 Jon Schoorlemmer
Jon Schoorlemmer Raquel Pérez-Palacios
Raquel Pérez-Palacios María Climent
María Climent Diana Guallar1†
Diana Guallar1† Pedro Muniesa
Pedro Muniesa- 1Regenerative Medicine Program, Instituto Aragonés de Ciencias de la Salud, Zaragoza, Spain
- 2ARAID Foundation, Zaragoza, Spain
- 3Departamento de Anatomía, Embriología y Genética Animal, Facultad de Veterinaria, Universidad de Zaragoza, Zaragoza, Spain
About half of the mammalian genome is occupied by DNA sequences that originate from transposable elements. Retrotransposons can modulate gene expression in different ways and, particularly retrotransposon-derived long terminal repeats, profoundly shape expression of both surrounding and distant genomic loci. This is especially important in pre-implantation development, during which extensive reprograming of the genome takes place and cells pass through totipotent and pluripotent states. At this stage, the main mechanism responsible for retrotransposon silencing, i.e., DNA methylation, is inoperative. A particular retrotransposon called muERV-L/MERVL is expressed during pre-implantation stages and contributes to the plasticity of mouse embryonic stem cells. This review will focus on the role of MERVL-derived sequences as controlling elements of gene expression specific for pre-implantation development, two-cell stage-specific gene expression, and stem cell pluripotency, the epigenetic mechanisms that control their expression, and the contributions of the pluripotency marker REX1 and the related Yin Yang 1 family of transcription factors to this regulation process.
Introduction
Transposable elements (TEs) are DNA sequences with the ability to move from one chromosomal location to another. They were discovered by McClintock (1), who was awarded the 1983 Nobel Prize in Physiology or Medicine for this work. To explain color patterns in maize kernels, she postulated that gene expression might be regulated by “controlling elements” that could jump around the genome [reviewed by Fedoroff (2)]. Initial drafts of both human and mouse genomes indicated that about half of the mammalian genomes are occupied by DNA sequences that originate from TE (3). Posterior estimates are even higher up to two-thirds (4). Among the TEs figure the endogenous retroviral elements (ERVs), which make up about 10% of mammalian genomes. As opposed to exogenous retrovirus, ERVs are an integral component of the genome in all cells of an organism. ERVs are the remnants of ancient retroviral infections of the germline that have produced stable integrations in the genome, which are therefore passed on to the offspring in a Mendelian fashion (5, 6). Expression of ERV is usually repressed by mechanisms dependent on DNA methylation (7), and deregulation of human ERV (HERV) contributes to disease (8). In contrast to other mammals like mouse and cat, there exists no definitive proof for the presence of infectious HERV particles, although ERV might be mobile even in human (9). Therefore, HERV might contribute to tumor development as retroviruses do (8), through HERV-encoded transcripts, or by regulating expression of genes nearby or even at a distance. Indeed, the expression of HERVs has been linked to a variety of tumors (8, 10), although HERV has not presently been identified as an etiological factor in tumor development.
Pluripotent self-renewing embryonic stem (ES) cells can be derived from the inner cell mass (ICM) of the pre-implantation blastocyst. Cells from earlier embryos (zygotes until eight-cell embryos) differentiate into both fetal and placental cell types. This ability is called totipotency as opposed to the more restricted differentiation potential of pluripotent ICM cells, which only contribute to embryonic tissue (11, 12). ES cells in culture are heterogeneous (13, 14), and several of their characteristics can best be understood in the context of successive changes occurring during pre-implantation development, as the cells transition between states that resemble cellular identities at distinct stages of pre-implantation development. Mouse ES cells display special epigenetic features such as a loose chromatin structure and altered DNA methylation, which underlie their cell type-specific properties (15). Similar to the pre-implantation embryo, mouse ES cells express several kinds of ERV, in line with the absence of DNA methylation. Among the ERV expressed is MERVL, whose expression actually peaks in two-cell stage embryos (16). Importantly, MERVL expression in mouse ES cells is restricted to a subpopulation of the cells with special characteristics as defined by gene expression and differentiation potential normally found in cells of two-cell embryos (17). Degenerated cis-acting elements derived from MERVL elements are also essential to pluripotency-related phenomena, i.e., re-activation of the second X-chromosome (18, 19), and we speculate on its role in developmental potency and differentiation.
