 Stacey Ann N. D’mello1
Stacey Ann N. D’mello1 Jack U. Flanagan2,3
Jack U. Flanagan2,3 Taryn N. Green1
Taryn N. Green1 Euphemia Y. Leung2
Euphemia Y. Leung2 Marjan E. Askarian-Amiri2Wayne R. Joseph2Michael R. McCrystal4,5Richard J. Isaacs6James H. F. Shaw7Christopher E. Furneaux8Matthew J. During9,10
Marjan E. Askarian-Amiri2Wayne R. Joseph2Michael R. McCrystal4,5Richard J. Isaacs6James H. F. Shaw7Christopher E. Furneaux8Matthew J. During9,10 Graeme J. Finlay2
Graeme J. Finlay2 Bruce C. Baguley2
Bruce C. Baguley2 Maggie L. Kalev-Zylinska1,11*
Maggie L. Kalev-Zylinska1,11*- 1Department of Molecular Medicine and Pathology, School of Medical Sciences, University of Auckland, Auckland, New Zealand
- 2Auckland Cancer Society Research Centre, University of Auckland, Auckland, New Zealand
- 3Maurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, Auckland, New Zealand
- 4Department of Clinical Oncology, Auckland District Health Board, Auckland, New Zealand
- 5Canopy Cancer Care, Mercy Hospital, Auckland, New Zealand
- 6Regional Cancer Treatment Service, Palmerston North Public Hospital, Palmerston North, New Zealand
- 7Oncology Surgery Ltd., Auckland, New Zealand
- 8Department of Neurosurgery, Auckland District Health Board, Auckland, New Zealand
- 9Department of Molecular Virology, Immunology and Medical Genetics, Neuroscience and Neurological Surgery, Ohio State University, Columbus, OH, USA
- 10Centre for Brain Research, University of Auckland, Auckland, New Zealand
- 11LabPlus Haematology, Auckland District Health Board, Auckland, New Zealand
Previous whole-exome sequencing has demonstrated that melanoma tumors harbor mutations in the GRIN2A gene. GRIN2A encodes the regulatory GluN2A subunit of the glutamate-gated N-methyl-d-aspartate receptor (NMDAR), involvement of which in melanoma remains undefined. Here, we sequenced coding exons of GRIN2A in 19 low-passage melanoma cell lines derived from patients with metastatic melanoma. Potential mutation impact was evaluated in silico, including within the GluN2A crystal structure, and clinical correlations were sought. We found that of 19 metastatic melanoma tumors, four (21%) carried five missense mutations in the evolutionarily conserved domains of GRIN2A; two were previously reported. Melanoma cells that carried these mutations were treatment-naïve. Sorting intolerant from tolerant analysis predicted that S349F, G762E, and P1132L would disrupt protein function. When modeled into the crystal structure of GluN2A, G762E was seen to potentially alter GluN1–GluN2A interactions and ligand binding, implying disruption to NMDAR functionality. Patients whose tumors carried non-synonymous GRIN2A mutations had faster disease progression and shorter overall survival (P < 0.05). This was in contrast to the BRAF V600E mutation, found in 58% of tumors but showing no correlation with clinical outcome (P = 0.963). Although numbers of patients in this study are small, and firm conclusions about the association between GRIN2A mutations and poor clinical outcome cannot be drawn, our results highlight the high prevalence of GRIN2A mutations in metastatic melanoma and suggest for the first time that mutated NMDARs impact melanoma progression.
Introduction
The genomic revolution of recent years has led to substantial advances in the cataloging of mutations in melanoma, most notable of which have been activating mutations in BRAF, NRAS, and KIT genes (1). The presence of these mutations helps to guide treatment with RAF, MEK, and KIT inhibitors, but they do not predict disease progression or survival of patients (2). In general, the usefulness of molecular biomarkers in determining prognosis in melanoma remains limited.
Mutations in GRIN2A have been reported in up to a third of melanoma samples (3), although there is wide variation between studies (4–7) and no data on their clinical relevance. The GRIN2A gene, located on chromosome 16p13.2, encodes the GluN2A protein, a regulatory subunit of the glutamate-gated N-methyl-d-aspartate receptor (NMDAR) (8, 9). NMDARs are best known for their roles in the brain; hence, the finding of GRIN2A mutations in melanoma had been unexpected. Nevertheless, NMDARs have attracted attention for their potential contribution in cancer due to the effects on cell death, survival, and migration (10, 11). Excessive NMDAR activation overloads the cell with calcium and leads to cell death (12). On the other hand, normal NMDAR activity promotes cell survival through the phosphatidylinositol 3-kinase (PI3-K) and extracellular signal-regulated kinase (ERK) signaling pathways (13). In addition, NMDAR effects on cell migration may affect tumor spread in tissue (10). Current knowledge on the NMDARs in the context of melanoma is limited, although expression of GluN2A in both normal and malignant melanocytes has been demonstrated (14, 15). NMDAR inhibitors reduce migration and proliferation of melanoma cells in vitro (15).
In response to the previously published exome sequencing data (3), we have investigated the prevalence of GRIN2A mutations in 19 low-passage metastatic melanoma cell lines out of over a 100 developed in our institution, and retrospectively correlated the presence of GRIN2A mutations with patient outcome.
Materials and Methods
Patients and Tumor Material
Low-passage melanoma cell lines derived from 19 patients with metastatic melanoma treated at two independent national sites were used in this study (Table S1 in Supplementary Material). Written informed consent was obtained from all participants prior to enrolment; all study procedures were approved by Northern A Health and Disability Human Ethics Committee. This was not a clinical trial, and study procedures did not affect patient management in any way. Patients underwent skin, lymph node, or distant organ biopsies for diagnosis, staging, or treatment, as required clinically. Excess tissue was used to establish melanoma cell lines, as described (16). Cell lines for sequencing were chosen randomly out of over a 100 previously established in our center; all were passaged <30 times. Cells were grown in 25 or 75 cm2 flasks to 75% confluency in a low-oxygen humidified incubator (37°C, 5% O2, and 5% CO2 in nitrogen). Cultures were maintained in Alpha-modified Minimal Essential Medium (Sigma-Aldrich, Saint Louis, MO, USA) containing 5% fetal bovine serum (Life Technologies, Carlsbad, CA, USA), penicillin (100 units ml−1), streptomycin (100 μg ml−1) (Sigma-Aldrich), insulin (5 μg ml−1), transferrin (5 μg ml−1), and sodium selenite (5 ng ml−1) (Roche Applied Science, San Diego, CA, USA). Melanoma cells were sub-cultured weekly to maintain them in a proliferative state.
Normal human epidermal melanocytes were used as control cells (HEMa-LP, Life Technologies); we did not have access to non-tumor tissue from patients. Melanocytes were cultured in Medium 254 (M-254-500; Life Technologies) supplemented with Human Melanocyte Growth Supplement (Life Technologies). Cultures were maintained in a humidified incubator (5% CO2 in air) at 37°C. Melanocytes were sub-cultured every 4 weeks.
