 Hae Min Jeong
Hae Min Jeong Mi Jeong Kwon
Mi Jeong Kwon Young Kee Shin
Young Kee Shin- 1Laboratory of Molecular Pathology and Cancer Genomics, College of Pharmacy, Seoul National University, Seoul, South Korea
- 2College of Pharmacy, Kyungpook National University, Daegu, South Korea
- 3Research Institute of Pharmaceutical Sciences, College of Pharmacy, Kyungpook National University, Daegu, South Korea
- 4Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul, South Korea
- 5Advanced Institutes of Convergence Technology, Suwon, South Korea
- 6Bio-MAX Institute, Seoul National University, Seoul, South Korea
Like other cancers, most gynecologic cancers are caused by aberrant expression of cancer-related genes. Epigenetics is one of the most important gene expression mechanisms, which contribute to cancer development and progression by regulating cancer-related genes. Since the discovery of differential gene expression patterns in cancer cells when compared with normal cells, extensive efforts have been made to explore the origins of abnormal gene expression in cancer. Epigenetics, the study of inheritable changes in gene expression that do not alter DNA sequence is a key area of this research. DNA methylation and histone modification are well-known epigenetic mechanisms, while microRNAs and alternative splicing have recently been identified as important regulators of epigenetic mechanisms. These mechanisms not only affect specific target gene expression but also regulate the functioning of other epigenetic mechanisms. Moreover, these diverse epigenetic regulations occur simultaneously. Epigenetic regulation of gene expression is extraordinarily complicated and all epigenetic mechanisms to be studied at once to determine the exact gene regulation mechanisms. Traditionally, the contribution of epigenetics to cancer is thought to be mediated through the inactivation of tumor suppressor genes expression. But recently, it is arising that some oncogenes or cancer-promoting genes (CPGs) are overexpressed in diverse type of cancers through epigenetic derepression mechanism, such as DNA and histone demethylation. Epigenetic derepression arises from diverse epigenetic changes, and all of these mechanisms actively interact with each other to increase oncogenes or CPGs expression in cancer cell. Oncogenes or CPGs overexpressed through epigenetic derepression can initiate cancer development, and accumulation of these abnormal epigenetic changes makes cancer more aggressive and treatment resistance. This review discusses epigenetic mechanisms involved in the overexpression of oncogenes or CPGs via epigenetic derepression in gynecologic cancers. Therefore, improved understanding of these epigenetic mechanisms will provide new targets for gynecologic cancer treatment.
Introduction
Cancer is a heterogeneous disease caused by uncontrollable cell division (1). Early research recognized cancer as a genetic disorder of proliferation-related genes, but recent studies have approached cancer multidirectionally, examining resistance to cell death, angiogenesis, invasion, metastasis, and other properties (2). Many genes related to these properties can be roughly divided into two groups: tumor suppressor genes (TSGs) and oncogenes or cancer-promoting genes (CPGs) (3, 4). As the name implies, TSGs have cancer inhibitory roles, and their functions are lost in cancer cells. Conversely, oncogenes or CPGs have the potential to cause cancer, and they are overexpressed in many cancers. Both TSGs and oncogenes or CPGs are abnormally expressed in diverse cancers via various mechanisms, and each gene has a specific function according to the characteristics of specific cancers. Cancer develops from both genetic and epigenetic mutations (5), and progression is more severe when such mutations accumulate, interrupting the normal function of cancer-related genes and inducing resistance to chemotherapy that makes cancer treatment more difficult. Such mutations may be inherited from parents or acquired during life as a result of diverse environmental factors such as chemical and hormone exposure, diet, alcohol use, smoking, and inflammation (6–8). Epigenetics is the key mediator connecting environmental factors to gene regulation systems (9–11). Moreover, a cohort study of twins in Europe has revealed that environmental factors are major causes of cancer, and inherited genetic factors are minor contributors (8). Therefore, the major driving force of cancer development is clearly acquired mutations or epigenetic alterations caused by environmental factors rather than inherited genetic mutations. So far, many research results have revealed a significant role of epigenetic regulation in gynecologic cancer. Aberrant gene expression caused by epigenetic mechanisms is mediated in two main ways. One is epigenetic silencing of TSGs, and the other is epigenetic derepression of oncogenes or CPGs (12). Some recent studies have shown that oncogenes or CPGs are overexpressed in human cancers through epigenetic derepression mechanisms, suggesting a significant role for these mechanisms in human cancers (13–16). Moreover, these mechanisms are tightly connected to one another, so it is important that integrated analysis of genetic/epigenetic mechanisms at the same time. Many researchers are trying to identify the significant epigenetic derepression mechanisms underlying the development of gynecologic cancers. This review article will discuss the diverse epigenetic derepression mechanisms related to aberrant gene expression in gynecologic cancer and their potency as targets for gynecologic cancer therapy.
Gynecologic Cancer
Gynecologic cancer includes any cancer that occurs in the female reproductive organs. There are five frequently occurring gynecologic cancers: ovarian, cervical, endometrial (uterine), vaginal, and vulvar. Gynecologic cancers usually have high mortality rates, because it is difficult to detect the cancer in early stage (17). Therefore, convenient diagnostic strategies for early detection of gynecologic cancers are needed. Human papillomavirus (HPV) is the major cause of most of these cancers. Especially, HPV-16 and HPV-18 cause approximately 70% of cervical cancers (18), reflecting the severe carcinogenic properties of these HPV types.
Ovarian cancer is classified into three major types according to the histology of the tumor. Epithelial ovarian cancer is the predominant type, and the other types, sex cord stromal tumors and germ cell tumors, account for <10% of malignant ovarian cancers (19). Among epithelial ovarian cancers, 80% are serous adenocarcinoma and the remainder are mucinous, endometrioid, or clear cell carcinoma (20). Epithelial ovarian cancer is generally believed to originate in the ovarian epithelium, but a new hypothesis that high-grade serous ovarian cancer (HGS-OvCa) originates in the fimbrial cells of fallopian tubes rather than the ovarian epithelium has recently arisen (21). Mutation of breast cancer 1 and 2, early onset (BRCA1 and BRCA2) are known to influence ovarian cancer development (22) and can be used as diagnostic or prognostic factors (23). Recently, the cancer genome atlas (TCGA) project has also found that approximately 10% of HGS-OvCa patients have BRCA1/2 germline mutations. In addition, nearly all HGS-OvCa samples show TP53 mutation (24). Serum cancer antigen 125 level also has significant correlation with tumor stage, grade, and histologic type, making it a useful prognostic factor in epithelial ovarian cancer (25, 26). It is also a widely used monitoring tool for assessing the effectiveness of therapy and asymptomatic recurrence rates at follow-up (27). Other type of cancers also show abnormal genetic/epigenetic status. Some genes are abnormally regulated by epigenetic mechanisms in cervical cancer (28–30), suggesting that epigenetics may be important in cervical cancer carcinogenesis. In case of endometrioid cancer, genetic mutations of KRAS, phosphate and tensin homolog (PTEN), and beta-catenin (CTNNB1) are associated with the initiation of endometrioid carcinomas (31). Moreover, endometrial cancer also displays genetic mutations of diverse TSGs including TP53, CTNNB1, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), PTEN, and protein phosphatase 2 and regulatory subunit A alpha (PPP2R1A) (32–34). Vaginal cancer and vulvar cancer are relatively rare gynecologic cancer (35), so the genetic/epigenetic mutations are not studied well.
Cancer Epigenetics
Epigenetics refers to the study of inheritable alterations in gene expression without accompanying changes in genomic DNA sequence, and it explains variations in gene expression originating from the same genetic information (36). Among the various factors related to cancer initiation and development, epigenetic alteration is critical because it causes global aberrant gene expression (37). More than 300 genes related to cancer cell properties are epigenetically deregulated in various human cancers including those of breast, gastric, ovarian, and prostate (38, 39). This number is expected to increase rapidly as epigenetic studies of cancer continue and techniques advance.
DNA Methylation in Cancer
The best-known epigenetic mechanism is DNA methylation, which usually represses downstream gene expression by changing local chromatin structure (40–42). Human tumors were initially discovered to have global DNA hypomethylation and local DNA hypermethylation patterns (43), and since then, many researchers have undertaken studies of the relationship between DNA methylation and cancer. As normal cells progress to invasive cancer cells, their overall DNA methylation levels decrease, whereas CpG Island hypermethylation and alteration of histone modification patterns accumulate gradually (36). TSG inactivation by promoter regions hypermethylation or DNA hypomethylation of highly repeated DNA regions are found in diverse cancer types. In addition, DNA hypomethylation in promoter regions has recently been discovered to derepress some CPGs or proto-oncogenes that are repressed in normal cells (12). Tahiliani et al. (44) have recently shown that tet methylcytosine dioxygenase (TET) 1 can convert 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) in a 2-oxoglutarate- and Fe (II)-dependent manner. 5-hmC is then actively demethylated via additional mechanisms including glycosylation and base excision repair. DNA demethylation of CpG Islands in promoter regions can reactivate downstream genes. Some genes repressed by DNA hypermethylation can be derepressed through DNA demethylation, and the latter plays a critical role in gene expression mechanisms in stem cell differentiation (45) or neural memory and immune systems (46). Among these properties, the pluripotency of stem cells is closely related to TET proteins by NANOG-dependent manner (47, 48). TET proteins also associated with diverse type of cancers. In breast cancer, TET1 demethylates the promoter region of homeobox A (HOXA) genes to induce the expression of HOXA7, 9, resulting tumor growth and metastasis suppression (49). In case of hepatocellular carcinoma, it is suggested that 5-hmC may be used as prognostic marker and decreased TET1 is underlying mechanism of 5-hmC loss (50).
