 Tünde Szatmári
Tünde Szatmári Katalin Dobra
Katalin Dobra- Department of Laboratory Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
Proteoglycans (PGs) and in particular the syndecans are involved in the differentiation process across the epithelial-mesenchymal axis, principally through their ability to bind growth factors and modulate their downstream signaling. Malignant tumors have individual proteoglycan profiles, which are closely associated with their differentiation and biological behavior, mesenchymal tumors showing a different profile from that of epithelial tumors. Syndecan-1 is the main syndecan of epithelial malignancies, whereas in sarcomas its expression level is generally low, in accordance with their mesenchymal phenotype and highly malignant behavior. This proteoglycan is often overexpressed in adenocarcinoma cells, whereas mesothelioma and fibrosarcoma cells express syndecan-2 and syndecan-4 more abundantly. Increased expression of syndecan-1 in mesenchymal tumors changes the tumor cell morphology to an epithelioid direction whereas downregulation results in a change in shape from polygonal to spindle-like morphology. Although syndecan-1 plays major roles on the cell-surface, there are also intracellular functions, which are not very well studied. On the functional level, syndecan-1 affects mesenchymal tumor cell proliferation, adhesion, migration and motility, and the effect varies with the different domains of the core protein. Syndecan-1 may exert stimulatory or inhibitory effects, depending on the concentration of various mitogens, enzymes, and signaling molecules, the ratio between the shed and membrane-associated syndecan-1 and histological grade of the tumour. Growth factor signaling seems to be delicately controlled by regulatory loops involving the syndecan expression levels and their sulfation patterns. Overexpression of syndecan-1 modulates the biosynthesis and sulfation of heparan sulfate and it also affects the expression of other PGs. On transcriptomic level, syndecan-1 modulation results in profound effects on genes involved in regulation of cell growth
Syndecan Structure
Syndecans are transmembrane proteoglycans (PGs) composed of a core protein to which growth factor binding glycosaminoglycan (GAG) side chains are attached. The syndecan family consists of four members. Syndecan-1 is the major syndecan of epithelial cells (1), syndecan-2 is present mainly on cells of mesenchymal origin (2), syndecan-3 is primarily found in neuronal tissue and cartilage (3, 4), and syndecan-4 is ubiquitously expressed (5, 6). The protein cores of syndecans consist of a highly conserved C-terminal cytoplasmic domain, a single-pass transmembrane domain and a large N-terminal extracellular domain (7, 8) (Figure 1). The ectodomain carries up to five GAG chains, and syndecan-1 from different tissues display different GAG types comprising heparan sulfate (HS) and chondroitin-sulfate (CS) of varying length and fine structure (9) (Figure 2).
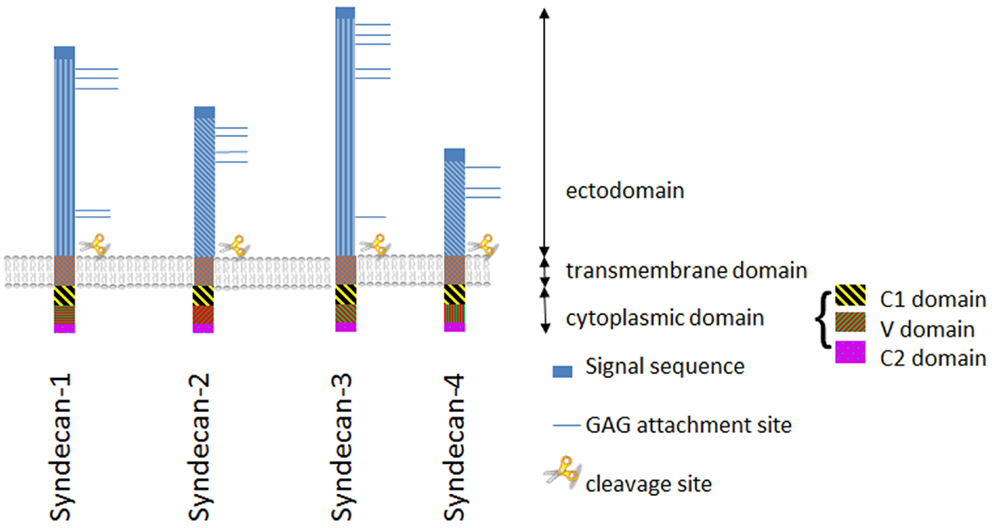
Figure 1. The syndecan family. Schematic illustration of structurally related syndecan genes, showing the two subfamilies of syndecans: syndecan-1 and -3, and syndecans -2 and -4, respectively. The extracellular domain is highly variable with the exception of the GAG attachment sites and the proteolytic cleavage site near the plasma membrane. In contrast the endo- and transmembrane domains are well preserved.
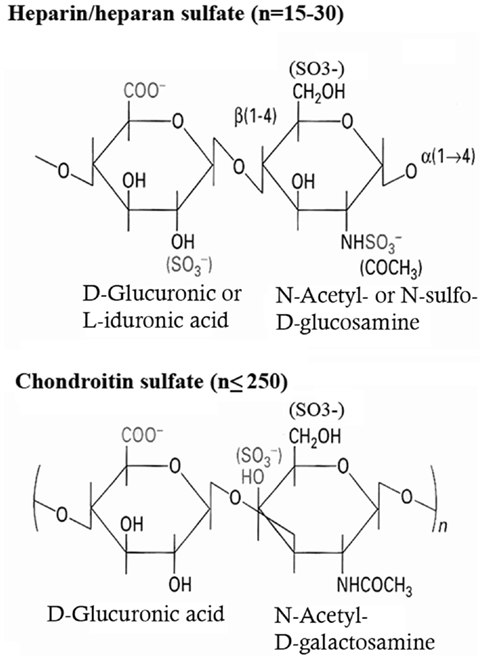
Figure 2. Biochemical structure of the repeating disaccharide units of heparan sulfate and chondroitin-sulfate.
Both HS and CS are attached to serine residues via the same linkage sequence (Xylose-Galactose-Galactose-Glucuronic Acid). Following the synthesis of this sequence, the first hexosamine derivative – N-acetyl-glucosamine (GlcNac) or N-acetyl-galactosamine (GalNac) – is added; this step being the decisive for the type of GAG subsequently formed. The basic GAG chain for both HS and CS then extends in the Golgi by further repetitive addition of glucuronic acid (GlcA) and a hexosamine derivative, which for HS is GlcNAc and for CS GalNAc. The resulting GAG is thus built up of repeating disaccharide units, consisting of an uronic acid and a hexosamine derivative (10).
Subsequently this basic structure is modified by a series of reactions (epimerization, deacetylation, sulfation), which occur in a tissue-specific manner. Particularly in HS, this generates a vast diversity of the fine structure and hence alters the capacity of this GAG to bind to other structures. These modification reactions thus represent one way to regulate the protein binding capacity of PGs.
Shedding of Syndecans from the Cell Surface
Syndecans are usually present on the cell surface (1, 7, 11), but they can also be released by the action of sheddases or accumulate in the cell nucleus (12); in the tumor stroma (13), and in body fluids (14–17). Shedding of the ectodomain is biologically important, converting the cell-bound syndecan to a soluble active ligand. Syndecan-1 shedding is regulated by matrix metalloproteinases (MMPs), including MMP-7, MMP-9, membrane-bound metalloproteinases (MT-MMP1), and a disintegrin and metalloproteases (ADAM10, ADAM17) (18–22). They act by proteolytical cleavage of the juxtamembrane site of the core protein. The mechanism of shedding is currently not completely understood (23), but recently a new mechanism where MMP-9 enhances syndecan-1 shedding via suppression of miR-494 was described (24). Accelerated shedding is mediated by MMPs (21), Rab-5 (25), growth factors (GFs) (26), heparanase, and HS (27, 28). It is also known that FGF-2 activates MMP-7 mediated shedding (29) and heparanase accelerate MMP-9 mediated shedding of syndecan-1 (27). Cell surface HSPGs can themselves participate in regulation of metalloproteinases, anchoring them to the cell surface via the GAG chains (23), and particularly for syndecan-1 it was demonstrated that the HS chains on the core protein suppresses shedding (28), giving an additional explanation on the above mentioned role of heparanase in this process.
The released ectodomain carries intact GAG chains, thus it has preserved ability to modulate growth factor responses and biological processes. Experimental studies have shown that membrane-bound and soluble forms of syndecan-1 have opposing effects on cancer cell functions (19, 30–33). High level of shed syndecan-1 is associated with infection, inflammation, and cancer. Recently it was found that chemotherapy can induce shedding of syndecan-1, particularly via ADAMs and this shed syndecan-1 being functionally active, leads to establishment of a more aggressive phenotype in case of relapse (34).
Soluble syndecan-1 binds pro-angiogenic factors like VEGF or FGF, activates them, creating a chemotactic gradient, and by this promotes endothelial cell’s invasion and angiogenesis. Besides, it activates the integrins αVβ3 and αVβ5 (35), which are also important for angiogenesis (36) and regulates the association of IGF1R to αVβ3 integrin, essential in endothelial cell migration (37). The pro-angiogenic effect of syndecan-1 was shown in myeloma (27), medulloblastoma (24), and in a variety of tumors of epithelial origin like endometrial cancer (38), breast cancer (39), and in stromal fibroblasts of breast cancer (32), but the effects of syndecan-1 in the angiogenesis of mesenchymal tumors is largely unknown.
Understanding of the importance of syndecan-1 shedding might help to resolve the seemingly contradictory expression levels documented in various malignancies.
