 Clare L. Scott1
Clare L. Scott1 Marc A. Becker
Marc A. Becker Goli Samimi
Goli Samimi- 1Walter and Eliza Hall Institute of Medical Research, Royal Melbourne Hospital and The Royal Women’s Hospital, Melbourne, VIC, Australia
- 2Division of Medical Oncology, Mayo Clinic, Rochester, MN, USA
- 3The Kinghorn Cancer Centre, St Vincent’s Clinical School, Garvan Institute of Medical Research, Sydney, NSW, Australia
Despite increasing evidence that precision therapy targeted to the molecular drivers of a cancer has the potential to improve clinical outcomes, high-grade epithelial ovarian cancer (OC) patients are currently treated without consideration of molecular phenotype, and predictive biomarkers that could better inform treatment remain unknown. Delivery of precision therapy requires improved integration of laboratory-based models and cutting-edge clinical research, with pre-clinical models predicting patient subsets that will benefit from a particular targeted therapeutic. Patient-derived xenografts (PDXs) are renewable tumor models engrafted in mice, generated from fresh human tumors without prior in vitro exposure. PDX models allow an invaluable assessment of tumor evolution and adaptive response to therapy. PDX models have been applied to pre-clinical drug testing and biomarker identification in a number of cancers including ovarian, pancreatic, breast, and prostate cancers. These models have been shown to be biologically stable and accurately reflect the patient tumor with regards to histopathology, gene expression, genetic mutations, and therapeutic response. However, pre-clinical analyses of molecularly annotated PDX models derived from high-grade serous ovarian cancer (HG-SOC) remain limited. In vivo response to conventional and/or targeted therapeutics has only been described for very small numbers of individual HG-SOC PDX in conjunction with sparse molecular annotation and patient outcome data. Recently, two consecutive panels of epithelial OC PDX correlate in vivo platinum response with molecular aberrations and source patient clinical outcomes. These studies underpin the value of PDX models to better direct chemotherapy and predict response to targeted therapy. Tumor heterogeneity, before and following treatment, as well as the importance of multiple molecular aberrations per individual tumor underscore some of the important issues addressed in PDX models.
Introduction
Cell lines and archival tumor tissue have provided a platform for discovery and validation of novel therapeutic targets in epithelial ovarian cancer (OC). However, despite increasing evidence that precision therapy targeted to the molecular driver(s) of a tumor has the potential to impact overall survival (1), patients with high-grade epithelial OC are currently treated with a “one-size fits all” approach, without consideration of molecular phenotype or biomarkers of response that could better inform treatment. Pre-clinical models to predict those patients who will benefit from targeted therapy are imperative to implement effective precision therapy strategies. Classic cell line-derived xenograft models have provided invaluable mechanistic insight toward the key signaling pathways and oncogenic drivers of OC tumorigenesis, malignant progression, and chemotherapeutic resistance. The translational potential of models, generated from either human OC, many years ago, with scant histo-pathologic data about the source OC tumor from which they were derived or from human ovarian surface epithelial (OSE) cell lines, remains questionable (2). In both cases, the cell lines used to generate xenografts have been expanded in vitro, and as such, have likely acquired significant alterations in morphology, motility, and proliferation that do not necessarily reflect the physiologic state of the tumor (3). More importantly, recent evidence suggests that the ovarian surface may not be the origin of “ovarian cancer” (4, 5).
The majority of epithelial OC are serous in sub-type (50% of OC) and display Fallopian tube-like or “endosalpingeal” characteristics. Endometrioid OC (20% of OC) and mucinous OC (10% of OC) represent additional epithelial sub-types displaying features of epithelia from other Mullerian tract (developmental female genital tract) organs, resembling endometrial and endocervical epithelia, respectively (6). Two main phenotypic groupings of human EOC have been described. Type I EOC includes low-grade, mucinous, and clear cell cancers, with progression identifiable from adenoma-borderline-cancer. Type II EOC comprises lethal, high-grade serous (HG-SOC), endometrioid, and undifferentiated EOC. Although previously thought not to have recognizable precursor lesions (7), the current consensus indicates an association with early lesions being found in the distal Fallopian tube in carriers of BRCA1/2 mutations (5, 8). The Fallopian tubes are derived from the Mullerian ducts (also of mesodermal origin) and consist of muscular, ciliated, and secretory epithelia (9). Fallopian tube cancer and primary Peritoneal cancer (the latter is derived from the coelomic epithelium, as is the OSE), behave in a clinically similar fashion to serous EOC and while often studied together, distinct molecular differences are evident (10, 11).
The OSE has long been postulated to be the source of putative cancer initiating cells for epithelial “ovarian” cancer (12), with an alternative origin postulated to derive from the distal ends of the Fallopian tube and malignant or pre-malignant cells migrating to and settling on contiguous OSE (5, 8). A proportion of malignant lesions originate in the Fallopian tube and may potentially metastasize to the OSE (13). The OSE persists as a single layer of squamous to cuboidal epithelium that covers the ovary (14), is derived from coelomic epithelium (of mesenchymal origin), has a basement membrane and, unusual for a surface epithelium, can undergo epithelial-mesenchymal transition (EMT) (15). The OSE has been described as a facultative stem cell niche, with cells retained within this niche maintaining pluripotency and expressing markers typical of a stem cell-like quiescent state (12). However, it is possible that the OSE provides a suitable “niche” for the development of “ovarian” cancer and that the majority of HG-SOC in fact derive from secretory cells from the fallopian tube (16). Many reports of OC xenografts and patient-derived xenografts (PDXs) have mixed together OC sub-types, which may have very different implications for cell of origin or treatment approaches. By not taking into account sub-type, the likelihood of deriving useful information is greatly diminished.