Histone Modification in Cancer
Histone modification is also major epigenetic mechanisms. Each histone modification can affect the overall structure of chromatin, and therefore it can also affect gene expression by changing the condensation of DNA or recruiting effector molecules that control downstream gene expression. However, unlike DNA methylation, histone modification is directly associated with both gene activation and gene repression according to individual modifications or specific modified genomic regions (51). Moreover, histone modification can positively or negatively affect other modifications by cross-interactions (52). Aberrant histone modification causes abnormal expression of cancer-related genes by changing DNA structure. Cancer cells are characterized by the loss of active histone marks in the promoter regions of TSGs or loss of repressive marks in subtelomeric DNA and other DNA repeats, which make the chromatin structure more flexible (53).
MicroRNA and Alternative Splicing in Cancer
Gene expression also can be regulated at the post-transcriptional level by microRNA (miRNA). miRNA is a small non-coding RNA that is normally composed of 20–22 nucleotides. Most miRNAs inhibit the translation of mRNA to protein by binding to the 3′-untranslated region of target mRNAs through imperfect complementary binding. More than 1400 miRNAs related to almost every cellular function, including proliferation, differentiation, and development, have been identified in humans (54–56). Proper miRNA regulation is also broken in cancers. Many factors affect miRNA biogenesis, including miRNA genomic localization, transcriptional regulation, processing steps, and post-transcriptional modification (57). Abnormal control of these steps causes overall miRNA dysregulation, which leads to aberrant cancer-related genes expression at post-transcription level. For example, miR-21 and miR-17-92 clusters are representative “oncomiRs” and their expression is enriched in various cancers (58, 59). Conversely, tumor suppressor miRNAs (TSmiRNAs) have also been found. The let-7/miR-98 families play roles in both apoptosis and cell proliferation pathways (60), and the miR-141/200 families are highly associated with epithelial-to-mesenchymal transition (EMT) or chemosensitivity (61, 62). It is now clear that alternative splicing is an important gene expression mechanism and emerging evidence indicates that alternative splicing regulates not only splicing machinery, but also chromatin structure and cancer development (63–65). Serine/arginine-rich splicing factor 1 (SRSF1) is a typical alternative splicing regulator that shows proto-oncogenic properties. The overexpression of SRSF1 transforms fibroblasts into sarcomas (66) and makes mammary epithelial cells more antiapoptotic by regulating BIM and BIN1 splicing isoforms (67). Interestingly, some genes including Wilms tumor suppressor gene (WT1) (68) and Bcl-x (69, 70) have both properties of oncogene or tumor suppressor gene according to their splicing isoform.
Mutual Interaction among Epigenetic Mechanisms in Cancer
All of these epigenetic mechanisms are tightly connected to one another and compose the overall gene regulation system. Therefore, integrated analysis of diverse epigenetic mechanisms is essential to understanding the gene expression regulation system in its entirety. The miR-29 family, including miR-29a, miR-29b, and miR-29c, is representative miRNA that interacts with other epigenetic mechanisms. The expression of miR-29b is regulated by both histone modification (71) and DNA methylation (72). On the contrary, recent studies have shown that miR-29a is closely correlated with DNMT proteins, suggesting that the miR-29 family has an important role in overall epigenetic mechanisms (73–75). miR-7/miR-218 can modify DNA methylation and histone modification status by decreasing homeobox B3 (HOXB3) expression (76), and miR-28/miR-505 can affect alternative splicing through SRSF1 inhibition (77). With these recent findings, integrated analysis of epigenetic studies has gradually progressed through the efforts of diverse groups. TCGA is a substantial cooperative project investigating genome-wide epigenetic alterations in various cancers. Creighton et al. (78) have profiled miRNA expression in 489 high-grade serous ovarian adenocarcinomas in the TCGA database and analyzed their widespread effects on gene expression. The results show a reverse correlation between miRNA and mRNA level that corresponds with canonical interaction of miRNA and mRNA. Integrated analysis of gene amplification and gene expression (79); gene expression, DNA methylation, and miRNA expression (80); DNA copy number, DNA methylation, and mRNA expression (81) have also been performed recently. These studies demonstrate the importance of integrating diverse types of epigenetic data to understand cancer biology more thoroughly.
Integrated Epigenetic Derepression Mechanisms in Cancer
Until recently, the focus of cancer epigenetics has been the repression of TSG expression, and many reports have discussed the repression by DNA hypermethylation or decreases in active histone marks in promoter regions. The concept of epigenetic derepression mechanisms in cancer was first introduced by Feinberg and Vogelstein in 1983 (43). They found that the promoter regions of two human growth hormone γ-globin genes are methylated in normal tissues but hypomethylated in malignant colon cancers overexpressing both genes. Since then, many studies have found that a number of oncogenes or CPGs are abnormally increased in several cancers by epigenetic derepression. These genes are involved in critical functions in cancer cells including proliferation, DNA repair, angiogenesis, cell migration, metastasis, and chemoresistance (82). Therefore, determination of the molecular mechanisms underlying the overexpression of oncogenes or CPGs is necessary in the study of cancer, and epigenetic derepression may be a key mechanism.
The inactivation of TSGs is mediated through TSG promoter region DNA hypermethylation and repressive histone modification including H2A ubiquitination (83), H3K9me2, H3K9me3, H3K27me2, H3K27me3 (84, 85), and H4K20me3 (86), resulting in low TSG expression. Moreover, aberrant overexpression of oncomiRs, which is caused by DNA hypomethylation and active histone modification including H2Bub (83), H3K4me1, H3K4me2, H3K4me3 (87, 88), H3K79me1, H3K79me2, H3K79me3 (84, 89), H3Ac, and H4Ac (90) degrades TSG mRNAs or inhibits translation of mRNA, increasing TSG repression. Conversely, the activation of CPGs via DNA hypomethylation and active histone modification facilitates the transcription of these genes. Furthermore, DNA hypermethylation and repressive histone modification of TSmiRNA gene promoters reduce expression, derepressing CPGs normally repressed by TSmiRNAs. These diverse epigenetic changes occur throughout the genome at the same moment, resulting in the overexpression of oncogenes or CPGs and downregulation of TSGs. Ultimately, the sum of these epigenetic aberrations contributes to cancer development, progression, and treatment resistance (Figure 1).
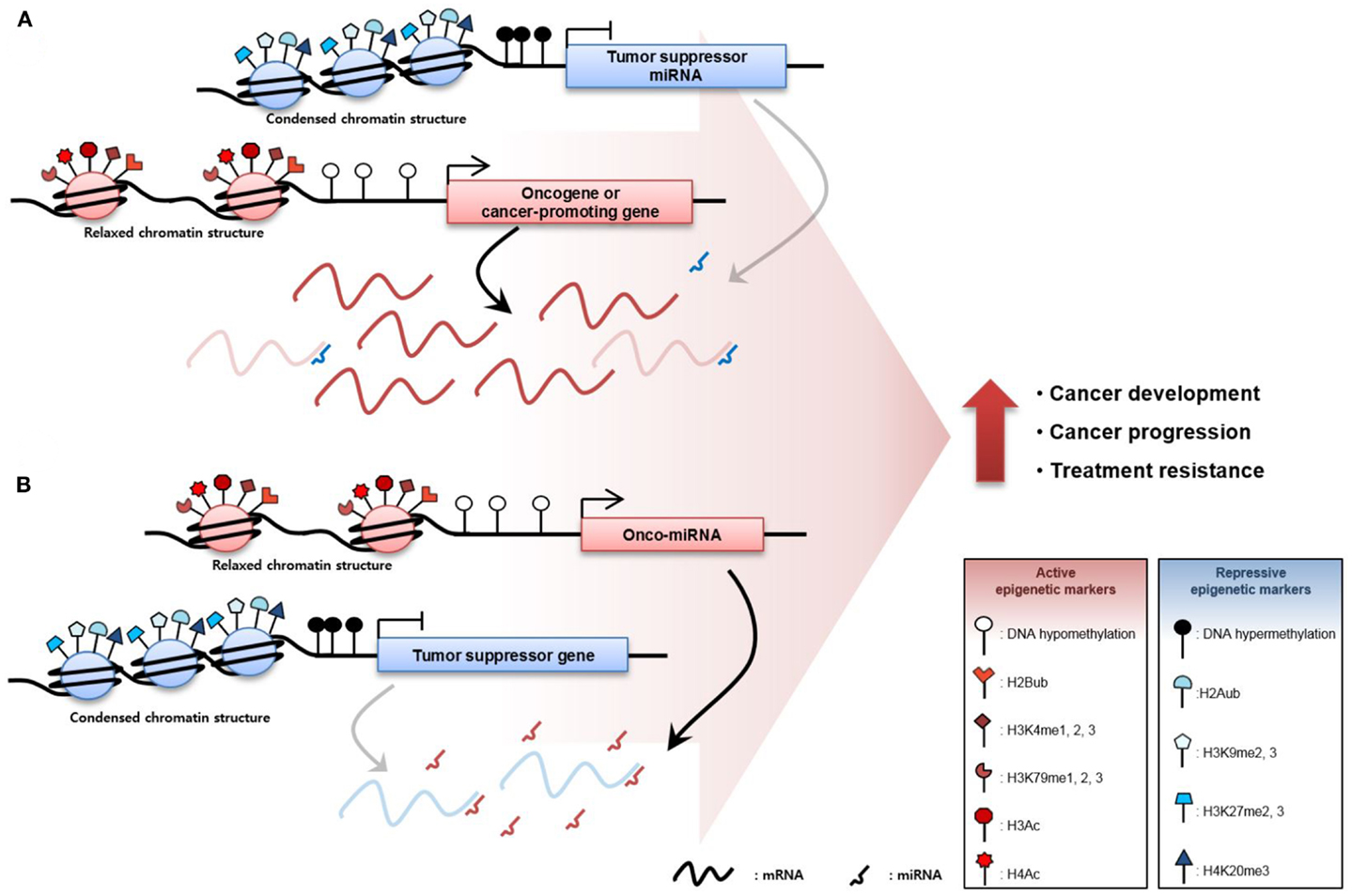
Figure 1. Epigenetic derepression mechanisms in cancer. (A) Oncogenes or cancer-promoting genes (CPGs), are overexpressed through DNA hypomethylation or gain of active histone modification such as histone 2B ubiquitination, mono-, di-, tri-methylation of the fourth lysine in the histone 3 tail (H3K4me1, H3K4me2, and H3K4me3), mono-, di-, tri-methylation of the 79th lysine in the histone 3 tail (H3K79me1, H3K79me2, and H3K79me3), histone 3 acetylation (H3Ac), and histone 4 acetylation (H4Ac) in the promoter region of the gene. Oncogene or CPG expression can be increased through suppression of tumor suppressor micro RNAs (TSmiRNAs). TSmiRNAs, which degrade the messenger RNA of the oncogene or CPG, are repressed through epigenetic mechanisms such as DNA hypermethylation and gain of repressive histone tri-methylation of histone 2A ubiquitination, di-, tri-methylation of the 9th lysine in the histone 3 tail (H3K9me2 and H3K9me3), di-tri-methylation of the 27th lysine in the histone 3 tail (H3K27me2 and H3K27me3), and tri-methylation of the 20th lysine in the histone 4 tail (H4K20me3). (B) Tumor-suppressor genes (TSGs) are abnormally downregulated in cancer via gain of repressive epigenetic markers. In addition, TSGs are inactivated by oncomiRs, which degrade TSGs. Ultimately, increased oncogene or CPGs and decreased TSGs expression contribute to cancer development, cancer progression, and resistance to cancer therapy.
