 Laura Zinzi
Laura Zinzi Elena Capparelli
Elena Capparelli Mariangela Cantore
Mariangela Cantore Marialessandra Contino
Marialessandra Contino Marcello Leopoldo
Marcello Leopoldo Nicola Antonio Colabufo*
Nicola Antonio Colabufo*- Dipartimento di Farmacia – Scienze del Farmaco, Università degli Studi di Bari “A. Moro”, Bari, Italy
Multidrug resistance (MDR) is a complex phenomenon principally due to the overexpression of some transmembrane proteins belonging to the ATP binding cassette (ABC) transporter family. Among these transporters, P-glycoprotein (P-gp) is mostly involved in MDR and its overexpression is the major cause of cancer therapy failure. The classical approach used to overcome MDR is the co-administration of a P-gp inhibitor and the classic antineoplastic drugs, although the results were often unsatisfactory. Different classes of P-gp ligands have been developed and, among them, Tariquidar has been extensively studied both in vitro and in vivo. Although Tariquidar has been considered for several years as the lead compound for the development of P-gp inhibitors, recent studies demonstrated it to be a substrate and inhibitor, in a dose-dependent manner. Moreover, Tariquidar structure–activity relationship studies were difficult to carry out because of the complexity of the structure that does not allow establishing the role of each moiety for P-gp activity. For this purpose, SMALL molecules bearing different scaffolds such as tetralin, biphenyl, arylthiazole, furoxane, furazan have been developed. Many of these ligands have been tested both in in vitro assays and in in vivo PET studies. These preliminary evaluations lead to obtain a library of P-gp interacting agents useful to conjugate chemotherapeutic agents displaying reduced pharmacological activity and appropriate small molecules. These molecules could get over the limits due to the antineoplastic-P-gp inhibitor co-administration since pharmacokinetic and pharmacodynamic profiles are related to a dual innovative drug.
Introduction
Human ATP binding cassette (ABC) transporters belong to a family of 49 genes classified in seven subfamilies (A–G) (1, 2).
Some of these transporters are involved in multidrug resistance (MDR) such as ABC-B1 (P-glycoprotein, P-gp), ABC-G2 (breast cancer resistance protein, BCRP), and ABC-C1-6 (MDR associated proteins, MRP1-6) (3).
Multidrug resistance is a complex phenomenon that limits the efficacy of chemotherapeutic treatment. Some tumors are intrinsically resistant to pharmacological therapy, while others, initially sensitive to chemotherapy, become resistant during the treatment. Resistance to anticancer drugs is due to several factors such as pharmacokinetic, tumor micro-environmental changes, or cancer cell-specific factors that occur at different levels:
– increased drug efflux or decreased drug influx;
– drug inactivation;
– drug target modification;
– apoptosis evasion.
The first of these mechanisms is mediated by plasma membrane transporters such as P-gp.
Several strategies were suggested for reversing MDR and, among them, the co-administration of anticancer drugs with an ABC transporter inhibitor has been proposed to improve the bioavailability of chemotherapeutic agents (4, 5).
Among MDR pumps, P-gp is one of the most studied because of the broadest substrate specificity and the widest tissues and organs distribution such as liver, intestine, brain, and kidneys (6). This transporter actively effluxes several compounds from cells and, being overexpressed in tumor cells exerting a significant effect on the bioavailability, distribution, and activity of many drugs, especially those used in the cancer treatment (7).
P-glycoprotein is a 170-kDa phosphorylated glycoprotein encoded by MDR1 gene. Structurally, P-gp contains 12 transmembrane helices organized in 2 membrane spanning domains (MSDs), each containing 6 transmembrane helices and 2 nucleotide-binding domains (NBDs) responsible for ATP binding (3, 8) (Figure 1).
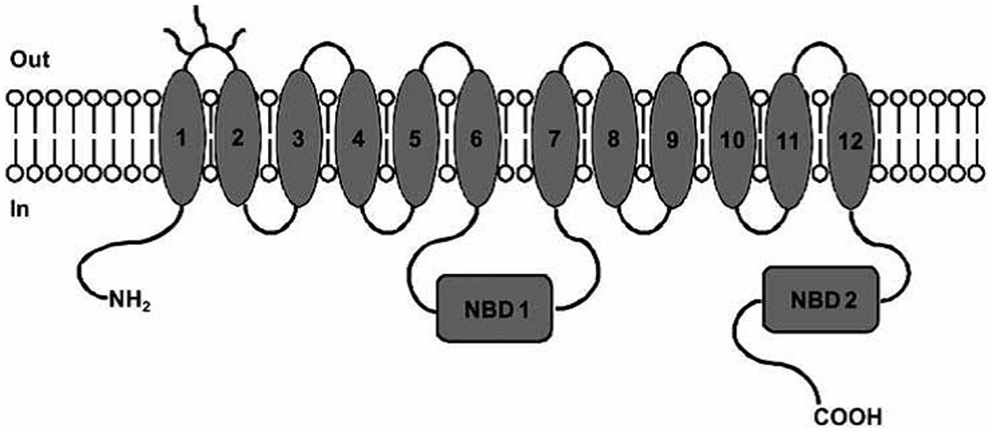
Figure 1. P-gp structure: MSDs and NBDs. Picture reported in Ref. (6).
This protein uses ATP hydrolysis as the energy source for the translocation of several structurally unrelated molecules (9). This suggests the presence of different binding sites (10, 11). Indeed, four distinct interacting binding sites have been identified in P-gp structure (Figure 2). Sites I–II are assigned for the binding of substrates, site III is for the modulators, and site IV binds the inhibitors. It has been hypothesized that the binding site of inhibitors is folded to inhibit the ATP binding and so the pump, although binds the substrate, cannot extrude it. The four binding sites are able to allosterically communicate in a negative heterotropic manner and the binding to one of these sites switches the other sites to a low-affinity conformation (9).
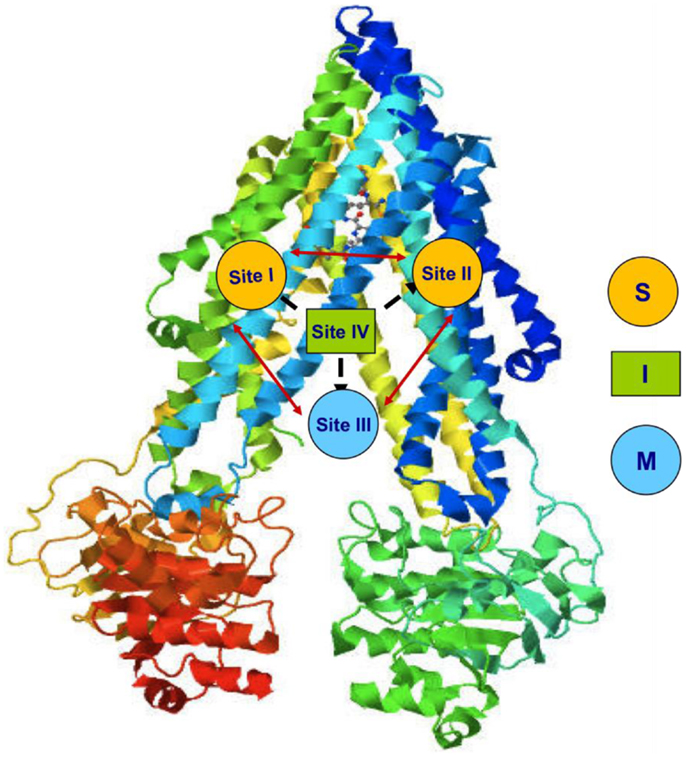
Figure 2. P-gp binding sites (S: substrate, M: modulator, I: inhibitor).
Several models have been proposed for P-gp efflux: (1) pore, (2) flippase, (3) hydrophobic vacuum cleaner, and (4) two-cylinder engine (12, 13).
In the pore model, drugs binding P-gp to the cytosol are transported out of cells through a channel created by protein.
In the flippase model, P-gp links the drugs that are transported from the inner to the outer compartment of the plasma membrane against a concentration gradient.
In the hydrophobic vacuum cleaner model, molecules, recognized by P-gp in the lipid bilayer, enter into the protein from the membranous site and exit through the central cavity.
In the two-cylinder engine model, it has been hypothesized that P-gp contains two drug-binding sites, in which each half-transporter has its own drug carrier (14).