Furthermore, traditional in vivo pre-clinical models do not accurately recapitulate the complexity and heterogeneity of patient tumors (17). As each tumor’s molecular phenotype impacts prognosis and response to treatment, detailed genomic annotation of each xenograft is necessary for comprehensive evaluation of targeted therapies. Xenografts derived from a cell line originating from OC have to undergo extensive selection. More often these lines reflect the in vitro culture system and are devoid of the complex pathology and molecular attributes of the original patient tumor. A compelling example of this discordant phenomenon was reported in a study involving 41 cell lines, each of which rarely contained BRCA1 and/or BRCA2 mutations (18). The authors concluded that the use of these cell lines for xenograft studies would not accurately reflect the patient population. Moreover, xenografts derived from cell lines cultured from potentially irrelevant tissues (e.g., non-surface epithelium), may be even more flawed as models of human HG-SOC.
Xenografts may be derived directly from patient tissue without prior in vitro culture (PDXs), in which tumor tissue excised at the time of surgery is immediately transplanted into immune-deficient mice. Importantly, digestion of tumor material using protocols known to involve harsh cell dissociation buffers may inadvertently strip the cell surface of molecules integral toward in vivo cell–cell interactions. Alternatively, methods such as mincing of tumor fragments or the use of whole fragments may more closely model the heterogeneity of clinical disease. PDXs derived from non-digested OC can provide extremely flexible models for pre-clinical analysis of novel therapeutics (19). Primary OC and resultant serial PDX can be histologically assessed for known diagnostic and prognostic markers and characterized by molecular techniques including genome sequencing. These PDX models can therefore be extensively annotated and serve as powerful models for pre-clinical studies of targeted therapeutic strategies, thus bridging the gap between lab bench discoveries and clinical translation. As such, there has been an increase in characterization and application of PDX models for drug screening across a range of cancers [reviewed in Ref. (17)].
Thus, major concerns regarding OC PDX literature to date are as follows: numerous papers lack detail regarding histologic sub-type, molecular phenotype, a detailed description of the methods used to generate and maintain the PDX, limited genomic characterization has been performed (e.g., CGH or CNV analysis), and the stability of various phenotypes over successive generations is noteworthy. As a result, a substantial barrier to the study of OC is the paucity of translationally (e.g., transient in vitro primary cell lines) and clinically (e.g., archived tissues from retrospective analyses) relevant models, thereby highlighting the salient need for an alternative, clinically relevant means to rapidly translate results from bench-to-bedside. The development of personalized PDX models, with each patient having a PDX generated across her disease progression (primary tumor, metastasis, recurrence) and stage of treatment (prior to treatment, at relapse), with availability of source biospecimens (germline DNA, serum, frozen, and FFPE tissue, etc.) and prospective clinical annotations could overcome many of the current hurdles (e.g., the dependence on isolation/digestion and subsequent amplification in vitro prior to establishment and testing in animals). These PDX models recapitulate primary patient tumors (e.g., formation of bowel metastases, obstruction, ascites, etc.), reproducibly engraft, retain the molecular and gross phenotypic characteristics of the donor OC patient, can be accurately monitored for tumor detection and progression (e.g., gross tumor palpation, calipers, ultrasound-guided imaging, etc.) and represent a practical and highly translatable medium to study the effects of both standard chemotherapy and precision targeted therapeutics.
Methodology
As previously noted, standard OC xenografts are derived from established, highly annotated, and widely available cell lines. While OSE models are commonly utilized and have become a mainstay workhorse to investigators, their uncommon origin (e.g., murine-derived) brings into question functional significance. For example, the ID8 cell line was originally developed as a syngeneic mouse model to study the early molecular and immune events related to ovarian carcinogenesis (20). As a result, greater attention toward patient-derived OC models has been expended.
Patient-derived xenograft (digested) have played a key role toward the study of the cancer stem cell (CSC) niche and identified tumor-initiating cells (TICs) as key players in primary patient ovarian xenografts (21). The frequency of TICs represents an intrinsic property of the primary patient tumor. However, the integrity of the TIC landscape (e.g., proportion of CD133 positive versus negative cells) is altered in PDX models. It is plausible that the extensive ex vivo digestion prior to PDX engraftment is the confounding source of TIC PDX discrepancies.
Patient-derived xenograft (fragments) are generated by sectioning of fresh tumor tissue and engrafting (1–2 × 1–3 mm3) pieces either subcutaneously or orthotopically into immuno-deficient mice (e.g., NOD-SCID IL2Rγ−/−). Engraftment rates for this generation (T1) range from 25 to 80% depending on tumor type (22), and growth usually takes 2–6 months. Once the T1 PDX tumor has reached ∼700–1500 mm3 (23), it is harvested and directly re-transplanted for expansion in later serial generations (T2, T3) which are used for in vivo drug response, biomarker studies and generating cell lines for additional drug response and molecular studies. Alternatively, the fresh patient tumor can be minced and cryo-preserved in DMSO for later thawing and transplantation, thus ensuring the renewability of the resource. For molecular comparisons, the original patient tumor and the PDX models can undergo extensive histo-pathological and genomic analysis. In addition, for OC PDX models, the T1 PDX can be analyzed for platinum response and homologous recombination (HR) activity, which are key clinical indicators of drug response.