These epigenetic mechanisms work not only in gene promoter regions but also in non-promoter regions, which are highly repeated DNA sequence regions. Specifically, highly repeated regions occupy nearly half of the genome, and these regions are silenced by DNA hypermethylation to maintain DNA integrity and stability (91). Decreases in DNA methylation in these repeated regions induce genomic alteration, resulting in the genome-wide instability usually observed in cancers (Figure 2) (92). For example, LINE-1 repeat is a representative repeated sequence in the human genome that is usually hypomethylated in various cancers (93–95). Moreover, LINE-1 hypomethylation is related to poor prognosis in various cancers including HCC (96), gastric cancer (97), and multiple myeloma (98).
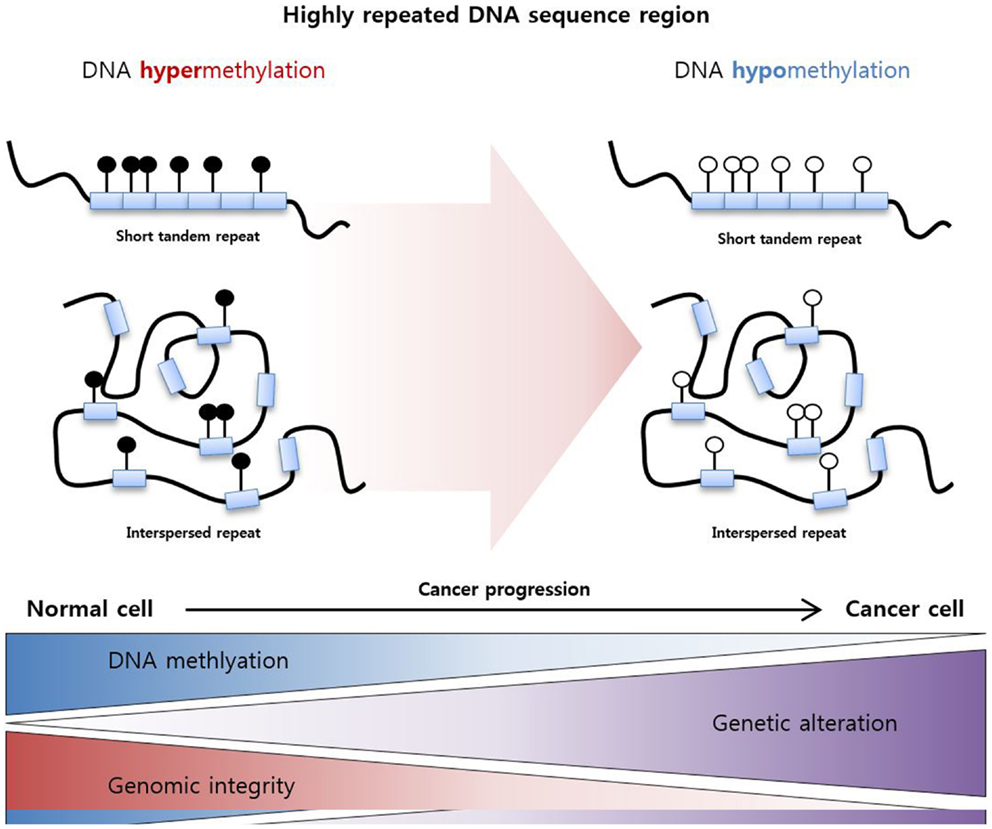
Figure 2. DNA hypomethylation in a highly repeated DNA sequence region. Highly repeated sequence regions are mainly categorized as tandem repeats and interspersed repeats. These regions are highly methylated in normal cells to maintain genomic integrity, and hypomethylation of the regions increases genetic alterations and genomic instability, resulting cancer development.
Epigenetic Derepression in Gynecologic Cancer
In gynecologic cancer, Chan et al. (99) initially showed that BRCA2 expression is increased in ovarian cancer via promoter region DNA hypomethylation. Since the publication of these studies, a number of groups have investigated epigenetic derepression mechanisms in various gynecologic cancers (Table 1). Our group also have recently demonstrated that the CPGs CLDN3 and CLDN4 are overexpressed in ovarian cancer in association with decreases in repressive histone marks (13).
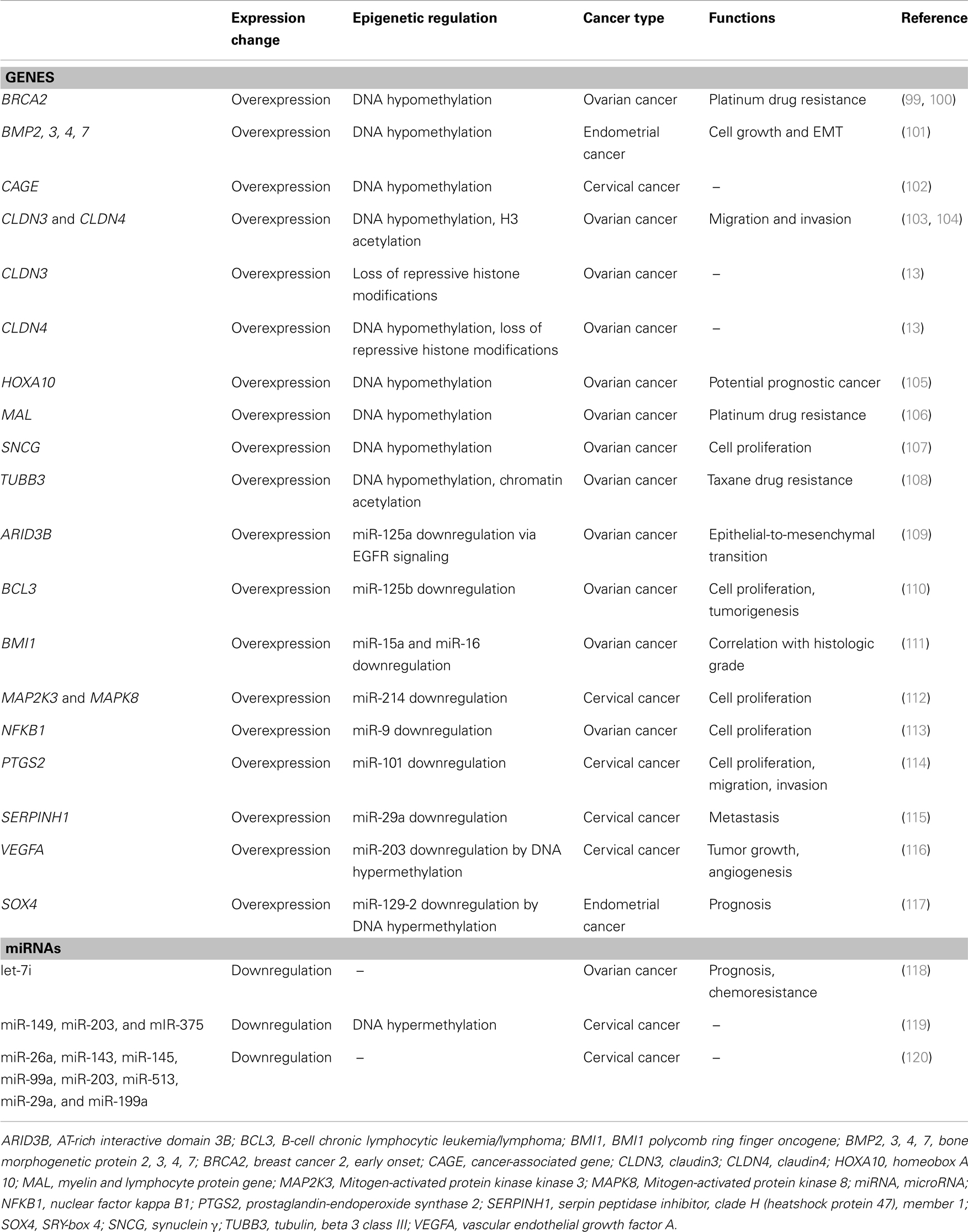
Table 1. Genes or miRNAs aberrantly regulated via epigenetic derepression mechanisms in gynecologic cancer.
Epigenetic Derepression in Ovarian Cancer
Candidate oncogene synuclein γ (SNCG) is known to stimulate cell proliferation and metastasis in several cancers. Gupta et al. (107) have shown that SNCG is overexpressed in ovarian cancer cells through hypomethylation of the CpG Island located in exon 1. Furthermore, SNCG expression is restored in some cell lines with low endogenous SNCG expression levels through treatment with the DNA demethylating agent 5-aza-CdR, supporting the tumorigenic role of DNA hypomethylation in ovarian cancer. Homeobox A 10 (HOXA10) is also regulated by DNA methylation in ovarian cancer. HOXA10 has crucial functions in receptivity, embryo implantation, and decidualization. It is overexpressed in ovarian cancers, and its gene promoter regions are hypomethylated, which may be a potential prognostic factor in the disease (105). DNA hypomethylation also contributes to chemoresistance in gynecologic cancer. The myelin and lymphocyte protein gene (MAL) is among the most highly expressed genes in ovarian cancer. Its expression is also regulated by promoter region DNA methylation, and it is largely demethylated in primary ovarian cancer tissues and ovarian cancer cell lines (106). In a study by Lee et al. (106), MAL expression patterns showed significant reverse correlation with platinum resistance, and the DNA methylation status of the MAL promoter region can be used as a marker for platinum drug sensitivity and as a therapeutic target in ovarian cancer.