Sub-Cellular Localization of Syndecan-1 in the Cell Nucleus
Syndecan-1 translocates to the cell nucleus of various tumor cells, including malignant mesothelioma, fibrosarcoma, neuroblastoma, breast- and lung adenocarcinoma, and multiple myeloma (12, 40–42). This translocation is tubulin dependent and the same transport route operates also for FGF-2 but not for the FGF Receptor. The minimal structural requirement for nuclear translocation is the RMKKK sequence at the cytoplasmic tail of syndecan-1 serving as a nuclear localization signal (NLS) (43). The nuclear translocation correlates to the differentiation and proliferation of certain tumor cells. One compound that seems to modulate the level of nuclear syndecan-1 in several tumor types is heparanase when simultaneously present in the nucleus (41, 44). High level of heparanase implies low levels of nuclear syndecan-1 and increased histon acetyl transferase (HAT) activity leading to enhanced transcription of VEGF and MMP-9, both known to drive an aggressive tumor phenotype (42). Furthermore, syndecan-1 restoration diminishes the nuclear HAT activity, providing a mechanistic link and establishing syndecan-1 as a powerful inhibitor of HAT driven gene transcription.
Accumulating evidences suggest that the localization of syndecan-1 might be crucial for its function and the nuclear translocation adds additional complexity which needs to be further addressed in the context of variously differentiated tumor components.
Tissue- and Tumor Specific Expression of Syndecan-1
Each syndecan is expressed in highly regulated cell-, tissue-, and developmental stage specific manner (8, 45). The syndecan-profile of different tissues, hence of different tumor types, differs greatly between mesenchymal and epithelial tumors.
Similar to the normal epithelial cells, syndecan-1 is overexpressed in epithelial malignancies. In dedifferentiated tumor components and mesenchymal tumors its expression is, however, lower than in the parental tissue. Changes in syndecan-1 level can have remarkable consequences for tumor cell behavior. The expression of this PG can associate to disease stage, tumor differentiation grade, and prognosis of the tumor, though the extent and even the direction of the association varies from one tumor type to another (46).
The mechanisms by which syndecan-1 regulates tumor cell behavior are complex, and depend, at least partly, upon the interplay between tumors and the surrounding matrix. The expression of syndecan-1 and its role as a stimulatory or inhibitory factor probably depends upon the concentration of various mitogens, enzymes, and signaling molecules that are specific for each cancer type and histologic grade. By interacting with such factors, this PG modulates cancer cell proliferation, adhesion, migration, and angiogenesis.
Syndecan-1 as Diagnostic and Prognostic Marker
Syndecan-1 is used as a standard diagnostic biomarker in multiple myeloma (47) and it is highly expressed in various human cancers (48) comprising pancreatic (49) and breast cancer (50). Hovewer, low cell surface syndecan-1 level is associated with a poor prognosis as demonstrated by immunohistochemistry in lung cancer (51), renal carcinomas (52), head and neck cancer (53), and in colorectal cancer (54, 55). In squamous cell carcinoma of the tonsil the level of syndecan-1 was found lower than in the benign keratoacanthoma and it correlated inversely with the proliferative index (56). In carcinoma of the uterine cervix expression of syndecan-1 is associated with histological differentiation grade but not with clinical outcome (57). Thus, syndecan-1 seems to have antithetic roles in different cancer types having inhibitory role on tumor formation and progression in many different epithelial malignancies but also promoting the growth of others (48, 58). The shed syndecan-1 can act opposing compared to cell-surface syndecan-1, since potentially it is able to sequester the GFs and other HS-binding soluble factors from the microenvironment of the tumor cell. Accordingly, the levels of shed syndecan-1 in serum correlate with a less favorable prognosis in lung cancer (16), lymphoma (59), myeloma (15), hepatocellular carcinoma (60), and glioma (61).
The tumor stroma has an important role in mediating tumor cell proliferation and invasiveness, leading to formation of metastases. In most tumor types the tumor stroma is abundant in matrix PGs, particularly versican, lumican, and fibromodulin, and this suggests an important role of stromal PGs in controlling tumor progression (58). The effects of shed syndecan-1 in the stroma is in majority of tumor types in contrast to those of cell-surface syndecan-1, the abundance of syndecan-1 in the tumor stroma being a negative prognostic factor (58) and it correlates to a more aggressive phenotype (33, 50, 62). Consequently, stromal syndecan-1 promotes breast epithelial cell proliferation (13); and in gastric cancer, ovarian cancer (63), and oral carcinoma (64) was associated with poor outcome (65). Similarly, in colorectal cancer immunoreactivity to syndecan-1 could be seen in both the tumor epithelium and stroma, whereas the normal colonic mucosa was negative for syndecan-1 (55). In basal cell carcinoma the opposite effect could be observed, where the stromal immunoreactivity of syndecan-1 inversely correlated to aggressiveness (66). Taken together, syndecan-1 seems to have an important role for epithelial-stromal interactions and a syndecan-1 dependent reciprocal feedback-loop has been proposed (67).
Syndecan-1 in Mesenchymal Tumors
In mesenchymal cells the syndecan-1 level is usually low, but it is elevated transiently during embryonal morphogenesis (68–72) concomitant with a loss of syndecan-1 in the adjacent epithelium. Given this low syndecan-1 level in mesenchymal tumors, the expression, and function of syndecan-1 is far less studied than in carcinomas. The most extensively studied mesenchymal tumors addressing syndecan-1 expression are malignant mesothelioma and fibrosarcoma. Tumor cells forming this PG, however, have also been found in epithelioid components of biphasic sarcoma, thymoma, synovial sarcoma, leiomyosarcoma, gastrointestinal stromal tumors, and schwannomas (73, 74). Furthermore, a recent study revealed that in bone metastasis of soft tissue sarcoma syndecan-1 expression is elevated and it correlates to expression of several growth signaling molecules (75).
Malignant Mesothelioma
Cell-surface expression of syndecan-1 is relatively low in malignant mesothelioma compared to epithelial malignancies, however, its expression relates to epithelioid differentiation thus correlating to better prognosis (76), and it is reduced or lost in the sarcomatoid phenotype. Malignant mesothelioma cells also synthesize syndecan-2 and -4 and these syndecans, less often expressed in carcinomas, are especially abundant in the epithelioid phenotype (77). Thus syndecan-1 and syndecan-2 has been proposed as biomarkers to distinguish malignant mesothelioma from metastatic adenocarcinoma (78, 79).
Epithelial-mesenchymal transition is a characteristic feature of malignant mesothelioma (80) and in vitro model systems can be generated to mimic mesothelioma differentiation (81, 82). The mesothelium itself has a remarkable plasticity and a potential to generate other cell-types (83), whereas the mesothelioma has the ability to trans-differentiate across the epithelial-mesenchymal axis and this has prognostic significance (84, 85). This ability to switch from epithelial to mesenchymal phenotype involves a simultaneous downregulation of epithelial markers including syndecan-1 and E-cadherin (46, 86, 87).
The tumor microenvironment and growth factor gradients have a considerable effect on mesothelioma morphology and by modulating the serum composition of cell cultures the morphological changes of mesothelioma cells mimics various differentiation states in vitro (82). Molecular characterization reveals specific proteoglycan profiles and distinct molecular signatures for the epithelioid and sarcomatoid phenotypes, respectively (77, 88, 89). Experimentally induced overexpression of syndecan-1 in mesenchymal tumors changes the tumor cell morphology in an epithelioid direction (90), whereas downregulation results in a change in shape of cells from polygonal to spindle-like (77). At the same time, such overexpression inhibits tumor growth (90) and migration (43) of malignant mesothelioma cells simultaneously with enhanced cell adhesion.
Fibrosarcoma
Fibrosarcomas are relatively rare malignant mesenchymal tumors, originating from fibroblasts, with an abundant extracellular matrix, rich in PGs. Though the amount of syndecan-1 is usually low in fibrosarcoma, some samples and cell lines can express also this PG (73, 90). Different studies show that this expression can modulate the proliferation, migration, and the malignant potential of tumor cells. Similar to carcinoma cells, however, the effects are cell-type dependent and seem to be governed by the spatio-temporal expression of syndecan-1. Variously differentiated tumor components behave differently: the proliferation and migration of a sarcomatoid fibrosarcoma cells is inhibited (43, 90) whereas in an epithelioid fibrosarcoma cell line is enhanced upon syndecan-1 overexpression (91, 92) in collaboration with syndecan-2 (92). In fibrosarcoma cells the location of syndecan-1 seems to be crucial. While cell-membrane-bound syndecan inhibited migration on collagen, the membrane type 1 metalloprotease (MT1-MMP) mediated shedding enhanced it (19).
The Role of Syndecan-1 in Signaling
Syndecan-1 exerts mainly its functions via the HS chains, which ligate to a wide range of proteins, including heparin-binding GFs and their corresponding receptors, comprising FGFs, VEGF, Wnt, and HGF (27, 33, 93, 94). This ability to bind GFs is dependent on the steric orientation of the sulfate and carboxyl groups in the GAG chains. When simultaneously binding to both the growth factor and its receptor, HS stabilizes the complex, thus acting as a signaling co-receptor (11, 95). Most studies dealing with the effect of syndecans on signaling, have been performed in carcinomas or hematological malignancies (33, 96–98); thus the function of syndecan-1 is less studied in mesenchymal cells.