We will review how MERVL sequences drive gene expression restricted to different states of developmental potency in mouse stem cells. These contributions depend on epigenetic regulation mechanisms exerted by protein complexes that modify or read histone modifications, particularly Polycomb complexes and demethylase complexes. REX1 is a pluripotency-associated nuclear protein that binds MERVL elements in vivo, and a contribution of REX1 to silencing of MERVL has been demonstrated (20). REX1 reunites additional characteristics consistent with a role in orchestrating how regulatory complexes interact with and shape MERVL-dependent transcription. We present a hypothetical model incorporating this potential role for REX1. As studies to determine the role of HERV -driven processes for human ES cells (HESCs) are ongoing, we will briefly review differences with the mouse and explain outstanding questions.
Apart from pluripotent cells, several TEs including ERV are active and expressed in the germline, in pre-implantation embryos, and in the placenta (21, 22). These are exactly the tissues relatively devoid of DNA methylation (23), similar to the epigenetic chromatin state in many tumors (24), which is also characterized by widespread DNA hypomethylation. At present, there is little information on the potential activity of other repressing mechanisms toward TE silencing, apart from the main methylation-dependent mechanism (7). However, we suggest that the epigenetic mechanism operative in ES cells described here may carry out such functions. We hope that a better understanding of the DNA-binding factors and their interactions with chromatin modifying regulators may advance future understanding of the relationship between HERV regulation and tumor prevention. We therefore point out the activity of similar mechanisms in human cells and consider their potential relevance for tumor formation and/or progression.
Transposable Elements and Endogenous Retroviral Elements
Transposable elements exist as either DNA transposons that directly jump from one location to another or as so-called retrotransposons that use an RNA intermediate (which in turn is retro-transcribed into DNA before reinsertion into the genome). Retrotransposons can be further divided into long terminal repeat (LTR)-containing TEs (LTR retrotransposons and ERV) and non-LTR retrotransposons LINE and SINE (long and short interspersed nuclear elements).
Long terminal repeats are generated during the reverse transcription step. LTRs recruit the cellular transcription factors (TFs) in charge of proviral transcription and produce the 5′ and 3′ ends of the transcripts (25). In mammals all LTR transposons are related to ERV, which are considered (defective) descendents of ancient retroviral infections of the germline.
Animal retroviral diseases (i.e., Jaagsiekte in sheep) were already described at the turn of the nineteenth century, and particles derived from ERV were identified in the late 1960s in birds and mice. Also, in different mammalian species abundant expression of ERV in placenta and trophoblastic cells had been known since the 1970s (26–28). Reverse transcriptase (RT) assays and electron microscopy were used to identify additional ERV particles, and ERV-reactive antibodies were searched for in sera and other body fluids. The understanding of ERV was limited however, as neither the integration of RNA in the genome, nor non-Mendelian genetics were widely accepted concepts at the time.
Much was learned about ERV in the course of studies on tumor-producing virus in chicken, especially avian endogenous leukosis virus (ALV) and the closely related Rous sarcoma virus (RSV), using a combination of virological and immunological methods common at the time or developed for this purpose [reviewed by Weiss (29)]. Neutralizing sera against envelope proteins were available and serological tests were developed for “group-specific antigen” (GAG) common to serotypes. This allowed the detection of GAG protein in non-infected animals, suggesting the endogenous presence of this viral protein. Mendelian transmission of virally derived characteristics was established in appropriate crosses (i.e., between Gag-positive and negative inbred lines). Furthermore, the use of nucleic acid hybridization allowed for a positive identification of endogenous copies very similar to virus-derived RNA. HERV was first discovered in normal brain tissue (30), and similar to other mammalian species the human placenta was shown to be permissive to the expression of HERV.
As repeated DNA elements, ERVs were included in the category of “junk” DNA and long considered not relevant. More detailed analysis has further been hampered by cross-hybridization among related elements, and both element-specific probes and amplification assays have been scarce for a long time. Extensive sequencing projects that have delivered complete mouse and human genomes, have finally provided tools and information to appreciate the extension and importance to the genome of TE and ERV in particular (21).