Sequencing
DNA was isolated using a PureLink Genomic DNA kit (Life Technologies), according to the manufacturer’s instructions. Concentrations and purity of DNA were determined using a nanodrop spectrophotometer (ND-1000, NanoDrop, ThermoFisher Scientific, Rockford, IL, USA). Twelve coding exons of GRIN2A (numbered 3–14), including their flanking intronic regions, were sequenced using Sanger method. The sequencing primers were as previously reported (3), except for 7Reverse (5′-GCAGGCCCTTTGTCTGAGTA-3′) and 8Forward (5′-CCTTGCATCCAGGTGGTC-3′), which we designed using Primer3web version 4.0.0 software1 to reduce interference from polyA sequences located in the intron between GRIN2A exons 7 and 8. PCR conditions for all primers are provided in Table 1. PCR reactions were performed in a final volume of 25 μl 1× PCR buffer containing 50–100 ng DNA, 0.3 μM forward and reverse primers each, 5 U Expand High Fidelity Enzyme mix (Roche Applied Science), 0.2 mM deoxynucleoside triphosphates, and 1.5 mM MgCl2. Bovine serum albumin (3 ng μl−1; Life Technologies) was used to counteract PCR inhibition where required. The correct sizes of the amplicons were confirmed on 2% agarose gels with DNA visualized with GelRed (Biotium, Hayward, CA, USA). DNA concentrations were estimated against Low-Mass Ladder (Life technologies). Sequencing was performed in two directions using ABI Prism 3730xl Genetic Analyzer with the ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction kit, version 3.1 (Applied Biosystems, Foster City, CA, USA).
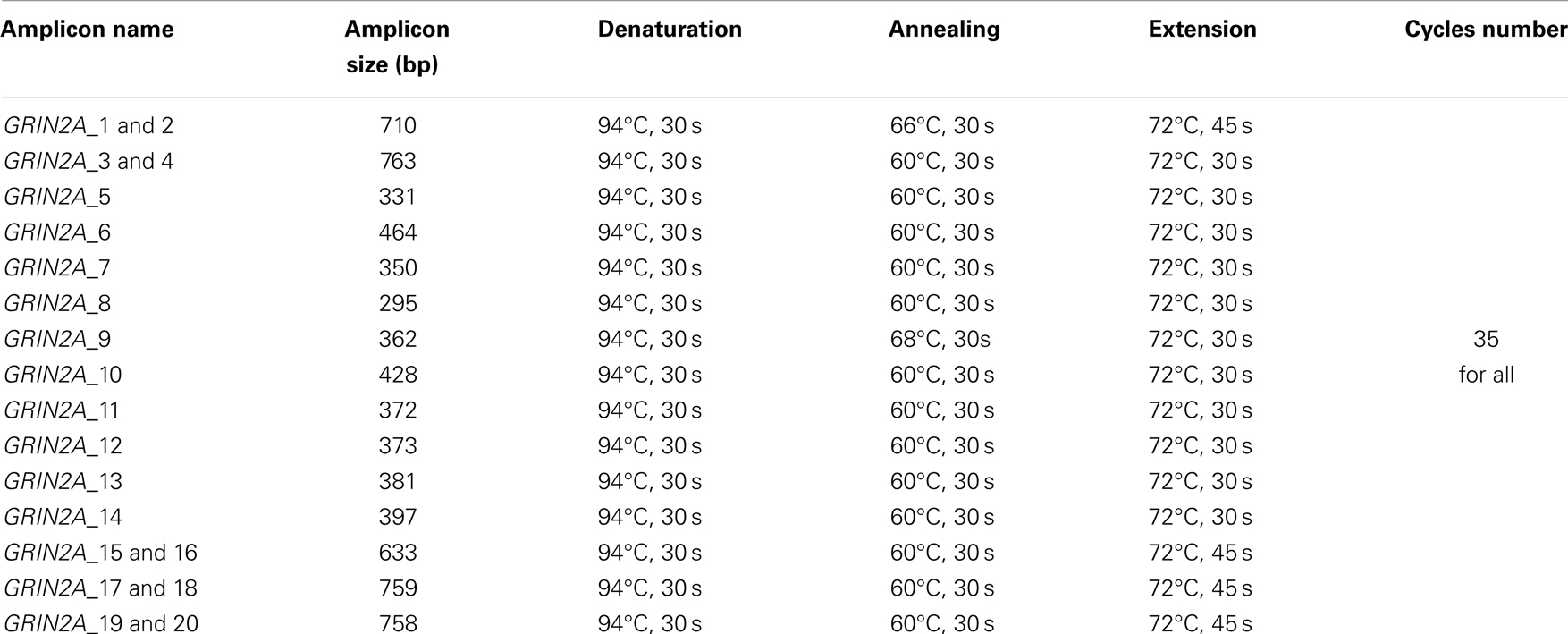
Table 1. PCR cycling conditions used to amplify GRIN2A sequences.
The BRAF V600E mutation status had been determined previously for most cell lines (17). For the remaining samples, Cobas® 4800 BRAF Mutation Test (Roche Molecular Systems, Pleasanton, CA, USA) was used.
Sequence Analysis and Prediction of Mutation Impact
Exon sequences of GRIN2A were analyzed by reference to human GRIN2A (GenBank accession number NG_011812; RefSeqGene number GI: 226492187) using Geneious Pro 5.6.4 software (Biomatters, Auckland, New Zealand). To help predict if amino acid substitutions would affect protein function, Sorting Intolerant from Tolerant (SIFT) analysis of mutations was performed2 (18), applying UniProt SWISS-PROT 57.15 database. Catalog of somatic mutations in cancer (COSMIC)3 and MelanomaDB (19) databases were interrogated to search for GRIN2A mutations previously found in melanoma. Common germline single nucleotide polymorphisms (SNPs) in GRIN2A were eliminated using the NCBI database of SNPs4 (20).
Modeling Mutation Impact
The G762E mutation was modeled into the X-ray crystal structure of the GluN1–GluN2A S1S2 heterodimer using Modeller 9.1 (21) and the Protein Data Bank entry 2A5T (22) as the template. Ten models were constructed using two rounds of optimization with the slow autoschedule and molecular dynamics refinement options; all other settings were kept at default values. The models were superimposed on the 2A5T heterodimer using PyMol (23) and inspected visually.
Collection of Clinical Data and Statistical Analysis
Data on antecedent primary melanoma, disease progression, and treatment were obtained from a retrospective review of patient medical records. Melanoma staging was determined clinically based on the criteria designated by the American Joint Committee on Cancer (24). In this system, patients with lymph node metastases are designated as stage III; stage IV disease is defined by the presence of distant organ metastases. Overall survival is shown as the length of time from the initial diagnosis of melanoma to death. Considering that patient groups were small in this study, we presented quantitative data as median (range) (Table 2) to best demonstrate sample distribution. To compare groups, cross-tabulations with significance tests were performed for data in categories. Analysis for mean differences between groups was performed using one-way ANOVA. Differences in time to event data were analyzed for the effect of GRIN2A mutation status using the survival analysis method (log-rank test). Kaplan–Meier curves were generated to plot time to events (such as development of lymph node or distant organ metastases, or death) for individual patients (dot plots are provided as Supplemental Material). Statistical analysis was conducted using IBM SPSS Statistics software package for Windows, version 19.0 (Chicago, IL, USA) (25). P values <0.05 were considered statistically significant.