Other studies have demonstrated integrated mechanisms of DNA methylation and histone modification related to epigenetic derepression in ovarian cancer. CLDN3 and CLDN4, which are separated by only 60 kb on chromosome 7, are transcribed in opposite directions and highly overexpressed in ovarian cancer. These tight junction proteins reportedly promote the migration and invasion of ovarian epithelial cells (121), and abnormal overexpression of CLDN3 and CLDN4 is caused by the simultaneous action of DNA methylation and histone modification. For example, Honda et al. (103, 104) have reported that DNA demethylation and increased H3 acetylation at the Sp1 binding site are critical factors in the overexpression of CLDN3 and CLDN4 (103, 104). Kwon et al. (13) have also shown the importance of simultaneous epigenetic changes in CLDN3 and CLDN4 overexpression. They have found that CLDN3 and CLDN4 repression in normal ovarian cells is associated with “bivalent” histone modification, and simultaneous increase in H3K4me3 and decrease in H4K20me3 and H3K27me3 work together to induce CLDN3 and CLDN4 expression levels (13). Interestingly, DNA methylation is inversely correlated only with the expression of CLDN4, not with CLDN3 expression. The region analyzed in this study contained Sp1 binding sites, but the cell lines are completely different. Therefore, the individual characteristics of each cell line may be responsible for this difference. With regard to chemoresistance, tubulin, beta 3 class III (TUBB3) overexpression has been reported as a primary mechanism of taxane drug resistance in diverse cancers. Epigenetic study of 66 primary ovarian tumors and 3 ovarian cancer cell lines has revealed that DNA methylation and chromatin acetylation are partly associated with TUBB3 overexpression in ovarian cancer (108).
Similar to TSGs, some TSmiRNAs are also downregulated in ovarian cancers. The let-7 miRNA family, which was the first reported group of TSmiRNAs in cancer, represses a number of oncogenic proteins such as KRAS, high mobility group AT-hook 2 (HMGA2), and v-myc myelocytomatosis viral oncogene homolog (MYC). Yang et al. (118) have identified chemotherapy response-related miRNAs in ovarian cancer using miRNA expression microarray. Among them, let-7i expression level is significantly reduced in chemotherapy-resistant patients, and decreased let-7i expression is associated with shorter progression-free survival in late-stage ovarian cancer patients. Therefore, let-7i has a tumor suppressive function related to resistance to chemotherapy, and target genes of let-7i may be related to chemoresistance.
A common role of miRNAs is the repression of target gene expression at the post-transcription level. Therefore, aberrantly decreased miRNA expression enhances the expression of many target genes, some of which may contribute to cancer development in diverse ways. EMT usually occurs during embryonic development and contributes to tumor migration, invasion, and metastasis in cancer. Dahl et al. (109) have shown that aberrantly low expression of miR-125a in ovarian cancer is caused by epidermal growth factor receptor signaling, which leads to EMT through increased levels of AT-rich interactive domain 3B (ARID3B) expression. These results demonstrate that miRNAs may be critical mediators in sequential regulatory systems from signaling pathways to cancer phenotypes. Another miR-125 family member, miR-125b, also has tumor suppressive functions in ovarian cancer. Guan et al. (110) have found that the expression of miR-125b is decreased in ovarian cancer, causing overexpression of proto-oncogene B-cell chronic lymphocytic leukemia/lymphoma (BCL3), which regulates cell proliferation and tumorigenesis. In addition, the major transcription factors nuclear factor kappa B1 and BMI1 polycomb ring finger oncogene (BMI1) are overexpressed in ovarian cancer through aberrant downregulation of miR-9, miR-15a, and miR-16. Both miR-125 members contribute to cancer development by stimulating cell proliferation in specific ways (111, 113).
Epigenetic Derepression in Cervical Cancer
In cervical cancer, cancer-associated gene (CAGE) shows hypomethylated patterns in its promoter region (102). Specifically, the promoter region of CAGE is frequently hypomethylated in diverse types of cancer including breast, lung, and stomach cancers. A study by Lee et al. (102), which examined tissues samples from normal patients and more than 40 cervical cancer patients, demonstrated the significantly high correlation between CAGE promoter hypomethylation and cervical cancer and suggested that the DNA methylation pattern in the CAGE gene may have diagnostic utility. Prostaglandin-endoperoxide synthase 2 (PTGS2) expression is also epigenetically derepressed in cervical cancer by loss of miR-101 expression. Exogenous overexpression of miR-101 decrease cell proliferation, migration, and invasion via inhibition of PTGS2 expression (114). Similarly to this, many recent studies find that epigenetic derepression of oncogenes or CPGs by miRNA downregulation in cervical cancer. Mitogen-activated protein kinase kinase 3 (MAP2K3), mitogen-activated protein kinase 8 (MAPK8) and miR-214 (112), Serpin peptidase inhibitor, clade H (heatshock protein 47), member 1 (SERPINH1) and miR-29a (115), vascular endothelial growth factor A (VEGFA), and miR-203 (116) and so on.
As with protein-coding genes, the expression of miRNA is also regulated by DNA methylation. Wilting et al. (119) have reported that miR-149, miR-203, and miR-375 are located within hypermethylated CpG Islands with decreased miRNA expression in cervical cancer, indicating that hypermethylated miRNAs may be candidate markers in the detection of cancerous lesions. Moreover, a recent studies has found a panel of six miRNAs, which are aberrantly repressed by DNA hypermethylation (122), and eight miRNAs that show decreased expression levels in atypical dysplasia and cervical cancer compared with those in normal cervix cells (120), suggesting a significant regulatory role for miRNA in cancer development.
Epigenetic Changes in Endometrial, Vaginal, and Vulvar Cancer
Gynecologic cancers other than ovarian and cervical cancer have not been researched as thoroughly, but some studies have reported results related to epigenetic derepression mechanisms in these cancers as well. Bone morphogenetic protein (BMP) family contributes to aggressive growth and EMT, and it’s expression is induced by promoter DNA hypomethylation in endometrial cancer (101). SRY-box 4 (SOX4) is often overexpressed in diverse cancers, including prostate, liver, lung, and bladder cancer, with poor prognostic features (123–126). In endometrial cancer, SOX4 expression is upregulated through silencing of miR-129-2, which is mediated by DNA hypermethylation of miR-129-2 (117). Huang et al. (117) have found that aberrant SOX4 expression is correlated with shorter patient survival time and is also related to microsatellite instability and the repression of mutL homolog 1 (MLH1) via DNA hypermethylation, suggesting that miR-129-2 contributes to endometrial cancer by deactivating DNA repair systems.
No direct evidence of epigenetic derepression mechanisms have been reported in vulvar cancer, but Samartzis et al. (127) have recently reported the differences in class I HDAC expression patterns between VIN and vulvar squamous cell cancer (VSCC). Using tissue microarray, they discovered that class I HDACs are highly expressed in VIN and VSCC, but HDAC2 expression in VIN is higher than that in VSCC, and HDAC3 is more frequently expressed in VSCC. Therefore, class I HDACs appear to stimulate vulvar cancer, suggesting specific roles for HDAC2 and HDAC3 during cancer progression. These results showed no clear epigenetic derepression mechanisms but provide clues about the epigenetic regulatory role of histone acetylation changes in vulvar cancer through analysis of HDAC expression patterns.
Diagnostic and Prognostic Utility of Epigenetic Alterations in Gynecologic Cancer
As mentioned above, abnormal gene expression in cancer is mainly caused by epigenetic mechanisms that contribute to cancer development. Therefore, knowledge of underlying epigenetic mechanisms can be used to identify prognostic markers or develop therapeutic targets of epigenetic drugs or small interfering RNA therapies. Many studies have reported the potential usefulness of epigenetic changes in the diagnosis and prognosis of gynecologic cancers (Table 2).
