity and diabetes (36, 123–125).
Sunlight and vitamin D
Results correlating vitamin D from sun exposure and PC are inconsistent. Some studies associated sun exposure with reduced PC death rates, suggesting that vitamin D might protect against PC (37–39). Other studies also assumed that dietary vitamin D and vitamin D derived from both diet and sunlight exposure might be protective (40, 41). In an additional study, no positive association between 25(OH)D and PC was found, although the previous finding correlating increased PC risk with low residential UVB exposure could be confirmed (42). However, more recently, no correlation of low levels of vitamin D and PC was found and it was even suggested that high vitamin D levels are associated with twofold increased risk of PC risk (43).
Diabetes
Diabetes mellitus is both risk factor for PC and as new onset diabetes a potential early disease sign (126). At diagnosis about 25% of PC patient suffer from diabetes mellitus another 40% are pre-diabetic (126, 127). Patients with long-term (≥5 years) diabetes type II have a 50% increased risk of PC compared with non-diabetic individuals (44). Recently performed studies provide evidence for elevated PC risk with diabetes independent on duration (45) and suggest that hyperglycemia, abnormal glucose metabolism, and insulin resistance correlate with increased risk of PC (46–49). Increased PC risk has also been reported among individuals with type I diabetes (50). Moreover, T3cDM referred to as secondary diabetes was evidenced as major subset of diabetes potentially accounting for the highest risk of PC in particular in patients with chronic pancreatitis. Further, T3cDM was pronounced as consequence of PC in at least 30% of patients (128). Additional preliminary data suggest, that GCKR rs780094, a single-nucleotide polymorphism related to diabetes, is associated with PC risk (129).
Other Risk Factors
Infections and other medical conditions
Several studies demonstrated increased PC risk among people with chronic hepatitis B (51), hepatitis C (52), and Helicobacter pylori (53) infections. In addition, a history of cholecystectomy (54) or partial gastrectomy (55) as well as other medical conditions including cystic fibrosis (56) and periodontal disease (57) were associated with increased PC risk. Recent studies suggested a slightly increased risk of PC for people with non-O blood groups (i.e., blood groups A, AB, and B) whereas blood group O was less frequent in patients with PC. However, the mechanisms behind this carcinogenesis association are still unclear (58, 59).
Pancreatitis
Several studies provide evidence for a strong association between long-standing chronic pancreatitis and PC. Importantly, pancreatitis is also considered an early indicator of PC (60, 130). An even sixfold increased risk of PC among patients with chronic pancreatitis after excluding PC was reported in a review study (60). The risk correlates with the duration of recurrent pancreatitis and chronic inflammation (61). An even higher risk was found in patients with rare types of pancreatitis, such as hereditary pancreatitis and tropical pancreatitis. The assessed life-time risk of PC in individuals with hereditary pancreatitis is with about 40% high, reaching approximately 75% in paternal inheritance pattern (62). The lag of time between pancreatitis diagnosis and PC onset is usually about 10–20 years. Despite these positive correlations chronic pancreatitis as risk for PC is still uncommon as only about 4% of chronic pancreatitis patient will develop PC within 20 years of diagnosis (63).
Genetic Risk
Although the majority of PC appears to be sporadic, around 10% of PC cases are attributable to inherited genetic factors (64, 65). Inherited predisposition to PC is currently classified in three distinct clinical settings: first, tumor predisposition syndromes such as hereditary breast and ovarian cancer, Peutz–Jeghers Syndrome (PJS), familial-atypical multiple mole melanoma (FAMMM), pancreatic-melanoma cancer syndrome (PCMS), or Li–Fraumeni-syndrome, etc., which are characterized by a clinical phenotype other than PC, but known to correlate with an increased risk of PC; second, hereditary pancreatitis and cystic fibrosis, in which genetically determined early changes of the pancreas might predispose to the development of PC; third, familial pancreatic cancer (FPC) which refers to families with two or more first-degree relatives (FDRs) with PC that do not fulfill the criteria for another inherited predisposition syndrome (95, 131, 132). The term FPC is used in the case of three or more relatives of any degree (133).
Family history
Accumulating evidence links family history with PC (67–70). Based on strict inclusion criteria a familial aggregation of PC was reported to be only 2.7 and 1.9% in two prospective studies from Sweden and Germany, respectively (134, 135). The life-time PC risk of 1.3–1.5% in the general population is low (9, 71), but for individuals with a family history of PC the risk can increase dramatically two- to threefold (71). Thereby, the risk stratification depends on the number of affected family members and the relationships between at risk individuals (70, 72). The risk has been estimated to be 6.4-fold greater in individuals with two FDRs with PC (life-time risk 8–12%) (73) and 32-fold greater in individuals with three or more FDRs with PC (life-time risk 40%) (66, 70, 73). Moreover, the risk is increased if a FDR is diagnosed with PC before age 50 (66).
Hereditary cancer syndromes, genetically determined early changes of the pancreas and FPC
Interestingly, PC occurs frequently in excess of expectance in several hereditary cancer syndromes, which are associated with specific germ-line gene mutations. These syndromes show an increased risk for PC varying from 5 up to 40% (136). Clinically defined familial cancer syndromes are extremely rare in the general population accounting only for a small number of FPC (112, 137). For example: familial breast cancer mutations in the BRCA2 gene, associated with a 3- to 10-fold increased risk of PC, account for increasing frequency and the highest proportion (5–17%) of known causes of inherited PC (74–76). A relative risk (RR) of 3.5 (95% CI 1.87–6.58) for PC in BCRA2 gene mutation carriers was observed (138, 139), whereas for BRCA1 gene mutation only a modestly increased RR of 2.3 for PC was demonstrated by a cohort study (140). More recently, an approximately doubled risk for PC was found in female BRCA carriers with a standardized IR for BRCA1 of 2.55 and for BRCA2 of 2.13 (141). The PJS, usually caused by germ-line mutations in the STK11/LKB1 gene, has a 132-fold increased risk for developing PC and shows a cumulative life-time-risk of 11–36% up to age 65–70 among affected individuals (71, 77, 78). A recent study proposed a cumulative risk for PC in individuals with PJS of 26% at age 70 years and a RR of 76 (95% CI 36–160) (79). Hereditary non-polyposis colorectal cancer (HNPCC or Lynch syndrome), associated with mismatch repair genes (MLH1, MSH2, MSH6, PMS2) mutations (132) has an estimated life-time risk of 3.7% for developing PC (8.6-fold higher risk) (65, 86, 142). FAMMM-PC syndrome, an autosomal dominant disease with variable penetrance and linked to mutations in the CDKN2A tumor-suppressor gene (143, 144), is associated with an approximately 13- to 22-fold increased risk of PC (80, 81). The estimated cumulative risk of developing PC in putative mutation carriers by age 75 years was 17% (82). Despite extensive study, germ-line p16 mutations in PC have not been found in the absence of any manifestation of familial-atypical multiple molmelanoma. More recently, Roberts et al. showed in a cohort of 166 familial PC probands that at least 2.4% (4/166) of familial pancreatic cases could be explained by deleterious ataxia-telangiectasia (ATM) mutations (89). Thus, the risk for PC was found to be increased with a RR of 2.41 (95% CI 0.34–17.1) (90). Familial adenomatous polyposis (FAP) linked to mutations in the APC tumor suppressor gene has also been associated with an increased risk for PC. A RR of 4.46 (95% CL 1.2–11.4) in polyposis patients and at risk relatives could be observed (88). Further, the Li–Fraumeni syndrome, a cancer predisposition syndrome featuring germ-line mutation of the p53 tumor suppressor gene, is characterized by a high incidence of a variety of cancers diagnosed at young ages. PC seems to be moderately associated with Li–Fraumeni (83) as only 1.3% of all cancers in Li–Fraumeni patients are PC (84). The expected risk of developing PC for p53 mutation carriers compared to the general population is increased with a RR of 7.3 (85). Another inherited genetic risk correlates with cystic fibrosis, characterized by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene which is associated with chronic idiopathic pancreatitis (145). A twofold increased risk for PC before the age of 60 years (94) and a RR of 5.3 (95% CI 2.4–10.1) for patients with CFTR carrier status was proposed (56). Hereditary pancreatitis associated with mutations in the cationic trypsinogen gene, PRSS1 (143), SPINK1, PRSS2, and CTRC, also lead to an increased risk ranging between 26- and 70-fold compared to the general population, with a cumulative risk of 40–55% by age 70 for developing PC (60, 91–93). Aside from the previously discussed hereditary cancer syndromes and genetically determined early changes of the pancreas, which are not accounting for many cases of FPC, only few other causative germ-line mutation have yet been reported in FPC: BRCA2 mutations were found to be causative for 15% of FPC in the EUROPAC study, even in the absence of breast cancer (75), for 17% of FPC as reported by the Hopkins group (76) and for 3% of FPC as proposed by the FaPaCa study (95). Interestingly, unclassified variants of BRCA2 mutations of unknown clinical importance were detected in 8.6% of cases in the FaPaCa study (98). In addition, the BRCA2-interacting protein PALB2 was identified as a PC susceptibility gene (146) with germ-line mutations found in up to 5% of patients with FPC (95–97). Further, oncogenic mutation P239S in the Palladin (PALLD) gene, found in a linkage analysis of one large FPC family, has been proposed to be an additional major PC susceptibility gene (99). However, in the analysis from EUROPAC and FaPaCa families neither linkage nor any PALLD mutation was found (100). For other tumor suppressor genes such as MAP2K4, MADH4 (SMAD4/DAC4), ACVR1B (ALK4, activin receptor type 1B) known to undergo germ-line or somatic genetic inactivation in clinically sporadic PC, no germ-line mutations could be identified in any of the FPC kindreds tested (76). Despite, previous linkage to PC none or non-deleterious germ-line mutations in RNASEL (147), STK11 (148), CHEK2 (149), and NOD2 (150) genes could be found in the FaPaCa collection study (147–150). Additionally, CDKN2a mutations could only be detected in PCMS or FAMMM-PC families (151), as none of the CDKN2a mutations were identified in FPC families without melanoma (98, 152). In summary, only BRCA2, PALB2, and PALLD germ-line mutations could be observed in FPC families, potentially predisposing to PC. Importantly, the major gene (s) responsible for the inheritance patterns of PC remain to be identified.
Potential Therapeutic Targets
As current chemotherapeutic regimens could not provide substantial survival benefit with a clear increase in overall survival, several new approaches to significantly improve the clinical outcome of PC are required. Thereby, four main target groups for novel compounds can be stratified: first, downstream signaling cascades [RAS, IGFR, mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK), Akt, phosphatidylinositol 3-kinase (PI3K), mammalian target of rapamycin (mTOR), Notch, signal transducer and activator of transcription 3 gene (STAT3)] within the tumors cells; second, desmoplasia and stromal response [sonic hedgehog (Shh), transforming growth factor β (TGFβ) PEGPH20 hyaluronidase]; third, tumor microenvironment and immune response [CD40L, cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) antibodies, L10-interleukin-2 (IL-2) fusion product]; and fourth, vasculature and angiogenesis (153) (Figure 1).