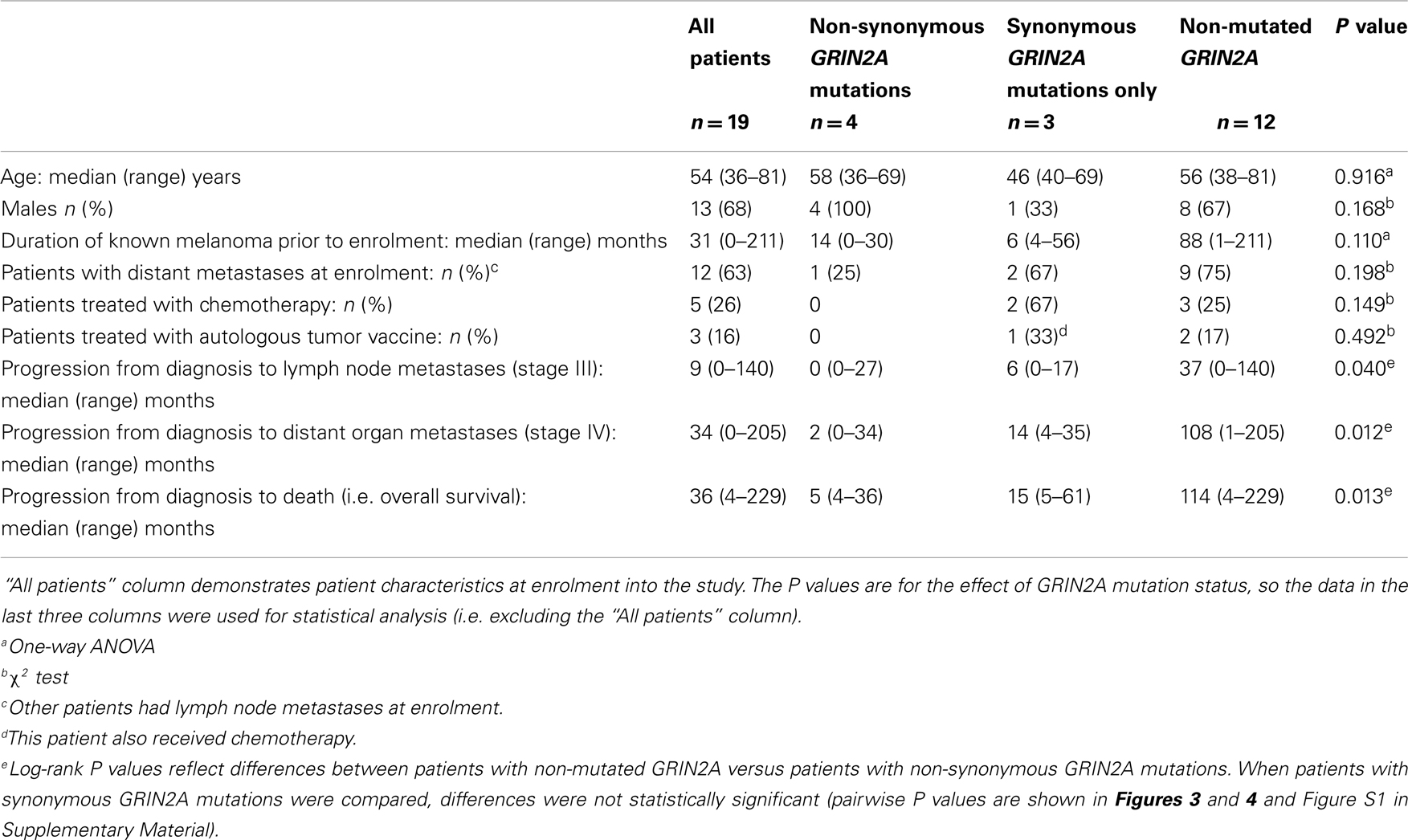
Table 2. Clinical characteristics for all patients, according to the presence or absence of GRIN2A mutations in tumor-derived cell lines.
Results
Nineteen low-passage melanoma cell lines, derived from 19 patients with metastatic melanoma, were used in this study to sequence coding exons of GRIN2A, together with their flanking intronic regions. Patient characteristics at enrolment into the study are shown in Table 2 (“All patients” column). Apart from two patients who presented with either bulky or disseminated disease and entered this study on presentation, other patients had a prior history of skin melanoma dating back a median of 34 (1–211) months. Seventeen patients had primary skin lesions in sun-exposed areas; in the other two, primary skin lesions remained occult. At the time of enrolment (between 1989 and 2010), 12 patients (63%) had metastatic melanoma in distant organs (stage IV disease) and 7 patients (37%) had lymph node involvement (stage III disease). All patients were managed surgically and with regional radiotherapy. Five patients also received chemotherapy – three had POC (procarbazine, vincristine, and lomustine), one temozolomide, and one DTIC (dacarbazine) – with the number of chemotherapy cycles ranging from one to six. Autologous tumor vaccine was used in three patients (in one, after an unsuccessful course of chemotherapy). One patient also received an experimental vascular disrupting agent within a phase I clinical trial, as well as interferon α. No patient received BRAF inhibitors. Treatment was the decision of a clinical team, independent of research procedures. One patient remained free of melanoma 12 years after nodal recurrence. The other 18 patients have all died of their disease. The median survival time was 36 (4–229) months.
GRIN2A Mutations and Predicted Impact on Protein Structure
Sequencing of GRIN2A was conducted using cell lines derived from tumors spread to lymph nodes in 12 patients (63%), distant organs (brain, small bowel, ascites, or lung) (five patients; 26%), or locoregional metastases (two patients; 11%). All cell lines used in this study were passaged <30 times, and were generated from treatment-naïve tumors, except for NZM 017 and NZM 040, which were derived from patients who received prior immunotherapy or chemotherapy, respectively (Table S1 in Supplementary Material).
Of 19 tumor samples tested, four (21%) carried five non-synonymous mutations in GRIN2A (Figure 1; Table 3). Tumors that carried these mutations were treatment-naïve, excluding therapy effect. All non-synonymous mutations were missense: three clustered in exon 14 (G889E, P1132L, P1133S), and the other two in exon 5 (S349F) and exon 12 (G762E) (Figure 1). These locations corresponded with the evolutionarily conserved domains in the GluN2A protein: C-terminal, N-terminal, and the S2 segment, respectively (Figure 1). The S2 segment forms the ligand binding domain, and the intracytoplasmic C-terminus is involved in intracellular signaling and interactions with the cytoskeleton. Two mutations, G889E and P1132L, were previously reported [Ref. (3, 17), respectively]. SIFT analysis predicted that S349F, G762E, and P1133S would deleteriously affect protein function (Table 3).