The translocation mechanism of P-gp was blocked by inhibitors activity (12).
Indeed, the initial step of the translocation process is the binding of drugs to an high-affinity site and simultaneously the binding of ATP to the NBDs. Drug and ATP binding are coupled to the ATP hydrolysis and two ATP molecules are needed for the turnover; the first molecule is responsible for drug translocation and the second is needed to set the transporter in the basal state (Figure 3).
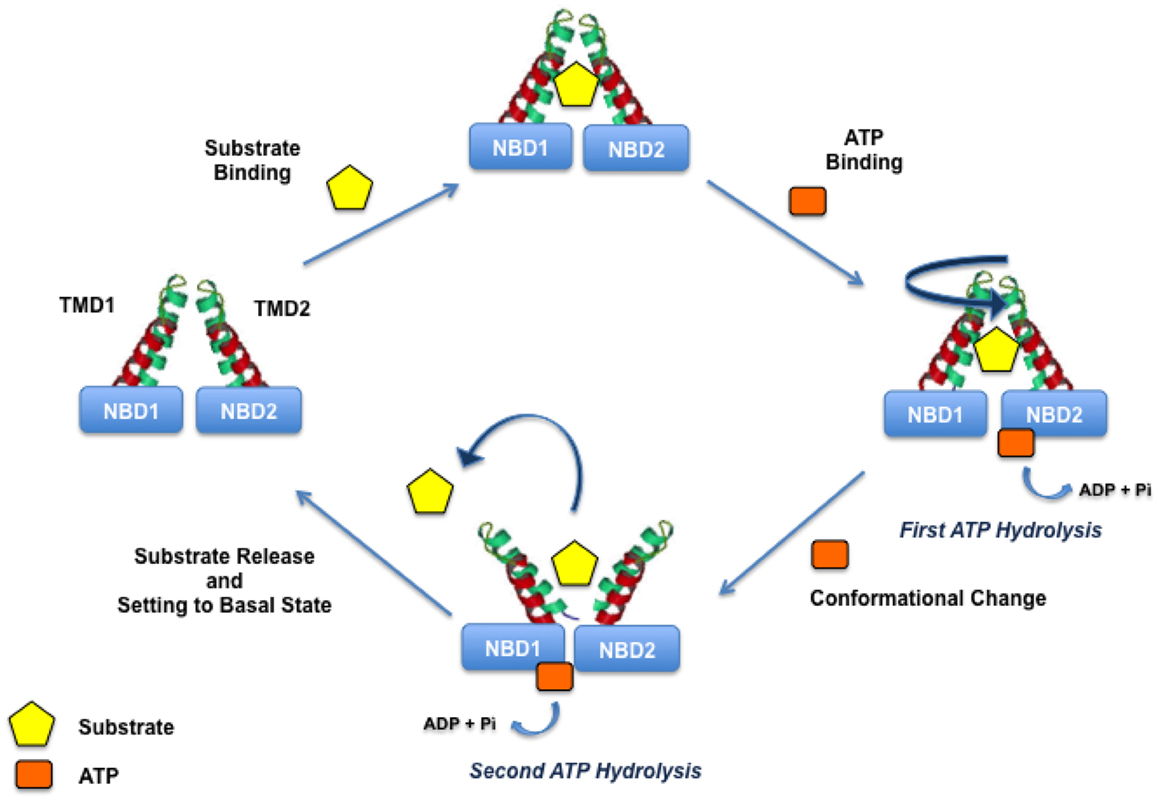
Figure 3. P-gp translocation mechanism.
Compounds interacting with P-gp have been classified into three categories: substrates, inhibitors, and modulators (Figure 4).
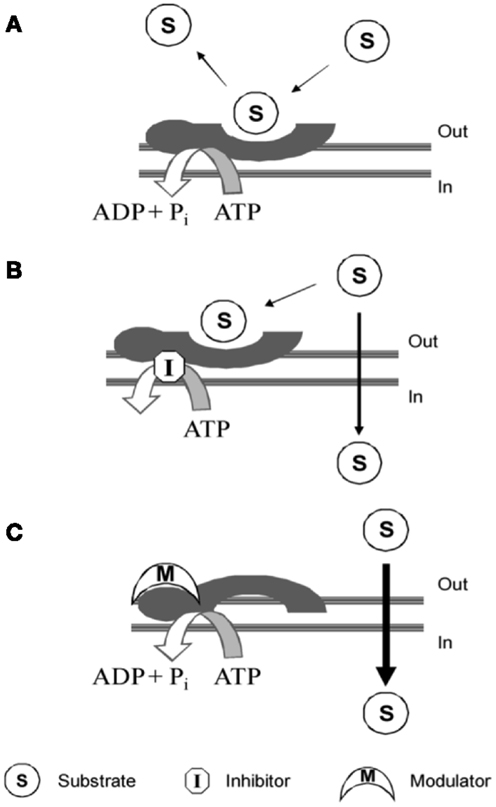
Figure 4. P-gp interacting mechanism of: (A) substrate; (B) inhibitor; (C) modulator. Picture reported in Journal of Medicinal Chemistry (2010) 53:1883–97.
Substrates are molecules that are actively transported by the protein and therefore have a higher concentration outside the cell with respect to the cytosol (10).
High substrate concentration causes a block of the pump by saturating the substrate-binding sites and in literature, this finding led to a mistake in terms of intrinsic P-gp-interacting mechanism (10).
Modulators modify substrate-binding site through a negative allosteric mechanism. Imaging studies with radiotracers demonstrated that modulators are able to alter the substrate-binding site in a non-competitive manner, modifying the maximal receptor density (Bmax) but not the dissociation equilibrium constant (Kd). Therefore, it suggests that allosteric communication between substrate- and modulator-binding sites exists (10).
Inhibitors block the translocation activity of P-gp by interfering with the ATP binding to NBD. However, although different mechanisms, substrates, modulators, and inhibitors could exert the same final biological effect restoring cell sensitivity to chemotherapeutic agents.
Biological Assays
The characterization of P-gp-interacting mechanism of drugs is an important task in the development of P-gp ligands and it is performed by specific biological in vitro assays (15) (Figure 5).
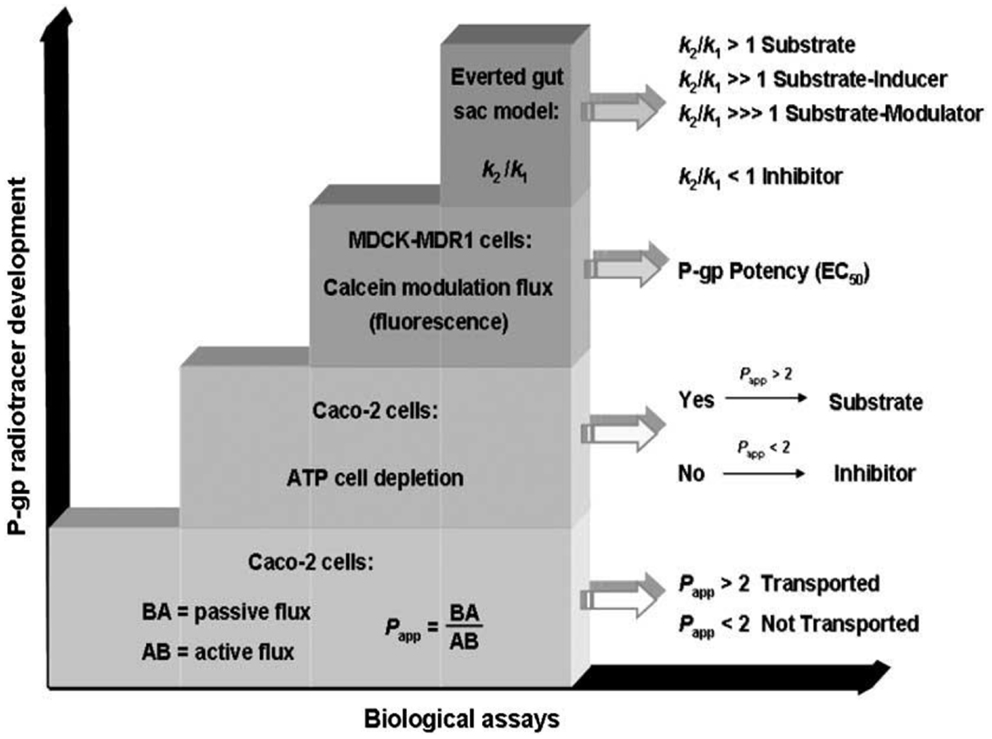
Figure 5. Characterization of P-gp ligands (k1: influx constant; k2: efflux constant). Picture reported in Ref. (16).