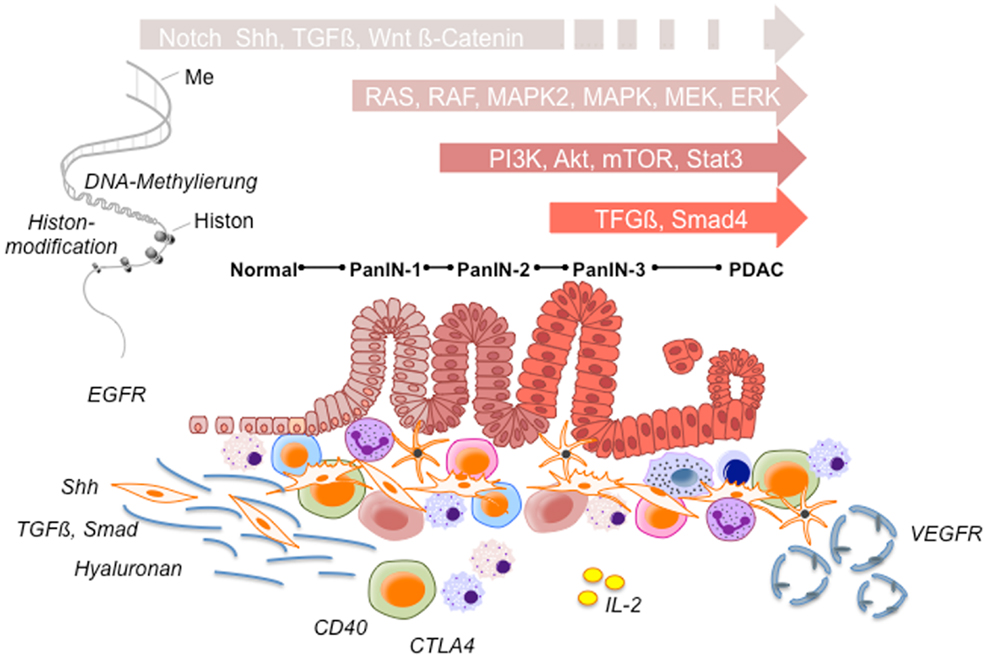
Figure 1. Potential therapeutic targets. During pancreatic cancer development a variety of signaling pathways participate in multiple stages of pancreatic tumorigenesis from early precursor lesions, histologically defined as pancreatic intraepithelial neoplastic lesions (PanINs 1–3 lesions) to advanced ductal pancreatic cancer. These histopathological changes are accompanied by infiltrating immune cells and an increasing desmoplastic stromal response. According to significantly involved signaling pathways tumor cell survival, angiogenesis, invasion, desmoplasia, and tumor immune response are affected, respectively. Ensuing alterations together with epigenetic changes are strongly involved in promoting tumor progression and chemotherapy resistance, and thus provide potential therapeutic targets in pancreatic cancer.
Signal-Transduction Pathways
The Ras, MAP2K, and MEK pathway
Activated K-Ras, mutated mainly at codon 12 but also less frequent at codons 13 and 61, is found in a high percentage of PC cases (154, 155). Oncogenic K-Ras is known to be involved in the initiation or early phase of pancreatic tumorigenesis. K-Ras, a member of the Ras family of genes, encodes membrane-bound GTP-binding proteins and gets activated by signaling partners, such as the epidermal growth factor receptor (EGFR). Mutation in K-Ras locks Ras proteins in an activated state, resulting in a continuous induction of downstream signaling cascades, such as the rapidly accelerated fibrosarcoma (Raf), MAP2K, mitogen-activated protein kinases (MAPK), and the PI3K–Akt cascades. Consequently, a variety of cellular processes, including transcription, translation, cell cycle progression, cell survival, and motility, are triggered. Several treatment approaches (156–158) targeting the Ras pathway seem to be promising for reaching a favorable outcome. Moreover, new molecules targeting downstream kinases of mutant K-Ras are currently investigated to overcome K-Ras induced drug resistance: MAP2K, though its inhibition has been only partly effective in PC therapy (159, 160) or MEK, known as one of the most significant downstream kinase of K-Ras signaling. As MEK mediates K-Ras induced effects on proliferation and survival, its inhibition might represent an innovative therapeutic target especially in tumors carrying an activating K-Ras mutation. A variety of small molecule inhibitors developed and validated in vitro for their therapeutic efficiency are under current investigation in clinical trials.
The EGFR, VEGFR, and IGFR-1 pathway
Tyrosine kinase receptors such as EGFR and vascular endothelial growth factor receptor (VEGFR) stimulated upon binding EGF and VEGF promote cell proliferation, survival, and angiogenesis. Therapeutic approaches inhibiting EGFR and VEGFR with monoclonal antibodies and small molecule inhibitors have been evaluated and approved for clinical use in several solid tumors (161). EGFR, a transmembrane receptor tyrosine kinase of the ErbB family, homo- or hetero-dimerizes with other ErB family members upon binding to its ligands. Thereby, a phosphorylation of tyrosine residues in its intracellular domain occurs, leading to recruitment of intracellular proteins causing downstream signaling events through Ras/Raf/MEK/MAPK, PI3K–AKT, and the STAT family of proteins. However, EGFR can be inappropriately activated due to its overexpression, activating mutations, overexpression of receptor ligands, or loss of their negative regulatory pathways. Importantly, overexpression of EGFR, which occurs in approximately 90% of PC (162) and its ligands EGF are both frequently observed in PC (163, 164). Inhibition of EGFR with the small molecule inhibitor erlotinib was in 2005 the first and until now the only Food and Drug Administration (FDA) approved targeted therapy for clinical use, providing only small survival benefit (165). The disappointing results are intriguing: EGFR signals upstream of K-Ras and hence, its inhibition should have almost no effect on downstream K-Ras driven oncogenic signals. Therefore, it is speculated, that the frequent activating K-Ras mutation acting downstream of EGFR accounts for the only minor impact of this inhibitor in PC. In addition, several therapeutic approaches for VEGF (166–168) have all pointed out to be ineffective in PC. These negative results of all antiangiogenetic approaches tested so far might be due to the largely hypovascular cancer surrounding stroma. Recent reported data suggest that decreasing the stromal density by the inhibition of stromal signaling pathways, e.g., Shh, might lead to enhanced intratumoral perfusion improving drug delivery and thus the efficacy of chemotherapy (169). Although, the therapeutic approaches for EGFR and VEGF showed only marginal effects, several novel targets within the receptor tyrosine kinase signaling cascades (e.g., for the IGF1R pathway) are under current investigation. IGF1R is constitutively overexpressed in 64% of PC (170). IGFR promotes survival and antiapoptotic effects in tumor cells through both K-Ras-dependent and independent downstream signaling cascades including the PI3K–Akt, MAPK, and STAT pathways, thus providing a promising new drug target in K-Ras mutated tumors (171, 172).
PI3K/Akt signaling pathway
Phosphatidylinositol 3-kinase/Akt signaling pathway, which is involved in cell proliferation, survival, resistance to apoptosis, angiogenesis, and invasion, represents another significant therapeutic target in PC (173). Signaling through receptor tyrosine kinases such as EGFR/IGF1R activate PI3K, which in turn activates Akt, thereby inducing multiple downstream targets, including mTOR and the transcription factor NFκB. Activation of the PI3K/AKT signaling pathway is found in 59% of PC (174) representing an independent negative prognostic factor in PC (175). Additionally, abnormal expression of the phosphatase and tensin homolog (PTEN), which normally inactivates PI3K, is frequently found in PC (176) – just as the overexpression of high mobility group A1 (HMGA1), an architectural transcription factor (177). Both alterations activate PI3K–Akt signaling, most likely responsible for the resistance to gemcitabine (178) and respective to HMGA1 suggested as promising target for therapeutic intervention (179, 180). More recently, cell-autonomous PI3K and 3-phosphoinositide-dependent protein kinase 1 (PDK1), but not Craf, were found to be key effectors of oncogenic K-Ras specifically in pancreas, suggesting PI3K/PDK1 as a new target for therapeutic approaches (181). Currently, therapeutic interventions targeting simultaneous inhibition of MEK/extracellular signal-regulated kinase (ERK) and PI3K/AKT signaling with the aim to overcome drug resistance mediated by upstream mutations of Ras and/or Raf are tested in advanced PC carrying K-Ras, N-Ras, and/or B-Raf mutations.
mTOR signaling pathway
Mammalian target of rapamycin, a serine/threonine kinase like Akt, activated by PI3K/Akt signaling and capable of regulating gene transcription and cell proliferation, represents another potential promising target. However, using mTOR inhibitors monotherapy provided only marginal benefit in patients with gemcitabine-refractory metastatic PC (182). In vitro studies showed evidence, that prolonged exposure of PC cells to mTOR inhibitors promote insulin receptor substrate-PI3K interactions, thereby inducing a paradoxically enhanced Akt phosphorylation and cyclin D1 expression (183). Based on these results, trials testing the effect of mTOR inhibition in combination with EGFR inhibitors were initiated and are currently ongoing. Intriguingly, the antidiabetic drug, metformin, is capable to inhibit the mTOR pathway by activating the AMP-activated protein kinase (AMPK) that negatively regulates mTOR activity via phosphorylation and stabilization of the tumor suppressor gene TSC2 (184). Recent, studies, suggest that the use of metformin is associated with a decreased risk of developing cancers (185, 186). Based on these findings, tumor cell metabolism as a potential therapeutic approach has gained rising interest. Several preclinical studies are ongoing to define modalities for earlier detection of PC and new therapeutic targets (187) via inducing changes in cellular metabolism.
STAT3 signaling pathway
STAT signaling pathway plays a role in cell proliferation, survival, motility, invasion, adhesion, angiogenesis, and inflammation (188). The classical STAT tyrosine phosphorylation is mediated by the Janus kinase (JAK) family of tyrosine kinases, which in turn is activated by cytokine and growth factor receptors (189, 190). In the pancreas, STAT3 is dispensable for normal development, however, in the majority of PC a constitutive STAT3 activation due to phosphorylation of Tyr705 can be found (191–193), suggesting STAT3 as a potential therapeutic target in PC. STAT3 is required for the development of the earliest pre-malignant pancreatic lesions, acinar-to-ductal metaplasia, and pancreatic intraepithelial neoplasia (PanIN) (194). More recently, IL-6 transsignaling-dependent activation of STAT3/Socs3 was found to be required for promoting PanIN progression and pancreatic ductal adenocarcinoma (PDAC) (195). Importantly, acute inactivation of STAT3 resulted in inhibition of PC initiation as previously shown in a genetically engineered mouse model (196). The important role of STAT3 in driving PDAC progression at multiple stages of pancreatic tumorigenesis in vivo suggests, that a pharmacological inhibition of the JAK/Stat pathway might be a promising therapeutic target.
Poly (ADP-ribose) polymerase pathway
Poly (ADP-ribose) polymerase (PARP) includes a family of nuclear protein enzymes, which are involved in a variety of cellular processes mainly mediating DNA damage response and apoptosis. However, only few PC patients including FPC patients carrying BRCA mutations or other defects in homologous repair might significantly respond to PARP inhibitors in combination with DNA damaging agents (197). Currently, the PARP inhibitor olaparib in combination with cisplatin and irinotecan, and also another PARP inhibitor, veliparib, in combination with different drugs are evaluated in PC (153). A recently performed in vitro study demonstrated that the PARP-1 inhibitor Rucaparib acts as a chemoradiosensitizer in BRCA2-deficient and -proficient PC cells (198). Moreover, in vitro using human PC cells and in vivo using a murine model, a novel function of PARP-1 in regulating the extrinsic apoptosis machinery, and also an interference combining PARP-1 inhibitors with death receptor agonists for PC therapy was proposed (199).
RET proto-oncogene (rearranged during transfection) pathway
The proto-oncogene RET, encoding a receptor tyrosine kinase, together with the glial derived neurotrophic factor (GDNF) were found strongly expressed in PC (200) and significantly correlated with invasion and survival after surgical resection (201). GDNF binds to the receptor tyrosine kinase and mediates through the MAPK pathway proliferation and invasiveness in PC (202, 203). A previous study demonstrated that glucose concentration-dependent expression of GDNF and RET in human PC cells correlates with alterations of cell proliferation, suggesting GDNF and RET as hyperglycemia underlying mechanism inducing PC progression (204). Further, neuroinvasive pancreatic carcinoma were found to higher express GDNF receptors RET and GRFα1 compared with normal tissue. Treating mice systemically with pyrazolopyrimidine-1, a tyrosine kinase inhibitor targeting the RET pathway, resulted in suppressed nerve invasion toward the spinal cord and prevented paralysis in mice, suggesting RET targeted therapy as a potential therapy directed against nerve invasion in PC (205). By applying the anti-RET antibody or RET siRNA in vitro to human PC cells the effect of GDNF on cell invasion was abrogated confirming a RET mediated GDNF effect (200). Further, a G691S RET polymorphism correlating with enhanced invasiveness was found unregulated in some human PC cells and in 37% of primary PC, representing a somatic mutation associated with PC (200). Although substantial clinical studies are missing, RET might provide a potential target for anti-invasive therapy in PC (206).