PDX Models in Ovarian Cancer
Patient-derived xenograft models have been applied to pre-clinical drug testing and biomarker identification in a number of cancers including pancreatic cancer (24), NSCLC (25, 26), melanoma (27), breast cancer (28, 29), and prostate cancer (30). As reviewed by Tentler et al. comprehensive genomic analysis including sequencing, expression, and copy number, have demonstrated that PDX models maintain overall global gene expression and activity as the source tumor (17). In OC, PDX models have been developed that accurately reflect the patient tumor and have successfully been used to examine drug response and effects of targeted treatment (22).
Some of the earliest applications of OC PDXs in studies of drug response were reported by the Repasky group. They developed 20 different PDX models in severe combined immuno-deficient (SCID) mice. Histo-pathologic and in situ hybridization analyses were carried out to confirm similarity to the source tumor. While all implanted PDXs eventually formed tumors, 65% (13/20) of them reached 1–2 cm within 2–6 months and were further expanded. Three of the later generation PDX models developed metastases and two developed ascites, representing clinical progression of the disease (31). The group then applied their subcutaneous PDX models in two separate studies to examine the effects of IL-12 and Flt-3 ligand on ovarian tumor growth. Following engraftment, PDX mice were treated with either placebo or IL-12 (32), or placebo or Flt-3 ligand (33), and tumor volume was measured over time. Treatment with IL-12 or Flt-3 ligand resulted in decreased tumor growth compared to control-treated mice, with increased NK cells and necrosis in the tumors of IL-12 or Flt-3 ligand treated mice. These findings suggest an immunologic reaction in response to IL-12 and Flt-3 ligand, supporting their potential therapeutic roles in the treatment of OC (32, 33).
Ghamande et al. followed up these studies by examining the effect of CD40 ligand therapy, previously shown to decrease growth in OC cells, on CD40 receptor-positive PDX serous OC models (34). PDX mice with subcutaneous or intra-abdominal tumors were treated with vehicle or increasing concentrations of recombinant CD40 ligand and tumor growth was assessed over time. Tumor growth in both locations was decreased following as little as one cycle of treatment, regardless of concentration. In addition, once tumors were excised following treatment, histological analysis revealed disruption of tissue architecture and increased fibrosis and apoptosis, providing further insight into the mechanism of therapy. Furthermore, the authors utilized these PDX models to examine the effect of combination therapy using standard chemotherapeutic agents and CD40 ligand therapy, further demonstrating an augmented effect when both drugs were used in treatment of CD40-positive tumors (34). These studies highlight the utility of PDX models in evaluating drug efficacy and mechanism of action.
While a majority of HG-SOC patients initially respond to first-line treatment (generally, a platinum drug in combination with a taxane), a large proportion eventually relapse and develop platinum-resistant disease. OC PDX models can be useful for screening drug sensitivity, which in turn provides guidance for clinical management of the patient who presents with recurrent disease. Kolfschoten et al. established 15 subcutaneous OC PDX models and examined sensitivity to standard chemotherapy (35). They reported that response rates in the PDX models correlated with those in OC patients (e.g., 40% of PDXs responded to cisplatin while 48% of patients respond). As detoxification by glutathiones has been demonstrated to render cells resistant to platinum treatment, the authors also investigated the glutathione-based mechanisms involved in the development of resistance. They measured levels of glutathione and glutathione-related enzymes in the PDX models and related them to drug response. They identified a correlation between glutathione reductase activity and efficacy of chemotherapeutic agents cisplatin and cyclophosphamide, suggesting that glutathione-related enzymes may be useful as predictors of drug sensitivity (35). These findings speak to the value of PDX models for expanding in vitro findings of drug response and relating them to patient tumors.
Because HG-SOC patients frequently develop resistance to platinum-based chemotherapy, it is imperative to identify novel therapies with efficacy toward tumors with de novo or acquired resistance. In an effort to investigate the efficacy of lurbinectedin, a new DNA binding drug, Vidal et al. generated serous PDX models by engrafting primary tumor tissue directly onto the mouse ovary surface (36). They included tumors with cisplatin sensitivity, as well as a tumor selected for acquired cisplatin resistance by repeated in vivo exposure. They reported a high correlation of histo-pathologic features between the patient and the platinum-sensitive and -resistant PDX tumors. The platinum-sensitive PDX displayed a dose-dependent response to cisplatin treatment, characterized by significant tumor volume reduction. Interestingly, 30–50% of treated mice relapsed at 6 months following treatment, and histo-pathologic features of the relapsed tumors were similar to the un-treated xenografts (36). As expected, cisplatin treatment did not significantly inhibit tumor growth in the cisplatin-resistant PDX model compared to control-treated mice. However, lurbinectedin treatment alone significantly decreased tumor growth in both cisplatin-sensitive and -resistant PDX models, and lurbinectedin in combination with cisplatin was more effective than either drug alone (36). Additional studies demonstrated an increase in apoptosis and mitotic catastrophe in lurbinectedin-treated PDX mice, providing further insight into its mechanism of action. Thus, drug-resistant PDX models can be used to identify therapies that may be effective in patients with tumors resistant to standard agents.
