 Richard J. Epstein
Richard J. Epstein- Laboratory of Genome Evolution & Informatics, The Kinghorn Cancer Centre, and Clinical Informatics & Research Centre, Department of Oncology, St Vincent’s Hospital, UNSW Clinical School, Sydney, NSW, Australia
The notion that targeted drugs can unplug gain-of-function tumor pathways has revitalized pharmaceutical research, but the survival benefits of this strategy have so far proven modest. A weakness of oncogene-blocking approaches is that they do not address the problem of cancer progression as selected by the recessive phenotypes of genetic instability and apoptotic resistance which in turn arise from loss-of-function – i.e., undruggable – defects of caretaker (e.g., BRCA, MLH1) or gatekeeper (e.g., TP53, PTEN) suppressor genes. Genetic instability ensures that rapid cell kill is balanced by rapid selection for apoptotic resistance and hence for metastasis, casting doubt on the assumption that cytotoxicity (“response”) remains the best way to identify survival-enhancing drugs. In the absence of gene therapy, it is proposed here that caretaker-defective (high-instability) tumors may be best treated with low-lethality drugs inducing replicative (RAS-RAF-ERK) arrest or dormancy, causing “stable disease” rather than tumorilytic remission. Gatekeeper-defective (death-resistant) tumors, on the other hand, may be best managed by combining survival (PI3K-AKT-mTOR) pathway blockade with metronomic or sequential pro-apoptotic drugs.
Introduction
Tolstoy’s Anna Karenina begins, “All happy families are alike, but all unhappy families are unhappy in their own special ways.” This is a literary way of saying that there are more ways for complex systems to go wrong than to remain right, and helps to explain why cancer remains the most challenging of human diseases. It also hints at why current approaches to drug development continue to yield frustratingly marginal benefits (1).
Common cancers arise from the progressive accumulation of common genetic errors, most of which subvert the function of normal cell-regulatory genes. If a germline defect in one of these regulatory genes predisposes to familial or heritable cancers, the nomenclature “tumor suppressor gene” has often been used. In 1997 Kinzler and Vogelstein noted that most tumor suppressor genes fall into just two functional categories: “caretaker” genes that repair DNA and maintain genetic stability, or “gatekeeper” genes that regulate cell-cycle progression and apoptosis (2). This semantic dichotomy is too simple (3), of course, given that genetic instability is exacerbated by gatekeeper gene defects that permit survival of cells which would otherwise self-destruct, whereas apoptotic resistance is worsened by caretaker defects that impair sensing of potentially lethal insults by the afferent limb of the DNA damage response (4). Nonetheless, as argued below, the potential utility of this model (5, 6) – contrasting as it does with more complex but less user-friendly models of cancer biology (7) – has not yet been exploited in clinical practice or research.
The ability of a cell to engage in oncogenic oversignaling implies selection for a pre-existing suppressor gene defect, given that normal cells with intact control pathways typically succumb to cell death as a result of constitutive hyperstimulation (8). For this reason alone, cancer treatment strategies focused solely on “driver” pathway inhibition seem likely to fail – for no sooner is the proverbial plug extracted from the driver pathway than the underlying apoptotic gene defect permits selection for heterologous pathway upregulation and/or additional oncogenic events, manifesting as a rapidly proliferative (high Ki67) tumor outgrowth reflecting the suppressor gene mutation burden (9). This problem is made even worse by coexisting caretaker defects that speed selection and cell adaptation – a useful coping mechanism for germline (species) evolution (10), but yet another therapeutic hurdle for restoring phenotypic stability to growing neoplasms.
A further impediment to the vision of personalized cancer medicine is that the heterogeneity of molecular defects within tumors far exceeds the existing range of targeted drugs. Broader characterizations of dominant tumorigenic pathway dysfunction, reflecting the relative overactivity of major signaling cascades – those mediated by RAS-ERK (replication) vs. PI3K-AKT (survival) signaling – could usefully guide clinicians as to best treatment decisions; with regard to the latter pathways, for example, whether to prioritize replication arrest and thus slow progression of genetic instability, or instead to focus on apoptotic sensitization by blocking mTOR upregulation originating from, say, PIK3CA mutations or heregulin- and insulin-related oversignaling (11–13). An example is detailed in our recent report of a patient with refractory progressive colorectal cancer which was mismatch-repair (MMR)-deficient, KRAS wild-type, and BRAFV600E-mutant, who appeared to benefit from a small-molecule BRAF inhibitor (14) only when an epidermal growth factor receptor (EGFR) inhibitor was co-prescribed to block this interfering anti-apoptotic pathway (15).