Gastrin
Gastrin, a peptide hormone, secreted by G cells in the gastric antrum and duodenum, acts as a growth factor for PC (207) and is expressed together with CCK-BR (the gastrin and cholecystokinin B receptor) and its precursors in 23, 95, 55–91% of PCs, respectively (208). Recent basic research studies demonstrated that knockdown of gastrin gene expression either by stable shRNA transfection (209) in human PC cells or by transient siRNA treatment (210) in gastric cancer cells as well as siRNA treatment of human BxPC-3 PC xenografts in mice results in suppressed proliferation and enhanced apoptosis. While for the orally active inhibitor Z-360 basical research studies were promising (211) and in combination with gemcitabine well tolerated by patients with advanced PC (212), substantial clinical data are missing. Other studies using the selective CCK-BR antagonist gastrazole demonstrated an improvement in patient overall survival however with no significant benefit over 5-fluorouracilin standard therapy in PC (213). Similarly, the use of gastrimmune, an immunogen, stimulating the formation of antibodies against gastrin 17 and its precursors and thus inhibiting the gastrin pathway was not effective in a phase III trial in advanced PC (214, 215).
Cyclooxygenase-2
COX consists of two isoforms converting arachidonic acid into prostaglandins, which is subsequently metabolized to prostaglandin E2 (PGE2), PGF2α, PGD2, and other eicosanoids. While COX-1 is constitutively expressed in many tissues exhibiting a homeostatic role, cyclooxygenase-2 (COX-2) is regulated by growth factors, cytokines, and tumor promoters. Although COX-2 expression is up regulated in 90% of pancreatic PC its impact in PC development is complex and rather unclear (216). A variety of mitogenic signaling pathways and molecules mediating invasiveness, angiogenesis, resistance to apoptosis, immunosuppression, the production of free radicals and peroxidation of procarcinogens to carcinogens are involved (217). Studies suppressing COX-2 by using NASIDs demonstrated suppression of proliferation in PC cells and angiogensis in in vivo and in vitro models (217, 218). A variety of phase II studies using Gemcitabine plus celecoxib in advanced PC were inconsistent in findings and provided only partly survival benefit (219–221). However a phase II study in advanced PC patients combining celecoxib, gemcitabine, and irinotecan resulted next to significantly improved median survival of 13 months and a 1-year survival of 64%, in improved pain and quality of life (222). More recently, apricoxib, a novel COX-2 inhibitor in phase II clinical trials, was found to significantly enhances the efficacy of gemcitabine/erlotinib in PC and promoting vascular normalization and reversed EMT (223). A further phase III trial of gemcitabine, celecoxib, and curcumin in patients with advance inoperable PC is still ongoing.
Immune Response
Over decades, the majority of research efforts had focused on molecular pathways crucial for tumor growth and maintenance thereby largely neglecting local and systemic immune response. Only recently, accumulating evidence highlighted the importance of cancer immunity, leading to enhanced tumor resistance and progression in PC (224). Currently, immunotherapies, which stimulate host immune response culminating in extensive tumor destruction (225), are under active preclinical and clinical investigations. A hallmark of PC is a distinct peritumoral stroma and an immunosuppressive tumor microenvironment rich in inflammatory cells such as T cells, macrophages, myeloid suppressor cells, etc. These immune cells, inhibit anti-tumor immunity, and enhance tumor resistance, progression, and tumor chemotherapeutic resistance, thus providing a promising target for a variety of immunotherapies (226, 227).
Cytotoxic T-lymphocyte-associated antigen 4
Cytotoxic T-lymphocyte-associated antigen 4 is an inhibitory signal produced by activated T cells to regulate and limit the immune response. CTLA4 can be blocked by specific antibodies such as ipilimumab or tremelimumab resulting in sustained anti-tumor immuneresponse by activated T cells. CTLA4 antibodies were the first of this class of immunotherapeutics that achieved U.S. FDA approval. In contrast, to a previously successful trial for advanced melanoma, Ipilimumab as a single agent seems to be ineffective for the treatment of advanced PC (228).
CD40
Another promising target is CD40, a member of the tumor necrosis factor receptor superfamily. CD40 activation can reverse immune suppression and drive anti-tumor T cell responses by mediating the “licensing” of antigen-presenting cells (229). CD40 agonists are suggested to mediate both T cell-dependent and T cell-independent immune mechanisms of tumor regression in mice and humans. Thereby, T cell-independent mechanisms seem, particularly in PC, to be linked to the re-education of tumor-promoting macrophages and stromal involution (229, 230).
Immune-cytokines
Cytokines, especially IL-2, characterized as one of the most potent anti-tumor cytokines, are a potential therapeutic approach in PC. Systemic application of IL-2 due to its toxicity has failed. Local therapy on the other hand seems promising (231). To guide IL-2 to the tumor site, a tumor-selective human single-chain Fv antibody fragment L19 antibody (232) which binds with high affinity to extradomain B (ED-B) of fibronectin, one of the most tumor-selective antigens associated with neoangiogenesis and tumor growth (233, 234), can be used. The resulting L19-IL-2 fusion product might be an attractive concept to enhance therapeutic effects of IL-2 by directly conjugating IL-2 to the tumor site (232).
Stromal Reaction
A hallmark of PC is the extensive peritumoral stroma and desmoplasia consisting of a variety of cellular components such as stellate cells, activated fibroblasts, and inflammatory cells, surrounded by extracellular matrix (235, 236). The stroma represents up to 90% of the tumor volume. In the past, therapeutic approaches mainly targeted tumor cells. Only recently, intense stroma has been recognized as a barrier surrounding tumor cells (236) and was hypothesized to contribute to inefficient drug delivery and chemoresistance in PC (169, 236).
Sonic hedgehog pathway
Sonic hedgehog pathway is known to be one of the most dominant signaling cascades contributing to enhanced desmoplasia by affecting differentiation and motility of human pancreatic stellate cells and fibroblasts thereby, influencing tumor growth in PC (237). Activation of the Shh pathway is managed by two transmembrane proteins namely tumor-suppressor PTC1 protein and oncogenic SMO protein. SMO is normally suppressed by PTC1, however, inactivating mechanisms, such as mutation of PTC1 or the binding of hedgehog proteins to PTC1, lead to continuous SMO activation and transcriptional responses. Shh promotes additional pro-tumorigenic effects by mediating cell cycle, cell survival, angiogenesis, or interference with activated K-Ras. Shh is expressed in about 70% of human PC (238). Previously, the hedgehog inhibitor IPI926, binding SMO, applied in a genetic mouse model resulted in a significant depletion of tumor-associated stroma (169). However, a trial investigating hedgehog inhibitor IPI926 in patients with advanced PC was terminated due to diminished overall survival in patients on the gemcitabine plus IPI926 arm. Another therapeutic target of the Shh pathway includes the transcription factor GLI1, which can be inhibited by miRNA (239).
Transforming growth factor β
Another pathway involved in stromal reaction is the TGFβ-dependent signaling cascade (240). TGFβ, a cytokine secreted by epithelial, endothelial, hematopoietic and mesenchymal cells, binds, and forms a heteromeric complex with the type I and II transforming growth factor β receptor (TGFBR), consequently triggering the phosphorylation of SMAD2 and 3. Cytoplasmic SMAD2/3 proteins form a complex with SMAD4, which translocates into the nucleus and activates gene transcription. However, TGFβ can also involve Ras, PI3K, and MAPK via pathways independent of SMAD. Mutations of the TGFBR1, TGFBR2, and SMAD4 genes are found in about 1, 4, and 50% of patients with PC, respectively (241). TGFβ is known to promote pro-invasive, pro-metastatic microenvironment playing a central role in stroma production, angiogenesis, and tumor-induced immunosuppression (242, 243). Inactivating mutations in the Smad4 gene and up regulation of the inhibitory Smad6 and 7 genes have been found in many PCs (244). Several drugs have been applied as therapeutic approach. Importantly, the antisense oligodeoxynucleotide trabedersen AP 12009 monotherapy, specifically inhibiting TGFβ2 expression, has shown a markedly enhanced survival in PC (245).
Hyaluronan
However, stromal-related signaling pathways such as TGFβ or Shh are not the only therapeutic possibilities; acellular matrix components can also be targeted. Hyaluronan, a non-sulfated glycosaminoglycan is highly abundant in the extracellular matrix of PC tissues (246, 247). To this effect, PEGylated human recombinant PH20 hyaluronidase (PEGPH20) leads to re-expansion of tumor blood vessels and increased concentration of gemcitabine within the tumor, resulting in significantly reduced tumor growth and enhanced survival as previously shown in a murine study (246).
Embryonic Signaling Pathways
Notch, Hedgehog, TGFβ, and Wntβ-catenin
Oncofetal signaling pathways are typically present during fetal development, but are also frequently reactivated in cancers enhancing tumor progression and mediating resistance to chemotherapy (248). Major signaling pathways that are genetically affected in PC (249) are Notch, Hedgehog, TGFβ, and Wntβ-catenin (249). As mentioned above, TGFβ and Hedgehog signaling pathway are mainly involved in stromal reactions. On the other hand, Wnt signaling is involved in normal embryonic development and homeostatic self-renewal of a number of adult tissues. In the setting of cancer Wnt is involved in proliferation and anti-apoptosis. Aberrant activation of this pathway, due to gain-of-function mutation of activators or loss-of-function mutation of inhibitors of Wnt signaling could result in carcinogenesis and progression. Enhanced activation of Wnt signaling is found in 65% of PC (250). Importantly, Wnt signaling is induced by the hedgehog and SMAD4 signaling pathway (251, 252), which should be considered for a combined therapeutic intervention. Wnt/β-catenin signaling in PC might also be involved in chemoresistance (253) and metastasis (254), thus representing a promising therapeutic approach. The main function of the Notch signaling appears to be maintenance of pancreatic progenitor-like cells in an undifferentiated state by promoting their survival and persistence (255), similar to its function in embryogenesis (256). The Notch ligand and its receptor are highly expressed in PC compared with normal epithelial tissue (257). Activation of Notch, similar to K-Ras has been implicated in development of PanINs and in the initiation, progression, and maintenance of invasive PC suggesting inhibition of Notch signaling as a promising therapeutic strategy in this malignancy (258–260).
Epigenetic Changes
DNA methylation and histone acetylation
Epigenetic alterations are heritable with no changes in DNA sequence, and can be reversed thus representing targets for therapeutic interventions. Several epigenetic mechanisms affecting gene expression at the chromatin level are involved in carcinogenesis and tumor progression in PC such as DNA methylation associated with gene silencing and histone acetylation associated with activation of gene transcription (261). Histone acetylation leads to a reversal of positive charge on the histones, resulting in euchromatin, a loosened chromatin structure, providing transcriptionally active DNA. Histone deacetylases (HDAC) are capable to reverse this relaxation thereby reducing transcription of genes, among them several potential tumor suppressor genes. Several members of the HDAC family are highly expressed in PC, as HDAC2 and 6, thereby enhancing resistance to apoptosis (262, 263). Aberrant DNA methylation is an important cancer hallmark and associated with gene silencing. Generally, in cancer cell DNA is considered to be hypomethylated, which is associated with genomic instability and transcription of silenced transposable sequences (264). However, CpG islands found in the promoter regions of tumor suppressor genes, commonly undergo DNA hypermethylation, resulting in gene silencing, thereby promoting tumor development and progression (264). Thus, DNA hypermethylation of tumor suppressor genes might be a promising therapeutic target.