In addition to subcutaneous and intra-peritoneal (IP) engraftments, OC PDX models have been established in mice by sub-renal capsule xenografts, allowing for follicle maturation. Lee et al. have demonstrated a high take-rate (95%) in sub-renal capsule PDX models, including low- or moderate-grade OC tissues that are typically difficult to engraft in subcutaneous or IP models (37). The authors compared histo-pathologic features in the original patient tumor, pre-graft tissue, and post-graft tissue and found no architectural or cytological differences, nor any major differences in immunomarker expression including CK20, CK7, or WT-1 (87–91% overall concordance). This group then investigated five individual sub-renal PDX models for drug response and genetic stability over subsequent passages (38). The authors analyzed the primary tumor and corresponding PDX by array CGH and reported similar gene copy numbers, with the primary tumors consistently clustering with their matching PDX. Furthermore, there was no significant difference in copy number changes between the primary tumor and corresponding PDX (38). These findings further support the accurate reflection of the patient tumor in PDX models. Furthermore, the high engraftment rate of sub-renal capsule PDX models may provide the opportunity to investigate the differences in tumor progression between low- and high-grade ovarian tumors.
As most HG-SOC tumors present at advanced stage, following peritoneal dissemination, IP PDX models are useful for investigation of tumor progression and metastasis. Bankert et al. generated IP PDX models from five different OC patients to examine metastasis and the microenvironment of human ovarian tumors (39). In these mice, tumor growth and spread reflect the patterns that occur clinically whereby tumors grew on surfaces within the peritoneal cavity including the omentum, spleen, ovaries, pancreas, and liver. In addition, these PDX mice formed distended abdomens with ascites fluid containing viable tumor cells, and CA-125 was present in their ascites and blood. Thus PDX OC models not only accurately reflect the histo-pathologic features of the tumor, but also present with clinically relevant disease, making them excellent models to investigate tumor progression.
Patient-derived xenograft models generated from patient ascites may also be useful for investigation of tumor progression and metastasis, and are readily transplanted. Ascites-derived PDX models have been used to characterize genome-wide chromosomal aberrations in BRCA1-mutated tumors (40). In addition, Stewart et al. generated ascites-derived PDX models to identify and characterize ovarian TICs (21), and found that these cells are molecularly heterogeneous across different tumors. Direct comparisons of PDX models generated from primary tumors and associated ascites would be helpful to determine whether ascites-derived PDX models accurately reflect the heterogeneity of the solid tumor. Not all patients develop ascites during their clinical course, suggesting that PDX derived only from ascites may not reflect the full disease spectrum.
PDX Models to Test Targeted Therapy
A targeted therapy currently under clinical investigation in OC treatment is inhibition of poly(ADP-ribose) polymerase (PARP), which targets cells with HR defects. PARP inhibition leads to accumulation of single-strand breaks, which generates double-strand breaks in DNA at replication forks. While double-strand breaks are effectively repaired in normal cells by HR repair, cells with deficiencies in BRCA1/2 use error-prone mechanisms resulting in chromosomal instability and cell death (41). Germline mutations in BRCA1/2 are present in 17% of HG-SOC cases (42) and in 25% of HG-SOC patients under the age of 50 (43). In addition, loss of BRCA function by genetic or epigenetic processes has been reported in 50% of HG-SOC cases (44). This high frequency of BRCA deficiency makes HG-SOC patients ideal candidates for PARP inhibition. PARP inhibitors have been shown in vitro and in Phase I/II clinical trials to be an effective treatment in some BRCA-deficient tumors, although it is still unclear why all patients with BRCA1/2 mutations do not respond to PARP inhibitors (45). Furthermore, a proportion of those who do respond eventually progress and thus studies are still necessary to determine the mechanism of resistance to PARP inhibitors. HG-SOC PDX models, generated from tumors before and after treatment, are extremely useful to better understand the mechanisms of therapeutic response and resistance.
The Wang group further analyzed two of their sub-renal PDX models that carried alterations in BRCA1 and BRCA2 (38). In one case, DNA sequencing revealed a germline mutation in exon 2 of BRCA1, as well as loss of heterozygosity. In the other case, promoter hypermethylation of BRCA1 was identified in the primary tumor, as well as a sequence variant in intron 2 of BRCA2. All of these alterations were maintained in the xenograft tumor. In assessing drug response in these models, tumor volume was decreased in carboplatin/paclitaxel-treated versus control-treated mice; however treatment with the PARP inhibitor PJ34 did not affect tumor growth in the BRCA models, despite decreased PAR expression in these tumors (38). It is unclear whether the lack of response was due to poor potency of this PARP inhibitor or due to additional tumor biology causing PARP inhibitor resistance.
In a more recent study to investigate targeted therapy, Kortmann et al. established PDX models from a BRCA wild-type and a BRCA2 germline-mutated HG-SOC tumor to examine response to the PARP inhibitor, olaparib (46). First generation (T1) xenografts were analyzed following daily olaparib treatment of 50 mg/kg for 4 weeks. Immunohistochemical analysis of the BRCA2 mutated model demonstrated decreased tumor cell proliferation and increased numbers of dead cells following olaparib treatment, while wild-type tumor characteristics were not affected. Moreover, olaparib treatment significantly decreased tumor volume in the BRCA2 mutant PDX while having no effect on the BRCA wild-type PDX (46). These studies demonstrate the value of PDX models for characterizing response to targeted therapy.