We have recently shown that overexpression of syndecan-1 in a malignant mesothelioma cell line influences a multitude of signaling pathways. These effects are not limited to cell-surface receptors but also influence their downstream effectors (99). The PDGF and FGF family members were downregulated, while their receptors were upregulated, whereas both the growth factor and its receptor were enhanced in EGF signaling. These changes in growth factor expression were accompanied by a deregulation of kinase cascade (ERK/MAPK, JNK, and p38/MAPK) and downstream transcription factors comprising MYC, FOS, JNK, and JUN which all were inhibited. On the other hand ETS-1 was upregulated due to syndecan-1 overexpression and inhibited when this PG was silenced. The direction or the magnitude of the effect seem to be cell-type specific, and does not allow direct extrapolation to other cells. Thus, ETS-1 was reported in colon carcinoma as inversely correlated to the level of syndecan-1 (100). Hence, growth factor signaling seems to be delicately controlled by the syndecan expression level, probably involving autoregulatory loops.
Syndecan-1 Regulates the Expression of Enzymes Involved in Heparan Sulfate Biosynthesis and the Proteoglycan Profile
The HS chains are important in regulation of cancer cell behavior, different studies reported modified sulfation pattern of HS chains during cancer progression (101). Already 20 years ago it was assumed that the HS and PGs of a cell are subject of a coordinated regulation, and this regulation is critical for controlling cell behavior (102). Accumulating data support this, indicating a complex interplay between different proteins involved in the synthesis and turnover of heparan sulfate proteoglycans (HSPGs) (103, 104).
The original concept about HS biosynthesis highlights its specificity, proposing that it is a regulated, hierarchical process, comprising steps in a defined order, depending on each other. The enzymes N-deacetylase/N-sulfotransferase (NDST) replace the acetyl group of GlcNac with a sulfate group. As the substrate for NDSTs are the chains polymerized by exostosins (EXT), to which NDSTs add sulfate groups deriving from 3′-Phosphoadenosine-5′-phosphosulfate (PAPS), their activity depends on PAPS synthases and on EXTs as well. This N-sulfation is a key step for the consecutive 2-, 6-, and 3-O-sulfations, as the 2-O, 6-O, and 3-O sulfotransferases (2-OST, 6-OST, and 3-OST, respectively) add sulfate groups to the respective positions of disaccharide units in a strictly regulated order. This succession of events can explain why some of the HS chains are extensively modified, while others could remain totally unprocessed (10). The fact that the structure of HS chains correlates to the cell-type from where they are originated, rather than the core proteins which they bind, also points to a controlled expression of the HS biosynthetic enzymes. The mechanism behind synthesis of defined non-random HS sequences, so important for specific interactions, is still much of a mystery.
Proteoglycan and heparan sulfate biosynthesis are critical for development, morphogenesis, and organogenesis. Studies using different model organisms with one or more HS biosynthetical genes knocked out, show distinct severe developmental disorders and phenotypic deficiencies (105). The absence of any of these enzymes has serious implications in the morphogenesis and development of these organisms. Despite the tight regulation, there is a high degree of plasticity in the sulfation pattern, as a result of this flexible HS biosynthesis (106). Some observations does not support the model for HS biosynthesis where one enzyme creates the substrate for the next step, indicating that this order is not so strict, exemplified by the presence of 6-O-sulfation in HS lacking N-sulfate groups (107). These enzymes may interact as evidenced by studies in different model organisms (fruit fly, nematode, zebra fish, mouse) where knockdown of one of these enzymes is followed by direct and indirect effects, affecting the other enzymes from the HS biosynthetic machinery [for review see (108)]. Experiments with mice lacking NDST1 and/or NDST2 has shown that the HS N-sulfation is not limited by the total amount of active NDST enzymes (109); in mice deficient in C5 epimerase the HS N-sulfation is increased (110); and compensatory effects of 6-O-sulfation for 2-O sulfation were noticed (111, 112). Another study points out that 6-OST acts at the internal N/sulfoglcosamine and non-reducing terminal N-sulfoglucosamine but not N-acetylglucosamine in vitro, while in vivo all these residues are sulfated, indicating a coupled reaction of the enzymes (105).
Recently, it was hypothesized that instead of the total amount of an enzyme in one stage of the HS synthesis the critical step of the regulation may be the assembly of enzyme complexes of the cell (109). The idea was raised that the different biosynthetic enzymes form also physical complexes (10). Indeed, several studies have shown physical interactions of these enzymes: polymerases EXT1 and EXT2 (113, 114) as well as NDST1 and EXT2 (109, 115) form a complex and similarly, 2-OST and the epimerase co-localize in Golgi and interact physically (116), although this regulation is not always coordinated (117).
When trying to disclose the regulation of HS biosynthesis, we have to consider also the fact that in mammals almost all biosynthetic enzymes have more than one isoforms and the substrate specificities of different isoforms largely overlap. Moreover some of the genes encoding HS biosynthetic enzymes are regulated posttranslationally (10, 118), giving further complexity to the process.
Beside the above mentioned dynamic co-operativity of the HS biosynthetic enzymes in the Golgi, the interplay between the sulfatases (SULF-1 and SULF-2) and the synthesizing enzymes also play a role in the formation and function of the complex heparanome (108). The cell surface associated extracellular sulfatases remove 6-O sulfate groups from well-defined regions of the mature HS chains (119, 120). Loss of SULF-1 and/or SULF-2 results in different HS chain composition in vivo. In SULF-1 knockout cells, in addition to the increase in 6-O sulfated disaccharides, 2-O sulfation, and N-O sulfation decreased in small, but significant extent (121). Moreover, upon SULF-2 knockdown, SULF-1 is able to compensate its effect, while double knockouts showed synergistic co-operativity, resulting in a supraadditive effect and increased amount of 6-O disaccharides (108).
There are also evidences that HS and CS biosynthesis affects each other, possibly by sharing the same linkage regions; the absence of one allowing the other to substitute. Furthermore, they share the common PAPS pool for sulfation (117).
The regulation of these enzymes implies a “balanced hierarchy” between their activity and expression, finally resulting in a complex, cell-type specific sulfation pattern (108). Many pieces of this puzzle and the dynamic interplay still remain to be elucidated. The picture is complex, as the interactions are built up by both negative and positive feedback loops which can be depicted in a network (122), where each member reciprocally affects multiple actions of the other members of the network. Thus changes in the expression of one gene will affect the whole HS biosynthetic machinery of the cell.
A threefold overexpression of syndecan-1 in malignant mesothelioma largely influenced the whole transcriptome often with a much higher deregulation of individual genes than the syndecan-1 itself (99). Thus it seems that also other, post transcriptional or epigenetic mechanisms might contribute to these effects. One way of this regulation seems to involve the sulfation pattern of the HS chains. Syndecan-1 overexpression caused a significant alteration in the expression level of several enzymes involved in HS biosynthesis, metabolism, and turnover. In this setting EXT1 and NDST1 were downregulated along with deregulation of 2-O, 6-O sulfotransferases and the two PAPS synthases, responsible for the synthesis of the sulfate donor PAPS (99) (Table 1). Furthermore, SULF-1 was highly downregulated, though, the level of SULF-2 was not affected.
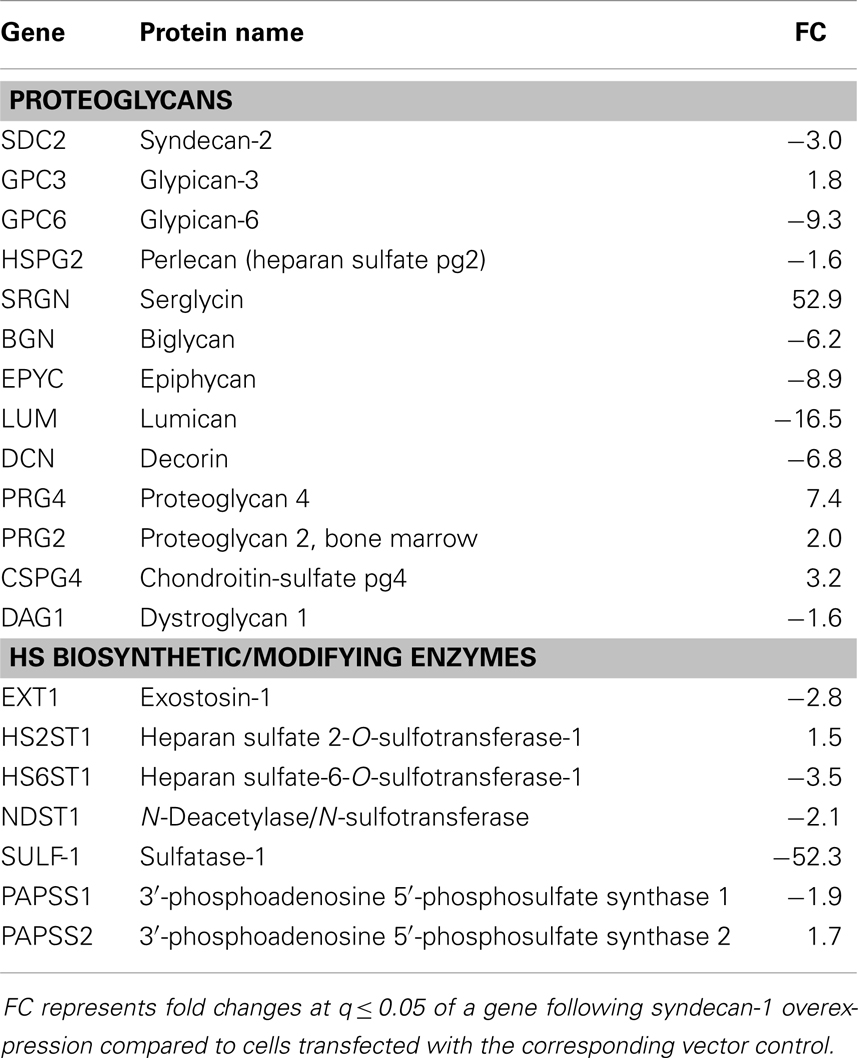
Table 1. List of genes encoding proteoglycans and heparan sulfate biosynthetic enzymes affected by syndecan-1 overexpression in a mesothelioma cell line.