A wide range of methodologies has been used to characterize the P-gp interaction. These methods employ intact cells or purified protein and a combination of different approaches is often required to identify the mechanism of interaction.
The identification of the P-gp-interacting mechanism is performed by the combination of three biological assays:
– determination of the apparent permeability (Papp);
– ATP cell depletion;
– inhibition of the P-gp-mediated transport of a fluorescent probe (Calcein or Rhodamine);
– everted gut sac model.
Apparent Permeability Determination
Apparent permeability (Papp) is a pharmacokinetic parameter that is determined in Caco-2 cells system, a cell monolayer model suitable for the study of the passive and active transport through the biological membranes. Indeed, in this system the Basolateral–Apical flux (B → A), representative of passive diffusion, and Apical–Basolateral flux (A → B), representative of active P-gp-modulated transport, are determined.
The BA/AB ratio is useful to identify P-gp inhibitors (BA/AB <2), P-gp substrates (BA/AB from 18 to 20), or P-gp modulators (BA/AB ranging from 2 to 18).
ATP Cell Depletion
This assay, performed in Caco-2 cell monolayer and in Madin–Darby Canine Kidney cells (MDCK) overexpressing P-gp, permits to establish if the compound is able to deplete ATP. Substrates activate ATPase whereas inhibitors are not transported unchanging the ATP cell level.
Inhibition of Calcein-AM Transport
This assay is useful to determine the potency (EC50) of P-gp ligands and is performed in MDCK cells, stably transfected for P-gp overexpression (MDCK-MDR1). The assay is carried out using a non-fluorescent prodrug, the acetoxymethyl ester of calcein (calcein-AM), which is a P-gp substrate. In the presence of a P-gp modulator, calcein-AM diffuses into the cytosol where it is hydrolyzed to the fluorescent dye calcein, that is not a P-gp substrate and since hydrophilic, it cannot diffuse through the membrane (17). In this assay also Rhodamine may be employed as a probe although calcein-AM is more useful because Rhodamine displays good cell permeability and therefore, its fluorescence determination at stationary state is more complex than calcein-AM.
The Everted Gut Sac Model
This assay is an ex vivo method to study the P-gp-mediated intestinal absorption of drugs and their interactions with CYP450 enzymes (18, 19). This double information (the effect of P-gp-mediated transport and CYP450-metabolizing activity) is obtained since the everted gut sac assay is performed on isolated rat ileum where CYP450 enzymes and P-gp are present.
This combined study is needed because inhibitors and substrates may display overlapping activities toward CYP450 enzymes and the P-gp pump (20).
In this method, the transport of a known P-gp radiolabeled or fluorescent substrate, in the absence and presence of a P-gp-interacting agent, is evaluated. The flux of a P-gp substrate such as Rhodamine 125, from serosal to mucosal compartment and vice versa, is represented by the efflux (k′2) and influx rate constants (k′1), respectively. These determinations are carried out in the presence of a P-gp-interacting agent to determine k″2 and k″1, the efflux and influx constants of the tested substrate after P-gp interaction.
P-gp Substrates, Modulators, Inhibitors
The most important studied P-gp ligands are classified in three different categories.
P-gp Substrates
This class is the most extensively studied and Verapamil and N-desmethyl-loperamide (Chart 1) are to date the gold standard of this class of compounds.
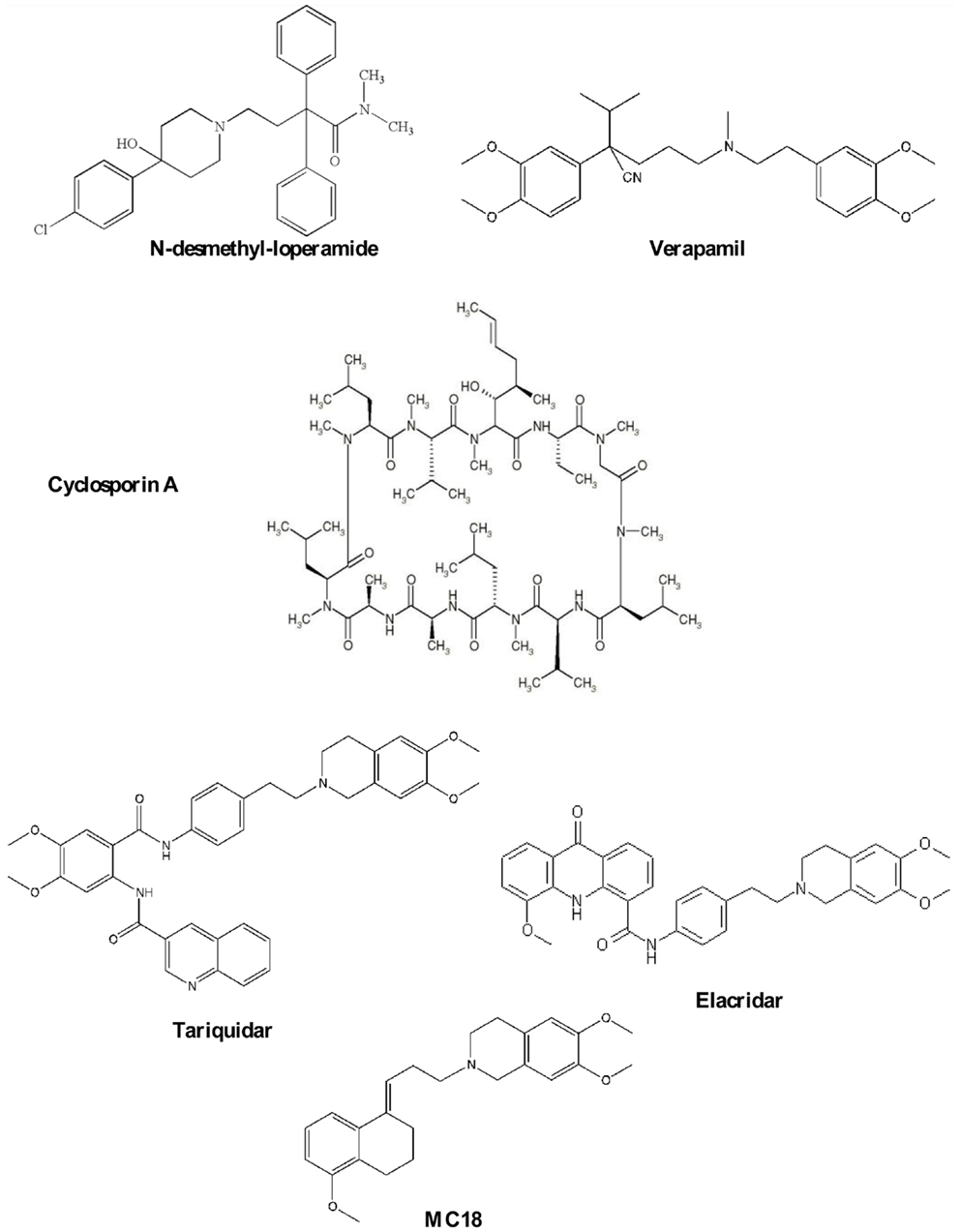
Chart 1. P-gp ligands.
Verapamil, a calcium channel blocker, was found to reverse MDR (21) and it has become the reference compound for developing other P-gp substrates. It saturates the pump at high doses and therefore, it is a potential ligand for reversing MDR in co-administration with antineoplastic agent for different types of cancer (21, 22). However, verapamil cannot be employed because of toxic cardiovascular side effects. Despite this, the radiolabeled compound, 11C-verapamil, has been developed to visualize P-gp function and to date, it is considered to be the reference substrate for imaging P-gp activity (23). However, verapamil is quickly metabolized by CYP450 enzymes giving radiometabolites, some of which are themselves P-gp substrates (24).
N-desmethyl-loperamide (dLop) is the major metabolite of Loperamide and at low concentrations it acts as substrate while at high concentrations, as reported for verapamil, dLop saturates the pump (25). Also, dLop has been radiolabeled and used for imaging P-gp in vivo by PET analysis (26).