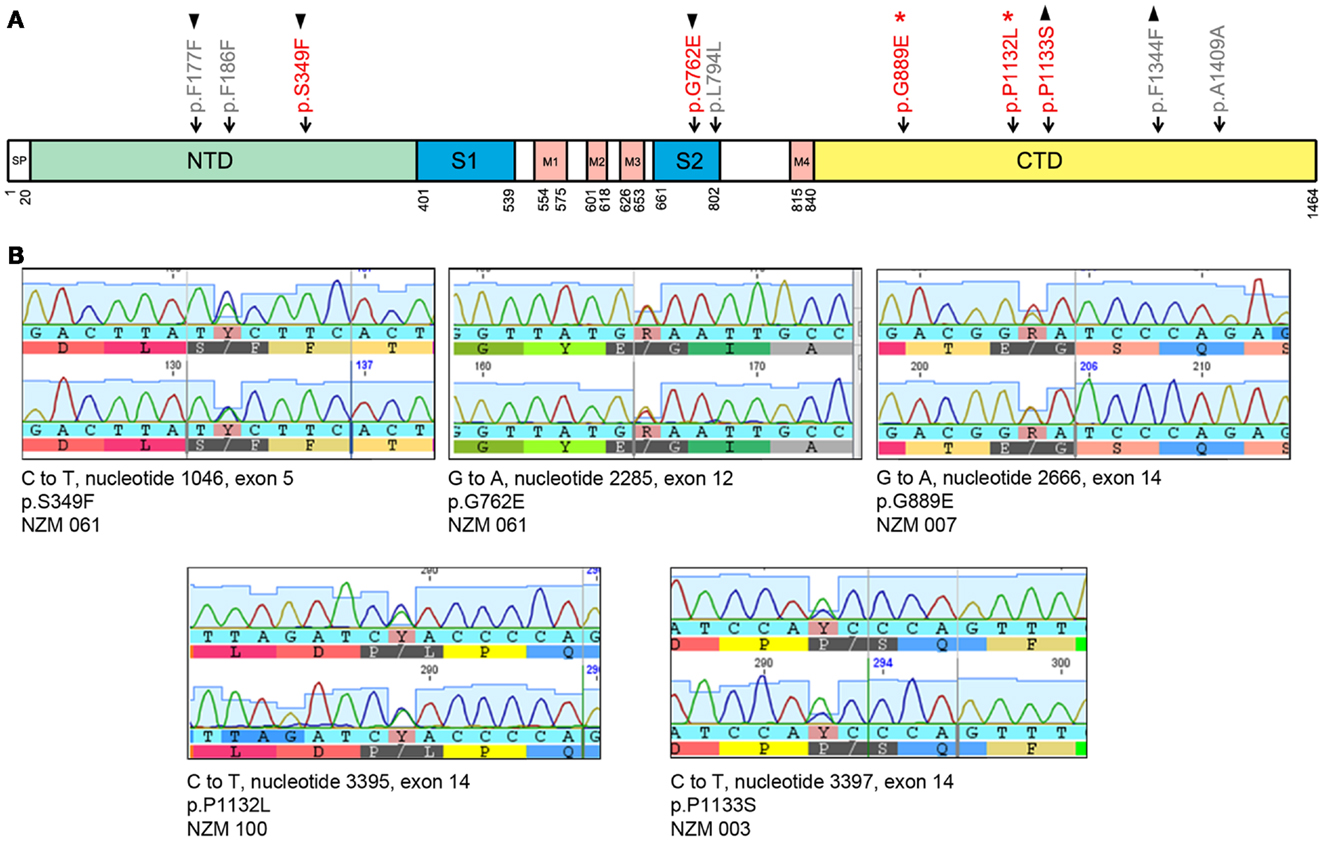
Figure 1. GRIN2A mutations in melanoma cell lines. (A) Schematic of the GluN2A protein together with the non-synonymous (in red) and synonymous (in gray) mutations in GRIN2A. GRIN2A cDNA is 4392 nucleotides long and can be divided into sections encoding evolutionarily conserved domains in the GluN2A protein. The first and last amino acid residues of the SP, S1, S2, and M1–M4 domains are numbered. Symbols ▴ and ▾ mark mutations that coincided in the same tumor samples; *marks mutations reported previously. Abbreviations: SP, signal peptide; NTD, N-terminal domain; S1 and S2 segments form the glutamate-binding domain; M1–M4 transmembrane segments form the ion channel pore; CTD, C-terminal domain. (B) Sanger sequencing output of non-synonymous mutations in GRIN2A. Each panel represents a non-synonymous mutation detected. Nucleotides are labeled below their respective peaks. Corresponding amino acid sequences are indicated.
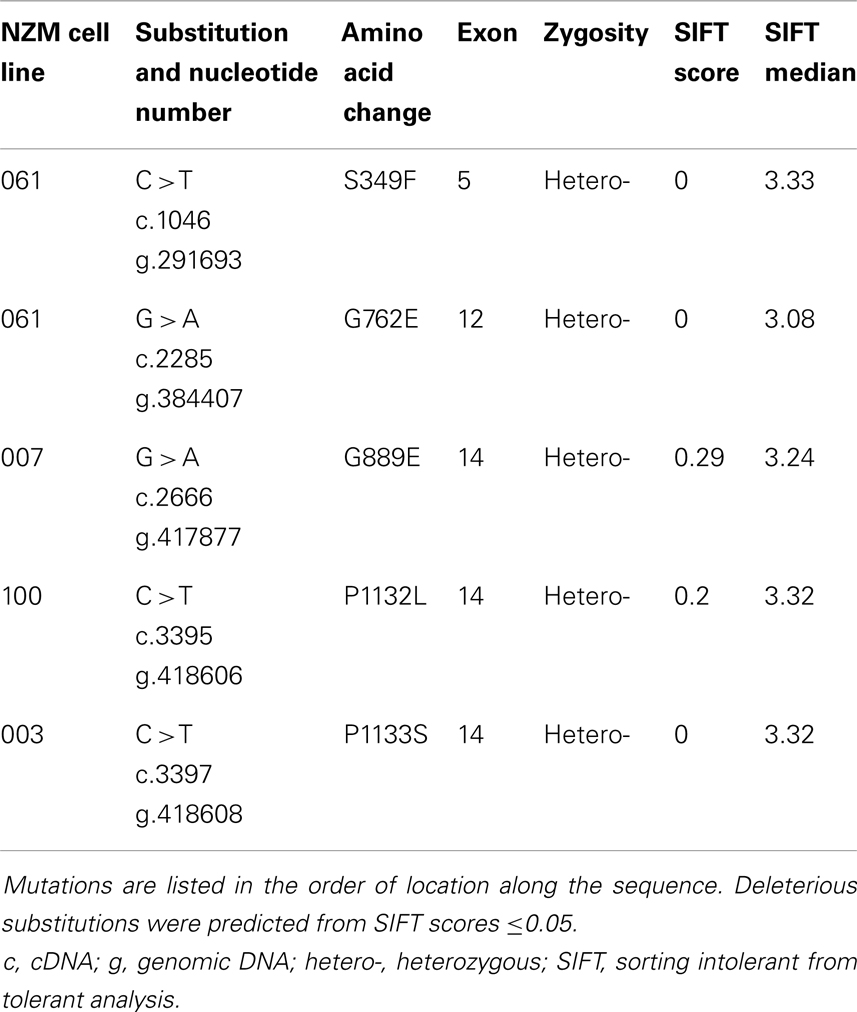
Table 3. Non-synonymous mutations in GRIN2A.