Telomerase
Telomeres are located at the end of chromosomes and normally decrease with each cell division limiting the lifespan of the cells. However, in a variety of malignant neoplasms including PC telomerase is strongly activated, ensuring unlimited proliferation by adding TTAGGG repeat at the end of the chromosome. Telomerase activity was found to be present in pancreatic juice of patients with PC (265) and overexpressed in 95% of PC thus providing a reliable target in PC therapy (266). Although a phase I/II trial of GV1001, a telomerase peptide vaccine designed to prime the immune system to recognize telomerase has shown promising results (267) an interim analysis of a recently performed phase III trial in 520 people with PC showed no survival benefit. Latterly, as the previous trial failed, GV1001 is tested in a phase III trial in combination with gemcitabine and capecitabine in locally advanced and metastatic PC (268). However the results of theses trial are pending.
Conclusion
Although the exact causes driving PC initiation and progression are still unclear this review aims to summarize most important established and modifying risk factors underlying PC development. It further elucidates signaling pathways already involved in therapeutic approaches or considerable as new therapeutic targets treating PC patients. In particular, signaling pathways mediating desmoplastic stromal response and tumor immunity, largely been neglected and attracted attention only recently, might provide promising therapeutic targets for future therapy. As current therapies failed to significantly impair PC progression and improve cancer patient survival, new therapeutic approaches, and clinical studies are strongly required.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Akt, protein kinase B; CTLA4, cytotoxic T-lymphocyte-associated antigen 4; COX-2, cyclooxygenase-2; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; FAMMM, familial-atypical multiple mole melanoma; FAP, familial adenomatous polyposis; FDR, first-degree relatives; FPC, familial pancreatic cancer; HDAC, histone deacetylases; HMGA1, high mobility group A1; HNPCC, hereditary non-polyposis colorectal cancer; IGFR-1, insulin-like growth factor receptor-1; IL-2, interleukin-2; JAK, Janus kinase; MAPK, mitogen-activated protein kinases; MEK, mitogen-activated protein/extracellular signal-regulated kinase kinase; mTOR, mammalian target of rapamycin; PanIN, pancreatic intraepithelial neoplasia; PARP, poly (ADP-ribose) polymerase; PC, pancreatic cancer; PCMS, pancreatic-melanoma cancer syndrome; PDAC, pancreatic ductal adenocarcinoma; PDK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphatidylinositol 3-kinase; PJS, Peutz–Jeghers syndrome; PTEN, phosphatase and tensin homolog; Raf, rapidly accelerated fibrosarcoma; RET proto-oncogene, rearranged during transfection; RR, relative risk; Shh, sonic hedgehog; STAT3, signal transducer and activator of transcription 3 gene; TGFβ, transforming growth factor β; TGFBR, transforming growth factor β receptor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
References
1. Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2013. Ann Oncol (2013) 24(3):792–800. doi: 10.1093/annonc/mdt010
2. World Health Organization. World Health Organization Statistical Information System. WHO Mortality Database (2012). Available from: http://www3.who.int/whosis/menu.cfm
3. GLOBOCAN. GLOBOCAN 2008 (IARC) Section of Cancer Information (2008). Available from: http://globocan.iarc.fr/factsheets/populations/
factsheet.asp?uno=900
4. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer (2010) 127(12):2893–917. doi:10.1002/ijc.25516
5. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC Cancer Base No. 10 [Internet] (2010). Available from: http://globocan.iarc.fr
6. Bosetti C, Bertuccio P, Negri E, La Vecchia C, Zeegers MP, Boffetta P. Pancreatic cancer: overview of descriptive epidemiology. Mol Carcinog (2012) 51(1):3–13. doi:10.1002/mc.20785
7. Hariharan D, Saied A, Kocher HM. Analysis of mortality rates for pancreatic cancer across the world. HPB (Oxford) (2008) 10(1):58–62. doi:10.1080/13651820701883148
8. American Cancer Society (ACS). Global Cancer Facts & Figures. 2nd ed. Atlanta: American Cancer Society (2011).
10. Howlader N, Noone AM, Krapcho M, Garshell J, Neyman N, Altekruse SF, et al. SEER Cancer Statistics Review, 1975-2010. Bethesda (MD): National Cancer Institute (2013). Available from: http://seer.cancer.gov/csr/1975_2010
11. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi:10.3322/caac.21166
12. Bilimoria KY, Bentrem DJ, Ko CY, Ritchey J, Stewart AK, Winchester DP, et al. Validation of the 6th edition AJCC pancreatic cancer staging system: report from the national cancer database. Cancer (2007) 110(4):738–44.
13. Cooperman AM. Pancreatic cancer: the bigger picture. Surg Clin North Am (2001) 81(3):557–74. doi:10.1016/S0039-6109(05)70143-2
14. Ni X, Yang J, Li M. Imaging-guided curative surgical resection of pancreatic cancer in a xenograft mouse model. Cancer Lett (2012) 324(2):179–85. doi:10.1016/j.canlet.2012.05.013
15. Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol (2010) 7(3):163–72. doi:10.1038/nrclinonc.2009.236
16. Silverman DT, Hoover RN, Brown LM, Swanson GM, Schiffman M, Greenberg RS, et al. Why do Black Americans have a higher risk of pancreatic cancer than White Americans? Epidemiology (2003) 14(1):45–54. doi:10.1097/00001648-200301000-00013
17. Parkin DM. 1. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010. Br J Cancer (2011) 105(Suppl 2):S2–5. doi:10.1038/bjc.2011.474
18. Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg (2008) 393(4):535–45. doi:10.1007/s00423-007-0266-2
19. Parkin DM, Boyd L, Walker LC. 16. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010. Br J Cancer (2011) 105(Suppl 2):S77–81. doi:10.1038/bjc.2011.489
20. Berrington de Gonzalez A, Sweetland S, Spencer E. A meta-analysis of obesity and the risk of pancreatic cancer. Br J Cancer (2003) 89(3):519–23. doi:10.1038/sj.bjc.6601140
21. Arslan AA, Helzlsouer KJ, Kooperberg C, Shu XO, Steplowski E, Bueno-de-Mesquita HB, et al. Anthropometric measures, body mass index, and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium (PanScan). Arch Intern Med (2010) 170(9):791–802. doi:10.1001/archinternmed.2010.63
22. Aune D, Greenwood DC, Chan DS, Vieira R, Vieira AR, Navarro Rosenblatt DA, et al. Body mass index, abdominal fatness and pancreatic cancer risk: a systematic review and non-linear dose-response meta-analysis of prospective studies. Ann Oncol (2012) 23(4):843–52. doi:10.1093/annonc/mdr398
23. Luo J, Margolis KL, Adami HO, LaCroix A, Ye W, Women’s Health Initiative InvestigatorsAdami. Women’s health initiative investigators. Obesity and risk of pancreatic cancer among postmenopausal women: the Women’s Health Initiative (United States). Br J Cancer (2008) 99(3):527–31. doi:10.1038/sj.bjc.6604487
24. Tramacere I, Scotti L, Jenab M, Bagnardi V, Bellocco R, Rota M, et al. Alcohol drinking and pancreatic cancer risk: a meta-analysis of the dose-risk relation. Int J Cancer (2010) 126(6):1474–86. doi:10.1002/ijc.24936
25. Michaud DS, Vrieling A, Jiao L, Mendelsohn JB, Steplowski E, Lynch SM, et al. Alcohol intake and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium (PanScan). Cancer Causes Control (2010) 21(8):1213–25. doi:10.1007/s10552-010-9548-z
26. Larsson SC, Wolk A. Red and processed meat consumption and risk of pancreatic cancer: meta-analysis of prospective studies. Br J Cancer (2012) 106(3):603–7. doi:10.1038/bjc.2011.585
27. Anderson KE, Mongin SJ, Sinha R, Stolzenberg-Solomon R, Gross MD, Ziegler RG, et al. Pancreatic cancer risk: associations with meat-derived carcinogen intake in the prostate, lung, colorectal, and ovarian cancer screening trial (PLCO) cohort. Mol Carcinog (2012) 51(1):128–37. doi:10.1002/mc.20794
28. Jansen RJ, Robinson DP, Frank RD, Stolzenberg-Solomon RZ, Bamlet WR, Oberg AL, et al. Meat-related mutagens and pancreatic cancer: null results from a clinic-based case-control study. Cancer Epidemiol Biomarkers Prev (2013) 22(7):1336–9. doi:10.1158/1055-9965.EPI-13-0343
29. Jansen RJ, Robinson DP, Stolzenberg-Solomon RZ, Bamlet WR, de Andrade M, Oberg AL, et al. Fruit and vegetable consumption is inversely associated with having pancreatic cancer. Cancer Causes Control (2011) 22(12):1613–25. doi:10.1007/s10552-011-9838-0
30. Jansen RJ, Robinson DP, Stolzenberg-Solomon RZ, Bamlet WR, de Andrade M, Oberg AL, et al. Nutrients from fruit and vegetable consumption reduce the risk of pancreatic cancer. J Gastrointest Cancer (2013) 44(2):152–61. doi:10.1007/s12029-012-9441-y
31. Larsson SC, Giovannucci E, Wolk A. Folate intake, MTHFR polymorphisms, and risk of esophageal, gastric, and pancreatic cancer: a meta-analysis. Gastroenterology (2006) 131(4):1271–83. doi:10.1053/j.gastro.2006.08.010
32. Bao Y, Michaud DS, Spiegelman D, Albanes D, Anderson KE, Bernstein L, et al. Folate intake and risk of pancreatic cancer: pooled analysis of prospective cohort studies. J Natl Cancer Inst (2011) 103(24):1840–50. doi:10.1093/jnci/djr431
33. Vrieling A, Verhage BA, van Duijnhoven FJ, Jenab M, Overvad K, Tjonneland A, et al. Fruit and vegetable consumption and pancreatic cancer risk in the European prospective investigation into cancer and nutrition. Int J Cancer (2009) 124(8):1926–34. doi:10.1002/ijc.24134
34. Koushik A, Spiegelman D, Albanes D, Anderson KE, Bernstein L, van den Brandt PA, et al. Intake of fruits and vegetables and risk of pancreatic cancer in a pooled analysis of 14 cohort studies. Am J Epidemiol (2012) 176(5):373–86. doi:10.1093/aje/kws027
35. Turati F, Galeone C, Edefonti V, Ferraroni M, Lagiou P, La Vecchia C, et al. A meta-analysis of coffee consumption and pancreatic cancer. Ann Oncol (2012) 23(2):311–8. doi:10.1093/annonc/mdr331
36. Genkinger JM, Li R, Spiegelman D, Anderson KE, Albanes D, Bergkvist L, et al. Coffee, tea, and sugar-sweetened carbonated soft drink intake and pancreatic cancer risk: a pooled analysis of 14 cohort studies. Cancer Epidemiol Biomarkers Prev (2012) 21(2):305–18. doi:10.1158/1055-9965.EPI-11-0945-T
37. Grant WB. An ecologic study of cancer mortality rates in Spain with respect to indices of solar UVB irradiance and smoking. Int J Cancer (2007) 120(5):1123–8. doi:10.1002/ijc.22386
38. Boscoe FP, Schymura MJ. Solar ultraviolet-B exposure and cancer incidence and mortality in the United States, 1993-2002. BMC Cancer (2006) 6:264. doi:10.1186/1471-2407-6-264
39. Mohr SB, Garland CF, Gorham ED, Grant WB, Garland FC. Ultraviolet B irradiance and vitamin D status are inversely associated with incidence rates of pancreatic cancer worldwide. Pancreas (2010) 39(5):669–74. doi:10.1097/MPA.0b013e3181ce654d
40. Bao Y, Ng K, Wolpin BM, Michaud DS, Giovannucci E, Fuchs CS. Predicted vitamin D status and pancreatic cancer risk in two prospective cohort studies. Br J Cancer (2010) 102(9):1422–7. doi:10.1038/sj.bjc.6605658
41. Giovannucci E. Vitamin D and cancer incidence in the Harvard cohorts. Ann Epidemiol (2009) 19(2):84–8. doi:10.1016/j.annepidem.2007.12.002
42. Stolzenberg-Solomon RZ, Hayes RB, Horst RL, Anderson KE, Hollis BW, Silverman DT. Serum vitamin D and risk of pancreatic cancer in the prostate, lung, colorectal, and ovarian screening trial. Cancer Res (2009) 69(4):1439–47. doi:10.1158/0008-5472.CAN-08-2694
43. Stolzenberg-Solomon RZ, Jacobs EJ, Arslan AA, Qi D, Patel AV, Helzlsouer KJ, et al. Circulating 25-hydroxyvitamin D and risk of pancreatic cancer: cohort consortium vitamin D pooling project of rarer cancers. Am J Epidemiol (2010) 172(1):81–93. doi:10.1093/aje/kwq120