P-gp Modulators
Cyclosporin A (CsA, Chart 1), an immunosuppressant agent, is a P-gp modulator, widely used in vitro as a tool to study MDR because it restores the cell concentration of chemotherapeutic agents. In imaging studies, the co-administration of CsA with a radiolabeled P-gp substrate (27) has been performed to visualize the P-gp activity because it increases radiotracer cell uptake by modulating the P-gp-binding sites. However, CsA treatment enhances the uptake of the radioligand in all regions where P-gp is present including targeted and non-targeted tissues (2–15, 17–30).
P-gp Inhibitors
Elacridar (Chart 1) is a dual P-gp/BCRP ligand and can be orally administered. It was tested in combination with doxorubicin in patients with advanced solid tumors (31). At the recommended dose of doxorubicin, a pharmacologic hematologic toxicity was observed, mainly consisting of leukocytopenia and granulocytopenia.
Moreover, Elacridar was co-administrated with topotecan (32), a P-gp and BCRP substrate (Phase I) with unsatisfactory results (33). 11C-Elacridar is tested in vivo to evaluate the overexpression of P-gp and BCRP in human colon adenocarcinoma (33, 34).
Tariquidar (Chart 1), an anthranilic derivative, is the most potent P-gp ligand in nanomolar range. It has been co-administrated in clinical trials with chemotherapeutic agents for restoring the efficacy of therapy (35–38). Results were quite unsatisfactory because of poor selectivity against other ABC transporters that are not involved in MDR. Tariquidar has been evaluated in vivo for diagnosing breast tumors in animal model using (R)-11C-verapamil (35, 39, 40).
Recently, the suitability of 11C-tariquidar and 11C-elacridar for visualizing cerebral P-gp expression in healthy human subjects, in analogy to a previous preclinical study (16, 35), was investigated. However, 11C-tariquidar and 11C-elacridar displayed a “substrate-like in vivo behavior”; in particular, they are dual P-gp/BCRP substrates and these findings disagreed with in vitro results (41).
(E)-6,7-Dimethoxy-2-[3-(5-methoxy-3,4-dihydronaphthalen-1(2H)-ylidene)propyl]-1,2,3,4-tetrahydroisoquinoline, better known as MC18 (Chart 1), is a small molecule bearing tetralin moiety (42). To date, 11C-MC18 is the first P-gp inhibitor studied in vivo in PET studies. It displayed fourfold higher uptake in the target organs compared with 11C-tariquidar and 11C-elacridar (43).
Strategies to Revert MDR
The pivotal role of P-gp in MDR has stimulated the development of P-gp ligands able to reverse the resistance to a wide number of drugs. Hence, the need to design potent and selective P-gp inhibitors stimulated the development of small molecules on which structure–activity relationship (SAR) studies could be easily and better performed. The development of these compounds is depicted in Figure 6 and it is based on the synthesis of bioisosteres obtained through subsequent lead optimization studies.
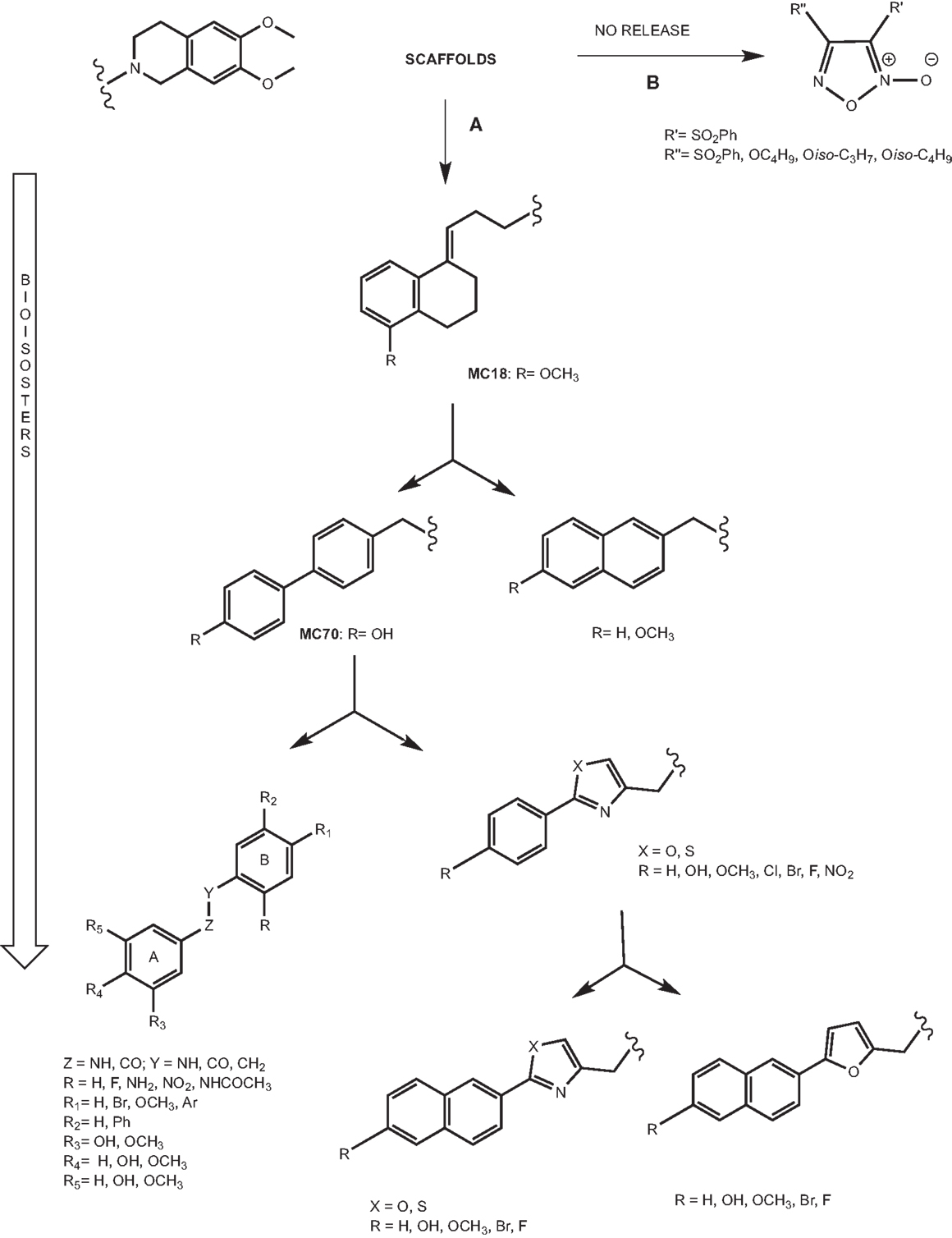
Figure 6. Scaffolds of versatile libraries. (A) Bioisosteric approach; (B) NO release ligands.
Small Libraries from Versatile Scaffolds
Tetralin derivatives
The lead compound of this class is MC18 (Figure 6A) (EC50 = 1.50 μM), bearing an (E)-double bond, a potent P-gp inhibitor (42). When the double bond shifts into the tetralin ring, the ligands are less potent than MC18 and are P-gp substrates. Moreover, the presence and the position of methoxy substituent on tetralin nucleus are important in terms of the potency and intrinsic activity. The saturated derivative, MC266 (EC50 = 6.35 μM), was the best P-gp substrate in this class. Therefore, the partial conformational restriction of spacer is involved in the P-gp-interacting mechanism. These two lead compounds, inhibitor and substrate respectively, have been 11C-radiolabeled and tested in vivo PET studies leading to significant and coherent results in comparison with the in vitro data previously reported (43).
Biphenyl and naphthyl derivatives
The conformational restriction of MC18 seems to be a requirement for improving P-gp-inhibitory activity. In order to evaluate this statement, the restriction of the spacer linking the non-basic moiety was tested in a series of molecules bearing two different fragments: 1,4-biphenyl and 2-naphthyl moieties (Figure 6A) (44).
In the biphenyl series, 4-biphenyl derivatives displayed the best activity in P-gp-inhibitory activity and among these compounds, the best result was obtained for MC70 (EC50 = 0.69 μM). In recent years, MC70 was extensively studied in order to confirm its P-gp-interacting activity considering its role to enhance the chemotherapeutic agent when co-administrated (45).
Although tetralin and biphenyl derivatives displayed high P-gp activity, they were active toward other ABC transporters such as BCRP and MRP1.