In addition to PARP inhibitors, signaling molecules make attractive targets to inhibit tumor growth in HG-SOC and other cancers. The Hedgehog (Hh) pathway promotes proliferation, regeneration, and differentiation in adult somatic tissues, and aberrant activation of the Hh pathway is associated with malignant transformation in several cancers. Combination treatment with standard chemotherapy plus Hh pathway inhibitors has been demonstrated to be effective against proliferation in basal cell cancer, medulloblastoma, and small cell lung cancer, amongst others (47). In OC, it has been reported that 20–50% of cases include Hh pathway activation (48). Furthermore, ectopic expression of Hh factors results in increased proliferation and motility of OC cells, while Hh inhibition impairs the growth of OC cell lines in vivo. Thus the Hh pathway may be a potential therapeutic target in OC treatment.
McCann et al. further examined the potential of Hh inhibitors as OC treatment using subcutaneous PDX models of serous OC (49). In this study, they treated a serous PDX model reported to have an activated Hh signaling pathway with the Hh pathway inhibitor cyclopamine. The cyclopamine-treated PDX mice had significantly decreased tumor volume compared to control-treated mice. In addition, the authors tested the efficacy of IPI-926, a derivative of cyclopamine that has increased oral bioavailability and potency and is currently in Phase I/II clinical trials, alone and in combination with standard first-line chemotherapy. In three different serous PDX models with activated Hh signaling pathways, Treatment with IPI-926 alone, or in combination, resulted in decreased tumor growth similar to results from chemotherapy alone, compared to controls (49). Interestingly, when the PDX mice were maintained on IPI-926 alone following combination treatment, tumor regression was retained for up to 50 days following initial therapy. These studies indicate the utility of PDX models in examining combination therapy as well as maintenance therapy, in clinically relevant models.
PDX Cohorts
One of the most useful features of PDX models is their renewability, providing a repository of xenografts, tissues, and cell lines for researchers to access, along with relevant clinical and molecular data (Table 1). These cohorts provide fully annotated, genome-specific PDX models as training and test sets, providing the opportunity to efficiently bring molecular targeted drugs into clinical trials for the treatment of OC. In order for these cohorts to be fully beneficial, comprehensive annotation is essential, particularly two major characterizations: (a) the methods utilized to generate and maintain the PDX models (e.g., source tissue to confirm HG-SOC origin, fragmentation or mincing versus digestion to limit selection and possibly allow retention of infiltrating stroma, no prior in vitro culture to ensure reflection of primary tumor, method of implantation, etc.); (b) histotype, molecular, and genomic characterization (mutation, gene expression, CGH, CNV analysis), as well as response to standard therapy, of the PDX models (Table 1). Upon complete characterization and annotation, this resource will greatly accelerate the development of newly targeted therapies and identification of predictive biomarkers in OC, further bridging the gap between laboratory-based discoveries of novel therapeutic targets and clinical care.
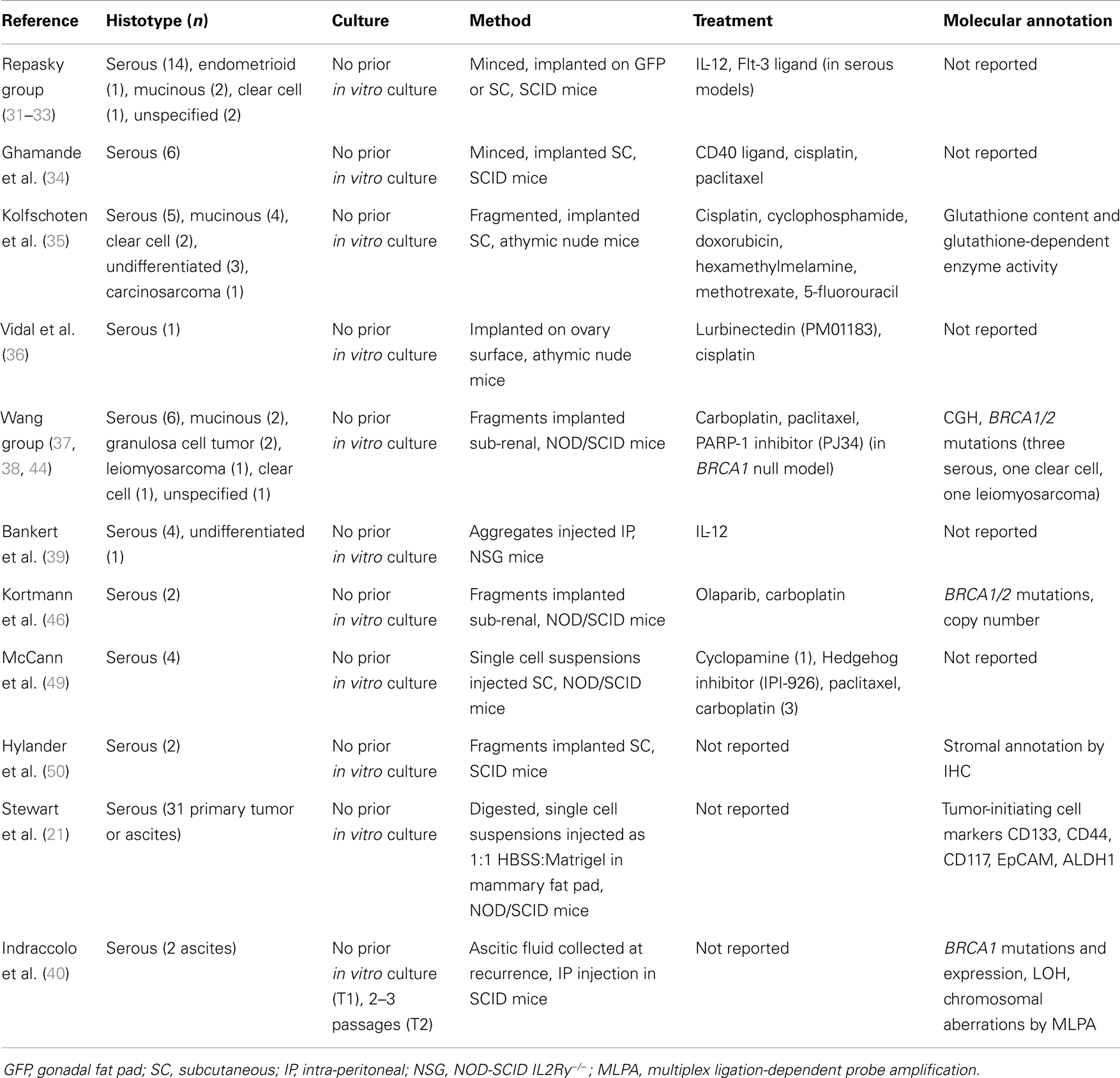
Table 1. Summary of ovarian cancer PDX models.