44. Huxley R, Ansary-Moghaddam A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer (2005) 92(11):2076–83. doi:10.1038/sj.bjc.6602619
45. Henry SA, Prizment AE, Anderson KE. Duration of diabetes and pancreatic cancer in a case-control study in the Midwest and the Iowa Women’s Health Study (IWHS) cohort. JOP (2013) 14(3):243–9. doi:10.6092/1590-8577/1317
46. Jee SH, Ohrr H, Sull JW, Yun JE, Ji M, Samet JM. Fasting serum glucose level and cancer risk in Korean men and women. JAMA (2005) 293(2):194–202. doi:10.1001/jama.293.2.194
47. Stolzenberg-Solomon RZ, Graubard BI, Chari S, Limburg P, Taylor PR, Virtamo J, et al. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA (2005) 294(22):2872–8. doi:10.1001/jama.294.22.2872
48. Stattin P, Bjor O, Ferrari P, Lukanova A, Lenner P, Lindahl B, et al. Prospective study of hyperglycemia and cancer risk. Diabetes Care (2007) 30(3):561–7. doi:10.2337/dc06-0922
49. Stocks T, Rapp K, Bjorge T, Manjer J, Ulmer H, Selmer R, et al. Blood glucose and risk of incident and fatal cancer in the metabolic syndrome and cancer project (me-can): analysis of six prospective cohorts. PLoS Med (2009) 6(12):e1000201. doi:10.1371/journal.pmed.1000201
50. Stevens RJ, Roddam AW, Beral V. Pancreatic cancer in type 1 and young-onset diabetes: systematic review and meta-analysis. Br J Cancer (2007) 96(3):507–9. doi:10.1038/sj.bjc.6603571
51. Hassan MM, Li D, El-Deeb AS, Wolff RA, Bondy ML, Davila M, et al. Association between hepatitis B virus and pancreatic cancer. J Clin Oncol (2008) 26(28):4557–62. doi:10.1200/JCO.2008.17.3526
52. El-Serag HB, Engels EA, Landgren O, Chiao E, Henderson L, Amaratunge HC, et al. Risk of hepatobiliary and pancreatic cancers after hepatitis C virus infection: a population-based study of U.S. veterans. Hepatology (2009) 49(1):116–23. doi:10.1002/hep.22606
53. Risch HA, Yu H, Lu L, Kidd MS. ABO blood group, Helicobacter pylori seropositivity, and risk of pancreatic cancer: a case-control study. J Natl Cancer Inst (2010) 102(7):502–5. doi:10.1093/jnci/djq007
54. Lin G, Zeng Z, Wang X, Wu Z, Wang J, Wang C, et al. Cholecystectomy and risk of pancreatic cancer: a meta-analysis of observational studies. Cancer Causes Control (2012) 23(1):59–67. doi:10.1007/s10552-011-9856-y
55. Gong Y, Zhou Q, Zhou Y, Lin Q, Zeng B, Chen R, et al. Gastrectomy and risk of pancreatic cancer: systematic review and meta-analysis of observational studies. Cancer Causes Control (2012) 23(8):1279–88. doi:10.1007/s10552-012-0005-z
56. Maisonneuve P, Marshall BC, Lowenfels AB. Risk of pancreatic cancer in patients with cystic fibrosis. Gut (2007) 56(9):1327–8. doi:10.1136/gut.2007.125278
57. Fitzpatrick SG, Katz J. The association between periodontal disease and cancer: a review of the literature. J Dent (2010) 38(2):83–95. doi:10.1016/j.jdent.2009.10.007
58. Wolpin BM, Kraft P, Gross M, Helzlsouer K, Bueno-de-Mesquita HB, Steplowski E, et al. Pancreatic cancer risk and ABO blood group alleles: results from the pancreatic cancer cohort consortium. Cancer Res (2010) 70(3):1015–23. doi:10.1158/0008-5472.CAN-09-2993
59. Pelzer U, Klein F, Bahra M, Sinn M, Dorken B, Neuhaus P, et al. Blood group determinates incidence for pancreatic cancer in Germany. Front Physiol (2013) 4:118. doi:10.3389/fphys.2013.00118
60. Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol (2010) 24(3):349–58. doi:10.1016/j.bpg.2010.02.007
61. Whitcomb DC. Inflammation and cancer V. chronic pancreatitis and pancreatic cancer. Am J Physiol Gastrointest Liver Physiol (2004) 287(2):G315–9. doi:10.1152/ajpgi.00115.2004
62. Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK Jr, Perrault J, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst (1997) 89(6):442–6. doi:10.1093/jnci/89.6.442
63. Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med (1993) 328(20):1433–7. doi:10.1056/NEJM199305203282001
64. Petersen GM, de Andrade M, Goggins M, Hruban RH, Bondy M, Korczak JF, et al. Pancreatic cancer genetic epidemiology consortium. Cancer Epidemiol Biomarkers Prev (2006) 15(4):704–10. doi:10.1158/1055-9965.EPI-05-0734
65. Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med (2009) 133(3):365–74. doi:10.1043/1543-2165-133.3.365
66. Brune KA, Lau B, Palmisano E, Canto M, Goggins MG, Hruban RH, et al. Importance of age of onset in pancreatic cancer kindreds. J Natl Cancer Inst (2010) 102(2):119–26. doi:10.1093/jnci/djp466
67. Lynch HT, Fitzsimmons ML, Smyrk TC, Lanspa SJ, Watson P, McClellan J, et al. Familial pancreatic cancer: clinicopathologic study of 18 nuclear families. Am J Gastroenterol (1990) 85(1):54–60.
68. Silverman DT, Schiffman M, Everhart J, Goldstein A, Lillemoe KD, Swanson GM, et al. Diabetes mellitus, other medical conditions and familial history of cancer as risk factors for pancreatic cancer. Br J Cancer (1999) 80(11):1830–7. doi:10.1038/sj.bjc.6690607
69. Tersmette AC, Petersen GM, Offerhaus GJ, Falatko FC, Brune KA, Goggins M, et al. Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clin Cancer Res (2001) 7(3):738–44.
70. Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res (2004) 64(7):2634–8. doi:10.1158/0008-5472.CAN-03-3823
71. Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, et al. International cancer of the pancreas screening (CAPS) consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut (2013) 62(3):339–47. doi:10.1136/gutjnl-2012-303108
72. Wang W, Chen S, Brune KA, Hruban RH, Parmigiani G, Klein AP. PancPRO: risk assessment for individuals with a family history of pancreatic cancer. J Clin Oncol (2007) 25(11):1417–22. doi:10.1200/JCO.2006.09.2452
73. Grover S, Syngal S. Hereditary pancreatic cancer. Gastroenterology (2010) 139(4):e1–2. doi:10.1053/j.gastro.2010.08.012
74. Couch FJ, Johnson MR, Rabe KG, Brune K, de Andrade M, Goggins M, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev (2007) 16(2):342–6. doi:10.1158/1055-9965.EPI-06-0783
75. Hahn SA, Greenhalf B, Ellis I, Sina-Frey M, Rieder H, Korte B, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst (2003) 95(3):214–21. doi:10.1093/jnci/95.3.214
76. Murphy KM, Brune KA, Griffin C, Sollenberger JE, Petersen GM, Bansal R, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res (2002) 62(13):3789–93.
77. van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol (2010) 105(6):1258–64. doi:10.1038/ajg.2009.725 author reply 65,
78. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology (2000) 119(6):1447–53. doi:10.1053/gast.2000.20228
79. Korsse SE, Harinck F, van Lier MG, Biermann K, Offerhaus GJ, Krak N, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. J Med Genet (2013) 50(1):59–64. doi:10.1136/jmedgenet-2012-101277
80. Lynch HT, Fusaro RM, Lynch JF, Brand R. Pancreatic cancer and the FAMMM syndrome. Fam Cancer (2008) 7(1):103–12. doi:10.1007/s10689-007-9166-4
81. de Snoo FA, Bishop DT, Bergman W, van Leeuwen I, van der Drift C, van Nieuwpoort FA, et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res (2008) 14(21):7151–7. doi:10.1158/1078-0432.CCR-08-0403
82. Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer (2000) 87(6):809–11. doi:10.1002/1097-0215(20000915)87:6<809::AID-IJC8>3.0.CO;2-U
83. Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene (2001) 20(34):4621–8. doi:10.1038/sj.onc.1204621
84. Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol (1997) 150(1):1–13.
85. Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FB, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet (2010) 47(6):421–8. doi:10.1136/jmg.2009.073429
86. Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol (2012) 30(9):958–64. doi:10.1200/JCO.2011.39.5590
87. Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA (2009) 302(16):1790–5. doi:10.1001/jama.2009.1529
88. Giardiello FM, Offerhaus GJ, Lee DH, Krush AJ, Tersmette AC, Booker SV, et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut (1993) 34(10):1394–6. doi:10.1136/gut.34.10.1394
89. Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov (2012) 2(1):41–6. doi:10.1158/2159-8290.CD-11-0194
90. Bakker JL, de Winter JP. A role for ATM in hereditary pancreatic cancer. Cancer Discov (2012) 2(1):14–5. doi:10.1158/2159-8290.CD-11-0318
91. Rebours V, Boutron-Ruault MC, Schnee M, Ferec C, Maire F, Hammel P, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol (2008) 103(1):111–9. doi:10.1111/j.1572-0241.2007.01597.x
92. Martin SP, Ulrich CD II. Pancreatic cancer surveillance in a high-risk cohort. Is it worth the cost? Med Clin North Am (2000) 84(3):739–47, xii–xiii. doi:10.1016/S0025-7125(05)70255-8
93. Lowenfels AB, Maisonneuve P, Whitcomb DC. Risk factors for cancer in hereditary pancreatitis. International Hereditary Pancreatitis Study Group. Med Clin North Am (2000) 84(3):565–73. doi:10.1016/S0025-7125(05)70240-6
94. McWilliams R, Highsmith WE, Rabe KG, de Andrade M, Tordsen LA, Holtegaard LM, et al. Cystic fibrosis transmembrane regulator gene carrier status is a risk factor for young onset pancreatic adenocarcinoma. Gut (2005) 54(11):1661–2. doi:10.1136/gut.2005.074534
95. Schneider R, Slater EP, Sina M, Habbe N, Fendrich V, Matthai E, et al. German national case collection for familial pancreatic cancer (FaPaCa): ten years experience. Fam Cancer (2011) 10(2):323–30. doi:10.1007/s10689-010-9414-x
96. Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet (2010) 78(5):490–4. doi:10.1111/j.1399-0004.2010.01425.x
97. Harinck F, Kluijt I, van Mil SE, Waisfisz Q, van Os TA, Aalfs CM, et al. Routine testing for PALB2 mutations in familial pancreatic cancer families and breast cancer families with pancreatic cancer is not indicated. Eur J Hum Genet (2012) 20(5):577–9. doi:10.1038/ejhg.2011.226
98. Slater EP, Langer P, Fendrich V, Habbe N, Chaloupka B, Matthai E, et al. Prevalence of BRCA2 and CDKN2a mutations in German familial pancreatic cancer families. Fam Cancer (2010) 9(3):335–43. doi:10.1007/s10689-010-9329-6
99. Pogue-Geile KL, Chen R, Bronner MP, Crnogorac-Jurcevic T, Moyes KW, Dowen S, et al. Palladin mutation causes familial pancreatic cancer and suggests a new cancer mechanism. PLoS Med (2006) 3(12):e516. doi:10.1371/journal.pmed.0030516
100. Slater E, Amrillaeva V, Fendrich V, Bartsch D, Earl J, Vitone LJ, et al. Palladin mutation causes familial pancreatic cancer: absence in European families. PLoS Med (2007) 4(4):e164. doi:10.1371/journal.pmed.0040164
101. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer (2013) 49(6):1374–403. doi:10.1016/j.ejca.2012.12.027
102. Andren-Sandberg A, Hoem D, Backman PL. Other risk factors for pancreatic cancer: hormonal aspects. Ann Oncol (1999) 10(Suppl 4):131–5. doi:10.1023/A:1008377928466
103. Jemal A, Simard EP, Xu J, Ma J, Anderson RN. Selected cancers with increasing mortality rates by educational attainment in 26 states in the United States, 1993-2007. Cancer Causes Control (2013) 24(3):559–65. doi:10.1007/s10552-012-9993-y
104. Lynch SM, Vrieling A, Lubin JH, Kraft P, Mendelsohn JB, Hartge P, et al. Cigarette smoking and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium. Am J Epidemiol (2009) 170(4):403–13. doi:10.1093/aje/kwp134
105. Bosetti C, Lucenteforte E, Silverman DT, Petersen G, Bracci PM, Ji BT, et al. Cigarette smoking and pancreatic cancer: an analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann Oncol (2012) 23(7):1880–8. doi:10.1093/annonc/mdr541
106. Anderson MA, Zolotarevsky E, Cooper KL, Sherman S, Shats O, Whitcomb DC, et al. Alcohol and tobacco lower the age of presentation in sporadic pancreatic cancer in a dose-dependent manner: a multicenter study. Am J Gastroenterol (2012) 107(11):1730–9. doi:10.1038/ajg.2012.288
107. Anderson K, Mack TM, Silverman DT. Cancer of the pancreas. In: Schottenfeld D, Fraumeni JF Jr, editors. Cancer Epidemiology and Prevention. 3rd ed. New York: Oxford University Press (2006). p. 721–62.