Aryloxazole and arylthiazole derivatives
Aryloxazole and arylthiazole derivatives (Figure 6A) were designed as cycloisosters to improve the P-gp-inhibitory activity and selectivity. The results demonstrated that aryloxazole and arylthiazole derivatives, designed as cycloisosteres of biphenyl derivative MC70, were found to be less potent than the reference compound in inhibiting P-gp (46). Indeed, these compounds were screened by SAR studies toward BCRP and MRP1 giving interesting structural determinants for these pumps as depicted (Figure 7). Finally, it was found that both aryloxazoles and arylthiazoles were P-gp substrates.
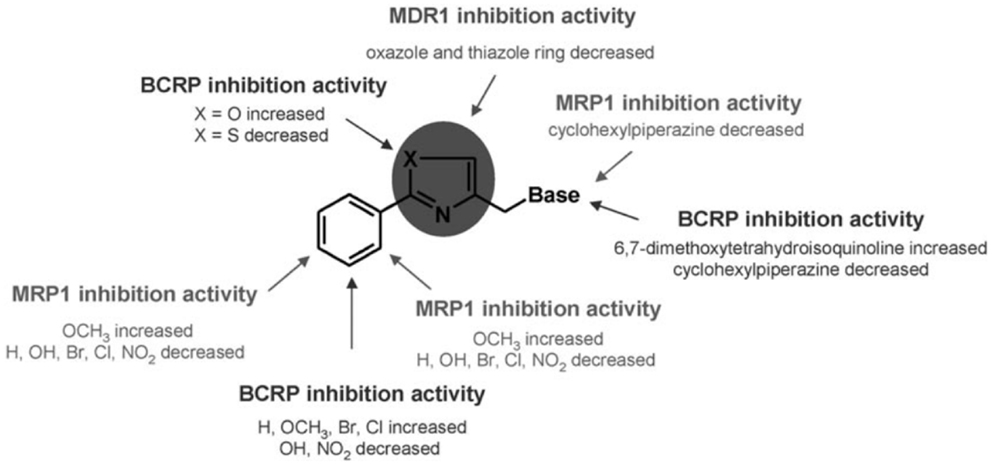
Figure 7. SAR studies of aryloxazole and arylthiazole derivatives. Picture reported in Ref. (47).
Furthermore, the aryl fragments were replaced by a naphthyl nucleus and three heteronuclei (oxazole, thiazole, and furyl) have been evaluated (47). The obtained results showed that the replacement of aryl nucleus with naphthyl moiety lead to obtain compounds with three different activity profiles:
1. P-glycoprotein inhibitors: unsubstituted oxazoles or bearing −F and −OH on the naphthyl fragment;
2. Unambiguous substrates (48): oxazole bearing −OCH3 on the naphthyl fragment and thiazole bearing −Br and −OCH3 on the naphthyl fragment.
3. Ambiguous substrates (48): oxazole bearing Br on the naphthyl fragment and unsubstituted thiazole or bearing Br on the naphthyl fragment.
Finally, all furyl derivatives were ambiguous substrates.
Galloyl-based derivatives
The 3,4,5-trihydroxybenzoyl and 3,4- and 3,5-dihydroxybenzoyl fragments (Figure 6A) have been employed as scaffolds for a set of ligands that are representative of pharmacophoric nucleus of tariquidar (49).
For this purpose, compounds have been divided into four different structural series that present:
1. variation at R,
2. variation at R1,
3. polyhydroxy derivatives,
4. pyrogallol 1-methyl ethers.
The benzamides of the first set showed good P-gp-inhibitory activity (IC50 ranging from 20 to 1.4 μM). These outcomes led to deepen the study with two approaches:
1. keeping fixed the 3,4,5-trimethoxy-N-(2-nitrophenyl)benzamide scaffold and introducing a series of R1 substituents in the 4-position of the aniline moiety;
2. desmethylating one or more methoxy groups belonging to A ring.
The evaluation of data suggested that, with the exception of the 4-bromo and 4-methoxy congeners, all compounds of this second set were potent and selective P-gp inhibitors. In particular, molecules bearing R = NO2 and in R1 H or benzo[1,3]dioxol-5-yl displayed submicromolar activity.
The screening of the gallamide derivatives indicated a moderate inhibitory potency for P-gp, independently with respect to the number and position of phenolic groups.
In the last series, all the pyrogallol-1-monomethyl ether derivatives showed moderate P-gp-inhibitory activity.
Taking into account that the most important inhibitory activity changes were mainly concerned with the structural modifications on B ring, the role of the amide function was evaluated by testing the corresponding anilide and amine. The amine was equipotent with respect to the other tested compounds toward P-gp, despite the drastic change in terms of planarity and conformational flexibility. By contrast, the anilide showed no inhibitory activity toward P-gp. The 3,4,5-trimethoxyamide derivatives displayed moderate inhibitory activity toward P-gp.
Furoxan derivatives
Another strategy to reverse MDR is the nitration of a tyrosine present in TM6 domain of P-gp (50). It was reported that furoxans are able to produce in situ NO interacting with a thiol group. In fact, furoxans (Figure 6B) have been developed on the base of their properties to induce NO release and this pharmacological effect is the mechanism of coronary dilators (51–53). Moreover, the correlation between a decreased NO synthesis and MDR onset in doxorubicin-sensitive and doxorubicin-resistant cells has been widely reported (54). For this reason, a series of furoxan derivatives was designed and tested in activity and selectivity toward ABC transporters. Firstly, diphenylfuroxan derivatives and 3- and 4-phenylfuroxan isomer pairs, bearing different substituents with stereo-selective and lipophilic properties, were evaluated. In particular, the compounds having electron-withdrawing substituent and high lipophilic group such as phenylsulfonyl, displayed the best P-gp activity. The evaluation of phenylsulfonylfuroxan isomer pairs, bis(phenylsulfonyl)furoxan derivatives, and 3-phenylsulfonyl substituted furoxans, bearing alkoxy groups at position 4, displayed that 3-phenylsulfonyl substituted furoxans were found to be P-gp inhibitors, and the 4-substituted ligands showed the best activity and selectivity. Indeed, the best results were obtained both for 3,4-diphenylsulfonyl derivative (EC50 = 3.0 μM) and for alkoxy derivatives such as n-Butoxy, iso-Propoxy, and iso-Butoxy (EC50 = 2.26, 2.15, and 2.23 μM, respectively) (55).
Dual Effect Drugs
Although the co-administration of a P-gp inhibitor and an antineoplastic agent could be considered a potential strategy to revert MDR, to date this approach was not clinically available because of pharmacokinetic limitations, in particular different apparent permeability, bioavailability, and metabolism. For this reason, others approaches were taken into account in the recent past:
– use of P-gp-targeted antibodies (56);
– encapsulation of anticancer drugs in liposomes (57);
– nanospheres able to circumvent MDR (58).
New innovative approaches to revert MDR could be: the development of molecules having a dual effect, potent cancer cell-killing agent and P-gp activity/expression inhibitor, and the collateral sensitivity (CS).
For example, some taxanes inhibited P-gp activity employing Rhodamine 123 as a fluorescent dye (59). These compounds could disclose new perspectives because they not only acted as cytotoxic agents but also inhibited the activity of P-gp efflux pump.
In another study (60), starting from Indirubin (a traditional Chinese medicine), the most potent derivative, PH II-7, has been evaluated. This compound showed antitumor activity inducing itself apoptosis and S phase cell cycle arrest, and in the meantime, it is not a P-gp substrate and so, high cell concentration of this compound was detected. The confocal microscopy displayed that this compound was not P-gp effluxed and significantly increased Adriamycin and Vincristine effect by reversing MDR.
Multitarget Drugs
Another strategy could be the design of multitarget drugs bearing scaffolds depicted in Figure 6, having antitumor and P-gp inhibitory activities. An example is the hybridization of the NO-donor furoxan scaffold with the anilinopyrimidine moiety present in Gefitinib, leading to phenylsulfonylfuroxan-anilinopyrimidine derivative (Figure 8). This compound displayed epidermal growth factor receptor (EGFR) inhibitory activity in the treatment of non-small-cell lung cancer (NSCLC) (61). It induced apoptosis in H1975 and HCC827 cells, inhibited EGFR downstream signaling in H1975 cells, and suppressed the nuclear factor-κB activation in H1975 cells. Furthermore, it released high levels of NO in H1975 cells but not in normal human cells, inducing apoptosis, inhibiting metastasis, and sensitizing tumor cells to chemotherapy by the inhibition of drug efflux transporters (51, 62). In this study, the two nuclei, furoxan and anilinopyrimidine, were separately evaluated for their effects. The activity showed by the two nuclei separately was lower than that exerted by the linked molecule. These results suggest that the antiproliferative activity of the compound might be attributed to the synergic effects of anilinopyrimidine and NO-donor moieties.