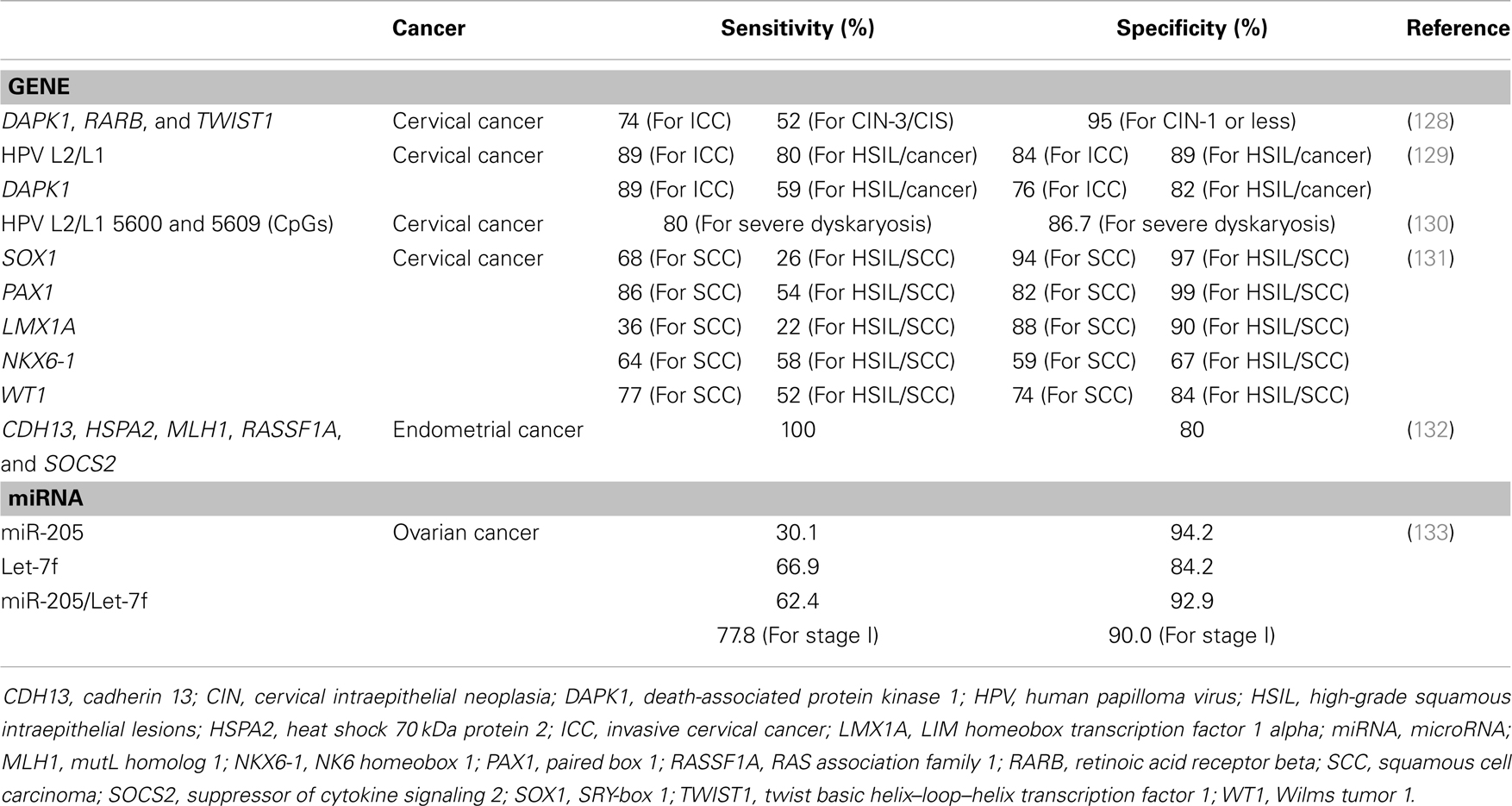
Table 2. Sensitivities and specificities of epigenetic changes for diagnosis and prognosis in gynecological cancer.
Epigenetic Alterations in Gynecologic Cancer Diagnosis
The most effective way to cure cancer is to detect it at its earliest stages and remove tumors completely before extensive cancer development. Therefore, accurate diagnostic methods are essential in cancer therapy. Clearly, the epigenetic status of cancer cells differs significantly from that of their normal cell counterparts, and some of these differences can be used as diagnostic factors in specific types of cancer. Feng et al. (128) have found that genes such as death-associated protein kinase 1 (DAPK1), retinoic acid receptor beta (RARB), and twist basic helix–loop–helix transcription factor 1 (TWIST1) might be useful markers in cervical intraepithelial neoplasia and invasive cervical cancer. DNA methylation patterns in the promoter regions of this three-gene panel efficiently distinguished cervical intraepithelial neoplasia from invasive cervical cancer, showing high specificity and sensitivity in exfoliated cell samples from cervical cancer patients. Lai et al. (131) have also identified six genes – SRY-box 1 (SOX1), paired box 1 (PAX1), LIM homeobox transcription factor 1 alpha (LMX1A), NK6 homeobox 1 (NKX6-1), Wilms tumor 1 (WT1), and one cut homeobox 1 (ONECUT1) – that show DNA hypermethylation in cervical cancer. Fiegl et al. (132) have developed a new strategy for the detection of endometrial cancer in cervicovaginal secretions by examining the expression of five genes – RASSF1A, MLH1, cadherin 13 (CDH13), heat shock 70 kDa protein 2 (HSPA2), and suppressor of cytokine signaling 2 (SOCS2). All endometrial cancer patient samples used in their study showed three or more hypermethylated genes among the five analyzed, and 99 of 109 non-endometrial cancer samples displayed no or fewer than three genes with DNA methylation.
Epigenetic Alterations in Gynecologic Cancer Prognosis
Epigenetic changes in cancer also provide prognostic information. In ovarian cancer, reduced expression of the methylation-controlled DNAJ gene (MCJ) owing to DNA hypermethylation increases chemotherapeutic drug resistance (134, 135). Interestingly, the CpG Island in exon 1 of MCJ is more critical than the promoter region CpG Island for gene repression. Moreover, high levels of DNA methylation in this region are significantly correlated with poor response to chemotherapy and worse prognosis. These results suggest that DNA hypermethylation in the exon region of MCJ may be a good marker for chemoresistance in ovarian cancer. Stem cell polycomb group targets show high DNA methylation level in cancers. DNA methylation of the polycomb group target homeobox A 11 (HOXA11) promoter is also increased in ovarian cancer specimens and strongly associated with poor prognosis, making it a potential prognostic biomarker in ovarian cancer (136). It is well-known that HPV, which is the major cause of cervical cancer, genomes DNA methylation increases with cancer progression. Very recent study suggests that combination of HPV L2/L1 and cellular DAPK1 DNA methylation can be used as prognostic biomarker in cervical cancer (129). Furthermore, DNA methylation of HPV L1/L2 nucleotide positions 5600 and 5609 highly correlates with cervical cancer grade (130), supporting prognostic function of HPV DNA methylation. Many TSGs in vulvar cancer, including tumor protein p73 (TP73), immunoglobulin superfamily member 4 (IGSf4), DAPK1, RASSF1A, RASSF2A, CDKN2A, thrombospondin 1 (TSP-1), MGMT, and others, are inactivated through promoter region DNA hypermethylation. Among these genes, TSP-1 shows a potential prognostic role in VSCC recurrence (137), and TP73 displays features that may serve as therapeutic biomarkers in vulvar cancer (138). DNA hypomethylation patterns may also be used as potential prognostic markers in several cancers. DNA demethylation in the promoter region of urokinase (139) and P-cadherin (140) increases the expression levels of both genes and is correlated with clinical outcomes in invasive breast cancer. DNA hypomethylation of certain repetitive sequences in HCC is also associated with disease recurrence, suggesting the diagnostic value of DNA hypomethylation (141). Pattamadilok et al. (142) have reported that decreased LINE-1 DNA methylation levels are related to advanced tumor grade, and patients with downregulated LINE-1 DNA methylation levels have poor overall survival. DNA hypomethylation of LINE-1 is an important process in ovarian cancer carcinogenesis and has potential for use as a prognostic marker in epithelial ovarian cancers. A recent work (133) has suggested that plasma miRNAs have the potential to be diagnostic and prognostic biomarkers in ovarian cancer. They found increased miR-205 and decreased let-7f levels in the plasma of early stage epithelial ovarian cancer patients and report that the combination of miR-205/let-7f is a powerful diagnostic factor in early-stage epithelial ovarian cancer. Because obtaining plasma from patients is convenient, miRNA has great potential to be a novel non-invasive biomarker.
Epigenetic Therapy in Gynecologic Cancer
The most fundamental way to cure cancer is to restore abnormal gene expression regulation systems. Epigenetic therapy may, therefore, occupy a significant portion of cancer treatment. Epigenetic therapy has been studied in diverse ways since the discovery of aberrant epigenetic regulation in cancer. A typical epigenetic drug is azacitidine, which is a chemical analog of cytosine nucleosides (143). This drug is mainly indicated to treat myelodysplastic syndrome, and it has received approval by the U.S. Food and Drug Administration. Because epigenetic studies of cancer have focused on the repression of TSGs through repressive epigenetic mechanisms, many epigenetic drugs (e.g., 5-aza-CdR) also target epigenetic reactive functions. However, in cancers caused by aberrant overexpression of oncogenes or CPGs through epigenetic derepression mechanisms, suppression of abnormal epigenetic derepression is a therapeutic target. As described above, many studies of epigenetic derepression in gynecologic cancers have been published recently. Therefore, new drugs that repress the expression of oncogenes or CPGs through epigenetic mechanisms might provide novel therapeutic approaches for treating specific cancers. For example, proto-oncogene BCL6 is reportedly repressed by the epigenetic drugs 5-aza-CdR and 4-phenylbutyric acid, which make chromatin structure more flexible to activate gene expression. In these cases, increased miR-127 facilitates the degradation of its target gene BCL6 through canonical RNA interference mechanisms, downregulating BCL6 expression (144).
The results of these studies are evidence of complex epigenetic regulation systems and suggest the potential use of epigenetic drugs that repress the expression of proto-oncogenes in the treatment of cancer. Another well-known TSmiRNA, the let-7 family, is also a potential candidate for cancer therapy. Wang et al. (145) have demonstrated that let-7i strongly lowers the expression of KRAS and HMGA2 in lung cancer cells and shows anti-cancer activities through inhibition of cell growth and migration. Because a single TSmiRNA can repress the expression of multiple CPGs, TSmiRNA therapy may have potent anti-cancer activity. Research on cancer therapy targeting epigenetic derepression remains to be completed, but the number of studies showing aberrant epigenetic derepression mechanisms in cancer development is rapidly increasing. Novel epigenetic drugs targeting derepressed oncogenes will be central in epigenetic cancer therapies.