108. Leenders M, Chuang SC, Dahm CC, Overvad K, Ueland PM, Midttun O, et al. Plasma cotinine levels and pancreatic cancer in the EPIC cohort study. Int J Cancer (2012) 131(4):997–1002. doi:10.1002/ijc.26452
109. Vrieling A, Bueno-de-Mesquita HB, Boshuizen HC, Michaud DS, Severinsen MT, Overvad K, et al. Cigarette smoking, environmental tobacco smoke exposure and pancreatic cancer risk in the European prospective investigation into cancer and nutrition. Int J Cancer (2010) 126(10):2394–403. doi:10.1002/ijc.24907
110. Chuang SC, Gallo V, Michaud D, Overvad K, Tjonneland A, Clavel-Chapelon F, et al. Exposure to environmental tobacco smoke in childhood and incidence of cancer in adulthood in never smokers in the European prospective investigation into cancer and nutrition. Cancer Causes Control (2011) 22(3):487–94. doi:10.1007/s10552-010-9723-2
111. Bertuccio P, La Vecchia C, Silverman DT, Petersen GM, Bracci PM, Negri E, et al. Cigar and pipe smoking, smokeless tobacco use and pancreatic cancer: an analysis from the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol (2011) 22(6):1420–6. doi:10.1093/annonc/mdq613
112. Cogliano VJ, Baan R, Straif K, Grosse Y, Lauby-Secretan B, El Ghissassi F, et al. Preventable exposures associated with human cancers. J Natl Cancer Inst (2011) 103(24):1827–39. doi:10.1093/jnci/djr483
113. Luo J, Ye W, Zendehdel K, Adami J, Adami HO, Boffetta P, et al. Oral use of Swedish moist snuff (snus) and risk for cancer of the mouth, lung, and pancreas in male construction workers: a retrospective cohort study. Lancet (2007) 369(9578):2015–20. doi:10.1016/S0140-6736(07)60678-3
114. IARC. Working Group on the Evaluation of Carcinogenic Risks to Humans. Smokeless Tobacco and Some Tobacco-Specific N-Nitrosamines. Lyon: IARC (2007).
115. Li D, Morris JS, Liu J, Hassan MM, Day RS, Bondy ML, et al. Body mass index and risk, age of onset, and survival in patients with pancreatic cancer. JAMA (2009) 301(24):2553–62. doi:10.1001/jama.2009.886
116. Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology (2013) 144(6):1199–209.e4. doi:10.1053/j.gastro.2013.02.007
117. Zhang J, Dhakal IB, Gross MD, Lang NP, Kadlubar FF, Harnack LJ, et al. Physical activity, diet, and pancreatic cancer: a population-based, case-control study in Minnesota. Nutr Cancer (2009) 61(4):457–65. doi:10.1080/01635580902718941
118. Heinen MM, Verhage BA, Goldbohm RA, Lumey LH, van den Brandt PA. Physical activity, energy restriction, and the risk of pancreatic cancer: a prospective study in the Netherlands. Am J Clin Nutr (2011) 94(5):1314–23. doi:10.3945/ajcn.110.007542
119. Nothlings U, Wilkens LR, Murphy SP, Hankin JH, Henderson BE, Kolonel LN. Body mass index and physical activity as risk factors for pancreatic cancer: the Multiethnic Cohort Study. Cancer Causes Control (2007) 18(2):165–75. doi:10.1007/s10552-006-0100-0
120. O’Rorke MA, Cantwell MM, Cardwell CR, Mulholland HG, Murray LJ. Can physical activity modulate pancreatic cancer risk? A systematic review and meta-analysis. Int J Cancer (2010) 126(12):2957–68. doi:10.1002/ijc.24997
121. Bao Y, Michaud DS. Physical activity and pancreatic cancer risk: a systematic review. Cancer Epidemiol Biomarkers Prev (2008) 17(10):2671–82. doi:10.1158/1055-9965.EPI-08-0488
122. Genkinger JM, Spiegelman D, Anderson KE, Bergkvist L, Bernstein L, van den Brandt PA, et al. Alcohol intake and pancreatic cancer risk: a pooled analysis of fourteen cohort studies. Cancer Epidemiol Biomarkers Prev (2009) 18(3):765–76. doi:10.1158/1055-9965.EPI-08-0880
123. Akgun S, Ertel NH. The effects of sucrose, fructose, and high-fructose corn syrup meals on plasma glucose and insulin in non-insulin-dependent diabetic subjects. Diabetes Care (1985) 8(3):279–83. doi:10.2337/diacare.8.3.279
124. Willett W, Manson J, Liu S. Glycemic index, glycemic load, and risk of type 2 diabetes. Am J Clin Nutr (2002) 76(1):274S–80.
125. Schulze MB, Manson JE, Ludwig DS, Colditz GA, Stampfer MJ, Willett WC, et al. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA (2004) 292(8):927–34. doi:10.1001/jama.292.8.927
126. Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, de Andrade M, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology (2008) 134(1):95–101. doi:10.1053/j.gastro.2007.10.040
127. Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM, Chari ST. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology (2008) 134(4):981–7. doi:10.1053/j.gastro.2008.01.039
128. Cui Y, Andersen DK. Diabetes and pancreatic cancer. Endocr Relat Cancer (2012) 19(5):F9–26. doi:10.1530/ERC-12-0105
129. Prizment AE, Gross M, Rasmussen-Torvik L, Peacock JM, Anderson KE. Genes related to diabetes may be associated with pancreatic cancer in a population-based case-control study in Minnesota. Pancreas (2012) 41(1):50–3. doi:10.1097/MPA.0b013e3182247625
130. Malka D, Hammel P, Maire F, Rufat P, Madeira I, Pessione F, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut (2002) 51(6):849–52. doi:10.1136/gut.51.6.849
131. Hruban RH, Petersen GM, Ha PK, Kern SE. Genetics of pancreatic cancer. From genes to families. Surg Oncol Clin N Am (1998) 7(1):1–23.
132. Allison DC, Piantadosi S, Hruban RH, Dooley WC, Fishman EK, Yeo CJ, et al. DNA content and other factors associated with ten-year survival after resection of pancreatic carcinoma. J Surg Oncol (1998) 67(3):151–9. doi:10.1002/(SICI)1096-9098(199803)67:3<151::AID-JSO2>3.0.CO;2-8
133. Lynch HT, Brand RE, Deters CA, Shaw TG, Lynch JF. Hereditary pancreatic cancer. Pancreatology (2001) 1(5):466–71. doi:10.1159/000055849
134. Bartsch DK, Kress R, Sina-Frey M, Grutzmann R, Gerdes B, Pilarsky C, et al. Prevalence of familial pancreatic cancer in Germany. Int J Cancer (2004) 110(6):902–6. doi:10.1002/ijc.20210
135. Hemminki K, Li X. Familial and second primary pancreatic cancers: a nationwide epidemiologic study from Sweden. Int J Cancer (2003) 103(4):525–30. doi:10.1002/ijc.10863
136. Aaij R, Adeva B, Adinolfi M, Adrover C, Affolder A, Ajaltouni Z, et al. Determination of f(s)/f(d) for 7 TeV pp collisions and measurement of the B0→D-K+ branching fraction. Phys Rev Lett (2011) 107(21):211801. doi:10.1103/PhysRevLett.107.211801
137. Landi S. Genetic predisposition and environmental risk factors to pancreatic cancer: a review of the literature. Mutat Res (2009) 681(2–3):299–307. doi:10.1016/j.mrrev.2008.12.001
138. Yilmaz EN, van Heek NT, van der Spoel JI, Bakker FC, Patka P, Haarman HJ. Myocardial contusion as a result of isolated sternal fractures: a fact or a myth? Eur J Emerg Med (1999) 6(4):293–5. doi:10.1097/00063110-199912000-00003
139. van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, Hoogerbrugge N, Verhoef S, Vasen HF, et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet (2005) 42(9):711–9. doi:10.1136/jmg.2004.028829
140. Thompson D, Easton DF. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst (2002) 94(18):1358–65. doi:10.1093/jnci/94.18.1358
141. Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer (2012) 107(12):2005–9. doi:10.1038/bjc.2012.483
142. Kastrinos F, Allen JI, Stockwell DH, Stoffel EM, Cook EF, Mutinga ML, et al. Development and validation of a colon cancer risk assessment tool for patients undergoing colonoscopy. Am J Gastroenterol (2009) 104(6):1508–18. doi:10.1038/ajg.2009.135
143. Ghaneh P, Costello E, Neoptolemos JP. Biology and management of pancreatic cancer. Gut (2007) 56(8):1134–52.
144. Efthimiou E, Crnogorac-Jurcevic T, Lemoine NR, Brentnall TA. Inherited predisposition to pancreatic cancer. Gut (2001) 48(2):143–7. doi:10.1136/gut.48.2.143
145. Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med (1998) 339(10):645–52. doi:10.1056/NEJM199809033391001
146. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science (2009) 324(5924):217. doi:10.1126/science.1171202
147. Bartsch DK, Fendrich V, Slater EP, Sina-Frey M, Rieder H, Greenhalf W, et al. RNASEL germline variants are associated with pancreatic cancer. Int J Cancer (2005) 117(5):718–22. doi:10.1002/ijc.21254
148. Grutzmann R, McFaul C, Bartsch DK, Sina-Frey M, Rieder H, Koch R, et al. No evidence for germline mutations of the LKB1/STK11 gene in familial pancreatic carcinoma. Cancer Lett (2004) 214(1):63–8. doi:10.1016/j.canlet.2004.06.012
149. Bartsch DK, Krysewski K, Sina-Frey M, Fendrich V, Rieder H, Langer P, et al. Low frequency of CHEK2 mutations in familial pancreatic cancer. Fam Cancer (2006) 5(4):305–8. doi:10.1007/s10689-006-7850-4
150. Nej K, Bartsch DK, Sina-Frey M, Rieder H, Hahn SA, Lubinski J. The NOD2 3020insC mutation and the risk of familial pancreatic cancer? Hered Cancer Clin Pract (2004) 2(3):149–50. doi:10.1186/1897-4287-2-3-149
151. Bartsch DK, Langer P, Habbe N, Matthai E, Chaloupka B, Sina M, et al. Clinical and genetic analysis of 18 pancreatic carcinoma/melanoma-prone families. Clin Genet (2010) 77(4):333–41. doi:10.1111/j.1399-0004.2009.01352.x
152. Bartsch DK, Sina-Frey M, Lang S, Wild A, Gerdes B, Barth P, et al. CDKN2A germline mutations in familial pancreatic cancer. Ann Surg (2002) 236(6):730–7. doi:10.1097/00000658-200212000-00005
153. Michl P, Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut (2013) 62(2):317–26. doi:10.1136/gutjnl-2012-303588
154. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell (1988) 53(4):549–54. doi:10.1016/0092-8674(88)90571-5
155. Remmers N, Bailey JM, Mohr AM, Hollingsworth MA. Molecular pathology of early pancreatic cancer. Cancer Biomark (2010) 9(1–6):421–40.