The massive downregulation of SULF-1 after syndecan-1 overexpression in malignant mesothelioma may constitute one mechanism by which syndecan-1 regulates cell growth, by modulating the growth factor binding properties of HSPGs (123) (Figure 3). Similarly, SULF-1 has a dual role in enhancing or inhibiting various growth factor signaling pathways and by that tumor cell proliferation: as it has a tumor suppressor role in the majority of carcinomas. SULF-1 is downregulated in many tumor types (124, 125), whereas in malignant mesothelioma and some other tumors it is overexpressed (124, 126). The mechanism of this dual effect has been ascribed to inhibition of the activity of FGF (127–129), HB-EGF (130), ERK-MAP, and AKT signaling pathways (131, 132). At the same time SULF-1 is known to promote WNT signaling (133, 134) and activates BMP/Noggin signaling (135). Currently it is hypothesized that cancers driven by WNT-1 signaling would likely be stimulated by SULF-1, whereas tumors depending on FGF-2 or HGF signaling as the most significant driving mechanism are inhibited (125, 136). In malignant mesothelioma the level of SULF-1 is elevated compared to the normal mesothelium and the Wnt pathway is also altered (137–139), thus we can hypothesize that SULF-1 downregulation contributes to inhibition of proliferation, however, the functional significance of these findings necessitates further investigations.
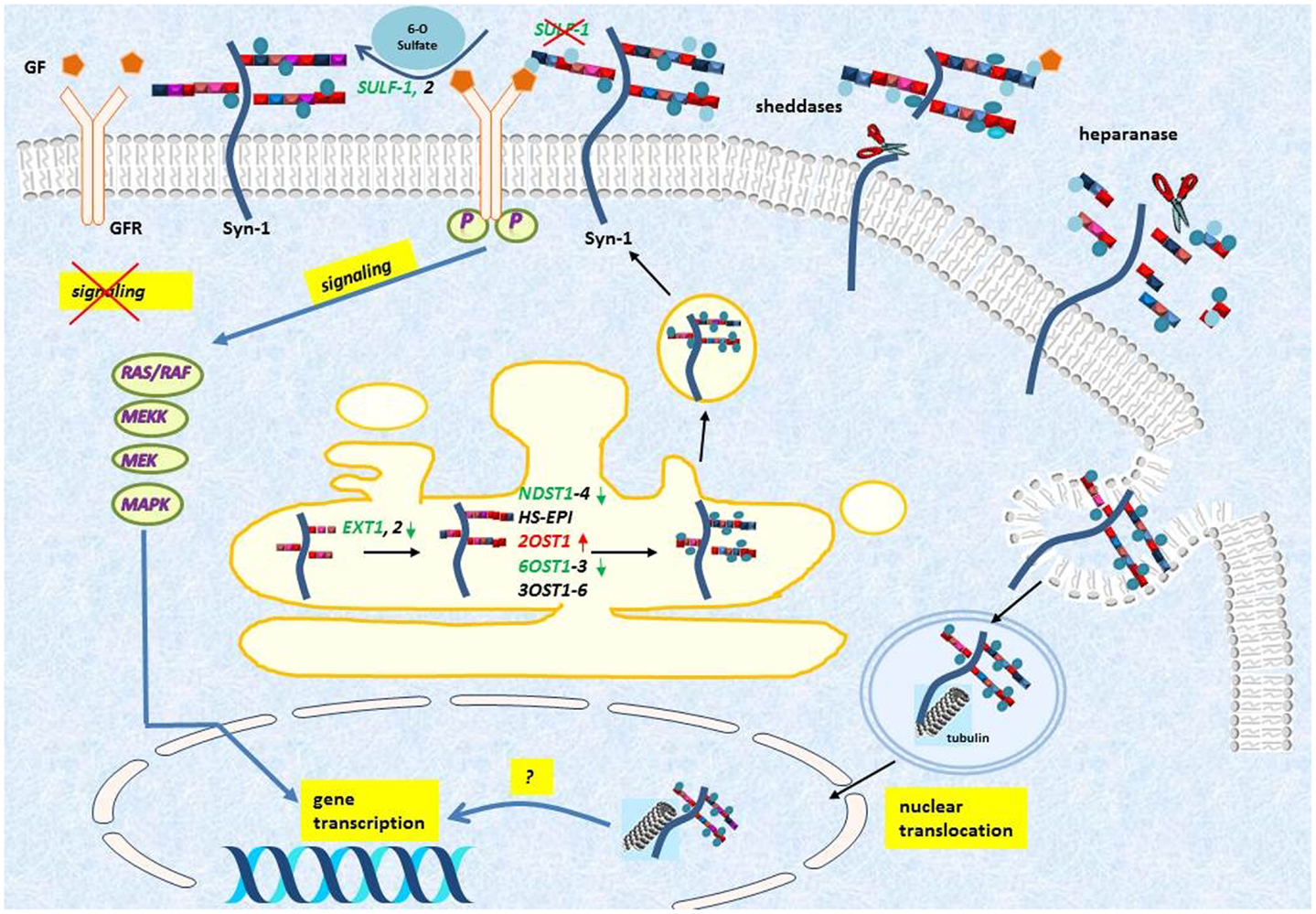
Figure 3. Syndecan-1 turnover and its effect on HS modifications in malignant mesothelioma. Syndecan-1 (Syn-1) is synthesized in Golgi and it is transported to cell-membrane where it acts as a co-receptor for various growth factors (GFs) and growth factor receptors (GFRs). The ectodomain is released from the cell-surface by the action of various enzymatic reactions collectively called sheddases, and the heparan sulfate can be further fragmented by the action of heparanases. The shedding results in a soluble molecule, which is still active and thereby can bind and sequester GFs. Syndecan-1 is also internalized and translocates to the nucleus in a tubulin dependent manner, but the function of this translocation is still incompletely understood. Upon syndecan-1 overexpression, several biosynthetic enzymes are modified including, EXT, exostosin; NDST, N-deacetylase/N-sulfotransferase; OST, O-sulfotransferase; HS-EPI, C5 epimerase; and they collectively lead to altered HS synthesis and sulfation pattern (Colors represent: red = upregulated and green = downregulated). The endosulfatase SULF-1, specifically removes the 6-O-sulfate groups from the HS chains, and thereby may inhibit growth factor signaling. Downregulation of SULF-1 by syndecan-1, detected at transcriptional level, may lead to modulation of downstream growth factor signaling.
Syndecan-1 not only regulates multiple levels of HS biosynthesis, but also coordinates the expression of various PGs and fine tunes their regulatory pathways. Experimental data suggest also a cooperation between the different members of the syndecan family (92). Syndecan-1 seems to control the expression of other HSPGs in mesenchymal tumors, although the effect varies in different cell-types and also in the same tumor with various differentiation. Overexpression of syndecan-1 resulted in a downregulation of syndecan-2 and upregulation of syndecan-4 in epithelioid mesothelioma cells (90, 99), whereas in epithelioid fibrosarcoma cell line syndecan-2 was upregulated (92), and in a sarcomatoid fibrosarcoma cell line syndecan-4 was downregulated (90).
This overexpression of syndecan-1 in malignant mesothelioma cells was also associated with considerable changes in expression of other HSPGs: glypican-3 was upregulated whereas glypican-6 and perlecan both were downregulated.
The fact that the expression of syndecan-1 can influence the whole proteoglycan pool is further supported by several independent studies, where a higher level of syndecan-1 is accompanied by perturbations in the proteoglycan profile and in the HS biosynthetic machinery. The cell-type specific nature of this process, however, has to be emphasized as shown also in breast cancer cells and glioblastoma (140, 141). Similar to malignant mesothelioma there seem to be a phenotype specific HSPG distribution in glioblastoma, the mesenchymal subgroup of glioblastomas typically having a worse prognosis (141).
Taken together, growth factor signaling seems to be delicately controlled by regulatory loops involving the syndecan-1 expression levels, its cellular localization and the sulfation pattern. Syndecan-1 itself regulates the expression of multiple PGs and coordinates the HS biosynthesis. Furthermore, modulation of syndecan-1 affects the biological behavior of mesenchymal tumor cells and this involves genes regulating cell growth, cell cycle progression, adhesion, migration, and extracellular matrix organization; orchestrated in a complex network of signaling pathways (99).
Targeting Syndecan-1 in Cancer
Syndecan-1 offers a multitude of possibilities for novel therapeutic approaches and targeted therapies. Therapeutic options should, however, consider that the syndecan-1 expression differs significantly from one tumor type to another and its effect is highly divergent comprising both anti-proliferative and pro-tumorigenic effects. Thus, in tumors with elevated syndecan-1 level such as multiple myeloma or breast adenocarcinoma, applicable approaches comprise anti-syndecan-1 antibodies, knockdown of syndecan-1, competitive inhibitors or anti-angiogenic agents (142–145). Synstatin, a short peptide that mimics a sequence of syndecan-1 extracellular domain seems to be a promising anti-angiogenic agent (35).
In contrast, in tumors of mesenchymal origin, and generally in tumors where syndecan-1 is downregulated, other approaches should apply. One interesting concept concerns possible growth inhibition by using soluble HS oligosaccharides or overexpression of syndecan-1 in mesenchymal tumors to hamper crucial biological responses including proliferation and migration (12, 43, 90, 146–148).