Limitations
Probably the most noted limitation of PDX models involves the use of immunocompromised mice, which may attenuate the impact of the tumor microenvironment on tumor growth and drug response. In addition, stromal components such as vasculature or secreted stromal factors are increasingly being targeted by novel therapies. Thus, it is imperative that PDX models recapitulate the heterogeneity of the patient tumor in order to accurately test these novel therapies. In a recent study, Hylander et al. investigated vascularization and stromal formation in 37 subcutaneous PDX models in SCID mice created from a range of tumor types, including ovary, pancreas, kidney, and colon cancers (50). Successfully engrafted tumors were histologically examined for stromal factors and blood vessels. Their findings demonstrated that at the first passage (15–25 weeks), tumors no longer contained human stromal factors or vasculature; indeed, the stromal fibroblasts and vessel markers within the tumor were of murine origin (50). Kinetic studies suggested that loss of human vascularization markers occurred within 3–9 weeks, depending on tumor type. The authors conclude that in PDX models in which tumors are engrafted directly into immunocompromised mice, tumor growth is supported by host stroma and vasculature, suggesting that studies of therapies targeting human stromal components may not be adequate in these models.
To overcome these challenges, various approaches have been applied and additional models have been generated with the aim of recapitulating the tumor microenvironment. Engraftment of whole, non-disrupted chunks of human tumor helps to preserve tumor microenvironment components including leukocytes, fibroblasts, extracellular matrix, and vasculature (51). In addition, the use of NOD-SCID IL2Rγ−/− mice provides improved PDX models for tumor–stromal interactions as they maintain tumor-associated leukocytes and stromal fibroblasts for up to 9 weeks after implantation (52). In the previously described IP OC PDX study by Bankert et al. performed in NOD-SCID IL2Rγ−/− mice, the authors identified functional human lymphocytes and fibroblasts in tumors from multiple organs within the peritoneal cavity up to 177 days following engraftment (39). These findings suggest that future PDX models would provide greater value if generated in NOD-SCID IL2Rγ−/− mice, particularly for studies involving drug response or microenvironment-targeting treatments.
Conclusion
The establishment of PDX models that recapitulate the complexity and genetic heterogeneity of HG-SOC will guide personalized cancer therapy and be invaluable toward establishing research priorities and strategies for developing new and more effective approaches to treatment in patients with recurrent OC. A repository of extensively characterized HG-SOC PDX models can be used for drug screening and discovery as well as biomarker development and testing. Furthermore, PDX models generated at initial diagnosis as well as at the time of recurrence will not only permit personalized treatment options, but in the long-term serve to enrich the recruitment and accrual of patients into early phase clinical trials.
Authors Contribution
Clare L. Scott, Marc A. Becker, Paul Haluska, and Goli Samimi all contributed to the writing and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Ovarian Cancer Group at the Garvan acknowledges support from the Gynecological Oncology (GO) Fund of the Royal Hospital for Women Foundation, Sydney, Australia. The Cancer Research Program at the Garvan acknowledges support from the National Health and Medical Research Council of Australia, Cancer Council NSW, Cancer Australia, Cure Cancer Australia Foundation, the Australian Cancer Research Foundation, The Petre Foundation and the RT Hall Trust. This work was supported by fellowships and grants from the Cancer Council Victoria (Sir Edward Dunlop Fellowship in Cancer Research to Clare L. Scott); the Victorian Cancer Agency (Clinical Fellowship to Clare L. Scott). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS and the Australian Cancer Research Foundation. The work was supported by the following: United States National Institute of Health Grant CA136393 (Paul Haluska), Mayo Clinic SPORE in Ovarian Cancer and DK07352 (Marc A. Becker), T32 Training Grant if Diabetes in Metabolism; Ovarian Cancer Research Fund OCRF258797 (Paul Haluska); and Ginkgo, LLC (Paul Haluska).