The Clinical Challenge of Suppressor Gene Defects
The crisis of blockbuster drug development in today’s omics-obsessed pharmaceutical industry (16) originates in part from commercial strategies that rely on unplugging “addicted” oncogene targets as a seductively simple solution to the cancer problem (1, 17). This approach may work for rare oncogene-expressing tumor types with low genetic instability, such as chronic myeloid leukemia or medullary thyroid cancer (18), but common cancers are complicated by a shifting balance: for every oncogenic driver pathway, there is a permissive spectrum of suppressor gene defects lurking in the molecular background which begin to erode survival gains as soon as signaling blockade is achieved. These suppressor defects are loss-of-function in type, and hence undruggable by standard pharmacologic approaches (19) which continue, for sound technical reasons, to focus on enzyme and/or receptor inhibition (20). As indicated in Table 1, clinical use of such drugs selects rapidly for a cascade of downstream control defects that accelerate both tumor resistance and disease progression (21).

Table 1. Sequence of steps in cancer drug development.
The challenge of networking a drug-based solution to this Humpty-Dumpty-like panoply of covert genetic errors (22) – not least in mutation-rich tumors such as smoking-related lung cancer, or inflammation-induced hepatocellular carcinoma – has long been consigned to the “too-hard basket” by clinicians and Big Pharma alike. The most immediate prospects for progress may well lie in the field of lifestyle-related cancers (e.g., breast, colon, prostate) where the burden of hard-wired gene defects is an order of magnitude lower than in carcinogen-dependent tumor types (23). This logic is illustrated by the observation that papillomavirus-associated oropharyngeal cancers, which are specifically initiated by viral E6/E7 oncoprotein blockade of p53 and pRb gatekeeper gene function, have a superior prognosis to smoking-induced cancers of the same anatomic site and morphology (24).
So where should we go from here? The caretaker/gatekeeper model of tumor progression provides a starting point. Consider, for example, tumors caused by mutations of caretaker gene function, such as MMR-defective colorectal cancer, whether sporadic or familial (25). These tumors are associated with numerous somatic mutations (23) consistent with their defining microsatellite instability (MSI); despite this, they are associated with better prognosis than stage-matched microsatellite-stable (MSS) tumors (26), consistent with a lack of major gatekeeper defects driving metastasis (27, 28). Adjuvant fluoropyrimidine chemotherapy appears of less benefit in MSI tumors (29) – partly reflecting the more favorable natural history of these cancers, to be sure, but plausibly also reflecting failure of misincorporated antimetabolites to trigger MMR and hence activate programed cell death (4) – whereas retrospective analyses have suggested that microenvironment-modulating drugs could selectively improve survival in this tumor subtype (30).
Conversely, germline mutations of the gatekeeper gene TP53 give rise in vivo to breast cancers which are HER2-overexpressing in ∼80% cases (31), supporting the view that apoptotic defects are a prerequisite for clonal outgrowth of such tumors (8), while also suggesting a clinical opportunity to reduce this defect and thus enhance chemosensitivity. Since the HER2 protein heterodimerizes preferentially with HER3 (32) – which, by virtue of numerous YXXM peptide motifs in its carboxyterminal tail (33), is a potent driver of the anti-apoptotic PI3K-AKT-mTOR pathway (34) – therapeutic inhibition of HER2-initiated signaling can be predicted to augment tumor cell kill by chemotherapy. This accords with experience in the clinic, where trastuzumab (Herceptin™) greatly increases chemotherapy efficacy (35) yet confers only minor clinical benefits when used as monotherapy (36, 37). Moreover, trastuzumab resistance is acquired during treatment via new activating PIK3CA mutations and/or PTEN losses (38–40), with drug sensitivity capable of being restored by downstream blockade of this pathway (41). This example illustrates how “undruggable” apoptotic defects (such as those mediated by mutant TP53) may be remedied by targeting more readily druggable heterologous pathway upregulation (such as mTOR signaling).
This approach could be extended to less well-defined clinical contexts by elucidating broad patterns of oncogenic pathway activation using phosphoproteomic fingerprinting (42). Genome sequencing analyses of lifestyle cancers have confirmed that the usual genetic stigmata of tumorigenic transformation comprise a small group of aberrations (43–46), consistent with the model of tumor suppressor gene loss proposed above: namely, anti-apoptotic dysfunctions affecting either TP53 (including those secondary to BRCA mutations (47) or PTEN; gain-of-function mutations affecting KRAS or PIK3CA; or MSI (flagged by low MMR expression on histochemistry) with or without activating BRAF mutations (48). If tomorrow’s clinicians can interpret this unambiguous molecular language, rational treatments may indeed become customizable for patients.