156. Toubaji A, Achtar M, Provenzano M, Herrin VE, Behrens R, Hamilton M, et al. Pilot study of mutant ras peptide-based vaccine as an adjuvant treatment in pancreatic and colorectal cancers. Cancer Immunol Immunother (2008) 57(9):1413–20. doi:10.1007/s00262-008-0477-6
157. Rudek MA, Khan Y, Goldsweig H, Donehower RC, Jimeno A, Shah P, et al. Integrated development of S-trans. Trans-farnesylthiosalicyclic acid (FTS, salisarib) in pancreatic cancer [abstract]. J Clin Oncol (2008) 26:a4626.
158. Rejiba S, Wack S, Aprahamian M, Hajri A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci (2007) 98(7):1128–36. doi:10.1111/j.1349-7006.2007.00506.x
159. Jimeno A, Rubio-Viqueira B, Amador ML, Grunwald V, Maitra A, Iacobuzio-Donahue C, et al. Dual mitogen-activated protein kinase and epidermal growth factor receptor inhibition in biliary and pancreatic cancer. Mol Cancer Ther (2007) 6(3):1079–88. doi:10.1158/1535-7163.MCT-06-0448
160. Takayama Y, Kokuryo T, Yokoyama Y, Nagino M, Nimura Y, Senga T, et al. MEK inhibitor enhances the inhibitory effect of imatinib on pancreatic cancer cell growth. Cancer Lett (2008) 264(2):241–9. doi:10.1016/j.canlet.2008.01.035
161. Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med (2009) 60:207–19. doi:10.1146/annurev.med.60.041807.132435
162. Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, et al. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets (2012) 13(6):802–10. doi:10.2174/138945012800564158
163. Korc M, Chandrasekar B, Yamanaka Y, Friess H, Buchier M, Beger HG. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest (1992) 90(4):1352–60. doi:10.1172/JCI116001
164. Bloomston M, Bhardwaj A, Ellison EC, Frankel WL. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig Surg (2006) 23(1–2):74–9. doi:10.1159/000093497
165. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol (2007) 25(15):1960–6. doi:10.1200/JCO.2006.07.9525
166. Kindler HL, Ioka T, Richel DJ, Bennouna J, Letourneau R, Okusaka T, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol (2011) 12(3):256–62. doi:10.1016/S1470-2045(11)70004-3
167. Goncalves A, Gilabert M, Francois E, Dahan L, Perrier H, Lamy R, et al. BAYPAN study: a double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann Oncol (2012) 23(11):2799–805. doi:10.1093/annonc/mds135
168. Gaya A, Tse V. A preclinical and clinical review of aflibercept for the management of cancer. Cancer Treat Rev (2012) 38(5):484–93. doi:10.1016/j.ctrv.2011.12.008
169. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324(5933):1457–61. doi:10.1126/science.1171362
170. Hakam A, Fang Q, Karl R, Coppola D. Coexpression of IGF-1R and c-Src proteins in human pancreatic ductal adenocarcinoma. Dig Dis Sci (2003) 48(10):1972–8. doi:10.1023/A:1026122421369
171. Momose I, Kunimoto S, Osono M, Ikeda D. Inhibitors of insulin-like growth factor-1 receptor tyrosine kinase are preferentially cytotoxic to nutrient-deprived pancreatic cancer cells. Biochem Biophys Res Commun (2009) 380(1):171–6. doi:10.1016/j.bbrc.2009.01.065
172. Rieder S, Michalski CW, Friess H, Kleeff J. Insulin-like growth factor signaling as a therapeutic target in pancreatic cancer. Anticancer Agents Med Chem (2011) 11(5):427–33. doi:10.2174/187152011795677454
173. Michl P, Downward J. Mechanisms of disease: PI3K/AKT signaling in gastrointestinal cancers. Z Gastroenterol (2005) 43(10):1133–9. doi:10.1055/s-2005-858638
174. Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer (2003) 89(11):2110–5. doi:10.1038/sj.bjc.6601396
175. Yamamoto S, Tomita Y, Hoshida Y, Morooka T, Nagano H, Dono K, et al. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res (2004) 10(8):2846–50. doi:10.1158/1078-0432.CCR-02-1441
176. Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene (2004) 23(53):8571–80. doi:10.1038/sj.onc.1207902
177. Abe N, Watanabe T, Masaki T, Mori T, Sugiyama M, Uchimura H, et al. Pancreatic duct cell carcinomas express high levels of high mobility group I(Y) proteins. Cancer Res (2000) 60(12):3117–22.
178. Liau SS, Whang E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin Cancer Res (2008) 14(5):1470–7. doi:10.1158/1078-0432.CCR-07-1450
179. Liau SS, Ashley SW, Whang EE. Lentivirus-mediated RNA interference of HMGA1 promotes chemosensitivity to gemcitabine in pancreatic adenocarcinoma. J Gastrointest Surg (2006) 10(9):1254–62. doi:10.1016/j.gassur.2006.06.011 discussion 63,
180. Trapasso F, Sarti M, Cesari R, Yendamuri S, Dumon KR, Aqeilan RI, et al. Therapy of human pancreatic carcinoma based on suppression of HMGA1 protein synthesis in preclinical models. Cancer Gene Ther (2004) 11(9):633–41. doi:10.1038/sj.cgt.7700745
181. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell (2013) 23(3):406–20. doi:10.1016/j.ccr.2013.01.023
182. Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol (2009) 27(2):193–8. doi:10.1200/JCO.2008.18.9514
183. Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer (2010) 10:368. doi:10.1186/1471-2407-10-368
184. Pollak M. Metformin and pancreatic cancer: a clue requiring investigation. Clin Cancer Res (2012) 18(10):2723–5. doi:10.1158/1078-0432.CCR-12-0694
185. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (2005) 330(7503):1304–5. doi:10.1136/bmj.38415.708634.F7
186. Bodmer M, Becker C, Meier C, Jick SS, Meier CR. Use of antidiabetic agents and the risk of pancreatic cancer: a case-control analysis. Am J Gastroenterol (2012) 107(4):620–6. doi:10.1038/ajg.2011.483
187. Regel I, Kong B, Raulefs S, Erkan M, Michalski CW, Hartel M, et al. Energy metabolism and proliferation in pancreatic carcinogenesis. Langenbecks Arch Surg (2012) 397(4):507–12. doi:10.1007/s00423-012-0933-9
188. Li H, Huang C, Huang K, Wu W, Jiang T, Cao J, et al. STAT3 knockdown reduces pancreatic cancer cell invasiveness and matrix metalloproteinase-7 expression in nude mice. PLoS One (2011) 6(10):e25941. doi:10.1371/journal.pone.0025941
189. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi:10.1126/science.8197455
190. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science (1994) 264(5155):95–8. doi:10.1126/science.8140422
191. Toyonaga T, Nakano K, Nagano M, Zhao G, Yamaguchi K, Kuroki S, et al. Blockade of constitutively activated Janus kinase/signal transducer and activator of transcription-3 pathway inhibits growth of human pancreatic cancer. Cancer Lett (2003) 201(1):107–16. doi:10.1016/S0304-3835(03)00482-8
192. Scholz A, Heinze S, Detjen KM, Peters M, Welzel M, Hauff P, et al. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology (2003) 125(3):891–905. doi:10.1016/S0016-5085(03)01064-3
193. Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett (2007) 251(2):199–210. doi:10.1016/j.canlet.2006.10.017
194. Miyatsuka T, Kaneto H, Shiraiwa T, Matsuoka TA, Yamamoto K, Kato K, et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev (2006) 20(11):1435–40. doi:10.1101/gad.1412806
195. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell (2011) 19(4):456–69. doi:10.1016/j.ccr.2011.03.009
196. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res (2011) 71(14):5020–9. doi:10.1158/0008-5472.CAN-11-0908
197. Fogelman DR, Wolff RA, Kopetz S, Javle M, Bradley C, Mok I, et al. Evidence for the efficacy of iniparib, a PARP-1 inhibitor, in BRCA2-associated pancreatic cancer. Anticancer Res (2011) 31(4):1417–20.
198. Porcelli L, Quatrale AE, Mantuano P, Leo MG, Silvestris N, Rolland JF, et al. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol Oncol (2013) 7(3):308–22. doi:10.1016/j.molonc.2012.10.002
199. Yuan K, Sun Y, Zhou T, McDonald J, Chen Y. PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin Cancer Res (2013) 19(17):4750–9. doi:10.1158/1078-0432.CCR-13-0516
200. Sawai H, Okada Y, Kazanjian K, Kim J, Hasan S, Hines OJ, et al. The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Cancer Res (2005) 65(24):11536–44. doi:10.1158/0008-5472.CAN-05-2843
201. Ito Y, Okada Y, Sato M, Sawai H, Funahashi H, Murase T, et al. Expression of glial cell line-derived neurotrophic factor family members and their receptors in pancreatic cancers. Surgery (2005) 138(4):788–94. doi:10.1016/j.surg.2005.07.007
202. Ketterer K, Rao S, Friess H, Weiss J, Buchler MW, Korc M. Reverse transcription-PCR analysis of laser-captured cells points to potential paracrine and autocrine actions of neurotrophins in pancreatic cancer. Clin Cancer Res (2003) 9(14):5127–36.
203. Sakamoto Y, Kitajima Y, Edakuni G, Sasatomi E, Mori M, Kitahara K, et al. Expression of Trk tyrosine kinase receptor is a biologic marker for cell proliferation and perineural invasion of human pancreatic ductal adenocarcinoma. Oncol Rep (2001) 8(3):477–84.
204. Liu H, Ma Q, Li J. High glucose promotes cell proliferation and enhances GDNF and RET expression in pancreatic cancer cells. Mol Cell Biochem (2011) 347(1–2):95–101. doi:10.1007/s11010-010-0617-0
205. Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP, et al. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J Natl Cancer Inst (2010) 102(2):107–18. doi:10.1093/jnci/djp456
206. Donahue TR, Hines OJ. CXCR2 and RET single nucleotide polymorphisms in pancreatic cancer. World J Surg (2009) 33(4):710–5. doi:10.1007/s00268-008-9826-z
207. Smith JP, Shih A, Wu Y, McLaughlin PJ, Zagon IS. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am J Physiol (1996) 270(5 Pt 2):R1078–84.