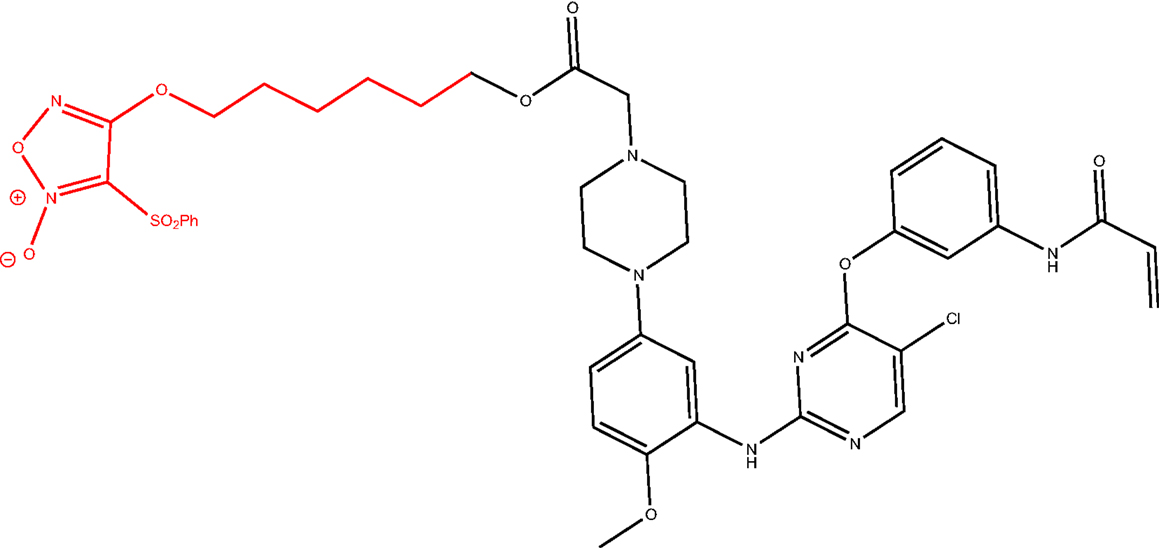
Figure 8. NO-donor furoxan moiety linked to Gefitinib-like scaffold.
In another study, a class of 4-substituted methoxybenzoylaryl-thiazoles (SMART, Figure 9) was evaluated (63). These compounds exhibited great potency in vitro and broad spectrum cellular cytotoxicity. The in vitro and in vivo evaluation of the anticancer properties of three SMART compounds demonstrated that they potently bound to the colchicine-binding site in tubulin, inhibited tubulin polymerization, arrested cancer cells in G2/M phase, and induced apoptosis. Moreover, these compounds were able to overcome MDR since they were found equally cytotoxic in a parent cell line (OVCAR-8) and in a MDR-positive cell line (NCI/ADR-RES).
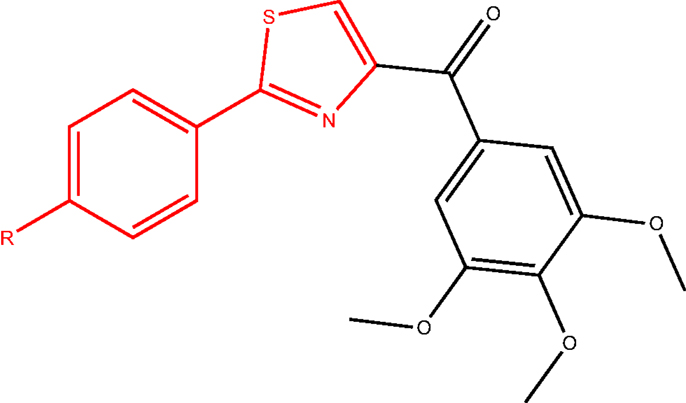
Figure 9. SMART ligand.
These findings demonstrated that some scaffolds such as furoxan and arylthiazole, already reported in our library as P-gp ligands, could be considered as the starting point to develop multitarget drugs. In these studies, the chemotherapeutic moiety is linked to furoxan (NO donor useful to revert MDR) or arylthiazole fragment (P-gp modulator), and the final effect was more potent that the single effect expected from each drug.
Collateral Sensitivity
An alternative potential approach to treat drug-resistant tumors is the CS where several compounds selectively kill MDR cells without affecting the non-resistant parental cells (64).
Different hypotheses have been proposed to better explain this mechanism:
– production of reactive oxygen species (ROS);
– energetic level changes;
– extrusion of essential endogenous substrates for cell survival;
– perturbation of cell membranes.
The first hypothesis takes into account that several CS agents are substrates of P-gp, stimulating ATPase activity and in the meantime, the substrate extrusion from the plasma membrane into the extracellular environment (65). Once back in the extracellular environment, substrates repeat this cycle and P-gp performs a process known as futile cycling to increase the ATP hydrolysis inducing oxidative stress. MDR cells initiate apoptosis when ROS levels overcome a certain limit. Two reported ligands inducing CS are siramesine and the P-gp substrate and σ2 agonist 9-[4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)butyl]-9H-carbazole that generate more ROS in the MCF7/Adr than in the MCF7 cell line (66).
Another hypothesis is that P-gp-expressing cells are more sensitive to changes in energy utilization. Indeed, several compounds that interfere with cellular metabolic pathways, such as glycolysis or oxidative phosphorylation, have been identified as MDR-selective agents. The glycolysis antimetabolite 2-deoxy-d-glucose (2-DG) seems to confirm this finding. Indeed, 2-DG activates apoptosis and selectively kills numerous MDR cell lines compared to drug-sensitive parental lines.
The extrusion hypothesis asserts that CS agents mediate cytotoxicity by stimulating, sensitizing, or facilitating the extrusion of endogenous essential components. This phenomenon is not reported in P-gp-expressing cells but it may be the case for MPR1-mediated CS.
Moreover, several CS agents alter membrane biophysical properties (67). Indeed, they induce membrane perturbation in P-gp-expressing cell lines, leading to the hypothesis that changes in membrane structure and fluidity contribute to CS (68). Pentazocine and verapamil are reported to reduce membrane fluidity in the colchicine-resistant B30 cell line.
Conclusion
To date the co-administration of a chemotherapeutic drug with a P-gp inhibitor, the encapsulation of anticancer drugs in liposomes, and the nanosphere formulation to reverse MDR failed for several reasons. This review aims to disclose new strategies in the design of multitarget drugs useful toward MDR. Interesting approaches are: (i) the development of drugs bearing a unique moiety responsible for the anticancer effect and MDR reversing activity; (ii) the design of molecules bearing different pharmacophores for multitarget activity; (iii) the evaluation of CS agents.
In the present review, we overviewed our P-gp ligands library to suggest new scaffolds that could be used to design multitarget drugs in accordance with the approaches already reported.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ross DD, Doyle LA. Mining our ABCs: pharmacogenomic approach for evaluating transporter function in cancer drug resistance. Cancer Cell (2004) 6:105–7. doi: 10.1016/j.ccr.2004.08.003
2. Pharm AN, Penchala S, Graf RA, Wang J, Huang Y. Multidrug Resistance: Biological and Pharmaceutical Advance in the Antitumour Treatment. Kerala: Research Signpost (2008). 19 p.
3. Gimenez-Bonafe P, Guillen Canovas A, Ambrosio S, Tortosa A, Perez-Tomas R. Multidrug Resistance: Biological and Pharmaceutical Advance in the Antitumour Treatment. Kerala: Research Signpost (2008). 63 p.
4. Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets (2006) 7:861–79. doi:10.2174/138945006777709593
5. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer (2002) 2:48–58. doi:10.1038/nrc706
6. Colabufo NA, Berardi F, Contino M, Niso M, Perrone R. ABC pumps and their role in active drug transport. Curr Top Med Chem (2009) 9:119–29. doi:10.2174/156802609787521553
7. German UA. P-glycoprotein – a mediator of multidrug resistance in tumour cells. Eur J Cancer (1996) 32A:927–44. doi:10.1016/0959-8049(96)00057-3
8. Gottesman MM, Ambudkar SV, Xia D. Structure of a multidrug transporter. Nat Biotechnol (2009) 27:546–7. doi:10.1038/nbt0609-546
9. Avendaño C, Menendez JC. Inhibitors of multidrug resistance to antitumor agents (MDR). Curr. Med Chem (2002) 9:159–93. doi:10.2174/0929867023371175
10. Martin C, Berridge G, Mistry P, Higgins C, Charlton P, Callaghan R. Drug binding sites on P-glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry (2000) 39:11901–6. doi:10.1021/bi000559b
11. Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, et al. Structure of P-glycoprotein reveals a molecular basis for polyspecific drug binding. Science (2009) 323:1718–22. doi:10.1126/science.1168750
12. Callaghan R, Ford RC, Kerr ID. The translocation mechanism of P-glycoprotein. FEBS Lett (2006) 580:1056–63. doi:10.1016/j.febslet.2005.11.083
13. Constantinides PP, Wasan KM. Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: in vitro/in vivo case studies. J Pharm Sci (2007) 96:235–48. doi:10.1002/jps.20780
14. vanVeen HW, Higgins CF, Konings WN. Multidrug transport by ATP binding cassette transporters: a proposed two-cylinder engine mechanism. Res Microbiol (2001) 152:365–74. doi:10.1016/S0923-2508(01)01208-6
15. Sharom FJ, Siarheyeva A. Multidrug Resistance: Biological and Pharmaceutical Advance in the Antitumour Treatment. Kerala: Research Signpost (2008). 261 p.
16. Colabufo NA, Berardi F, Perrone MG, Capparelli E, Cantore M, Inglese C, et al. Substrates, inhibitors and activators of P-glycoprotein: candidates for radiolabeling and imaging perspectives. Curr Top Med Chem (2010) 10:1703–14. doi:10.2174/156802610792928022
17. Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, et al. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos (2008) 36:268–75. doi:10.1124/dmd.107.017434
18. Arellano C, Philibert C, Vachoux C, Woodley J, Houin G. The metabolism of midazolam and comparison with other CYP enzyme substrates during intestinal absorption: in vitro studies with rat everted gut sacs. J Pharm Pharm Sci (2007) 10:26–36.
19. Berruet N, Sentenac S, Auchere D, Gimenez F, Farinotti R, Fernandez C. Effect of efavirenz on intestinal p-glycoprotein and hepatic p450 function in rats. J Pharm Pharm Sci (2005) 8:226–34.
20. Benet LZ, Cummins CL, Wu CY. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int J Pharm (2004) 277:3–9. doi:10.1016/j.ijpharm.2002.12.002
21. Cano-Gauci DF, Riordan JR. Action of calcium antagonists on multidrug resistant cells. Specific cytotoxicity independent of increased cancer drug accumulation. Biochem Pharmacol (1987) 36:2115–23. doi:10.1016/0006-2952(87)90139-0
22. Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. Multidrug Resistance: Biological and Pharmaceutical Advance in the Antitumour Treatment. Kerala: Research Signpost (2008). 141 p.
23. Luurtsema G, Molthoff CF, Schuit RC, Windhorst AD, Lammertsma AA, Franssen EJ. Evaluation of (R)-[11C]verapamil as PET tracer of P-glycoprotein function in the blood-brain barrier: kinetics and metabolism in the rat. Nucl Med Biol (2005) 32(1):87–93. doi:10.1016/j.nucmedbio.2004.06.007
24. Pauli-Magnus C, von Richter O, Burk O, Ziegler A, Mettang T, Eichelbaum M, et al. Characterization of the major metabolites of verapamil as substrates and inhibitors of P-glycoprotein. J Pharmacol Exp Ther (2000) 293:376–82.
25. Zoghbi SS, Liow JS, Yasuno F, Hong J, Tuan E, Lazarova N, et al. 11C-loperamide and its N-desmethylradiometabolite are avid substrates for brain permeability-glycoprotein efflux. J Nucl Med (2008) 49:649–56. doi:10.2967/jnumed.107.047308
26. Kreisl WC, Liow JS, Kimura N, Seneca N, Zoghbi SS, Morse CL, et al. P-glycoprotein function at the blood-brain barrier in humans can be quantified with the substrate radiotracer 11C-N-desmethyl-loperamide. J Nucl Med (2010) 51:559–66. doi:10.2967/jnumed.109.070151
27. Muzi M, Mankoff DA, Link JM, Shoner S, Collier AC, Sasongko L, et al. Imaging of cyclosporine inhibition of P-glycoprotein activity using 11C-verapamil in the brain: studies of healthy humans. J Nucl Med (2009) 50:1267–75. doi:10.2967/jnumed.108.059162
28. Ishiwata K, Kawamura K, Yanai K, Hendrikse NH. In vivo evaluation of P-glycoprotein modulation of 8 PET radioligands used clinically. J Nucl Med (2007) 48:81–7.
29. Syvänen S, Blomquist G, Sprycha M, Höglund AU, Roman M, Eriksson O, et al. Duration and degree of cyclosporin induced P-glycoprotein inhibition in the rat blood-brain barrier can be studied with PET. Neuroimage (2006) 32:1134–41. doi:10.1016/j.neuroimage.2006.05.047
30. Syvänen S, Hooker A, Rahman O, Wilking H, Blomquist G, Långström B, et al. Pharmacokinetics of P-glycoprotein inhibition in the rat blood-brain barrier. J Pharm Sci (2008) 97:5386–400. doi:10.1002/jps.21359
31. Planting AS, Sonneveld P, van der Gaast A, Sparreboom A, van der Burg ME, Luyten GP, et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother Pharmacol (2005) 55:91–9. doi:10.1007/s00280-004-0905-z
32. De Bruin M, Miyake K, Litman T, Robey R, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett (1999) 146:117–26. doi:10.1016/S0304-3835(99)00182-2
33. De Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res (2007) 13:6440–9. doi:10.1158/1078-0432.CCR-07-1335
34. Dörner B, Kuntner C, Bankstahl JP, Bankstahl M, Stanek J, Wanek T, et al. Synthesis and small-animal positron emission tomography evaluation of [11C]-elacridar as a radiotracer to assess the distribution of P-glycoprotein at the blood-brain barrier. J Med Chem (2009) 52:6073–82. doi:10.1021/jm900940f
35. Yamasaki T, Kawamura K, Hatori A, Yui J, Yanamoto K, Yoshida Y, et al. PET study on mice bearing human colon adenocarcinoma cells using [11C]GF120918, a dual radioligand for P-glycoprotein and breast cancer resistance protein. Nucl Med Commun (2010) 31(11):985–93. doi:10.1097/MNM.0b013e32833fbf87
36. Wanek T, Kuntner C, Bankstahl JP, Bankstahl M, Stanek J, Sauberer M, et al. A comparative small-animal PET evaluation of [11C]tariquidar, [11C]elacridar and (R)-[11C]verapamil for detection of P-glycoprotein-expressing murine breast cancer. Eur J Nucl Med Mol Imaging (2012) 39:149–59. doi:10.1007/s00259-011-1941-7
37. Stewart AJ, Mistry P, Dangerfield W, Bootle D, Baker M, Kofler B, et al. Antitumor activity of XR5944, a novel and potent topoisomerase poison. Anticancer Drugs (2001) 12:359–67. doi:10.1097/00001813-200104000-00009
38. Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D, et al. In vitro and in vivo reversal of P-glycoprotein mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res (2001) 61:749–58.
39. Stewart A, Steiner J, Mellows G, Laguda B, Norris D, Bevan P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin Cancer Res (2000) 6:4186–91.