Further support for the notion of suppressor-led therapeutics comes from studies showing restoration of hormone-sensitivity to breast (49) and prostate cancers (50) using mTOR pathway inhibition. PTEN deletions are amongst the commonest of all genetic lesions in hormone-dependent cancers (51), and activation of PIK3CA-inducible biomarkers correlates with both preclinical (52) and clinical response to the oral mTOR inhibitor everolimus (53). The logic of using a downstream inhibitor to block the consequences of an otherwise undruggable upstream gene defect is thus supported, reinforcing lessons learned from constitutive KRAS-mutant-activated colorectal cancer in which upstream RAS-ERK blockade by EGFR antibodies is ineffective (54). Different lesions upregulating the mTOR signaling pathway may have non-identical therapeutic implications (55), however, emphasizing that prediction of effective tumor targeting may come to involve more than a one-off genetic predictive assay.
Discovery of a “BRCA Attacker”?
Recent advances in the field of BRCA mutant cancers have likewise kindled fresh interest in the nascent field of suppressor-based therapeutics (56). Like MMR gene mutations, BRCA mutations are relatively common caretaker defects in the population at large, with this heterozygote frequency perhaps having been maintained by lethal epidemics such as bubonic plague (57). This raises the counterintuitive hypothesis that BRCA mutations may give rise to a population survival advantage under extreme environmental selection pressures – a hypothesis supported by an analysis of spontaneous abortions that showed an unexpected reduction in lifetime miscarriage frequency among BRCA mutant carriers (25.2%) compared to non-carriers (29.1%), correlating with a higher number of full-term pregnancies (2.15 vs. 1.94) (58). This is also consistent with our model of programed genetic instability, which posits that an evolutionarily conserved sequence-dependent (CpG-based) predisposition to germline caretaker gene mutation permits genomic plasticity and species adaptivity – i.e., positive selection facilitated by a mutator phenotype (59) – in response to changes in environmental stress (10). The mutability of caretaker genes such as BRCA1/2 may thus be a two-edged sword depending on the genomic context, with survival gains for an adapting species under apoptotic stress (47) ultimately overriding the minor mortality costs of cancer in older individuals due to somatic genetic instability.
Viewed from this evolutionary perspective, the discovery that therapeutic inhibition of poly(ADP-ribosyltransferase) polymerase (PARP) enzymes selectively enhances cytotoxicity in BRCA mutant tumor cells deficient in homologous recombination (60–62) – the novel paradigm of synthetic lethality (63) – merits cautious appraisal (64, 65). Like BRCA proteins (66), PARPs are implicated in the maintenance of genome stability (67), either by forming a “sugar plug” across DNA single-strand breaks when enzymatically cleaved during a potentially cell-lethal damage response, or else by blocking replication and thus enhancing repair when remaining bound to DNA while still intact. The problem of instability-induced resistance therefore remains pivotal to the clinical promise (i.e., the survival benefit – as distinct from tumor response) of PARP inhibitory therapy for BRCA mutant disease (68). Since both BRCA1/2 and PARP1/2 proteins are required for normal genetic stability, reduced (defective or inhibited) BRCA (69) and/or PARP function could plausibly accelerate resistance (70) and/or disease progression (68), thereby offsetting short term benefits of PARP1/2 blockade in certain subsets of BRCA mutant tumors (71).
Chromosomal instability (CIN) markers such as telomere allelic imbalance or quadriradial chromosomes indicate the “BRCAness” (i.e., genomic instability due to impaired error-free homologous recombination/repair) of tumors – even tumors lacking BRCA gene mutations, such as those with BRCA gene promoter methylation (72). Of note, such BRCAness correlates positively with tumor response rates to alkylating chemotherapy drugs like cisplatin – as indeed does PARP1/2 gene knockout in mice – yet negatively with responses to taxane-based chemotherapies (73). Similarly, the clinical observation that BRCA-mutant patients do not exhibit hypersensitivity to ionizing radiation (74) raises a more nuanced interpretation of this genotype than that of a damage sensitization predictor. Moreover, given that BRCAness implies a default (salvage) increase in error-prone DNA repair, such as must presumably be associated with greater genetic instability, high chemotherapy response rates of such tumors (75) may not necessarily yield overall survival benefit. Notwithstanding these caveats, the potential contribution of combined PARP inhibitor and alkylator chemotherapy will remain an important priority for clarification in the palliative context.
Putting Humpty Dumpty Back Together
The insights outlined above point to the emergence of an exciting new era in cancer management. For the foreseeable future, however, progress against common solid tumors is likely to remain incremental rather than transformational for the following reasons.
First, there will remain serious difficulties in rectifying complex loss-of-function molecular defects on a durable basis, especially in high-grade poorly differentiated carcinomas with heavy mutation loads affecting both caretakers and gatekeepers.