Cell surface HSPGs are also promising for efficient intracellular delivery of macromolecules across biological membranes (149–157) and offer encouraging possibilities of developing novel targeted treatments (158–161). The design of intracellular drug delivery, however, requires an increased understanding of the physiological processes that mediate cellular communication and transport across the plasma membrane.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge Prof. Anders Hjerpe for constructive suggestions and critical review of the manuscript.
References
1. Saunders S, Jalkanen M, O’Farrell S, Bernfield M. Molecular cloning of syndecan, an integral membrane proteoglycan. J Cell Biol (1989) 108(4):1547–56. doi: 10.1083/jcb.108.4.1547
2. Marynen P, Zhang J, Cassiman JJ, Van den Berghe H, David G. Partial primary structure of the 48- and 90-kilodalton core proteins of cell surface-associated heparan sulfate proteoglycans of lung fibroblasts. Prediction of an integral membrane domain and evidence for multiple distinct core proteins at the cell surface of human lung fibroblasts. J Biol Chem (1989) 264(12):7017–24.
3. Carey DJ, Evans DM, Stahl RC, Asundi VK, Conner KJ, Garbes P, et al. Molecular cloning and characterization of N-syndecan, a novel transmembrane heparan sulfate proteoglycan. J Cell Biol (1992) 117(1):191–201. doi:10.1083/jcb.117.1.191
4. Gould SE, Upholt WB, Kosher RA. Syndecan 3: a member of the syndecan family of membrane-intercalated proteoglycans that is expressed in high amounts at the onset of chicken limb cartilage differentiation. Proc Natl Acad Sci U S A (1992) 89(8):3271–5. doi:10.1073/pnas.89.8.3271
5. David G, van der Schueren B, Marynen P, Cassiman JJ, van den Berghe H. Molecular cloning of amphiglycan, a novel integral membrane heparan sulfate proteoglycan expressed by epithelial and fibroblastic cells. J Cell Biol (1992) 118(4):961–9. doi:10.1083/jcb.118.4.961
6. Kojima T, Shworak NW, Rosenberg RD. Molecular cloning and expression of two distinct cDNA-encoding heparan sulfate proteoglycan core proteins from a rat endothelial cell line. J Biol Chem (1992) 267(7):4870–7.
7. Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem (1999) 68:729–77. doi:10.1146/annurev.biochem.68.1.729
8. Xian X, Gopal S, Couchman JR. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res (2010) 339(1):31–46. doi:10.1007/s00441-009-0829-3
9. Sanderson RD, Bernfield M. Molecular polymorphism of a cell surface proteoglycan: distinct structures on simple and stratified epithelia. Proc Natl Acad Sci U S A (1988) 85(24):9562–6. doi:10.1073/pnas.85.24.9562
10. Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem (2002) 71:435–71. doi:10.1146/annurev.biochem.71.110601.135458
11. Carey DJ. Syndecans: multifunctional cell-surface co-receptors. Biochem J (1997) 327(Pt 1):1–16.
12. Brockstedt U, Dobra K, Nurminen M, Hjerpe A. Immunoreactivity to cell surface syndecans in cytoplasm and nucleus: tubulin-dependent rearrangements. Exp Cell Res (2002) 274(2):235–45. doi:10.1006/excr.2002.5477
13. Stanley MJ, Stanley MW, Sanderson RD, Zera R. Syndecan-1 expression is induced in the stroma of infiltrating breast carcinoma. Am J Clin Pathol (1999) 112(3):377–83.
14. Perrimon N, Bernfield M. Cellular functions of proteoglycans – an overview. Semin Cell Dev Biol (2001) 12(2):65–7. doi:10.1006/scdb.2000.0237
15. Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, et al. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood (2000) 95(2):388–92.
16. Joensuu H, Anttonen A, Eriksson M, Makitaro R, Alfthan H, Kinnula V, et al. Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res (2002) 62(18):5210–7.
17. Vassilakopoulos TP, Kyrtsonis MC, Papadogiannis A, Nadali G, Angelopoulou MK, Tzenou T, et al. Serum levels of soluble syndecan-1 in Hodgkin’s lymphoma. Anticancer Res (2005) 25(6C):4743–6.
18. Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell (2002) 111(5):635–46. doi:10.1016/S0092-8674(02)01079-6
19. Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem (2003) 278(42):40764–70. doi:10.1074/jbc.M306736200
20. Su G, Blaine SA, Qiao D, Friedl A. Membrane type 1 matrix metalloproteinase-mediated stromal syndecan-1 shedding stimulates breast carcinoma cell proliferation. Cancer Res (2008) 68(22):9558–65. doi:10.1158/0008-5472.CAN-08-1645
21. Brule S, Charnaux N, Sutton A, Ledoux D, Chaigneau T, Saffar L, et al. The shedding of syndecan-4 and syndecan-1 from HeLa cells and human primary macrophages is accelerated by SDF-1/CXCL12 and mediated by the matrix metalloproteinase-9. Glycobiology (2006) 16(6):488–501. doi:10.1093/glycob/cwj098
22. Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, et al. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem (2010) 285(1):555–64. doi:10.1074/jbc.M109.059394
23. Georges S, Heymann D, Padrines M. Modulatory effects of proteoglycans on proteinase activities. Methods Mol Biol (2012) 836:307–22. doi:10.1007/978-1-61779-498-8_20
24. Asuthkar S, Velpula KK, Nalla AK, Gogineni VR, Gondi CS, Rao JS. Irradiation-induced angiogenesis is associated with an MMP-9-miR-494-syndecan-1 regulatory loop in medulloblastoma cells. Oncogene (2013). doi:10.1038/onc.2013.151
25. Hayashida K, Stahl PD, Park PW. Syndecan-1 ectodomain shedding is regulated by the small GTPase Rab5. J Biol Chem (2008) 283(51):35435–44. doi:10.1074/jbc.M804172200
26. Subramanian SV, Fitzgerald ML, Bernfield M. Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J Biol Chem (1997) 272(23):14713–20. doi:10.1074/jbc.272.23.14713
27. Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, et al. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood (2010) 115(12):2449–57. doi:10.1182/blood-2009-07-234757
28. Ramani VC, Pruett PS, Thompson CA, DeLucas LD, Sanderson RD. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem (2012) 287(13):9952–61. doi:10.1074/jbc.M111.330803
29. Ding K, Lopez-Burks M, Sanchez-Duran JA, Korc M, Lander AD. Growth factor-induced shedding of syndecan-1 confers glypican-1 dependence on mitogenic responses of cancer cells. J Cell Biol (2005) 171(4):729–38. doi:10.1083/jcb.200508010
30. Nikolova V, Koo CY, Ibrahim SA, Wang Z, Spillmann D, Dreier R, et al. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis (2009) 30(3):397–407. doi:10.1093/carcin/bgp001
31. Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol (2012) 31(1):3–16. doi:10.1016/j.matbio.2011.10.001
32. Maeda T, Desouky J, Friedl A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene (2006) 25(9):1408–12. doi:10.1038/sj.onc.1209168
33. Su G, Blaine SA, Qiao D, Friedl A. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem (2007) 282(20):14906–15. doi:10.1074/jbc.M611739200
34. Ramani VC, Sanderson RD. Chemotherapy stimulates syndecan-1 shedding: a potentially negative effect of treatment that may promote tumor relapse. Matrix Biol (2013). doi:10.1016/j.matbio.2013.10.005
35. Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med (2009) 206(3):691–705. doi:10.1084/jem.20081278
36. Serini G, Napione L, Arese M, Bussolino F. Besides adhesion: new perspectives of integrin functions in angiogenesis. Cardiovasc Res (2008) 78(2):213–22. doi:10.1093/cvr/cvn045
37. Rapraeger AC, Ell BJ, Roy M, Li X, Morrison OR, Thomas GM, et al. Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between alphaVbeta3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J (2013) 280(10):2194–206. doi:10.1111/febs.12134
38. Oh JH, Lee HS, Park SH, Ryu HS, Min CK. Syndecan-1 overexpression promotes tumor growth and angiogenesis in an endometrial cancer xenograft model. Int J Gynecol Cancer (2010) 20(5):751–6. doi:10.1111/IGC.0b013e3181e02faa
39. Gotte M, Kersting C, Radke I, Kiesel L, Wulfing P. An expression signature of syndecan-1 (CD138), E-cadherin and c-met is associated with factors of angiogenesis and lymphangiogenesis in ductal breast carcinoma in situ. Breast Cancer Res (2007) 9(1):R8. doi:10.1186/bcr1641
40. Zong F, Fthenou E, Wolmer N, Hollosi P, Kovalszky I, Szilak L, et al. Syndecan-1 and FGF-2, but not FGF receptor-1, share a common transport route and co-localize with heparanase in the nuclei of mesenchymal tumor cells. PLoS One (2009) 4(10):e7346. doi:10.1371/journal.pone.0007346
41. Chen L, Sanderson RD. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS One (2009) 4(3):e4947. doi:10.1371/journal.pone.0004947
42. Purushothaman A, Hurst DR, Pisano C, Mizumoto S, Sugahara K, Sanderson RD. Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. J Biol Chem (2011) 286(35):30377–83. doi:10.1074/jbc.M111.254789
43. Zong F, Fthenou E, Mundt F, Szatmari T, Kovalszky I, Szilak L, et al. Specific syndecan-1 domains regulate mesenchymal tumor cell adhesion, motility and migration. PLoS One (2011) 6(6):e14816. doi:10.1371/journal.pone.0014816
44. Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL, et al. The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J (2013) 280(10):2294–306. doi:10.1111/febs.12168
45. Kim CW, Goldberger OA, Gallo RL, Bernfield M. Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns. Mol Biol Cell (1994) 5(7):797–805. doi:10.1091/mbc.5.7.797
46. Contreras HR, Ledezma RA, Vergara J, Cifuentes F, Barra C, Cabello P, et al. The expression of syndecan-1 and -2 is associated with Gleason score and epithelial-mesenchymal transition markers, E-cadherin and beta-catenin, in prostate cancer. Urol Oncol (2010) 28(5):534–40. doi:10.1016/j.urolonc.2009.03.018
47. Ridley RC, Xiao H, Hata H, Woodliff J, Epstein J, Sanderson RD. Expression of syndecan regulates human myeloma plasma cell adhesion to type I collagen. Blood (1993) 81(3):767–74.