Five synonymous mutations were also detected (Table 4), as well as four SNPs (Table 5); SNPs were excluded from further analysis. F186F synonymous mutation was found in the NZM 040 cell line derived from patient who received two cycles of POC chemotherapy 8 months prior to cell line derivation (Table S1 in Supplementary Material). In this instance, the possibility that therapy contributed to the presence of this mutation could not be ruled out.
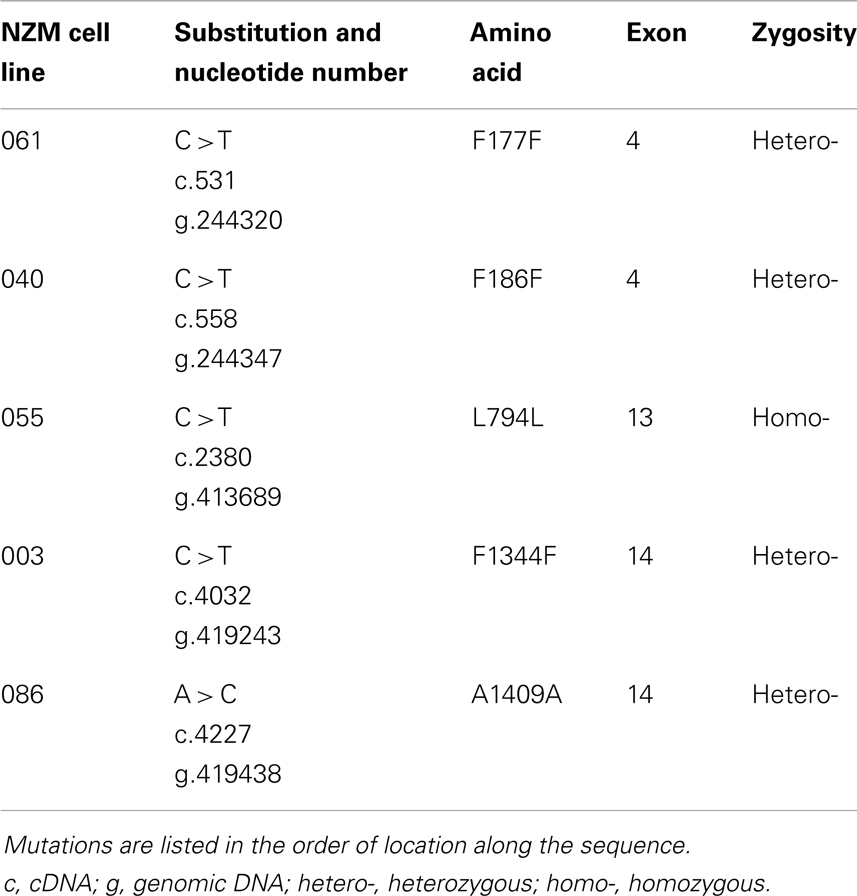
Table 4. Synonymous mutations in GRIN2A.
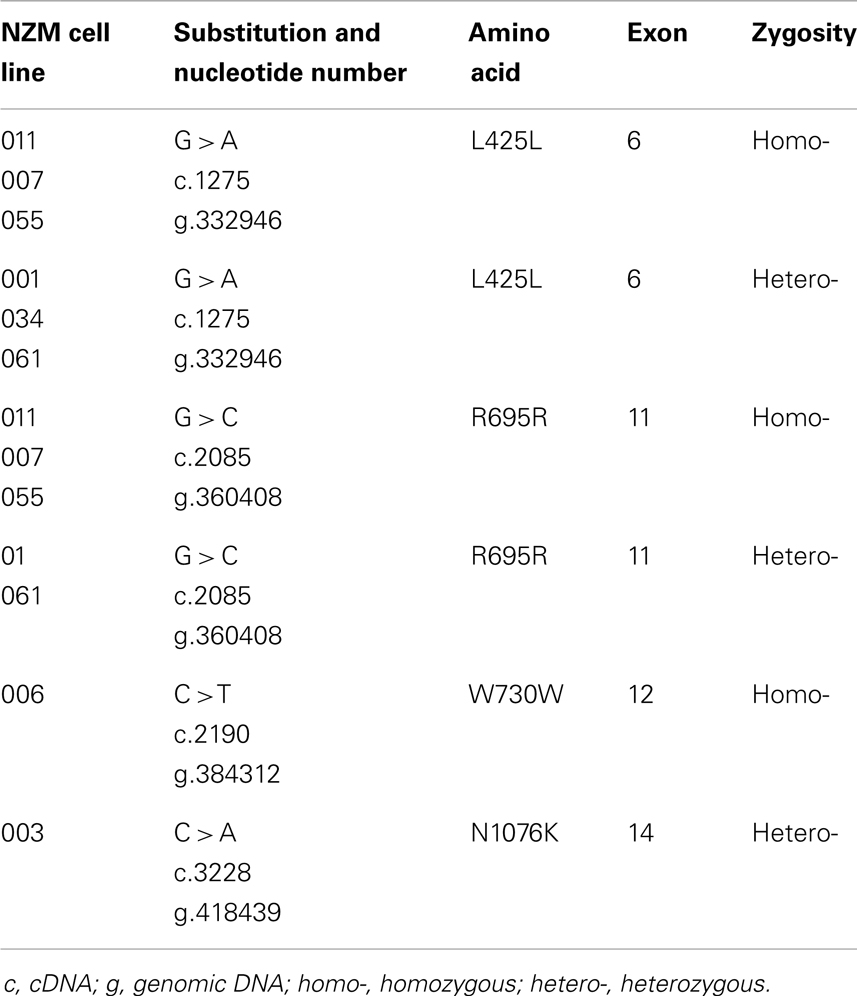
Table 5. GRIN2A SNPs in tumor-derived cell lines.