A second and related problem concerns the “moving goalposts” of the cancer problem, reflecting therapeutic frustration over time due to the target-evading double-hit of genetic instability and apoptotic resistance. Modern genomic fingerprinting assays provide an impressive molecular snapshot of malignant processes at any one time, but development of a more dynamic “molecular film” technology is now needed to predict disease biology as it affects treated patients in real time. An ideal management strategy will be to minimize tumor genomic instability by slowing cell replication to the point of dormancy – i.e., as distinct from current ablative strategies of killing the most apoptosis-sensitive tumor cells upfront, inadvertently driving Gompertzian growth and metastasis of the remaining resistant cells in the longer term (76). This strategy would seem most plausible in lifestyle-related cancers, which appear driven in part by environmentally sensitive epigenetic defects (77). Changing clinical trial strategies to focus less on response and more on disease stabilization is a key step in this direction, whereas false economies in pursuing commercially optimistic but biologically misguided designs need conscious avoidance.
Third, it is vital to be aware that cancer growth is regulated by numerous factors outside the tumor itself – e.g., metabolic, endocrine, immune, stromal – which cannot be analyzed or predicted by even the most comprehensive tumor-centric analyses. Cancer is not like an infection which can be cultured to determine its drug sensitivity in vitro; rather, it is a disease of a multicellular biological system in which breakdown of regulatory crosstalk between normal and transformed cells is the primary problem (78). Immune modulation may provide one such extra-tumoral approach to “immunogenic” malignancies such as renal cell carcinoma or melanoma (79), though this strategy could prove less relevant to lifestyle-related or smoking-related neoplasms. Deepening insights into the independent tumorilytic sequelae of dieting (reduced insulin axis survival signaling), fat reduction (reduced pro-inflammatory NFκB signaling), and exercise (increased pro-apoptotic AMPK pathway activation) may help lead the way toward this objective (80).
Similarly, the judicious adjuvant use of cytotoxic chemotherapy to induce stromal toxicity and thus trigger micrometastatic apoptosis due to paracrine loop disruption – for example, in the relatively unexplored context of high-grade prostate cancer – remains as rationally justified as more costly molecularly targeted initiatives. Indeed, it is becoming understood that toxicity can often be a reliable predictor of anticancer drug benefit (81, 82), consistent with a role for normal cell interaction in tumor viability and progression. Other drug classes that may not affect tumor response or survival in the metastatic setting – e.g., protease inhibitors, or antagonists of G-protein-coupled receptors (83) – could still provide first-in-class targets by which to block metastasis in the adjuvant setting, thus improving survival.
The implications of this paradigm shift for clinical practice could prove to be profound. If a given tumor, such as glioblastoma, is characterized by high apoptotic resistance but low genetic instability, treatment may be focused on lowering the apoptotic threshold, e.g., by using PI3K-AKT-mTOR inhibitors (84). On the other hand, if a tumor exhibits high genetic instability driving widespread metastasis – e.g., BRAF mutant melanoma – then reducing tumor replication by damping down RAS-ERK signaling could slow worsening instability (85). Additional examples illustrating this clinical research strategy are shown in Table 2.