48. Fears CY, Woods A. The role of syndecans in disease and wound healing. Matrix Biol (2006) 25(7):443–56. doi:10.1016/j.matbio.2006.07.003
49. Conejo JR, Kleeff J, Koliopanos A, Matsuda K, Zhu ZW, Goecke H, et al. Syndecan-1 expression is up-regulated in pancreatic but not in other gastrointestinal cancers. Int J Cancer (2000) 88(1):12–20. doi:10.1002/1097-0215(20001001)88:1<12::AID-IJC3>3.0.CO;2-T
50. Matsuda K, Maruyama H, Guo F, Kleeff J, Itakura J, Matsumoto Y, et al. Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer Res (2001) 61(14):5562–9.
51. Anttonen A, Heikkila P, Kajanti M, Jalkanen M, Joensuu H. High syndecan-1 expression is associated with favourable outcome in squamous cell lung carcinoma treated with radical surgery. Lung Cancer (2001) 32(3):297–305. doi:10.1016/S0169-5002(00)00230-0
52. Gokden N, Greene GF, Bayer-Garner IB, Spencer HJ, Sanderson RD, Gokden M. Expression of CD138 (Syndecan-1) in renal cell carcinoma is reduced with increasing nuclear grade. Appl Immunohistochem Mol Morphol (2006) 14(2):173–7. doi:10.1097/01.pai.0000168592.58721.7d
53. Anttonen A, Kajanti M, Heikkila P, Jalkanen M, Joensuu H. Syndecan-1 expression has prognostic significance in head and neck carcinoma. Br J Cancer (1999) 79(3–4):558–64. doi:10.1038/sj.bjc.6690088
54. Lundin M, Nordling S, Lundin J, Isola J, Wiksten JP, Haglund C. Epithelial syndecan-1 expression is associated with stage and grade in colorectal cancer. Oncology (2005) 68(4–6):306–13. doi:10.1159/000086969
55. Mitselou A, Skoufi U, Tsimogiannis KE, Briasoulis E, Vougiouklakis T, Arvanitis D, et al. Association of syndecan-1 with angiogenesis-related markers, extracellular matrix components, and clinicopathological features in colorectal carcinoma. Anticancer Res (2012) 32(9):3977–85.
56. Lammoglia-Ordiales L, Toussaint-Caire S, Contreras-Barrera M, Fonte-Avalos V, Rodriguez-Carreon AA, Rivera-Macias S, et al. Assessment of syndecan-1 (CD138) and Ki-67 expression for differentiating keratoacanthoma and squamous cell carcinoma. J Drugs Dermatol (2013) 12(3):e53–8.
57. Rintala M, Inki P, Klemi P, Jalkanen M, Grenman S. Association of syndecan-1 with tumor grade and histology in primary invasive cervical carcinoma. Gynecol Oncol (1999) 75(3):372–8. doi:10.1006/gyno.1999.5595
58. Garusi E, Rossi S, Perris R. Antithetic roles of proteoglycans in cancer. Cell Mol Life Sci (2012) 69(4):553–79. doi:10.1007/s00018-011-0816-1
59. Bodoor K, Matalka I, Hayajneh R, Haddad Y, Gharaibeh W. Evaluation of BCL-6, CD10, CD138 and MUM-1 expression in diffuse large B-cell lymphoma patients: CD138 is a marker of poor prognosis. Asian Pac J Cancer Prev (2012) 13(7):3037–46. doi:10.7314/APJCP.2012.13.7.3037
60. Nault JC, Guyot E, Laguillier C, Chevret S, Ganne-Carrie N, N’Kontchou G, et al. Serum proteoglycans as prognostic biomarkers of hepatocellular carcinoma in patients with alcoholic cirrhosis. Cancer Epidemiol Biomarkers Prev (2013) 22(8):1343–52. doi:10.1158/1055-9965.EPI-13-0179
61. Xu Y, Yuan J, Zhang Z, Lin L, Xu S. Syndecan-1 expression in human glioma is correlated with advanced tumor progression and poor prognosis. Mol Biol Rep (2012) 39(9):8979–85. doi:10.1007/s11033-012-1767-9
62. Yang N, Mosher R, Seo S, Beebe D, Friedl A. Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am J Pathol (2011) 178(1):325–35. doi:10.1016/j.ajpath.2010.11.039
63. Kusumoto T, Kodama J, Seki N, Nakamura K, Hongo A, Hiramatsu Y. Clinical significance of syndecan-1 and versican expression in human epithelial ovarian cancer. Oncol Rep (2010) 23(4):917–25.
64. Mathe M, Suba Z, Nemeth Z, Tatrai P, Fule T, Borgulya G, et al. Stromal syndecan-1 expression is an adverse prognostic factor in oral carcinomas. Oral Oncol (2006) 42(5):493–500. doi:10.1016/j.oraloncology.2005.10.003
65. Wiksten JP, Lundin J, Nordling S, Lundin M, Kokkola A, von Boguslawski K, et al. Epithelial and stromal syndecan-1 expression as predictor of outcome in patients with gastric cancer. Int J Cancer (2001) 95(1):1–6. doi:10.1002/1097-0215(20010120)95:1<1::AID-IJC1000>3.0.CO;2-5
66. Bayer-Garner IB, Dilday B, Sanderson RD, Smoller BR. Syndecan-1 expression is decreased with increasing aggressiveness of basal cell carcinoma. Am J Dermatopathol (2000) 22(2):119–22. doi:10.1097/00000372-200004000-00005
67. Maeda T, Alexander CM, Friedl A. Induction of syndecan-1 expression in stromal fibroblasts promotes proliferation of human breast cancer cells. Cancer Res (2004) 64(2):612–21. doi:10.1158/0008-5472.CAN-03-2439
68. Dias RA, Shibata S, Hashimoto-Uoshima M, Podyma-Inoue KA, Ishikawa I, Yanagishita M. Syndecan-1 expression during the formation of junctional epithelium. J Periodontol (2005) 76(5):696–704. doi:10.1902/jop.2005.76.5.696
69. Steer DL, Shah MM, Bush KT, Stuart RO, Sampogna RV, Meyer TN, et al. Regulation of ureteric bud branching morphogenesis by sulfated proteoglycans in the developing kidney. Dev Biol (2004) 272(2):310–27. doi:10.1016/j.ydbio.2004.04.029
70. Nakanishi T, Kadomatsu K, Okamoto T, Ichihara-Tanaka K, Kojima T, Saito H, et al. Expression of syndecan-1 and -3 during embryogenesis of the central nervous system in relation to binding with midkine. J Biochem (1997) 121(2):197–205.
71. Vainio S, Jalkanen M, Thesleff I. Syndecan and tenascin expression is induced by epithelial-mesenchymal interactions in embryonic tooth mesenchyme. J Cell Biol (1989) 108(5):1945–53. doi:10.1083/jcb.108.5.1945
72. Vainio S, Lehtonen E, Jalkanen M, Bernfield M, Saxen L. Epithelial-mesenchymal interactions regulate the stage-specific expression of a cell surface proteoglycan, syndecan, in the developing kidney. Dev Biol (1989) 134(2):382–91. doi:10.1016/0012-1606(89)90110-3
73. Orosz Z, Kopper L. Syndecan-1 expression in different soft tissue tumours. Anticancer Res (2001) 21(1B):733–7.
74. O’Connell FP, Pinkus JL, Pinkus GS. CD138 (syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am J Clin Pathol (2004) 121(2):254–63. doi:10.1309/617DWB5GNFWXHW4L
75. Conti A, Espina V, Chiechi A, Magagnoli G, Novello C, Pazzaglia L, et al. Mapping protein signal pathway interaction in sarcoma bone metastasis: linkage between rank, metalloproteinases turnover and growth factor signaling pathways. Clin Exp Metastasis (2013). doi:10.1007/s10585-013-9605-6
76. Kumar-Singh S, Jacobs W, Dhaene K, Weyn B, Bogers J, Weyler J, et al. Syndecan-1 expression in malignant mesothelioma: correlation with cell differentiation, WT1 expression, and clinical outcome. J Pathol (1998) 186(3):300–5. doi:10.1002/(SICI)1096-9896(1998110)186:3<300::AID-PATH180>3.0.CO;2-Q
77. Dobra K, Andang M, Syrokou A, Karamanos NK, Hjerpe A. Differentiation of mesothelioma cells is influenced by the expression of proteoglycans. Exp Cell Res (2000) 258(1):12–22. doi:10.1006/excr.2000.4915
78. Gulyas M, Hjerpe A. Proteoglycans and WT1 as markers for distinguishing adenocarcinoma, epithelioid mesothelioma, and benign mesothelium. J Pathol (2003) 199(4):479–87. doi:10.1002/path.1312
79. Saqi A, Yun SS, Yu GH, Alexis D, Taub RN, Powell CA, et al. Utility of CD138 (syndecan-1) in distinguishing carcinomas from mesotheliomas. Diagn Cytopathol (2005) 33(2):65–70. doi:10.1002/dc.20297
80. Fassina A, Cappellesso R, Guzzardo V, Dalla Via L, Piccolo S, Ventura L, et al. Epithelial-mesenchymal transition in malignant mesothelioma. Mod Pathol (2012) 25(1):86–99. doi:10.1038/modpathol.2011.144
81. Klominek J, Robert K-H, Hjerpe A, Wickström B, Gahrton G. Serum-dependent growth patterns of two, newly established human mesothelioma cell lines. Cancer Res (1989) 49:6118–22.