Conclusion
Abnormal expression of diverse genes involved in cancer-related properties such as cell growth or differentiation is a main causal factor in cancer development (146). Some critical genetic/epigenetic mutations can transform normal cell to cancer cell, and accumulation of cancer-related genes abnormal expression increases its severity. Epigenetic mechanisms are primary sources of this aberrant gene expression. Growing evidences have shown that epigenetic derepression mechanisms including DNA demethylation, gain of active histone modifications, TSmiRNA downregulation, oncogenic alternative splicing and hypomethylation of repeated sequences play critical roles in cancer development. These aberrant epigenetic changes occur in all stage of cancer, from early stages of carcinogenesis to cancer metastasis and resistance to therapy. As shown in above, a lot of studies have found that various epigenetic derepression mechanisms contribute progression of gynecologic cancers, and can be used as diagnostic, prognostic marker for gynecologic cancer therapy. Actually, these multiple epigenetic mechanisms work together to regulate gene expression system. Therefore, integrated analysis of diverse epigenetic factors is important in understanding the epigenetic derepression mechanism of oncogene or CPG expression in gynecologic cancer. Compared to the researches of TSGs epigenetic inactivation, epigenetic activation of CAGEs are not studied well. So, additional research on the activation of oncogenes or CPGs via epigenetic depression is essential for a comprehensive understanding of the epigenetic mechanisms of gynecologic cancer. Moreover, studies of epigenetic derepression will provide insight into cancer development mechanisms and improved therapeutic approaches for gynecologic cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2013M3C8A1075908) and by the Global Core Research Center (GCRC) grant (No. 2011-0030001) funded by the Korea government (MSIP).
References
1. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature (2001) 411:342–8. doi: 10.1038/35077213
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
3. Lee EY, Muller WJ. Oncogenes and tumor suppressor genes. Cold Spring Harb Perspect Biol (2010) 2(10):a003236. doi:10.1101/cshperspect.a003236
4. Oren M. The involvement of oncogenes and tumor suppressor genes in the control of apoptosis. Cancer Metastasis Rev (1992) 11:141–8. doi:10.1007/BF00048060
5. Sadikovic B, Al-Romaih K, Squire JA, Zielenska M. Cause and consequences of genetic and epigenetic alterations in human cancer. Curr Genomics (2008) 9:394–408. doi:10.2174/138920208785699580
6. Aguilera O, Fernandez AF, Munoz A, Fraga MF. Epigenetics and environment: a complex relationship. J Appl Physiol (2010) 109:243–51. doi:10.1152/japplphysiol.00068.2010
7. Boffetta P, Nyberg F. Contribution of environmental factors to cancer risk. Br Med Bull (2003) 68:71–94. doi:10.1093/bmp/ldg023
8. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med (2000) 343:78–85. doi:10.1056/NEJM200007133430201
9. Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr (2009) 21:243. doi:10.1097/MOP.0b013e32832925cc
10. Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutat Res (2011) 727:55–61. doi:10.1016/j.mrrev.2011.04.001
11. Widschwendter M, Jones A, Teschendorff AE. Epigenetics makes its mark on women-specific cancers – an opportunity to redefine oncological approaches? Gynecol Oncol (2013) 128:134–43. doi:10.1016/j.ygyno.2012.09.027
12. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics (2009) 1:239–59. doi:10.2217/epi.09.33
13. Kwon MJ, Kim SS, Choi YL, Jung HS, Balch C, Kim SH, et al. Derepression of CLDN3 and CLDN4 during ovarian tumorigenesis is associated with loss of repressive histone modifications. Carcinogenesis (2010) 31:974–83. doi:10.1093/carcin/bgp336
14. Kwon MJ, Kim SH, Jeong HM, Jung HS, Kim SS, Lee JE, et al. Claudin-4 overexpression is associated with epigenetic derepression in gastric carcinoma. Lab Invest (2011) 91:1652–67. doi:10.1038/labinvest.2011.117
15. Saito Y, Jones PM. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle (2006) 5:2220–2. doi:10.4161/cc.5.19.3340
16. Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature (2012) 481:329–34. doi:10.1038/nature10733
17. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin (2012) 62(1):10–29. doi:10.3322/caac.20138
18. Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet (2007) 370:890–907. doi:10.1016/S0140-6736(07)61416-0
19. Morgan RJ, Alvarez RD, Armstrong DK, Boston B, Burger RA, Chen L-M, et al. Epithelial ovarian cancer. J Natl Compr Canc Netw (2011) 9:82–113.
20. Li J, Fadare O, Xiang L, Kong B, Zheng W. Ovarian serous carcinoma: recent concepts on its origin and carcinogenesis. J Hematol Oncol (2012) 5:8–8. doi:10.1186/1756-8722-5-8
21. Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum Pathol (2011) 42:918–31. doi:10.1016/j.humpath.2011.03.003
22. Holschneider CH, Berek JS. Ovarian cancer: epidemiology, biology, and prognostic factors. Semin Surg Oncol (2000) 19:3–10. doi:10.1002/1098-2388(200007/08)19:1<3::AID-SSU2>3.0.CO;2-S
23. Alsop K, Fereday S, Meldrum C, Defazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol (2012) 30:2654–63. doi:10.1200/JCO.2011.39.8545
24. TCGA-ResearchNetwork. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474:609–15. doi:10.1038/nature10166
25. Cooper BC, Sood AK, Davis CS, Ritchie JM, Sorosky JI, Anderson B, et al. Preoperative CA 125 levels: an independent prognostic factor for epithelial ovarian cancer. Obstet Gynecol (2002) 100:59–64. doi:10.1016/S0029-7844(02)02057-4
26. Rustin GJ, Bast RC Jr, Kelloff GJ, Barrett JC, Carter SK, Nisen PD, et al. Use of Ca-125 in clinical trial evaluation of new therapeutic drugs for ovarian cancer. Clin Cancer Res (2004) 10:3919–26. doi:10.1158/1078-0432.CCR-03-0787
27. Pignata S, Cannella L, Leopardo D, Bruni GS, Facchini G, Pisano C. Follow-up with CA125 after primary therapy of advanced ovarian cancer: in favor of continuing to prescribe CA125 during follow-up. Ann Oncol (2011) 22(Suppl 8):viii40–4. doi:10.1093/annonc/mdr470
28. Duenas-Gonzalez A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer (2005) 4:38. doi:10.1186/1476-4598-4-38
29. Jeong DH, Youm MY, Kim YN, Lee KB, Sung MS, Yoon HK, et al. Promoter methylation of p16, Dapk, CDH1, and Timp-3 genes in cervical cancer: correlation with clinicopathologic characteristics. Int J Gynecol Cancer (2006) 16:1234–40. doi:10.1111/j.1525-1438.2006.00522.x
30. Su P, Lin Y, Huang R, Liao Y, Lee H, Wang H, et al. Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer. Oncogene (2012) 32(1):15–26. doi:10.1038/onc.2012.29
31. Matias-Guiu X, Catasus L, Bussaglia E, Lagarda H, Garcia A, Pons C, et al. Molecular pathology of endometrial hyperplasia and carcinoma. Hum Pathol (2001) 32:569–77. doi:10.1053/hupa.2001.25929
32. Fukuchi T, Sakamoto M, Tsuda H, Maruyama K, Nozawa S, Hirohashi S. Beta-catenin mutation in carcinoma of the uterine endometrium. Cancer Res (1998) 58:3526–8.
33. Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res (2005) 65:10669–73. doi:10.1158/0008-5472.CAN-05-2620
34. Risinger JI, Dent GA, Ignar-Trowbridge D, McLachlan JA, Tsao MS, Senterman M, et al. p53 gene mutations in human endometrial carcinoma. Mol Carcinog (1992) 5:250–3. doi:10.1002/mc.2940050403
35. Ghurani GB, Penalver MA. An update on vulvar cancer. Am J Obstet Gynecol (2001) 185:294–9. doi:10.1067/mob.2001.117401
37. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet (2002) 3:415–28. doi:10.1038/nrg816
38. Kanwal R, Gupta S. Epigenetics and cancer. J Appl Physiol (2010) 109:598–605. doi:10.1152/japplphysiol.00066.2010
39. Gronbaek K, Hother C, Jones PA. Epigenetic changes in cancer. APMIS (2007) 115:1039–59. doi:10.1111/j.1600-0463.2007.apm_636.xml.x
40. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb) (2010) 105:4–13. doi:10.1038/hdy.2010.54
41. Hervouet E, Vallette FM, Cartron PF. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics (2009) 4:487–99. doi:10.4161/epi.4.7.9883
42. Wade PA. Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene (2001) 20:3166–73. doi:10.1038/sj.onc.1204340
43. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature (1983) 301:89–92. doi:10.1038/301089a0
44. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324:930–5. doi:10.1126/science.1170116
45. Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell (2011) 9:193–204. doi:10.1016/j.stem.2011.08.007
46. Niehrs C, Schafer A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol (2012) 22:220–7. doi:10.1016/j.tcb.2012.01.002
47. Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, et al. Nanog-dependent function of TET1 and TET2 in establishment of pluripotency. Nature (2013) 495:370–4. doi:10.1038/nature11925
48. Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell (2011) 8:200–13. doi:10.1016/j.stem.2011.01.008
49. Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A (2013) 110:9920–5. doi:10.1073/pnas.1305172110
50. Liu C, Liu L, Chen X, Shen J, Shan J, Xu Y, et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One (2013) 8:e62828. doi:10.1371/journal.pone.0062828
51. Nephew KP, Balch C, Zhang S, Huang THM. Epigenetics and ovarian cancer. Cancer Treat Res (2010) 149:131–46. doi:10.1007/978-0-387-98094-2_6
52. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21:381–95. doi:10.1038/cr.2011.22
53. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet (2007) 8:286–98. doi:10.1038/nrg2005
54. Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res (2013) 73:473–7. doi:10.1158/0008-5472.CAN-12-3731
55. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer (2006) 6:857–66. doi:10.1038/nrc1997
56. Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol (2008) 9:219–30. doi:10.1038/nrm2347
57. Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol (2011) 223:102–15. doi:10.1002/path.2806
58. Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol (2010) 42:1348–54. doi:10.1016/j.biocel.2010.03.004
59. Selcuklu SD, Donoghue MT, Spillane C. miR-21 as a key regulator of oncogenic processes. Biochem Soc Trans (2009) 37:918–25. doi:10.1042/BST0370918
60. Wang Y, Lee CG. MicroRNA and cancer – focus on apoptosis. J Cell Mol Med (2009) 13:12–23. doi:10.1111/j.1582-4934.2008.00510.x
61. Feng B, Wang R, Chen LB. Review of miR-200b and cancer chemosensitivity. Biomed Pharmacother (2012) 66:397–402. doi:10.1016/j.biopha.2012.06.002
62. Kent O, Mendell J. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene (2006) 25:6188–96. doi:10.1038/sj.onc.1209913
63. Ghigna C, Moroni M, Porta C, Riva S, Biamonti G. Altered expression of heterogeneous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Res (1998) 58:5818–24.