208. Caplin M, Savage K, Khan K, Brett B, Rode J, Varro A, et al. Expression and processing of gastrin in pancreatic adenocarcinoma. Br J Surg (2000) 87(8):1035–40. doi:10.1046/j.1365-2168.2000.01488.x
209. Matters GL, Harms JF, McGovern CO, Jayakumar C, Crepin K, Smith ZP, et al. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas (2009) 38(5):e151–61. doi:10.1097/MPA.0b013e3181a66fdc
210. Grabowska AM, Berry CA, Hughes J, Bushell M, Willis AE, Watson SA. A gastrin transcript expressed in gastrointestinal cancer cells contains an internal ribosome entry site. Br J Cancer (2008) 98(10):1696–703. doi:10.1038/sj.bjc.6604326
211. Kawasaki D, Emori Y, Eta R, Iino Y, Hamano H, Yoshinaga K, et al. Effect of Z-360, a novel orally active CCK-2/gastrin receptor antagonist on tumor growth in human pancreatic adenocarcinoma cell lines in vivo and mode of action determinations in vitro. Cancer Chemother Pharmacol (2008) 61(5):883–92. doi:10.1007/s00280-007-0591-8
212. Meyer T, Caplin ME, Palmer DH, Valle JW, Larvin M, Waters JS, et al. A phase Ib/IIa trial to evaluate the CCK2 receptor antagonist Z-360 in combination with gemcitabine in patients with advanced pancreatic cancer. Eur J Cancer (2010) 46(3):526–33. doi:10.1016/j.ejca.2009.11.004
213. Chau I, Cunningham D, Russell C, Norman AR, Kurzawinski T, Harper P, et al. Gastrazole (JB95008), a novel CCK2/gastrin receptor antagonist, in the treatment of advanced pancreatic cancer: results from two randomised controlled trials. Br J Cancer (2006) 94(8):1107–15. doi:10.1038/sj.bjc.6603058
214. Shapiro J, Marshall J, Karasek P. G17DT + gemcitabine [Gem] versus placebo + Gem in untreated subjects with locally advanced, recurrent, or metastatic adenocarcinoma of the pancreas: results of a randomized, double-blind, multinational, multicenter study [abstract]. J Clin Oncol (2005) 23(4012).
215. Wong HH, Lemoine NR. Pancreatic cancer: molecular pathogenesis and new therapeutic targets. Nat Rev Gastroenterol Hepatol (2009) 6(7):412–22. doi:10.1038/nrgastro.2009.89
216. Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res (1999) 59(5):987–90.
217. Ding XZ, Hennig R, Adrian TE. Lipoxygenase and cyclooxygenase metabolism: new insights in treatment and chemoprevention of pancreatic cancer. Mol Cancer (2003) 2:10. doi:10.1186/1476-4598-2-10
218. Wei D, Wang L, He Y, Xiong HQ, Abbruzzese JL, Xie K. Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res (2004) 64(6):2030–8. doi:10.1158/0008-5472.CAN-03-1945
219. Xiong HQ, Hess KR, Kayaleh OR, Goodwin JW, Banerjee T, Sinclair SS, et al. A phase II trial of gemcitabine and celecoxib for metastatic pancreatic cancer [abstract]. J Clin Oncol (2005) 23(Suppl 1):4174.
220. Ferrari V, Valcamonico F, Amoroso V, Simoncini E, Vassalli L, Marpicati P, et al. Gemcitabine plus celecoxib (GECO) in advanced pancreatic cancer: a phase II trial. Cancer Chemother Pharmacol (2006) 57(2):185–90. doi:10.1007/s00280-005-0028-1
221. Dragovich T, Burris H III, Loehrer P, Von Hoff DD, Chow S, Stratton S, et al. Gemcitabine plus celecoxib in patients with advanced or metastatic pancreatic adenocarcinoma: results of a phase II trial. Am J Clin Oncol (2008) 31(2):157–62. doi:10.1097/COC.0b013e31815878c9
222. Kerr S, Campbell C, Legore K, Witters L, Harvey H, Lipton A. Phase II trial of gemcitabine and irinotecan plus celecoxib in advanced adenocarcinoma of the pancreas [abstract]. J Clin Oncol (2005) 23(Suppl 1):4155.
223. Kirane A, Toombs JE, Ostapoff K, Carbon JG, Zaknoen S, Braunfeld J, et al. Apricoxib, a novel inhibitor of COX-2, markedly improves standard therapy response in molecularly defined models of pancreatic cancer. Clin Cancer Res (2012) 18(18):5031–42. doi:10.1158/1078-0432.CCR-12-0453
224. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
225. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12(4):237–51. doi:10.1038/nrc3237
226. Dodson LF, Hawkins WG, Goedegebuure P. Potential targets for pancreatic cancer immunotherapeutics. Immunotherapy (2011) 3(4):517–37. doi:10.2217/imt.11.10
227. Koido S, Homma S, Takahara A, Namiki Y, Tsukinaga S, Mitobe J, et al. Current immunotherapeutic approaches in pancreatic cancer. Clin Dev Immunol (2011) 2011:267539. doi:10.1155/2011/267539
228. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (2010) 33(8):828–33. doi:10.1097/CJI.0b013e3181eec14c
229. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331(6024):1612–6. doi:10.1126/science.1198443
230. Vonderheide RH, Bajor DL, Winograd R, Evans RA, Bayne LJ, Beatty GL. CD40 immunotherapy for pancreatic cancer. Cancer Immunol Immunother (2013) 62(5):949–54. doi:10.1007/s00262-013-1427-5
231. Bubenik J, Den Otter W, Huland E. Local cytokine therapy of cancer: interleukin-2, interferons and related cytokines. Cancer Immunol Immunother (2000) 49(2):116–22. doi:10.1007/s002620050610
232. Wagner K, Schulz P, Scholz A, Wiedenmann B, Menrad A. The targeted immunocytokine L19-IL2 efficiently inhibits the growth of orthotopic pancreatic cancer. Clin Cancer Res (2008) 14(15):4951–60. doi:10.1158/1078-0432.CCR-08-0157
233. Alessi P, Ebbinghaus C, Neri D. Molecular targeting of angiogenesis. Biochim Biophys Acta (2004) 1654(1):39–49. doi:10.1016/j.bbcan.2003.08.001
234. Halin C, Zardi L, Neri D. Antibody-based targeting of angiogenesis. News Physiol Sci (2001) 16:191–4.
235. Michl P, Gress TM. Improving drug delivery to pancreatic cancer: breaching the stromal fortress by targeting hyaluronic acid. Gut (2012) 61(10):1377–9. doi:10.1136/gutjnl-2012-302604
236. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut (2011) 60(6):861–8. doi:10.1136/gut.2010.226092
237. Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res (2008) 14(19):5995–6004. doi:10.1158/1078-0432.CCR-08-0291
238. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature (2003) 425(6960):851–6. doi:10.1038/nature02009
239. Tsuda N, Mine T, Ioannides CG, Chang DZ. Synthetic microRNA targeting glioma-associated antigen-1 protein. Methods Mol Biol (2009) 487:435–49. doi:10.1007/978-1-60327-547-7_21
240. Hilbig A, Oettle H. Transforming growth factor beta in pancreatic cancer. Curr Pharm Biotechnol (2011) 12(12):2158–64. doi:10.2174/138920111798808356
241. Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res (1998) 58(23):5329–32.
242. Schnurr M, Duewell P. Breaking tumor-induced immunosuppression with 5’-triphosphate siRNA silencing TGFbeta and activating RIG-I. Oncoimmunology (2013) 2(5):e24170. doi:10.4161/onci.24170
243. Fuxe J, Karlsson MC. TGF-beta-induced epithelial-mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol (2012) 22(5–6):455–61. doi:10.1016/j.semcancer.2012.05.004
244. Shi Y, Hata A, Lo RS, Massague J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature (1997) 388(6637):87–93. doi:10.1038/40431
245. Oettle H, Hilbig A, Seufferlein T, Tsianakas A, Luger T, Schmid RM, et al. Phase I/II study with trabedersen (AP 12009) monotherapy for the treatment of patients with advanced pancreatic cancer, malignant melanoma, and colorectal carcinoma [abstract]. J Clin Oncol (2011) 29:(abstract 2513).
246. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62(1):112–20. doi:10.1136/gutjnl-2012-302529
247. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(3):418–29. doi:10.1016/j.ccr.2012.01.007
248. Heiser PW, Hebrok M. Development and cancer: lessons learned in the pancreas. Cell Cycle (2004) 3(3):270–2. doi:10.4161/cc.3.3.731
249. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (2008) 321(5897):1801–6. doi:10.1126/science.1164368
250. Zeng G, Germinaro M, Micsenyi A, Monga NK, Bell A, Sood A, et al. Aberrant Wnt/beta-catenin signaling in pancreatic adenocarcinoma. Neoplasia (2006) 8(4):279–89. doi:10.1593/neo.05607
251. Pasca di Magliano M, Biankin AV, Heiser PW, Cano DA, Gutierrez PJ, Deramaudt T, et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS One (2007) 2(11):e1155. doi:10.1371/journal.pone.0001155
252. Romero D, Iglesias M, Vary CP, Quintanilla M. Functional blockade of Smad4 leads to a decrease in beta-catenin levels and signaling activity in human pancreatic carcinoma cells. Carcinogenesis (2008) 29(5):1070–6. doi:10.1093/carcin/bgn054
253. Cui J, Jiang W, Wang S, Wang L, Xie K. Role of Wnt/beta-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des (2012) 18(17):2464–71. doi:10.2174/13816128112092464
254. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature (2012) 487(7408):510–3. doi:10.1038/nature11217
255. Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med (2013) 19(5):320–7. doi:10.1016/j.molmed.2013.03.003
256. Ristorcelli E, Lombardo D. Targeting Notch signaling in pancreatic cancer. Expert Opin Ther Targets (2010) 14(5):541–52. doi:10.1517/14728221003769895
257. Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell (2003) 3(6):565–76. doi:10.1016/S1535-6108(03)00140-5
258. Mazur PK, Einwachter H, Lee M, Sipos B, Nakhai H, Rad R, et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A (2010) 107(30):13438–43. doi:10.1073/pnas.1002423107
259. Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res (2009) 15(7):2291–301. doi:10.1158/1078-0432.CCR-08-2004
260. Mysliwiec P, Boucher MJ. Targeting Notch signaling in pancreatic cancer patients – rationale for new therapy. Adv Med Sci (2009) 54(2):136–42. doi:10.2478/v10039-009-0026-3
261. Omura N, Goggins M. Epigenetics and epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol (2009) 2(4):310–26.
262. Fritsche P, Seidler B, Schuler S, Schnieke A, Gottlicher M, Schmid RM, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut (2009) 58(10):1399–409. doi:10.1136/gut.2009.180711
263. Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr, Huang P, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res (2005) 65(24):11658–66. doi:10.1158/0008-5472.CAN-05-2370
264. Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene (2012) 31(13):1609–22. doi:10.1038/onc.2011.354
265. Ohuchida K, Mizumoto K, Ogura Y, Ishikawa N, Nagai E, Yamaguchi K, et al. Quantitative assessment of telomerase activity and human telomerase reverse transcriptase messenger RNA levels in pancreatic juice samples for the diagnosis of pancreatic cancer. Clin Cancer Res (2005) 11(6):2285–92. doi:10.1158/1078-0432.CCR-04-1581
266. Hiyama E, Kodama T, Shinbara K, Iwao T, Itoh M, Hiyama K, et al. Telomerase activity is detected in pancreatic cancer but not in benign tumors. Cancer Res (1997) 57(2):326–31.
267. Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen JA, Meo M, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer (2006) 95(11):1474–82. doi:10.1038/sj.bjc.6603437
Keywords: pancreatic ductal adenocarcinoma, risk factors, hereditary cancer syndromes, therapeutic targets, signal-transduction pathways, immune response, stroma reaction, epigenetic changes
Citation: Wörmann SM and Algül H (2013) Risk factors and therapeutic targets in pancreatic cancer. Front. Oncol. 3:282. doi: 10.3389/fonc.2013.00282
Received: 16 August 2013; Accepted: 03 November 2013;
Published online: 18 November 2013.
Edited by:
Yunfeng Cui, Tianjin Medical University, ChinaReviewed by:
Mirko Omejc, University Medical Centre Ljubljana, SloveniaYi Zhong, Johns Hopkins Medical Institutions, USA
Crystal Grant, Albany University, USA
Copyright: © 2013 Wörmann and Algül. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hana Algül, II. Medizinische Klinik, Klinikum rechts der Isar, Universität München, Ismaninger Str. 22, Munich 81675, Germany e-mail:aGFuYS5hbGd1ZWxAbHJ6LnR1bS5kZQ==