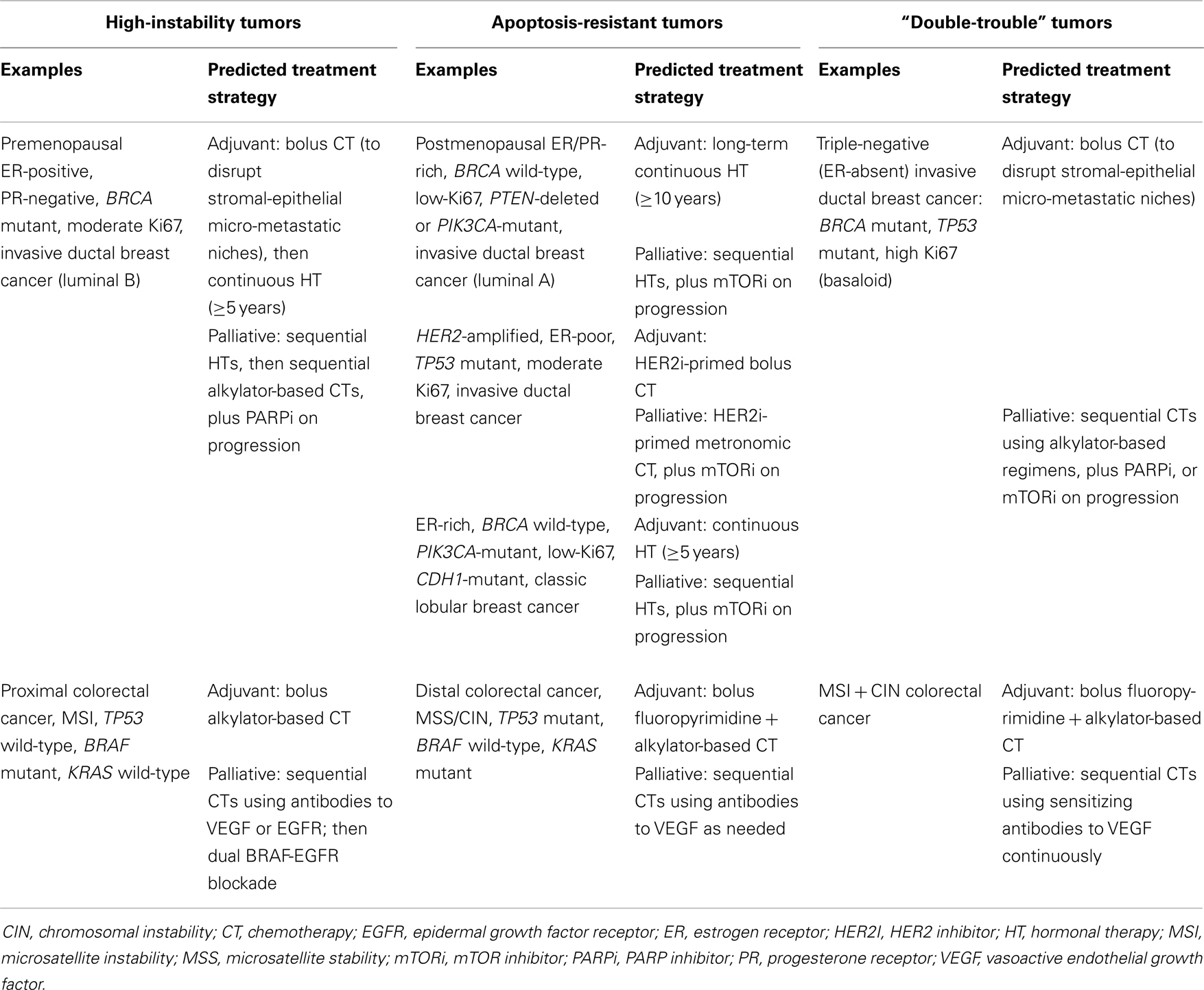
Table 2. Examples of tumor types differing in extent of caretaker/gatekeeper suppressor gene dysfunction, together with suggested therapeutic strategies.
Conclusion
There are now growing justifications for incorporating assessments of genetic instability and apoptotic resistance into therapeutic anticancer strategies and clinical trial designs. It is important to concede that not all tumors may lend themselves to this relatively simple classification, and more sophisticated modeling paradigms will undoubtedly be developed in the future. Even in the short term, however, greater awareness of these important phenotypic variables may improve the prospects for drug-induced disease control and survival gain in a significant subset of cancer patients.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CIN, chromosomal instability; CT, chemotherapy; EGFR, epidermal growth factor receptor; ER, estrogen receptor; HT, hormonal therapy; MMR, mismatch repair; MSI, microsatellite instability; MSS, microsatellite stability; PARP, poly(ADP-ribosyltransferase) polymerase; PR, progesterone receptor; VEGF, vasoactive endothelial growth factor.
References
1. Fojo T, Parkinson DR. Biologically targeted cancer therapy and marginal benefits: are we making too much of too little or are we achieving too little by giving too much? Clin Cancer Res (2010) 16(24):5972–80. doi:10.1158/1078-0432.CCR-10-1277
2. Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature (1997) 386(6627):761–67.
3. Zhao Y, Epstein RJ. Unexpected functional similarities between gatekeeper tumour suppressor genes and proto-oncogenes revealed by systems biology. J Hum Genet (2011) 56(5):369–76. doi:10.1038/jhg.2011.21
5. van Heemst D, den Reijer PM, Westendorp RG. Ageing or cancer: a review on the role of caretakers and gatekeepers. Eur J Cancer (2007) 43(15):2144–52. doi:10.1016/j.ejca.2007.07.011
6. Corcos D. Unbalanced replication as a major source of genetic instability in cancer cells. Am J Blood Res (2012) 2(3):160–9.
7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
8. Huang GC, Hobbs S, Walton M, Epstein RJ. Dominant negative knockout of p53 abolishes ErbB2-dependent apoptosis and permits growth acceleration in human breast cancer cells. Br J Cancer (2002) 86(7):1104–9. doi:10.1038/sj.bjc.6600219
9. Dictor M, Ehinger M, Mertens F, Akervall J, Wennerberg J. Abnormal cell cycle regulation in malignancy. Am J Clin Pathol (1999) 112(1 Suppl 1):S40–52.
10. Zhao Y, Epstein RJ. Programmed genetic instability: a tumor-permissive mechanism for maintaining the evolvability of higher species through methylation-dependent mutation of DNA repair genes in the male germ line. Mol Biol Evol (2008) 25(8):1737–49. doi:10.1093/molbev/msn126
11. Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer (2012) 19(6):R225–41. doi:10.1530/ERC-12-0203
12. Schmitt M, Walker MP, Richards RG, Bocchinfuso WP, Fukuda T, Medina D, et al. Expression of heregulin by mouse mammary tumor cells: role in activation of ErbB receptors. Mol Carcinog (2006) 45(7):490–505. doi:10.1002/mc.20180
13. Way TD, Lin JK. Role of HER2/HER3 co-receptor in breast carcinogenesis. Future Oncol (2005) 1(6):841–9. doi:10.2217/14796694.1.6.841
14. Connolly K, Brungs D, Szeto E, Epstein RJ. Anticancer activity of combination targeted therapy using cetuximab plus vemurafenib for refractory BRAFV600E-mutant metastatic colorectal carcinoma. Curr Oncol (in press).
15. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature (2012) 483(7387):100–3. doi:10.1038/nature10868
16. Epstein RJ. Growth of the Asian health-care market: global implications for the pharmaceutical industry. Nat Rev Drug Discov (2007) 6(10):785–92. doi:10.1038/nrd2360
17. Epstein RJ. Has discovery-based cancer research been a bust? Clin Transl Oncol (2013) 15(11):865–70. doi:10.1007/s12094-013-1071-8
18. Wells SA Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol (2012) 30(2):134–41. doi:10.1200/JCO.2011.35.5040
19. Simon R. Translational research in oncology: key bottlenecks and new paradigms. Expert Rev Mol Med (2010) 12:e32. doi:10.1017/S1462399410001638
20. Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin (2009) 59(2):111–37. doi:10.3322/caac.20003
21. Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res (2012) 72(2):527–36. doi:10.1158/0008-5472.CAN-11-3004
22. Epstein RJ, Leung TW. Reversing hepatocellular carcinoma progression by using networked biological therapies. Clin Cancer Res (2007) 13(1):11–7. doi:10.1158/1078-0432.CCR-06-1619
23. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339(6127):1546–58. doi:10.1126/science.1235122
24. Connolly K, Manders P, Earls P, Epstein RJ. Papillomavirus-associated squamous skin cancers following transplant immunosuppression: one Notch closer to control. Cancer Treat Rev (2014) 40(2):205–14. doi:10.1016/j.ctrv.2013.08.005
25. Lynch HT, Lynch PM. Colorectal cancer: update on the clinical management of Lynch syndrome. Nat Rev Gastroenterol Hepatol (2013) 10(6):323–4. doi:10.1038/nrgastro.2013.70
26. Stein A, Hiemer S, Schmoll HJ. Adjuvant therapy for early colon cancer: current status. Drugs (2011) 71(17):2257–75. doi:10.2165/11594490-000000000-00000
27. Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res (2003) 63(12):3386–94.
28. van Boxtel R, Kuiper RV, Toonen PW, van Heesch S, Hermsen R, de Bruin A, et al. Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am J Pathol (2011) 179(4):1616–22. doi:10.1016/j.ajpath.2011.06.036
29. Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol (2010) 28(20):3219–26. doi:10.1200/JCO.2009.27.1825
30. Pogue-Geile K, Yothers G, Taniyama Y, Tanaka N, Gavin P, Colangelo L, et al. Defective mismatch repair and benefit from bevacizumab for colon cancer: findings from NSABP C-08. J Natl Cancer Inst (2013) 105(13):989–92. doi:10.1093/jnci/djt140
31. Wilson JR, Bateman AC, Hanson H, An Q, Evans G, Rahman N, et al. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J Med Genet (2010) 47(11):771–4. doi:10.1136/jmg.2010.078113
32. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF III, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A (2003) 100(15):8933–8. doi:10.1073/pnas.1537685100
33. Smirnova T, Zhou ZN, Flinn RJ, Wyckoff J, Boimel PJ, Pozzuto M, et al. Phosphoinositide 3-kinase signaling is critical for ErbB3-driven breast cancer cell motility and metastasis. Oncogene (2012) 31(6):706–15. doi:10.1038/onc.2011.275
34. Hellyer NJ, Kim MS, Koland JG. Heregulin-dependent activation of phosphoinositide 3-kinase and Akt via the ErbB2/ErbB3 co-receptor. J Biol Chem (2001) 276(45):42153–61. doi:10.1074/jbc.M102079200
35. Pegram MD, Slamon DJ. Combination therapy with trastuzumab (Herceptin) and cisplatin for chemoresistant metastatic breast cancer: evidence for receptor-enhanced chemosensitivity. Semin Oncol (1999) 26(4 Suppl 12): 89–95.