64. Kornblihtt AR, Schor IE, ALL M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol (2013) 14(3):153–65. doi:10.1038/nrm3525
65. Lixia M, Zhijian C, Chao S, Chaojiang G, Congyi Z. Alternative splicing of breast cancer associated gene BRCA1 from breast cancer cell line. J Biochem Mol Biol (2007) 40:15–21. doi:10.5483/BMBRep.2007.40.1.015
66. Karni R, DE Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol (2007) 14:185–93. doi:10.1038/nsmb1209
67. Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol (2012) 19:220–8. doi:10.1038/nsmb.2207
68. Huff V. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer (2011) 11:111–21. doi:10.1038/nrc3002
69. Ladomery M. Aberrant alternative splicing is another hallmark of cancer. Int J Cell Biol (2013) 2013:463786. doi:10.1155/2013/463786
70. Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays (2006) 28:378–86. doi:10.1002/bies.20390
71. Chen K-C, Liao Y-C, Hsieh IC, Wang Y-S, Hu C-Y, Juo S-HH. OxLDL causes both epigenetic modification and signaling regulation on the microRNA-29b gene: novel mechanisms for cardiovascular diseases. J Mol Cell Cardiol (2012) 52:587–95. doi:10.1016/j.yjmcc.2011.12.005
72. Li Y, Kong D, Ahmad A, Bao B, Dyson G, Sarkar FH. Epigenetic deregulation of miR-29a and miR-1256 by isoflavone contributes to the inhibition of prostate cancer cell growth and invasion. Epigenetics (2012) 7:940–9. doi:10.4161/epi.21236
73. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A (2007) 104:15805–10. doi:10.1073/pnas.0707628104
74. Meunier L, Siddeek B, Vega A, Lakhdari N, Inoubli L, Bellon RP, et al. Perinatal programming of adult rat germ cell death after exposure to xenoestrogens: role of microRNA miR-29 family in the down-regulation of DNA methyltransferases and Mcl-1. Endocrinology (2012) 153:1936–47. doi:10.1210/en.2011-1109
75. Wang Y, Zhang X, Li H, Yu J, Ren X. The role of miRNA-29 family in cancer. Eur J Cell Biol (2012) 92(3):123–8. doi:10.1016/j.ejcb.2012.11.004
76. Li Q, Zhu F, Chen P. miR-7 and miR-218 epigenetically control tumor suppressor genes RASSF1A and Claudin-6 by targeting HoxB3 in breast cancer. Biochem Biophys Res Commun (2012) 424:28–33. doi:10.1016/j.bbrc.2012.06.028
77. Verduci L, Simili M, Rizzo M, Mercatanti A, Evangelista M, Mariani L, et al. MicroRNA (miRNA)-mediated interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J Biol Chem (2010) 285:39551–63. doi:10.1074/jbc.M110.114736
78. Creighton CJ, Hernandez-Herrera A, Jacobsen A, Levine DA, Mankoo P, Schultz N, et al. Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS One (2012) 7:e34546. doi:10.1371/journal.pone.0034546
79. Raeder MB, Birkeland E, Trovik J, Krakstad C, Shehata S, Schumacher S, et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to Myc-dependent cancers. PLoS One (2013) 8:e54873. doi:10.1371/journal.pone.0054873
80. La DE, Rica L, Urquiza JM, Gomez-Cabrero D, Islam AB, Lopez-Bigas N, et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun (2013) 41:6–16. doi:10.1016/j.jaut.2012.12.005
81. Kresse SH, Rydbeck H, Skarn M, Namlos HM, Barragan-Polania AH, Cleton-Jansen AM, et al. Integrative analysis reveals relationships of genetic and epigenetic alterations in osteosarcoma. PLoS One (2012) 7:e48262. doi:10.1371/journal.pone.0048262
82. Balch C, Huang THM, Brown R, Nephew KP. The epigenetics of ovarian cancer drug resistance and resensitization. Am J Obstet Gynecol (2004) 191:1552–72. doi:10.1016/j.ajog.2004.05.025
83. Cao J, Yan Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol (2012) 2:26. doi:10.3389/fonc.2012.00026
84. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell (2007) 129:823–37. doi:10.1016/j.cell.2007.05.009
85. Rosenfeld JA, Wang Z, Schones DE, Zhao K, Desalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics (2009) 10:143. doi:10.1186/1471-2164-10-143
86. Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev (2004) 18:1251–62. doi:10.1101/gad.300704
87. Benevolenskaya EV. Histone H3K4 demethylases are essential in development and differentiation. Biochem Cell Biol (2007) 85:435–43. doi:10.1139/O07-057
88. Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res (2007) 17:691–707. doi:10.1101/gr.5704207
89. Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol (2008) 28:2825–39. doi:10.1128/MCB.02076-07
90. Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem (2001) 70:81–120. doi:10.1146/annurev.biochem.70.1.81
91. Costa FF, Paixao VA, Cavalher FP, Ribeiro KB, Cunha IW, Rinck JA Jr, et al. Satr-1 hypomethylation is a common and early event in breast cancer. Cancer Genet Cytogenet (2006) 165:135–43. doi:10.1016/j.cancergencyto.2005.07.023
92. Widschwendter M, Jiang G, Woods C, Muller HM, Fiegl H, Goebel G, et al. DNA hypomethylation and ovarian cancer biology. Cancer Res (2004) 64:4472–80. doi:10.1158/0008-5472.CAN-04-0238
93. Hur K, Cejas P, Feliu J, Moreno-Rubio J, Burgos E, Boland CR, et al. Hypomethylation of long interspersed nuclear element-1 (Line-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut (2013). doi:10.1136/gutjnl-2012-304219
94. Pavicic W, Joensuu EI, Nieminen T, Peltomaki P. LINE-1 hypomethylation in familial and sporadic cancer. J Mol Med (Berl) (2012) 90:827–35. doi:10.1007/s00109-011-0854-z
95. Sunami E, DE Maat M, Vu A, Turner RR, Hoon DS. LINE-1 hypomethylation during primary colon cancer progression. PLoS One (2011) 6:e18884. doi:10.1371/journal.pone.0018884
96. Gao XD, Qu JH, Chang XJ, Lu YY, Bai WL, Wang H, et al. Hypomethylation of long interspersed nuclear element-1 promoter is associated with poor outcomes for curative resected hepatocellular carcinoma. Liver Int (2014) 34(1):136–46. doi:10.1111/liv.12264
97. Shigaki H, Baba Y, Watanabe M, Murata A, Iwagami S, Miyake K, et al. LINE-1 hypomethylation in gastric cancer, detected by bisulfite pyrosequencing, is associated with poor prognosis. Gastric Cancer (2012) 16(4):480–7. doi:10.1007/s10120-012-0209-7
98. Aoki Y, Nojima M, Suzuki H, Yasui H, Maruyama R, Yamamoto E, et al. Genomic vulnerability to LINE-1 hypomethylation is a potential determinant of the clinicogenetic features of multiple myeloma. Genome Med (2012) 4:101. doi:10.1186/gm402
99. Chan KYK, OZ Elik H, Cheung ANY, Ngan HYS, Khoo US. Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer. Cancer Res (2002) 62:4151–6.
100. Dann RB, Deloia JA, Timms KM, Zorn KK, Potter J, Flake DD II, et al. BRCA1/2 mutations and expression: response to platinum chemotherapy in patients with advanced stage epithelial ovarian cancer. Gynecol Oncol (2012) 125:677–82. doi:10.1016/j.ygyno.2012.03.006
101. Hsu YT, Gu F, Huang YW, Liu J, Ruan J, Huang RL, et al. Promoter hypomethylation of EpCAM-regulated bone morphogenetic protein gene family in recurrent endometrial cancer. Clin Cancer Res (2013) 19:6272–85. doi:10.1158/1078-0432.CCR-13-1734
102. Lee TS, Kim JW, Kang GH, Park NH, Song YS, Kang SB, et al. DNA hypomethylation of CAGE promotors in squamous cell carcinoma of uterine cervix. Ann N Y Acad Sci (2006) 1091:218–24. doi:10.1196/annals.1378.068
103. Honda H, Pazin MJ, Ji H, Wernyj RP, Morin PJ. Crucial roles of Sp1 and epigenetic modifications in the regulation of the CLDN4 promoter in ovarian cancer cells. J Biol Chem (2006) 281:21433–44. doi:10.1074/jbc.M603767200
104. Honda H, Pazin MJ, D’Souza T, Ji H, Morin PJ. Regulation of the CLDN3 gene in ovarian cancer cells. Cancer Biol Ther (2007) 6:1733–42. doi:10.4161/cbt.6.11.4832
105. Cheng W, Jiang Y, Liu C, Shen O, Tang W, Wang X. Identification of aberrant promoter hypomethylation of HOXA10 in ovarian cancer. J Cancer Res Clin Oncol (2010) 136:1221–7. doi:10.1007/s00432-010-0772-4
106. Lee PS, Teaberry VS, Bland AE, Huang Z, Whitaker RS, Baba T, et al. Elevated MAL expression is accompanied by promoter hypomethylation and platinum resistance in epithelial ovarian cancer. Int J Cancer (2010) 126:1378–89. doi:10.1002/ijc.24797
107. Gupta A, Godwin AK, Vanderveer L, Lu A, Liu J. Hypomethylation of the synuclein gamma gene CpG Island promotes its aberrant expression in breast carcinoma and ovarian carcinoma. Cancer Res (2003) 63:664–73.