References
1. Duffy MJ. The war on cancer: are we winning? Tumour Biol (2013) 34:1275–84. doi: 10.1007/s13277-013-0759-2
2. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun (2013) 4:2126. doi:10.1038/ncomms3126
3. Sale S, Orsulic S. Models of ovarian cancer metastasis: murine models. Drug Discov Today Dis Models (2006) 3:149–54. doi:10.1016/j.ddmod.2006.05.006
4. Piek JM, Van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol (2001) 195:451–6. doi:10.1002/path.1000
5. Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol (2006) 30:230–6. doi:10.1097/01.pas.0000180854.28831.77
6. Kurman RJ, Shih IeM. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol (2010) 34:433–43. doi:10.1097/PAS.0b013e3181cf3d79
7. Cho KR, Shih IeM. Ovarian cancer. Annu Rev Pathol (2009) 4:287–313. doi:10.1146/annurev.pathol.4.110807.092246
8. Piek JM, Verheijen RH, Kenemans P, Massuger LF, Bulten H, Van Diest PJ. BRCA1/2-related ovarian cancers are of tubal origin: a hypothesis. Gynecol Oncol (2003) 90:491. doi:10.1016/S0090-8258(03)00365-2
9. Nutu M, Weijdegard B, Thomas P, Thurin-Kjellberg A, Billig H, Larsson DG. Distribution and hormonal regulation of membrane progesterone receptors beta and gamma in ciliated epithelial cells of mouse and human fallopian tubes. Reprod Biol Endocrinol (2009) 7:89. doi:10.1186/1477-7827-7-89
10. Pectasides D, Pectasides E, Economopoulos T. Fallopian tube carcinoma: a review. Oncologist (2006) 11:902–12. doi:10.1634/theoncologist.11-8-902
11. Pentheroudakis G, Pavlidis N. Serous papillary peritoneal carcinoma: unknown primary tumour, ovarian cancer counterpart or a distinct entity? A systematic review. Crit Rev Oncol Hematol (2010) 75:27–42. doi:10.1016/j.critrevonc.2009.10.003
12. Bowen NJ, Walker LD, Matyunina LV, Logani S, Totten KA, Benigno BB, et al. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med Genomics (2009) 2:71. doi:10.1186/1755-8794-2-71
13. Mehra K, Mehrad M, Ning G, Drapkin R, McKeon FD, Xian W, et al. STICS, SCOUTs and p53 signatures; a new language for pelvic serous carcinogenesis. Front Biosci (Elite Ed) (2011) 3:625–34. doi:10.2741/e275
14. Gillett WR, Mitchell A, Hurst PR. A scanning electron microscopic study of the human ovarian surface epithelium: characterization of two cell types. Hum Reprod (1991) 6:645–50.
15. Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation. Endocr Rev (2009) 30:624–712. doi:10.1210/er.2009-0012
16. Crum CP, Drapkin R, Miron A, Ince TA, Muto M, Kindelberger DW, et al. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol (2007) 19:3–9. doi:10.1097/GCO.0b013e328011a21f
17. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol (2012) 9:338–50. doi:10.1038/nrclinonc.2012.61
18. Stordal B, Timms K, Farrelly A, Gallagher D, Busschots S, Renaud M, et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol (2013) 7:567–79. doi:10.1016/j.molonc.2012.12.007
19. Mourad WA, Vallieres E. Ex-vivo fine needle aspiration. A new method of xenografting non-small cell carcinoma of the lung. In vivo (1995) 9:149–54.
20. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis (2000) 21:585–91. doi:10.1093/carcin/21.4.585
21. Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci U S A (2011) 108:6468–73. doi:10.1073/pnas.1005529108
22. Elkas JC, Baldwin RL, Pegram M, Tseng Y, Slamon D, Karlan BY. A human ovarian carcinoma murine xenograft model useful for preclinical trials. Gynecol Oncol (2002) 87:200–6. doi:10.1006/gyno.2002.6819
23. Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc (2007) 2:247–50. doi:10.1038/nprot.2007.25
24. Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang XF, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res (2006) 12:4652–61. doi:10.1158/1078-0432.CCR-06-0113
25. Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res (2008) 14:6456–68. doi:10.1158/1078-0432.CCR-08-0138
26. Merk J, Rolff J, Becker M, Leschber G, Fichtner I. Patient-derived xenografts of non-small-cell lung cancer: a pre-clinical model to evaluate adjuvant chemotherapy? Eur J Cardiothorac Surg (2009) 36:454–9. doi:10.1016/j.ejcts.2009.03.054
27. Fiebig HH, Schuler J, Bausch N, Hofmann M, Metz T, Korrat A. Gene signatures developed from patient tumor explants grown in nude mice to predict tumor response to 11 cytotoxic drugs. Cancer Genomics Proteomics (2007) 4:197–209.
28. Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, De Cremoux P, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res (2007) 13:3989–98. doi:10.1158/1078-0432.CCR-07-0078
29. de Plater L, Lauge A, Guyader C, Poupon MF, Assayag F, De Cremoux P, et al. Establishment and characterisation of a new breast cancer xenograft obtained from a woman carrying a germline BRCA2 mutation. Br J Cancer (2010) 103:1192–200. doi:10.1038/sj.bjc.6605900
30. Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res (2005) 65:9611–6. doi:10.1158/0008-5472.CAN-05-0817
31. Xu Y, Silver DF, Yang NP, Oflazoglu E, Hempling RE, Piver MS, et al. Characterization of human ovarian carcinomas in a SCID mouse model. Gynecol Oncol (1999) 72:161–70. doi:10.1006/gyno.1998.5238
32. Silver DF, Hempling RE, Piver MS, Repasky EA. Effects of IL-12 on human ovarian tumors engrafted into SCID mice. Gynecol Oncol (1999) 72:154–60. doi:10.1006/gyno.1998.5239
33. Silver DF, Hempling RE, Piver MS, Repasky EA. Flt-3 ligand inhibits growth of human ovarian tumors engrafted in severe combined immunodeficient mice. Gynecol Oncol (2000) 77:377–82. doi:10.1006/gyno.2000.5782
34. Ghamande S, Hylander BL, Oflazoglu E, Lele S, Fanslow W, Repasky EA. Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res (2001) 61:7556–62.