36. Inoue K, Nakagami K, Mizutani M, Hozumi Y, Fujiwara Y, Masuda N, et al. Randomized phase III trial of trastuzumab monotherapy followed by trastuzumab plus docetaxel versus trastuzumab plus docetaxel as first-line therapy in patients with HER2-positive metastatic breast cancer: the JO17360 Trial Group. Breast Cancer Res Treat (2010) 119(1):127–36. doi:10.1007/s10549-009-0498-7
37. Nishimura R, Okumura Y, Arima N. Trastuzumab monotherapy versus combination therapy for treating recurrent breast cancer: time to progression and survival. Breast Cancer (2008) 15(1):57–64. doi:10.1007/s12282-007-0014-z
38. Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol (2010) 177(4):1647–56. doi:10.2353/ajpath.2010.090885
39. Dave B, Migliaccio I, Gutierrez MC, Wu MF, Chamness GC, Wong H, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol (2011) 29(2):166–73. doi:10.1200/JCO.2009.27.7814
40. Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res (2012) 18(24):6784–91. doi:10.1158/1078-0432.CCR-12-1785
41. Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol (2011) 29(23):3126–32. doi:10.1200/JCO.2010.32.2321
42. Lim YP, Diong LS, Qi R, Druker BJ, Epstein RJ. Phosphoproteomic fingerprinting of epidermal growth factor signaling and anticancer drug action in human tumor cells. Mol Cancer Ther (2003) 2(12):1369–77.
43. Haverty PM, Fridlyand J, Li L, Getz G, Beroukhim R, Lohr S, et al. High-resolution genomic and expression analyses of copy number alterations in breast tumors. Genes Chromosomes Cancer (2008) 47(6):530–42. doi:10.1002/gcc.20558
44. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol (2013) 63(5):920–6. doi:10.1016/j.eururo.2012.08.053
45. Eklof V, Wikberg ML, Edin S, Dahlin AM, Jonsson BA, Oberg A, et al. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer (2013) 108(10):2153–63. doi:10.1038/bjc.2013.212
46. Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature (2013) 497(7447):67–73. doi:10.1038/nature12113
47. Gasco M, Yulug IG, Crook T. TP53 mutations in familial breast cancer: functional aspects. Hum Mutat (2003) 21(3):301–6. doi:10.1002/humu.10173
48. Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst (2013) 105(15):1151–6. doi:10.1093/jnci/djt173
49. Beaver JA, Park BH. The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol (2012) 8(6):651–7. doi:10.2217/fon.12.49
50. Templeton AJ, Dutoit V, Cathomas R, Rothermundt C, Bartschi D, Droge C, et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur Urol (2013) 64(1):150–8. doi:10.1016/j.eururo.2013.03.040
51. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw (2013) 11(6):670–8.
52. Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res (2012) 18(6):1777–89. doi:10.1158/1078-0432.CCR-11-2123
53. Loi S, Michiels S, Baselga J, Bartlett JM, Singhal SK, Sabine VS, et al. PIK3CA genotype and a PIK3CA mutation-related gene signature and response to everolimus and letrozole in estrogen receptor positive breast cancer. PLoS One (2013) 8(1):e53292. doi:10.1371/journal.pone.0053292
54. Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, Eckhardt SG. KRAS mutations and susceptibility to cetuximab and panitumumab in colorectal cancer. Cancer J (2009) 15(2):110–3. doi:10.1097/PPO.0b013e31819e3202
55. Weigelt B, Warne PH, Downward J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene (2011) 30(29):3222–33. doi:10.1038/onc.2011.42
56. Martin SA, Hewish M, Lord CJ, Ashworth A. Genomic instability and the selection of treatments for cancer. J Pathol (2010) 220(2):281–9. doi:10.1002/path.2631
57. Moller P, Hagen AI, Apold J, Maehle L, Clark N, Fiane B, et al. Genetic epidemiology of BRCA mutations – family history detects less than 50% of the mutation carriers. Eur J Cancer (2007) 43(11):1713–7. doi:10.1016/j.ejca.2007.04.023
58. Friedman E, Kotsopoulos J, Lubinski J, Lynch HT, Ghadirian P, Neuhausen SL, et al. Spontaneous and therapeutic abortions and the risk of breast cancer among BRCA mutation carriers. Breast Cancer Res (2006) 8(2):R15. doi:10.1186/bcr1570
59. Oliver A, Mena A. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin Microbiol Infect (2010) 16(7):798–808. doi:10.1111/j.1469-0691.2010.03250.x
60. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434(7035):913–7. doi:10.1038/nature03443
61. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A (2008) 105(44):17079–84. doi:10.1073/pnas.0806092105
62. Kyle S, Thomas HD, Mitchell J, Curtin NJ. Exploiting the Achilles heel of cancer: the therapeutic potential of poly(ADP-ribose) polymerase inhibitors in BRCA2-defective cancer. Br J Radiol (2008) 81(Spec No 1):S6–11. doi:10.1259/bjr/99111297
63. Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle (2011) 10(8):1192–9. doi:10.4161/cc.10.8.15273
64. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol (2011) 5(4):387–93. doi:10.1016/j.molonc.2011.07.001
65. De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The elephant and the blind men: making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front Oncol (2013) 3:228. doi:10.3389/fonc.2013.00228
66. Deng CX, Scott F. Role of the tumor suppressor gene Brca1 in genetic stability and mammary gland tumor formation. Oncogene (2000) 19(8):1059–64. doi:10.1038/sj.onc.1203269
67. Wang Z, Wang F, Tang T, Guo C. The role of PARP1 in the DNA damage response and its application in tumor therapy. Front Med (2012) 6(2):156–64. doi:10.1007/s11684-012-0197-3
68. Chiarugi A. A snapshot of chemoresistance to PARP inhibitors. Trends Pharmacol Sci (2012) 33(1):42–8. doi:10.1016/j.tips.2011.10.001
69. Michalak EM, Jonkers J. Studying therapy response and resistance in mouse models for BRCA1-deficient breast cancer. J Mammary Gland Biol Neoplasia (2011) 16(1):41–50. doi:10.1007/s10911-011-9199-z
70. Montoni A, Robu M, Pouliot E, Shah GM. Resistance to PARP-inhibitors in cancer therapy. Front Pharmacol (2013) 4:18. doi:10.3389/fphar.2013.00018
71. Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell (2011) 20(6):797–809. doi:10.1016/j.ccr.2011.11.014
72. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer (2013) 108(10):2172–7. doi:10.1038/bjc.2013.144
73. Chalasani P, Livingston R. Differential chemotherapeutic sensitivity for breast tumors with “BRCAness”: a review. Oncologist (2013) 18(8):909–16. doi:10.1634/theoncologist.2013-0039
74. Pierce LJ, Strawderman M, Narod SA, Oliviotto I, Eisen A, Dawson L, et al. Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. J Clin Oncol (2013) 18(19):3360–9. doi:10.1016/j.critrevonc.2013.08.002
75. Rigakos G, Razis E. BRCAness: finding the Achilles heel in ovarian cancer. Oncologist (2012) 17(7):956–62. doi:10.1634/theoncologist.2012-0028
76. Demicheli R, Pratesi G, Foroni R. The exponential-Gompertzian tumor growth model: data from six tumor cell lines in vitro and in vivo. Estimate of the transition point from exponential to Gompertzian growth and potential clinical implications. Tumori (1991) 77(3):189–95.
77. Mathers JC, Strathdee G, Relton CL. Induction of epigenetic alterations by dietary and other environmental factors. Adv Genet (2010) 71:3–39. doi:10.1016/B978-0-12-380864-6.00001-8
78. Smithers DW. An attack on cytologism. Lancet (1962) 1(7228):493–9. doi:10.1016/S0140-6736(62)91475-7
79. Merelli B, Massi D, Cattaneo L, Mandala M. Targeting the PD1/PD-L1 axis in melanoma: biological rationale, clinical challenges and opportunities. Crit Rev Oncol Hematol (2013). doi:10.1016/j.critrevonc.2013.08.002
80. Campbell KL, Foster-Schubert KE, Makar KW, Kratz M, Hagman D, Schur EA, et al. Gene expression changes in adipose tissue with diet- and/or exercise-induced weight loss. Cancer Prev Res (2013) 6(3):217–31. doi:10.1158/1940-6207.CAPR-12-0212
81. Boston EA, Gaffney EA. The influence of toxicity constraints in models of chemotherapeutic protocol escalation. Math Med Biol (2011) 28(4):357–84. doi:10.1093/imammb/dqr004
82. Stintzing S, Kapaun C, Laubender RP, Jung A, Neumann J, Modest DP, et al. Prognostic value of cetuximab-related skin toxicity in metastatic colorectal cancer patients and its correlation with parameters of the epidermal growth factor receptor signal transduction pathway: results from a randomized trial of the GERMAN AIO CRC Study Group. Int J Cancer (2013) 132(1):236–45. doi:10.1002/ijc.27654
83. Epstein RJ. The CXCL12-CXCR4 chemotactic pathway as a target of adjuvant breast cancer therapies. Nat Rev Cancer (2004) 4(11):901–9. doi:10.1038/nrc1473
84. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science (2004) 304(5670):554. doi:10.1126/science.1096502
Keywords: tumor suppressor genes, genetic instability, apoptosis, carcinogenesis, drug development
Citation: Epstein RJ (2013) The unpluggable in pursuit of the undruggable: tackling the dark matter of the cancer therapeutics universe. Front. Oncol. 3:304. doi: 10.3389/fonc.2013.00304
Received: 10 October 2013; Paper pending published: 13 November 2013;
Accepted: 29 November 2013; Published online: 12 December 2013.
Edited by:
Alexander Levitzki, The Hebrew University of Jerusalem, IsraelReviewed by:
Hongying Wang, Chinese Academy of Medical Sciences, ChinaYun Dai, Virginia Commonwealth University, USA
Copyright: © 2013 Epstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard J. Epstein, The Kinghorn Cancer Centre, Level 6, 370 Victoria Street, Darlinghurst, Sydney, NSW 2010, Australia e-mail:cmVwc3RlaW5Ac3R2aW5jZW50cy5jb20uYXU=