82. Dobra K, Nurminen M, Hjerpe A. Growth factors regulate the expression profile of their syndecan co-receptors and the differentiation of mesothelioma cells. Anticancer Res (2003) 23(3B):2435–44.
83. Herrick SE, Mutsaers SE. Mesothelial progenitor cells and their potential in tissue engineering. Int J Biochem Cell Biol (2004) 36(4):621–42. doi:10.1016/j.biocel.2003.11.002
84. Schramm A, Opitz I, Thies S, Seifert B, Moch H, Weder W, et al. Prognostic significance of epithelial-mesenchymal transition in malignant pleural mesothelioma. Eur J Cardiothorac Surg (2010) 37(3):566–72. doi:10.1016/j.ejcts.2009.08.027
85. Kobayashi M, Huang CL, Sonobe M, Kikuchi R, Ishikawa M, Imamura N, et al. Snail expression is associated with a poor prognosis in malignant pleural mesotheliomas. Ann Thorac Surg (2013) 95(4):1181–8. doi:10.1016/j.athoracsur.2013.01.012
86. Sun D, McAlmon KR, Davies JA, Bernfield M, Hay ED. Simultaneous loss of expression of syndecan-1 and E-cadherin in the embryonic palate during epithelial-mesenchymal transformation. Int J Dev Biol (1998) 42(5):733–6.
87. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest (2009) 119(6):1429–37. doi:10.1172/JCI36183
88. Sun X, Dobra K, Bjornstedt M, Hjerpe A. Upregulation of 9 genes, including that for thioredoxin, during epithelial differentiation of mesothelioma cells. Differentiation (2000) 66(4–5):181–8. doi:10.1046/j.1432-0436.2000.660404.x
89. Sun X, Wei L, Liden J, Hui G, Dahlman-Wright K, Hjerpe A, et al. Molecular characterization of tumour heterogeneity and malignant mesothelioma cell differentiation by gene profiling. J Pathol (2005) 207(1):91–101. doi:10.1002/path.1810
90. Zong F, Fthenou E, Castro J, Peterfia B, Kovalszky I, Szilak L, et al. Effect of syndecan-1 overexpression on mesenchymal tumour cell proliferation with focus on different functional domains. Cell Prolif (2010) 43(1):29–40. doi:10.1111/j.1365-2184.2009.00651.x
91. Peterfia B, Hollosi P, Szilak L, Timar F, Paku S, Jeney A, et al. Role of syndecan-1 proteoglycan in the invasiveness of HT-1080 fibrosarcoma. Magy Onkol (2006) 50(2):115–20.
92. Peterfia B, Fule T, Baghy K, Szabadkai K, Fullar A, Dobos K, et al. Syndecan-1 Enhances Proliferation, Migration and Metastasis of HT-1080 Cells in Cooperation with Syndecan-2. PLoS One (2012) 7(6):e39474. doi:10.1371/journal.pone.0039474
93. Alexander CM, Reichsman F, Hinkes MT, Lincecum J, Becker KA, Cumberledge S, et al. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat Genet (2000) 25(3):329–32. doi:10.1038/77108
94. Seidel C, Borset M, Hjertner O, Cao D, Abildgaard N, Hjorth-Hansen H, et al. High levels of soluble syndecan-1 in myeloma-derived bone marrow: modulation of hepatocyte growth factor activity. Blood (2000) 96(9):3139–46.
95. Tumova S, Woods A, Couchman JR. Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol (2000) 32(3):269–88. doi:10.1016/S1357-2725(99)00116-8
96. Derksen PW, Keehnen RM, Evers LM, van Oers MH, Spaargaren M, Pals ST. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood (2002) 99(4):1405–10. doi:10.1182/blood.V99.4.1405
97. Filla MS, Dam P, Rapraeger AC. The cell surface proteoglycan syndecan-1 mediates fibroblast growth factor-2 binding and activity. J Cell Physiol (1998) 174(3):310–21. doi:10.1002/(SICI)1097-4652(199803)174:3<310::AID-JCP5>3.0.CO;2-R
98. Rahmoune H, Chen HL, Gallagher JT, Rudland PS, Fernig DG. Interaction of heparan sulfate from mammary cells with acidic fibroblast growth factor (FGF) and basic FGF. Regulation of the activity of basic FGF by high and low affinity binding sites in heparan sulfate. J Biol Chem (1998) 273(13):7303–10. doi:10.1074/jbc.273.13.7303
99. Szatmari T, Mundt F, Heidari-Hamedani G, Zong F, Ferolla E, Alexeyenko A, et al. Novel genes and pathways modulated by syndecan-1: implications for the proliferation and cell-cycle regulation of malignant mesothelioma cells. PLoS One (2012) 7(10):e48091. doi:10.1371/journal.pone.0048091
100. Pap Z, Pavai Z, Denes L, Kovalszky I, Jung J. An immunohistochemical study of colon adenomas and carcinomas: E-cadherin, Syndecan-1, Ets-1. Pathol Oncol Res (2009) 15(4):579–87. doi:10.1007/s12253-009-9157-x
101. Afratis N, Gialeli C, Nikitovic D, Tsegenidis T, Karousou E, Theocharis AD, et al. Glycosaminoglycans: key players in cancer cell biology and treatment. FEBS J (2012) 279(7):1177–97. doi:10.1111/j.1742-4658.2012.08529.x
102. Rapraeger AC. The coordinated regulation of heparan sulfate, syndecans and cell behavior. Curr Opin Cell Biol (1993) 5(5):844–53. doi:10.1016/0955-0674(93)90034-N
103. Phillips JJ. Novel therapeutic targets in the brain tumor microenvironment. Oncotarget (2012) 3(5):568–75.
104. Chin L, Meyerson M, Aldape K, Bigner D, Mikkelsen T, VandenBerg S, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi:10.1038/nature07385
105. Habuchi H, Habuchi O, Kimata K. Sulfation pattern in glycosaminoglycan: does it have a code? Glycoconj J (2004) 21(1-2):47–52. doi:10.1023/B:GLYC.0000043747.87325.5e
106. Gorsi B, Stringer SE. Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol (2007) 17(4):173–7. doi:10.1016/j.tcb.2007.02.006
107. Holmborn K, Ledin J, Smeds E, Eriksson I, Kusche-Gullberg M, Kjellen L. Heparan sulfate synthesized by mouse embryonic stem cells deficient in NDST1 and NDST2 is 6-O-sulfated but contains no N-sulfate groups. J Biol Chem (2004) 279(41):42355–8. doi:10.1074/jbc.C400373200
108. Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. The heparanome – the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol (2007) 129(2):290–307. doi:10.1016/j.jbiotec.2007.01.022
109. Ledin J, Ringvall M, Thuveson M, Eriksson I, Wilen M, Kusche-Gullberg M, et al. Enzymatically active N-deacetylase/N-sulfotransferase-2 is present in liver but does not contribute to heparan sulfate N-sulfation. J Biol Chem (2006) 281(47):35727–34. doi:10.1074/jbc.M604113200
110. Li JP, Gong F, Hagner-McWhirter A, Forsberg E, Abrink M, Kisilevsky R, et al. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking l-iduronic acid and in neonatal lethality. J Biol Chem (2003) 278(31):28363–6. doi:10.1074/jbc.C300219200
111. Merry CL, Bullock SL, Swan DC, Backen AC, Lyon M, Beddington RS, et al. The molecular phenotype of heparan sulfate in the Hs2st−/− mutant mouse. J Biol Chem (2001) 276(38):35429–34. doi:10.1074/jbc.M100379200
112. Kamimura K, Koyama T, Habuchi H, Ueda R, Masu M, Kimata K, et al. Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling. J Cell Biol (2006) 174(6):773–8. doi:10.1083/jcb.200603129
113. McCormick C, Duncan G, Goutsos KT, Tufaro F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci U S A (2000) 97(2):668–73. doi:10.1073/pnas.97.2.668
114. Senay C, Lind T, Muguruma K, Tone Y, Kitagawa H, Sugahara K, et al. The EXT1/EXT2 tumor suppressors: catalytic activities and role in heparan sulfate biosynthesis. EMBO Rep (2000) 1(3):282–6. doi:10.1093/embo-reports/kvd045
115. Presto J, Thuveson M, Carlsson P, Busse M, Wilen M, Eriksson I, et al. Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc Natl Acad Sci U S A (2008) 105(12):4751–6. doi:10.1073/pnas.0705807105
116. Pinhal MA, Smith B, Olson S, Aikawa J, Kimata K, Esko JD. Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc Natl Acad Sci U S A (2001) 98(23):12984–9. doi:10.1073/pnas.241175798
117. Kreuger J, Kjellen L. Heparan sulfate biosynthesis: regulation and variability. J Histochem Cytochem (2012) 60(12):898–907. doi:10.1369/0022155412464972
118. Aikawa J, Grobe K, Tsujimoto M, Esko JD. Multiple isozymes of heparan sulfate/heparin GlcNAc N-deacetylase/GlcN N-sulfotransferase. Structure and activity of the fourth member, NDST4. J Biol Chem (2001) 276(8):5876–82. doi:10.1074/jbc.M009606200
119. Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest (2001) 108(2):169–73. doi:10.1172/JCI13530
120. Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J (2006) 20(1):9–22. doi:10.1096/fj.05-4682rev
121. Lamanna WC, Baldwin RJ, Padva M, Kalus I, Ten Dam G, van Kuppevelt TH, et al. Heparan sulfate 6-O-endosulfatases: discrete in vivo activities and functional co-operativity. Biochem J (2006) 400(1):63–73. doi:10.1042/BJ20060848
122. Townley RA, Bulow HE. Genetic analysis of the heparan modification network in Caenorhabditis elegans. J Biol Chem (2011) 286(19):16824–31. doi:10.1074/jbc.M111.227926
123. Kato M, Wang H, Bernfield M, Gallagher JT, Turnbull JE. Cell surface syndecan-1 on distinct cell types differs in fine structure and ligand binding of its heparan sulfate chains. J Biol Chem (1994) 269(29):18881–90.