40. Bankstahl JP, Kuntner C, Abrahim A, Karch R, Stanek J, Wanek T, et al. Tariquidar-induced P-glycoprotein inhibition at the rat blood-brain barrier studied with (R)-11C-verapamil and PET. J Nucl Med (2008) 49:1328–35. doi:10.2967/jnumed.108.051235
41. Müllauer J, Karch R, Bankstahl JP, Bankstahl M, Stanek J, Wanek T, et al. Assessment of cerebral P-glycoprotein expression and function with PET by combined [11C]inhibitor and [11C]substrate scans in rats. Nucl Med Biol (2013) 40:755–63. doi:10.1016/j.nucmedbio.2013.05.005
42. Bauer M, Karch R, Zeitlinger M, Stanek J, Philippe C, Wadsak W, et al. Interaction of 11C-tariquidar and 11C-elacridar with P-glycoprotein and breast cancer resistance protein at the human blood-brain barrier. J Nucl Med (2013) 54(8):1181–7. doi:10.2967/jnumed.112.118232
43. Colabufo NA, Berardi F, Cantore M, Perrone MG, Contino M, Inglese C, et al. Small P-gp modulating molecules: SAR studies on tetrahydroisoquinoline derivatives. Bioorg Med Chem (2008) 16:362–73. doi:10.1016/j.bmc.2007.09.039
44. Van Waarde A, Ramakrishnan NK, Rybczynska AA, Elsinga PH, Berardi F, de Jong JR, et al. Synthesis and preclinical evaluation of novel PET probes for P-glycoprotein function and expression. J Med Chem (2009) 52:4524–32. doi:10.1021/jm900485a
45. Colabufo NA, Berardi F, Cantore M, Perrone MG, Contino M, Inglese C, et al. 4-Biphenyl and 2-naphthyl substituted 6,7-dimethoxytetrahydroisoquinoline derivatives as potent P-gp modulators. Bioorg Med Chem (2008) 16:3732–43. doi:10.1016/j.bmc.2008.01.055
46. Azzariti A, Quatrale AE, Porcelli L, Colabufo NA, Cantore M, Cassano G, et al. MC70 potentiates doxorubicin efficacy in colon and breastcancer in vitro treatment. Eur J Pharmacol (2011) 670:74–84. doi:10.1016/j.ejphar.2011.08.025
47. Colabufo NA, Berardi F, Perrone MG, Cantore M, Contino M, Inglese C, et al. Multi-drug-resistancereverting agents: 2-aryloxazole and 2-arylthiazole derivatives as potent BCRP or MRP1 inhibitors. ChemMedChem (2009) 4:188–95. doi:10.1002/cmdc.200800329
48. Colabufo NA, Contino M, Cantore M, Capparelli E, Perrone MG, Cassano G, et al. Naphthalenyl derivatives for hitting P-gp/MRP1/BCRP transporters. Bio Med Chem (2013) 21:1324–32. doi:10.1016/j.bmc.2012.12.021
49. Wanek T, Kuntner C, Banksthahl JP, Stanek J, Sauberer M, Mairinger S, et al. A comparative small-animal PET evaluation of [11C]tariquidar, [11C]elacridar and (R)-[11C]verapamil for detection of P-glycoprotein-expressing murine breast cancer. Eur J Nucl Med Mol Imaging. (2012) 39:149–59. doi:10.1007/s00259-011-1941-7
50. Pellicani RZ, Stefanachi A, Niso M, Carotti A, Leonetti F, Nicolotti O, et al. Potent galloyl-based selective modulators targeting multidrug resistance associated protein 1 and P-glycoprotein. J Med Chem (2012) 55:424–36. doi:10.1021/jm201305y
51. Feelisch M, Schonafinger K, Noack E. Thiol-mediated generation of nitric oxide accounts for the vasodilator action of furoxans. Biochem Pharmacol (1992) 44:1149–57. doi:10.1016/0006-2952(92)90379-W
52. Riganti C, Miraglia E, Viarisio D, Costamagna C, Pescarmona G, Ghigo D, et al. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inibhiting the drug efflux. Cancer Res (2005) 65:516–25.
53. Medana C, Ermondi G, Fruttero R, Di Stilo A, Ferretti C, Gasco A. Furoxans as nitric oxide donors. 4-Phenyl-3-furoxancarbonitrile: thiol-mediated nitric oxide release and biological evaluation. J Med Chem (1994) 37:4412–6. doi:10.1021/jm00051a020
54. Sorba G, Medana C, Fruttero R, Cena C, Di Stilo A, Galli U, et al. Water soluble furoxan derivatives as NO prodrugs. J Med Chem (1997) 40:463–9. doi:10.1021/jm970281u
55. Fruttero R, Crosetti M, Chegaev K, Guglielmo S, Gasco A, Berardi F, et al. Phenylsulfonylfuroxans as modulators of multidrug-resistance-associated protein-1 and P-glycoprotein. J Med Chem (2010) 53:5467–75. doi:10.1021/jm100066y
56. Mechetner E, Roninson IB. Efficient innhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc Natl Acad Sci USA (1992) 89:5824–8. doi:10.1073/pnas.89.13.5824
57. Thierry AR, Vige D, Coughlin SS, Belli JA, Dritschillo A, Rahman A. Modulation of doxorubicin resistance in multidrug-resistent cells by liposomes. FASEB J (1993) 7:572–9.
58. Cuvier C, Roblot-treupel L, Millot JM, Lizard G, Chevillard S, Manfait M, et al. Doxorubicin-loaded nanospheres bypass tumor cell multidrug resistance. Biochem Pharmacol (1992) 44:509–17.
59. Ferlini C, Distefano M, Pignatelli F, Lin S, Riva A, Bombardelli E, et al. Antitumour activity of novel taxanes that act at the same time as cytotoxic agents and P-glycoprotein inhibitors. Br J Cancer (2000) 83(12):1762–8. doi:10.1054/bjoc.2000.1500
60. Su Y, Cheng X, Tan Y, Hu Y, Zhou Y, Liu J, et al. Synthesis of a dual functional anti-MDR tumor agent PH II-7 with elucidations of anti-tumor effects and mechanisms. PLoS One (2012) 7(3):e32782. doi:10.1371/journal.pone.0032782
61. Han C, Huang Z, Zheng C, Wan L, Zhang L, Peng S, et al. Novel hybrids of (phenylsulfonyl)furoxan and anilinopyrimidine as potent and selective epidermal growth factor receptor inhibitors for intervention of non-small-cell lung cancer. J Med Chem (2013) 56:4738–48. doi:10.1021/jm400463q
62. Sullivan R, Graham CH. Chemosensitization of cancer bynitric oxide. Curr Pharm Des (2008) 14:1113–23. doi:10.2174/138161208784246225
63. Li CM, Wang Z, Lu Y, Ahn S, Narayanan R, Kearbey JD, et al. Biological activity of 4-substituted methoxybenzoyl-aryl-thiazole: an active microtubule inhibitor. Cancer Res (2011) 71:216–24. doi:10.1158/0008-5472.CAN-10-1725
64. Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol Sci (2009) 30:546–56. doi:10.1016/j.tips.2009.07.003
65. Laberge RM, Ambadipudi R, Georges E. P-glycoprotein (ABCB1) modulates collateral sensitivity of a multidrug resistant cell line to verapamil. Arch Biochem Biophys (2009) 491:53–60. doi:10.1016/j.abb.2009.09.012
66. Niso M, Abate C, Contino M, Ferorelli S, Azzariti A, Perrone R, et al. Sigma-2 receptor agonists as possible antitumor agents in resistant tumors: hints for collateral sensitivity. ChemMedChem (2013) 8:2026–2035. doi:10.1002/cmdc.201300291
67. Bech-Hansen NT, Till JE, Ling V. Pleiotropic phenotype of colchicines resistant CHO cells: cross-resistance and collateral sensitivity. J Cell Physiol (1976) 8:23–31. doi:10.1002/jcp.1040880104
Keywords: MDR, P-gp, dual effect, multitarget drugs, MDR reverting activity
Citation: Zinzi L, Capparelli E, Cantore M, Contino M, Leopoldo M and Colabufo NA (2014) Small and innovative molecules as new strategy to revert MDR. Front. Oncol. 4:2. doi: 10.3389/fonc.2014.00002
Received: 03 December 2013; Paper pending published: 08 December 2013;
Accepted: 03 January 2014; Published online: 21 January 2014.
Edited by:
Chiara Riganti, University of Turin, ItalyReviewed by:
Chiara Riganti, University of Turin, ItalyAttilio Di Piero, UMR5086 CNRS-University of Lyon, France
Copyright: © 2014 Zinzi, Capparelli, Cantore, Contino, Leopoldo and Colabufo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Antonio Colabufo, Dipartimento di Farmacia – Scienze del Farmaco, Università degli Studi di Bari “A. Moro”, Via Orabona 4, 70125 Bari, Italy e-mail:bmljb2xhYW50b25pby5jb2xhYnVmb0B1bmliYS5pdA==