We modeled the G762E missense substitution into the 3-dimensional X-ray crystal structure of the GluN1–GluN2A heterodimer, 2A5T (22) (Figure 2). This revealed that G762E was located in the distal “hinge” of the glutamate-binding clam-shell-like region of GluN2A. In this location, the mutated glutamate residue was seen to interact with K531 in the GluN1 protein interfacing GluN2A in this region. While K531 formed a hydrogen bond with the backbone carbonyl of F524 in GluN2A, its proximity to the mutated glutamate side-chain (G762E) indicated the potential for new electrostatic interactions between G762E (in GluN2A) and K531 (in GluN1) (Figure 2) with the ability to alter interactions between GluN2A and GluN1 subunits and consequently, impact NMDAR functionality. Conformational changes that developed to accommodate G762E could also affect ligand binding, as the residue preceding G762E (Y761) was part of the glutamate-binding site (Figure 2) (26).
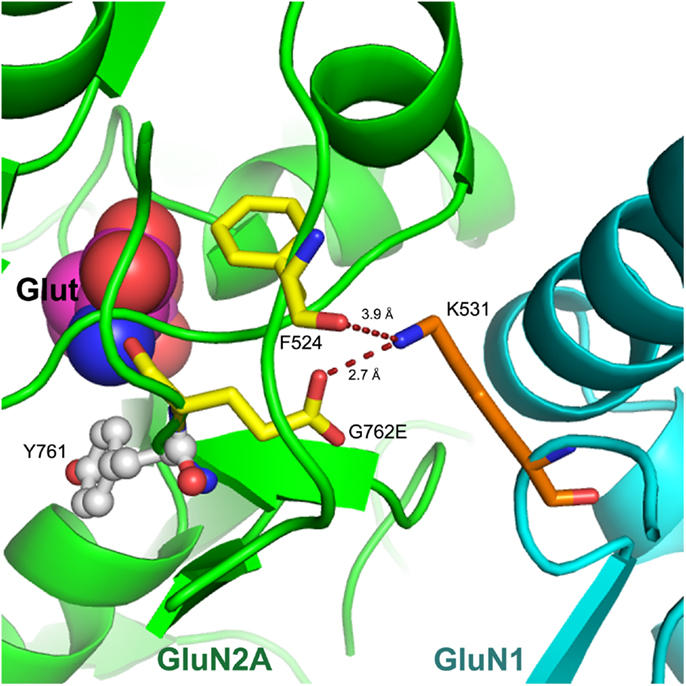
Figure 2. Model of G762E within the GluN1–GluN2A X-ray crystal structure. The S1S2 loop of GluN2A (in green) is shown interfacing with GluN1 (in cyan). A portion of the GluN2A agonist-binding site is on the left together with bound glutamate (Glut). G762E and F524 residues of GluN2A are 2.7 and 3.9 Å away from K531 in GluN1, respectively; potential new electrostatic interactions are indicated as dashed lines. Nitrogen atoms are in blue, and oxygen in red.
Clinical Associations of GRIN2A Mutations
There was no difference in age and gender between patients whose melanoma lines carried GRIN2A mutations and those who did not (Table 2). Two patients in this study presented with disseminated melanoma and both were found to carry non-synonymous mutations in GRIN2A. The other two patients with the non-synonymous GRIN2A mutations presented with skin lesions that spread to lymph nodes within 9 and 27 months, compared with a median of 37 (0–140) months for patients with non-mutated GRIN2A (P = 0.041; Table 2). Overall, patients with non-synonymous GRIN2A mutations had faster progression of melanoma from skin lesions to the involvement of lymph nodes (P = 0.04) or distant organs (P = 0.012), and shorter overall survival (P = 0.013) compared with patients with non-mutated GRIN2A (Table 2; Figures 3 and 4A). The BRAF V600E mutation was found in 11 of 19 (58%) tumor samples but in contrast to GRIN2A, its presence showed no correlation with overall survival (P = 0.963; Figure 4B; Table S1 in Supplementary Material).
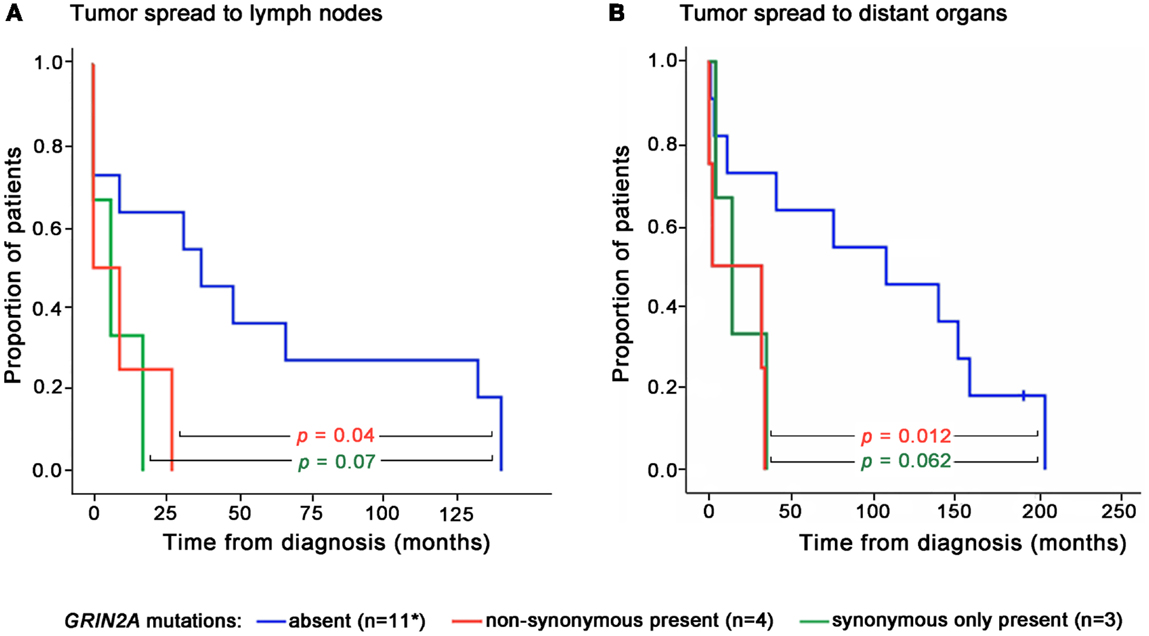
Figure 3. Times of disease progression from diagnosis to lymph node (A) or distant organ (B) metastases for individual patients according to the presence or absence of GRIN2A mutations. Levels of statistical significance are shown. *Progression data for one patient with non-mutated GRIN2A was not available.