124. Bret C, Moreaux J, Schved JF, Hose D, Klein B. SULFs in human neoplasia: implication as progression and prognosis factors. J Transl Med (2011) 9:72. doi:10.1186/1479-5876-9-72
125. Dai Y, Yang Y, MacLeod V, Yue X, Rapraeger AC, Shriver Z, et al. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem (2005) 280(48):40066–73. doi:10.1074/jbc.M508136200
126. Rosen SD, Lemjabbar-Alaoui H. Sulf-2: an extracellular modulator of cell signaling and a cancer target candidate. Expert Opin Ther Targets (2010) 14(9):935–49. doi:10.1517/14728222.2010.504718
127. Wang S, Ai X, Freeman SD, Pownall ME, Lu Q, Kessler DS, et al. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci U S A (2004) 101(14):4833–8. doi:10.1073/pnas.0401028101
128. Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem (1993) 268(32):23906–14.
129. Sugaya N, Habuchi H, Nagai N, Ashikari-Hada S, Kimata K. 6-O-sulfation of heparan sulfate differentially regulates various fibroblast growth factor-dependent signalings in culture. J Biol Chem (2008) 283(16):10366–76. doi:10.1074/jbc.M705948200
130. Lai J, Chien J, Staub J, Avula R, Greene EL, Matthews TA, et al. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem (2003) 278(25):23107–17. doi:10.1074/jbc.M302203200
131. Lai JP, Chien J, Strome SE, Staub J, Montoya DP, Greene EL, et al. HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene (2004) 23(7):1439–47. doi:10.1038/sj.onc.1207258
132. Lai JP, Chien JR, Moser DR, Staub JK, Aderca I, Montoya DP, et al. hSulf1 Sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology (2004) 126(1):231–48. doi:10.1053/j.gastro.2003.09.043
133. Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP Jr. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science (2001) 293(5535):1663–6. doi:10.1126/science.293.5535.1663
134. Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U, Emerson CP Jr. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol (2003) 162(2):341–51. doi:10.1083/jcb.200212083
135. Viviano BL, Paine-Saunders S, Gasiunas N, Gallagher J, Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J Biol Chem (2004) 279(7):5604–11. doi:10.1074/jbc.M310691200
136. Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS One (2007) 2(4):e392. doi:10.1371/journal.pone.0000392
137. He B, Lee AY, Dadfarmay S, You L, Xu Z, Reguart N, et al. Secreted frizzled-related protein 4 is silenced by hypermethylation and induces apoptosis in beta-catenin-deficient human mesothelioma cells. Cancer Res (2005) 65(3):743–8.
138. Lee AY, Raz DJ, He B, Jablons DM. Update on the molecular biology of malignant mesothelioma. Cancer (2007) 109(8):1454–61. doi:10.1002/cncr.22552
139. Fox S, Dharmarajan A. WNT signaling in malignant mesothelioma. Front Biosci (2006) 11:2106–12. doi:10.2741/1953
140. Fernandez-Vega I, Garcia O, Crespo A, Castanon S, Menendez P, Astudillo A, et al. Specific genes involved in synthesis and editing of heparan sulfate proteoglycans show altered expression patterns in breast cancer. BMC Cancer (2013) 13:24. doi:10.1186/1471-2407-13-24
141. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res (2011) 17(6):1603–15. doi:10.1158/1078-0432.CCR-10-2563
142. Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation ofcCellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med (2002) 195(1):125–33. doi:10.1084/jem.20011097
143. Orecchia P, Conte R, Balza E, Petretto A, Mauri P, Mingari MC, et al. A novel human anti-syndecan-1 antibody inhibits vascular maturation and tumour growth in melanoma. Eur J Cancer (2013) 49(8):2022–33. doi:10.1016/j.ejca.2012.12.019
144. Yang Y, MacLeod V, Dai Y, Khotskaya-Sample Y, Shriver Z, Venkataraman G, et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood (2007) 110(6):2041–8. doi:10.1182/blood-2007-04-082495
145. Rousseau C, Ruellan AL, Bernardeau K, Kraeber-Bodere F, Gouard S, Loussouarn D, et al. Syndecan-1 antigen, a promising new target for triple-negative breast cancer immuno-PET and radioimmunotherapy. A preclinical study on MDA-MB-468 xenograft tumors. EJNMMI Res (2011) 1(1):20. doi:10.1186/2191-219X-1-20
146. Fedarko NS, Ishihara M, Conrad HE. Control of cell division in hepatoma cells by exogenous heparan sulfate proteoglycan. J Cell Physiol (1989) 139(2):287–94. doi:10.1002/jcp.1041390210
147. Roghani M, Moscatelli D. Basic fibroblast growth factor is internalized through both receptor-mediated and heparan sulfate-mediated mechanisms. J Biol Chem (1992) 267(31):22156–62.
148. Cheng F, Petersson P, Arroyo-Yanguas Y, Westergren-Thorsson G. Differences in the uptake and nuclear localization of anti-proliferative heparan sulfate between human lung fibroblasts and human lung carcinoma cells. J Cell Biochem (2001) 83(4):597–606. doi:10.1002/jcb.1254
149. Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci U S A (1996) 93(22):12349–54. doi:10.1073/pnas.93.22.12349
150. Belting M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem Sci (2003) 28(3):145–51. doi:10.1016/S0968-0004(03)00031-8
151. Belting M, Mani K, Jonsson M, Cheng F, Sandgren S, Jonsson S, et al. Glypican-1 is a vehicle for polyamine uptake in mammalian cells: a pivital role for nitrosothiol-derived nitric oxide. J Biol Chem (2003) 278(47):47181–9. doi:10.1074/jbc.M308325200
152. Sandgren S, Wittrup A, Cheng F, Jonsson M, Eklund E, Busch S, et al. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J Biol Chem (2004) 279(17):17951–6. doi:10.1074/jbc.M311440200
153. Poon GM, Gariepy J. Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem Soc Trans (2007) 35(Pt 4):788–93. doi:10.1042/BST0350788
154. Wittrup A, Sandgren S, Lilja J, Bratt C, Gustavsson N, Morgelin M, et al. Identification of proteins released by mammalian cells that mediate DNA internalization through proteoglycan-dependent macropinocytosis. J Biol Chem (2007) 282(38):27897–904. doi:10.1074/jbc.M701611200
155. Wittrup A, Zhang SH, ten Dam GB, van Kuppevelt TH, Bengtson P, Johansson M, et al. ScFv antibody-induced translocation of cell-surface heparan sulfate proteoglycan to endocytic vesicles: evidence for heparan sulfate epitope specificity and role of both syndecan and glypican. J Biol Chem (2009) 284(47):32959–67. doi:10.1074/jbc.M109.036129
156. Letoha T, Keller-Pinter A, Kusz E, Kolozsi C, Bozso Z, Toth G, et al. Cell-penetrating peptide exploited syndecans. Biochim Biophys Acta (2010) 1798(12):2258–65. doi:10.1016/j.bbamem.2010.01.022
157. Wittrup A, Zhang SH, Belting M. Studies of proteoglycan involvement in CPP-mediated delivery. Methods Mol Biol (2011) 683:99–115. doi:10.1007/978-1-60761-919-2_8
158. Belting M, Sandgren S, Wittrup A. Nuclear delivery of macromolecules: barriers and carriers. Adv Drug Deliv Rev (2005) 57(4):505–27. doi:10.1016/j.addr.2004.10.004
159. Raja SM, Metkar SS, Honing S, Wang B, Russin WA, Pipalia NH, et al. A novel mechanism for protein delivery: granzyme B undergoes electrostatic exchange from serglycin to target cells. J Biol Chem (2005) 280(21):20752–61. doi:10.1074/jbc.M501181200
160. Belting M, Wittrup A. Macromolecular drug delivery: basic principles and therapeutic applications. Mol Biotechnol (2009) 43(1):89–94. doi:10.1007/s12033-009-9185-5
Keywords: syndecan-1, heparan sulfate, signaling, cancer, mesenchymal tumor
Citation: Szatmári T and Dobra K (2013) The role of syndecan-1 in cellular signaling and its effects on heparan sulfate biosynthesis in mesenchymal tumors. Front. Oncol. 3:310. doi: 10.3389/fonc.2013.00310
Received: 28 October 2013; Accepted: 04 December 2013;
Published online: 19 December 2013.
Edited by:
Elvira V. Grigorieva, Institute of Molecular Biology and Biophysics SB RAMS, RussiaReviewed by:
Markus A. N. Hartl, University of Innsbruck, AustriaSwapna Asuthkar, University of Illinois College of Medicine at Peoria, USA
Copyright: © 2013 Szatmári and Dobra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katalin Dobra, Department of Laboratory Medicine, Karolinska Institutet, Karolinska University Hospital F-46, SE-141 86 Stockholm, Sweden e-mail:a2F0YWxpbi5kb2JyYUBraS5zZQ==