35. Kolfschoten GM, Pinedo HM, Scheffer PG, Schluper HM, Erkelens CA, Boven E. Development of a panel of 15 human ovarian cancer xenografts for drug screening and determination of the role of the glutathione detoxification system. Gynecol Oncol (2000) 76:362–8. doi:10.1006/gyno.1999.5689
36. Vidal A, Munoz C, Guillen MJ, Moreto J, Puertas S, Martinez-Iniesta M, et al. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin Cancer Res (2012) 18:5399–411. doi:10.1158/1078-0432.CCR-12-1513
37. Lee CH, Xue H, Sutcliffe M, Gout PW, Huntsman DG, Miller DM, et al. Establishment of subrenal capsule xenografts of primary human ovarian tumors in SCID mice: potential models. Gynecol Oncol (2005) 96:48–55. doi:10.1016/j.ygyno.2004.09.025
38. Press JZ, Kenyon JA, Xue H, Miller MA, De Luca A, Miller DM, et al. Xenografts of primary human gynecological tumors grown under the renal capsule of NOD/SCID mice show genetic stability during serial transplantation and respond to cytotoxic chemotherapy. Gynecol Oncol (2008) 110:256–64. doi:10.1016/j.ygyno.2008.03.011
39. Bankert RB, Balu-Iyer SV, Odunsi K, Shultz LD, Kelleher RJ Jr, Barnas JL, et al. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS One (2011) 6:e24420. doi:10.1371/journal.pone.0024420
40. Indraccolo S, Tisato V, Agata S, Moserle L, Ferrari S, Callegaro M, et al. Establishment and characterization of xenografts and cancer cell cultures derived from BRCA1 -/- epithelial ovarian cancers. Eur J Cancer (2006) 42:1475–83. doi:10.1016/j.ejca.2006.01.057
41. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434:917–21. doi:10.1038/nature03445
42. TCGA. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474:609–15. doi:10.1038/nature10166
43. Alsop K, Fereday S, Meldrum C, Defazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian ovarian cancer study group. J Clin Oncol (2012) 30(21):2654–63.
44. Press JZ, De Luca A, Boyd N, Young S, Troussard A, Ridge Y, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer (2008) 8:17. doi:10.1186/1471-2407-8-17
45. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet (2010) 376:245–51. doi:10.1016/S0140-6736(10)60893-8
46. Kortmann U, McAlpine JN, Xue H, Guan J, Ha G, Tully S, et al. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clin Cancer Res (2011) 17:783–91. doi:10.1158/1078-0432.CCR-10-1382
47. McMillan R, Matsui W. Molecular pathways: the hedgehog signaling pathway in cancer. Clin Cancer Res (2012) 18:4883–8. doi:10.1158/1078-0432.CCR-11-2509
48. Bhattacharya R, Kwon J, Ali B, Wang E, Patra S, Shridhar V, et al. Role of hedgehog signaling in ovarian cancer. Clin Cancer Res (2008) 14:7659–66. doi:10.1158/1078-0432.CCR-08-1414
49. McCann CK, Growdon WB, Kulkarni-Datar K, Curley MD, Friel AM, Proctor JL, et al. Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PLoS One (2011) 6:e28077. doi:10.1371/journal.pone.0028077
50. Hylander BL, Punt N, Tang H, Hillman J, Vaughan M, Bshara W, et al. Origin of the vasculature supporting growth of primary patient tumor xenografts. J Transl Med (2013) 11:110. doi:10.1186/1479-5876-11-110
51. Bankert RB, Egilmez NK, Hess SD. Human-SCID mouse chimeric models for the evaluation of anti-cancer therapies. Trends Immunol (2001) 22:386–93. doi:10.1016/S1471-4906(01)01943-3
52. Simpson-Abelson MR, Sonnenberg GF, Takita H, Yokota SJ, Conway TF Jr, Kelleher RJ Jr, et al. Long-term engraftment and expansion of tumor-derived memory T cells following the implantation of non-disrupted pieces of human lung tumor into NOD-scid IL2Rgamma(null) mice. J Immunol (2008) 180:7009–18.
Keywords: ovarian cancer, patient-derived xenografts, pre-clinical models, targeted therapy, clinical trials
Citation: Scott CL, Becker MA, Haluska P and Samimi G (2013) Patient-derived xenograft models to improve targeted therapy in epithelial ovarian cancer treatment. Front. Oncol. 3:295. doi: 10.3389/fonc.2013.00295
Received: 30 September 2013; Accepted: 19 November 2013;
Published online: 04 December 2013.
Edited by:
Viive Maarika Howell, University of Sydney, AustraliaCopyright: © 2013 Scott, Becker, Haluska and Samimi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Goli Samimi, The Kinghorn Cancer Centre, Garvan Institute of Medical Research, 370 Victoria Street, Darlinghurst, Sydney, NSW 2010, Australia e-mail:Zy5zYW1pbWlAZ2FydmFuLm9yZy5hdQ==