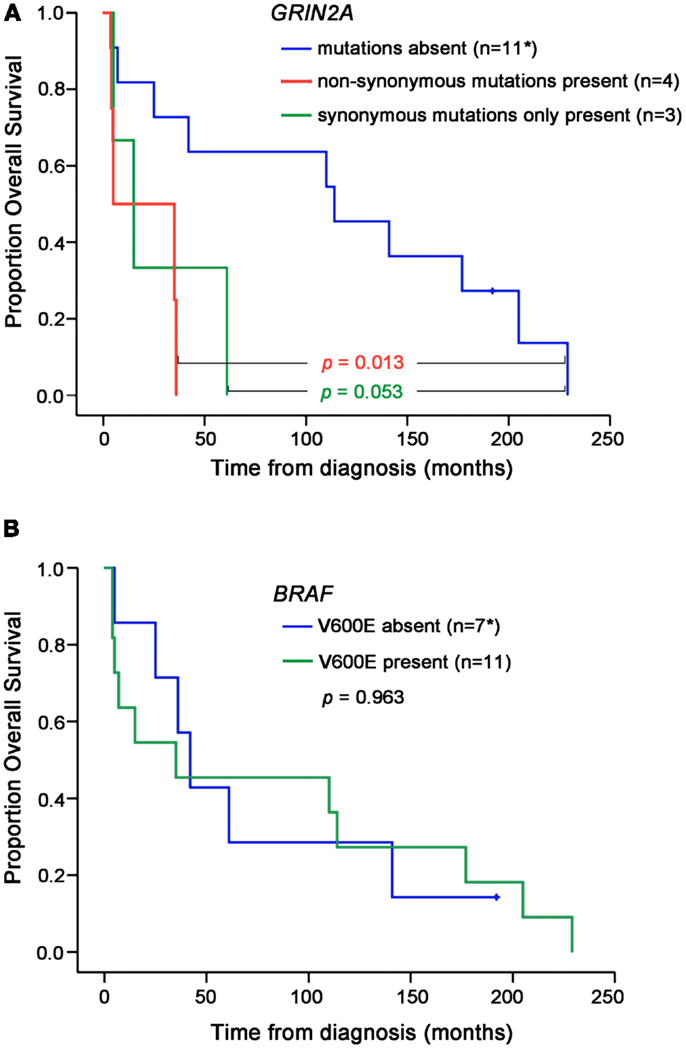
Figure 4. Overall survival according to the GRIN2A (A) or BRAF V600E (B) mutation status. Levels of statistical significance are shown. *Data for one patient with non-mutated GRIN2A (V600E absent) was not available.
Seven patients in this study received systemic therapy – five had chemotherapy and another two autologous tumor vaccine (Table 2). These were in addition to surgery and involved field radiotherapy used in all patients. When compared with patients managed with surgery and radiotherapy alone, the administration of systemic therapy did not change disease spread to lymph nodes (P = 0.849), distant organs (P = 0.499), or overall survival (P = 0.843).
Patients with isolated synonymous GRIN2A mutations (n = 3) displayed a trend for poorer outcome, compared with patients with non-mutated GRIN2A, but this was not statistically significant (Figures 3 and 4).
Discussion
This study has shown that cell lines derived from 4 of 19 (21%) patients with metastatic melanoma carried five missense mutations in GRIN2A. They occurred in three of the four evolutionarily conserved domains of the GluN2A subunit of the NMDAR: the N-terminal, glutamate-binding, and C-terminal domains. All missense mutations were found in treatment-naïve samples. The S349F, G762E, and P1132L substitutions were predicted by SIFT to disrupt protein function. When modeled into the crystal structure of GluN2A, the G762E substitution was seen to potentially alter GluN1–GluN2A interactions and ligand binding, implying disruption to the NMDAR functionality. Patients whose tumors carried non-synonymous mutations in GRIN2A had faster disease progression and shorter overall survival. Our findings suggest for the first time, to our knowledge, that GRIN2A mutations may drive melanoma progression.
Our results are in agreement with the seminal whole-exome sequencing work, where non-synonymous mutations in GRIN2A were found in 26% of melanoma samples (3). Other exome-wide sequencing projects detected GRIN2A mutations at lower frequencies (4–7). The true frequency of GRIN2A mutations in melanoma remains uncertain, but our results support the high prevalence of GRIN2A mutations in metastatic melanoma (3). Possible causes for differences between studies include the stage of tumors tested, or biological heterogeneity of tumors influenced by demographic, geographical, or environmental factors. Our study demonstrates for the first time that patients with GRIN2A mutations may have more aggressive disease. Considering that there is currently no reliable genetic biomarker that predicts melanoma progression, GRIN2A mutation testing may offer valuable prognostic information. Earlier detection of highly aggressive tumors could assist faster introduction of new therapies for melanoma patients. We propose that the GRIN2A mutation testing be incorporated in larger prospective studies for further evaluation of our findings.
Our work has obvious limitations. Patient numbers are small, and the clinical outcome was assessed retrospectively. We did not have access to non-diseased patient DNA to exclude germline polymorphisms (SNPs were excluded using online databases). Original tumor tissue is no longer available to confirm that these contained GRIN2A mutations found in cell lines, but an error in cell line authentication is extremely unlikely. Short tandem repeat profiling has been conducted on NZM cell lines to ensure authentication is possible in the future; however, profiles of original tumors are not available. Small sample size limits our conclusions, and confounders cannot be excluded. Nevertheless, the BRAF mutation status and systemic therapy had no effect on clinical outcome in these patients. Other possible limitations include the relatively late stage of patients at presentation and the selection of cell lines tested. Our success rate of establishing melanoma cell lines is at least 90%, but the possibility of bias toward melanomas that can be grown in culture cannot be ruled out.
Synonymous mutations in GRIN2A associated with poorer patient outcome; however, these observations were not statistically significant. Recent studies indicate that synonymous mutations may be important in cancer, primarily through mechanisms that affect RNA processing and protein translation (27, 28). Further work in this area will be required to clarify if synonymous GRIN2A mutations play a role in melanoma biology.
Our results have strong implications for basic research. The roles of NMDAR-mediated pathways in melanoma are still unknown and will require elucidation. Well-characterized melanoma cell lines with known mutations, such as those described in this manuscript, will be valuable tools to examine the mechanisms of action and consequences of specific GRIN2A mutations in melanoma tumors. We hypothesize that possible mechanisms through which G762E and other GRIN2A mutations interfere with the NMDAR include reduced NMDAR channel function and disturbed intracellular signaling downstream. Such effects would be most relevant under conditions of NMDAR overactivation, where excessive calcium uptake induces cell toxicity. The lack of NMDAR-mediated cell death could facilitate tumor progression. Our hypothesis is consistent with the previously suggested role for the NMDAR as a tumor suppressor (29). Other GluN subunits (if expressed in melanoma cells) could compensate for the GluN2A disruption or contribute additional functionality. NMDAR-mediated pro-cell-survival signaling could also provide oncogenic effects, in keeping with the functional dichotomy of the NMDAR (11).
In conclusion, our study suggests that non-synonymous mutations in GRIN2A are present in approximately 20% of patients with metastatic melanoma and associate with faster disease progression and shorter overall survival. The most direct clinical implication of our work is that GRIN2A mutation status may allow earlier detection and hence faster treatments of patients with aggressive tumors. Our data also imply that NMDAR may be a novel molecular modifier in melanoma; hence, further studies into its biological role should be pursued.