108. Izutsu N, Maesawa C, Shibazaki M, Oikawa H, Shoji T, Sugiyama T, et al. Epigenetic modification is involved in aberrant expression of class III beta-tubulin, TUBB3, in ovarian cancer cells. Int J Oncol (2008) 32:1227. doi:10.3892/ijo_32_6_1227
109. Dahl KDC, Dahl R, Kruichak JN, Hudson LG. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia (2009) 11:1208. doi:10.1593/neo.09942
110. Guan Y, Yao H, Zheng Z, Qiu G, Sun K. MiR-125b targets BCL3 and suppresses ovarian cancer proliferation. Int J Cancer (2011) 128:2274–83. doi:10.1002/ijc.25575
111. Bhattacharya R, Nicoloso M, Arvizo R, Wang E, Cortez A, Rossi S, et al. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res (2009) 69:9090–5. doi:10.1158/0008-5472.CAN-09-2552
112. Yang Z, Chen S, Luan X, Li Y, Liu M, Li X, et al. MicroRNA-214 is aberrantly expressed in cervical cancers and inhibits the growth of HeLa cells. IUBMB Life (2009) 61:1075–82. doi:10.1002/iub.252
113. Guo LM, Pu Y, Han Z, Liu T, Li YX, Liu M, et al. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-κB1. FEBS J (2009) 276:5537–46. doi:10.1111/j.1742-4658.2009.07237.x
114. Huang F, Lin C, Shi YH, Kuerban G. MicroRNA-101 inhibits cell proliferation, invasion, and promotes apoptosis by regulating cyclooxygenase-2 in Hela cervical carcinoma cells. Asian Pac J Cancer Prev (2013) 14:5915–20. doi:10.7314/APJCP.2013.14.10.5915
115. Yamamoto N, Kinoshita T, Nohata N, Yoshino H, Itesako T, Fujimura L, et al. Tumor-suppressive microRNA-29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol (2013) 43:1855–63. doi:10.3892/ijo.2013.2145
116. Zhu X, Er K, Mao C, Yan Q, Xu H, Zhang Y, et al. miR-203 suppresses tumor growth and angiogenesis by targeting VEGFA in cervical cancer. Cell Physiol Biochem (2013) 32:64–73. doi:10.1159/000350125
117. Huang YW, Liu JC, Deatherage DE, Luo J, Mutch DG, Goodfellow PJ, et al. Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res (2009) 69:9038–46. doi:10.1158/0008-5472.CAN-09-1499
118. Yang N, Kaur S, Volinia S, Greshock J, Lassus H, Hasegawa K, et al. MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res (2008) 68:10307–14. doi:10.1158/0008-5472.CAN-08-1954
119. Wilting SM, Verlaat W, Jaspers A, Makazaji NA, Agami R, Meijer CJ, et al. Methylation-mediated transcriptional repression of microRNAs during cervical carcinogenesis. Epigenetics (2013) 8(2):220–8. doi:10.4161/epi.23605
120. Pereira PM, Marques JP, Soares AR, Carreto L, Santos MA. MicroRNA expression variability in human cervical tissues. PLoS One (2010) 5:e11780. doi:10.1371/journal.pone.0011780
121. Agarwal R, D’Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res (2005) 65:7378–85. doi:10.1158/0008-5472.CAN-05-1036
122. Yao T, Rao Q, Liu L, Zheng C, Xie Q, Liang J, et al. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in cervical cancer. Virol J (2013) 10:175. doi:10.1186/1743-422X-10-175
123. Aaboe M, Birkenkamp-Demtroder K, Wiuf C, Sorensen FB, Thykjaer T, Sauter G, et al. SOX4 expression in bladder carcinoma: clinical aspects and in vitro functional characterization. Cancer Res (2006) 66:3434–42. doi:10.1158/0008-5472.CAN-05-3456
124. Liao YL, Sun YM, Chau GY, Chau YP, Lai TC, Wang JL, et al. Identification of SOX4 target genes using phylogenetic footprinting-based prediction from expression microarrays suggests that overexpression of SOX4 potentiates metastasis in hepatocellular carcinoma. Oncogene (2008) 27:5578–89. doi:10.1038/onc.2008.168
125. Liu P, Ramachandran S, ALI Seyed M, Scharer CD, Laycock N, Dalton WB, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res (2006) 66:4011–9. doi:10.1158/0008-5472.CAN-05-3055
126. Medina PP, Castillo SD, Blanco S, Sanz-Garcia M, Largo C, Alvarez S, et al. The Sry-HMG box gene, SOX4, is a target of gene amplification at chromosome 6p in lung cancer. Hum Mol Genet (2009) 18:1343–52. doi:10.1093/hmg/ddp034
127. Samartzis N, Imesch P, Dedes KJ, Samartzis EP, Fedier A, Fink D, et al. Expression pattern of class I histone deacetylases in vulvar intraepithelial neoplasia and vulvar cancer: a tissue microarray study. BMC Cancer (2011) 11:463. doi:10.1186/1471-2407-11-463
128. Feng Q, Balasubramanian A, Hawes SE, Toure P, Sow PS, Dem A, et al. Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst (2005) 97:273–82. doi:10.1093/jnci/dji318
129. Kalantari M, Osann K, Calleja-Macias IE, Kim S, Yan B, Jordan S, et al. Methylation of human papillomavirus 16, 18, 31, and 45 L2 and L1 genes and the cellular DAPK gene: considerations for use as biomarkers of the progression of cervical neoplasia. Virology (2014) 448:314–21. doi:10.1016/j.virol.2013.10.032
130. Bryant D, Tristram A, Liloglou T, Hibbitts S, Fiander A, Powell N. Quantitative measurement of human papillomavirus type 16 L1/L2 DNA methylation correlates with cervical disease grade. J Clin Virol (2013) 59(1):24–9. doi:10.1016/j.jcv.2013.10.029
131. Lai HC, Lin YW, Huang TH, Yan P, Huang RL, Wang HC, et al. Identification of novel DNA methylation markers in cervical cancer. Int J Cancer (2008) 123:161–7. doi:10.1002/ijc.23519
132. Fiegl H, Gattringer C, Widschwendter A, Schneitter A, Ramoni A, Sarlay D, et al. Methylated DNA collected by tampons – a new tool to detect endometrial cancer. Cancer Epidemiol Biomarkers Prev (2004) 13:882–8.
133. Zheng H, Zhang L, Zhao Y, Yang D, Song F, Wen Y, et al. Plasma miRNAs as diagnostic and prognostic biomarkers for ovarian cancer. PLoS One (2013) 8:e77853. doi:10.1371/journal.pone.0077853
134. Strathdee G, Davies B, Vass J, Siddiqui N, Brown R. Cell type-specific methylation of an intronic CpG Island controls expression of the MCJ gene. Carcinogenesis (2004) 25:693–701. doi:10.1093/carcin/bgh066
135. Strathdee G, Vass JK, Oien KA, Siddiqui N, Curto-Garcia J, Brown R. Demethylation of the MCJ gene in stage III/IV epithelial ovarian cancer and response to chemotherapy. Gynecol Oncol (2005) 97:898–903. doi:10.1016/j.ygyno.2005.03.023
136. Fiegl H, Windbichler G, Mueller-Holzner E, Goebel G, Lechner M, Jacobs IJ, et al. HOXA11 DNA methylation – a novel prognostic biomarker in ovarian cancer. Int J Cancer (2008) 123:725–9. doi:10.1002/ijc.23563
137. Guerrero D, Guarch R, Ojer A, Casas JM, Mendez-Meca C, Esteller M, et al. Differential hypermethylation of genes in vulvar cancer and lichen sclerosus coexisting or not with vulvar cancer. Int J Cancer (2011) 128:2853–64. doi:10.1002/ijc.25629
138. Stephen JK, Chen KM, Raitanen M, Grenman S, Worsham MJ. DNA hypermethylation profiles in squamous cell carcinoma of the vulva. Int J Gynecol Pathol (2009) 28:63–75. doi:10.1097/PGP.0b013e31817d9c61
139. Pakneshan P, Tetu B, Rabbani SA. Demethylation of urokinase promoter as a prognostic marker in patients with breast carcinoma. Clin Cancer Res (2004) 10:3035–41. doi:10.1158/1078-0432.CCR-03-0545
140. Paredes J, Albergaria A, Oliveira JT, Jeronimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res (2005) 11:5869–77. doi:10.1158/1078-0432.CCR-05-0059
141. Itano O, Ueda M, Kikuchi K, Hashimoto O, Hayatsu S, Kawaguchi M, et al. Correlation of postoperative recurrence in hepatocellular carcinoma with demethylation of repetitive sequences. Oncogene (2002) 21:789–97. doi:10.1038/sj.onc.1205124
142. Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Triratanachat S, Tresukosol D, et al. LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int J Gynecol Cancer (2008) 18:711–7. doi:10.1111/j.1525-1438.2007.01117.x
143. Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist (2005) 10:176–82. doi:10.1634/theoncologist.10-3-176
144. Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell (2006) 9:435–43. doi:10.1016/j.ccr.2006.04.020
145. Wang QZ, Lv YH, Gong YH, Li ZF, Xu W, Diao Y, et al. Double-stranded Let-7 mimics, potential candidates for cancer gene therapy. J Physiol Biochem (2012) 68:107–19. doi:10.1007/s13105-011-0124-0
Keywords: epigenetic derepression, gynecologic cancer, DNA methylation, histone modification, microRNA, epigenetic therapy
Citation: Jeong HM, Kwon MJ and Shin YK (2014) Overexpression of cancer-associated genes via epigenetic derepression mechanisms in gynecologic cancer. Front. Oncol. 4:12. doi: 10.3389/fonc.2014.00012
Received: 19 October 2013; Accepted: 20 January 2014;
Published online: 04 February 2014.
Edited by:
Ivan Garcia-Bassets, University of California San Diego, USAReviewed by:
Miriam Reuschenbach, University Hospital Heidelberg, GermanyReuven Reich, Hebrew University of Jerusalem, Israel
Deborah Marsh, Kolling Institute of Medical Research, Australia
Copyright: © 2014 Jeong, Kwon and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Young Kee Shin, Department of Pharmacy, College of Pharmacy, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, South Korea e-mail:eWtlZXNoaW5Ac251LmFjLmty