Author Contributions
Stacey Ann N. D’mello conducted experimental work, analyzed data, and wrote the manuscript together with Maggie L. Kalev-Zylinska; Jack U. Flanagan supervised structural modeling of mutation impact; Taryn N. Green, Euphemia Y. Leung, Marjan E. Askarian-Amiri, and Wayne R. Joseph provided advice and assisted experimental procedures; Michael R. McCrystal provided clinical advice; Richard J. Isaacs, James H. F. Shaw, and Christopher E. Furneaux contributed patient samples; Matthew J. During provided mentorship and advice; Graeme J. Finlay provided supervision and edited the manuscript; Bruce C. Baguley helped to design the study, provided supervision, and advice; Maggie L. Kalev-Zylinska designed the study, obtained and analyzed clinical data, provided supervision, and wrote the manuscript together with Stacey Ann N. D’mello.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sequencing was performed by Genetic Analysis Services at the University of Otago. Dr. Donald R. Love facilitated additional BRAF mutation testing at LabPlus, Auckland District Health Board. Alexander Trevarton advised on bioinformatics approaches and Nicholas Knowlton on statistical analysis. Funding: This study was supported by Auckland Medical Research Foundation (UOA3700909); Cancer Society (UOA3702269) and Glenn Family Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fonc.2013.00333/abstract
Figure S1. Summary of disease progression events for patients with no, synonymous, and non-synonymous mutations in GRIN2A. Data points for individual patients are shown; horizontal lines mark median values in each group. Levels of statistical difference between groups are shown. *Disease progression data for one patient with non-mutated GRIN2A were not available.
Table S1. Summary of NZM cell lines tested in this study, together with their mutation status and systemic therapy received by patients prior to cell line establishment.
Footnotes
References
1. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell (2012) 150:251–63. doi: 10.1016/j.cell.2012.06.024
2. Jang S, Atkins MB. Which drug, and when, for patients with BRAF- mutant melanoma? Lancet Oncol (2013) 14:e60–9. doi:10.1016/S1470-2045(12)70539-9
3. Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet (2011) 43:442–6. doi:10.1038/ng.810
4. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature (2012) 485:502–6. doi:10.1038/nature11071
5. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet (2012) 44:1006–14. doi:10.1038/ng.2359
6. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet (2012) 44:133–9. doi:10.1038/ng.1026
7. Stark MS, Woods SL, Gartside MG, Bonazzi VF, Dutton-Regester K, Aoude LG, et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat Genet (2012) 44:165–9. doi:10.1038/ng.1041
8. Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology (2009) 56:2–5. doi:10.1016/j.neuropharm.2008.06.063
9. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev (2010) 62:405–96. doi:10.1124/pr.109.002451
10. Li L, Hanahan D. Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell (2013) 153:86–100. doi:10.1016/j.cell.2013.02.051
11. North WG, Gao G, Memoli VA, Pang RH, Lynch L. Breast cancer expresses functional NMDA receptors. Breast Cancer Res Treat (2010) 122:307–14. doi:10.1007/s10549-009-0556-1
12. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch (2010) 460:525–42. doi:10.1007/s00424-010-0809-1
13. Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem (2002) 80:239–54. doi:10.1046/j.0022-3042.2001.00699.x
14. Hoogduijn MJ, Hitchcock IS, Smit NP, Gillbro JM, Schallreuter KU, Genever PG. Glutamate receptors on human melanocytes regulate the expression of MiTF. Pigment Cell Res (2006) 19:58–67. doi:10.1111/j.1600-0749.2005.00284.x
15. Song Z, He C-D, Liu J, Sun C, Lu P, Li L, et al. Blocking glutamate-mediated signalling inhibits human melanoma growth and migration. Exp Dermatol (2012) 21:926–31. doi:10.1111/exd.12048
16. Marshall ES, Matthews JH, Shaw JH, Nixon J, Tumewu P, Finlay GJ, et al. Radiosensitivity of new and established human melanoma cell lines: comparison of [3H]thymidine incorporation and soft agar clonogenic assays. Eur J Cancer (1994) 30A:1370–6. doi:10.1016/0959-8049(94)90188-0
17. Kim JE, Stones C, Joseph W, Leung E, Finlay G, Shelling A, et al. Comparison of growth factor signalling pathway utilisation in cultured normal melanocytes and melanoma cell lines. BMC Cancer (2012) 12:141. doi:10.1186/1471-2407-12-141
18. Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet (2006) 7:61–80. doi:10.1146/annurev.genom.7.080505.115630
19. Trevarton AJ, Mann MB, Knapp C, Araki H, Wren JD, Stones-Havas S, et al. MelanomaDB: a web tool for integrative analysis of melanoma genomic information to identify disease-associated molecular pathways. Front Oncol (2013) 3:184. doi:10.3389/fonc.2013.00184
21. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol (1993) 234:779–815. doi:10.1006/jmbi.1993.1626
22. Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature (2005) 438:185–92. doi:10.1038/nature04089
23. Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans (2009) 37:1147–60. doi:10.1042/BST0371147
24. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol (2009) 27:6199–206. doi:10.1200/JCO.2009.23.4799
26. Erreger K, Geballe MT, Kristensen A, Chen PE, Hansen KB, Lee CJ, et al. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-d-aspartate glutamate receptors. Mol Pharmacol (2007) 72:907–20. doi:10.1124/mol.107.037333
27. Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet (2011) 12:683–91. doi:10.1038/nrg3051
28. Gartner JJ, Parker SC, Prickett TD, Dutton-Regester K, Stitzel ML, Lin JC, et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc Natl Acad Sci U S A (2013) 110(33):13481–6. doi:10.1073/pnas.1304227110
Keywords: melanoma, GRIN2A, mutation, GluN2A, NMDAR, NMDA receptor, glutamate, prognosis
Citation: D’mello SAN, Flanagan JU, Green TN, Leung EY, Askarian-Amiri ME, Joseph WR, McCrystal MR, Isaacs RJ, Shaw JHF, Furneaux CE, During MJ, Finlay GJ, Baguley BC and Kalev-Zylinska ML (2014) Evidence that GRIN2A mutations in melanoma correlate with decreased survival. Front. Oncol. 3:333. doi: 10.3389/fonc.2013.00333
Received: 02 November 2013; Accepted: 30 December 2013;
Published online: 13 January 2014.
Edited by:
Sven Bilke, National Institutes of Health, USAReviewed by:
Josh Waterfall, National Institutes of Health, USAAnna DeFazio, University of Sydney at Westmead Millennium Institute, Australia
Copyright: © 2014 D’mello, Flanagan, Green, Leung, Askarian-Amiri, Joseph, McCrystal, Isaacs, Shaw, Furneaux, During, Finlay, Baguley and Kalev-Zylinska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maggie L. Kalev-Zylinska, Department of Molecular Medicine and Pathology, University of Auckland, 85 Park Road, Grafton, Auckland, ACM 1142, New Zealand e-mail:bS5rYWxldkBhdWNrbGFuZC5hYy5ueg==