As opposed to exogenous retrovirus, ERVs are an integral component of the genome in all cells of an organism (5, 6). Posterior to integration, re-activation, amplification, and transposition events have produced defective proviral derivatives of variable integrity dispersed throughout the genome. In the most extensive cases of genetic lesion/degeneration only isolated LTRs are left as molecular fossils of previous integrations. Autonomous ERV encode the canonical retroviral GAG (capsid and matrix protein), POL (RT; IN, integrase), and ENV (envelope proteins) (Figure 1). Although over the years ERVs have mutated and accumulated defects in the coding regions of some or all of their genes, some still have open reading frames (ORFs) and thus direct the expression of (a subset of) these proteins. Dependent on the amount of mutations acquired, autonomous elements can still recruit the necessary cellular machinery to produce infectious particles, while non-autonomous variants rely on related elements to do so. Although TEs are classified based on sequence similarity, each group consists of a range of similar but clearly distinguishable elements. Furthermore, within each group copy numbers and autonomy status differs between individual elements (5).
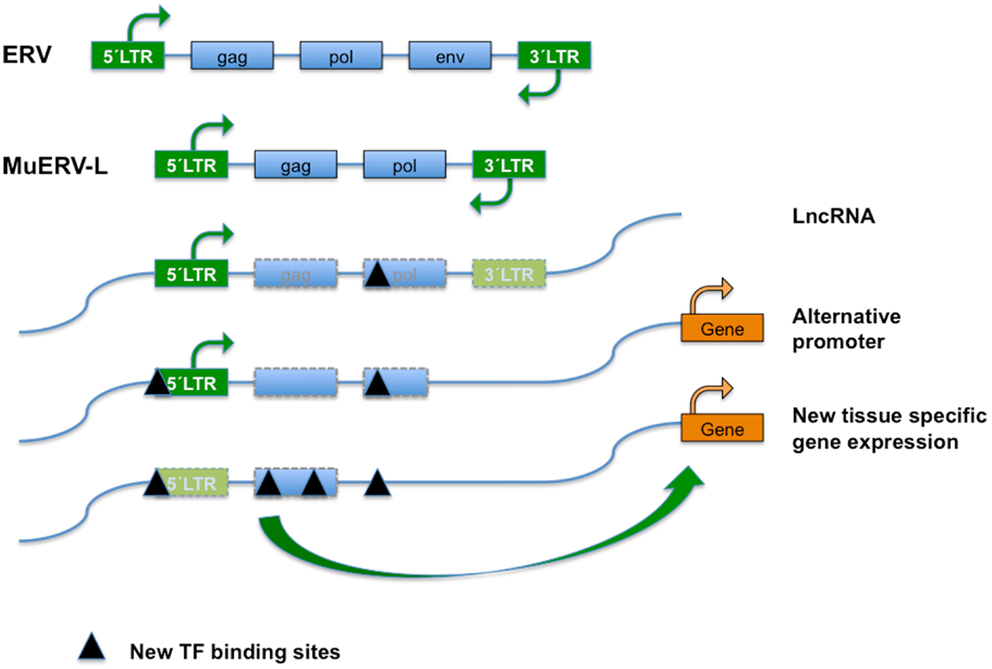
Figure 1. Endogenous retroviral elements (ERVs) result from the occasional infection of a germline cell by a retrovirus, whose (potentially) defective offspring is transmitted further as a provirus in a Mendelian fashion. ERVs typically encode the canonical retroviral proteins GAG (group-specific antigens; capsid and matrix protein), POL (RT, reverse transcriptase; IN, integrase), and ENV (envelope). The ERVs are divided into three classes based on the sequence conservation with active retrovirus; class I (eMLVs), class II (musD/ETn/IAPs), and class III (ERV-L). ERVs are flanked by LTRs (green boxes) of 300–1,200 nucleotides as a result of the retrotransposition event. Class III ERVs make up almost 5% of the mouse genome and consists of muERV-L/MERVL elements (depicted) and the non-autonomous mammalian apparent LTR retrotransposons (MaLRs). The muERV-L family members have Gag and Pol genes but no detectable Env, a dUTPase gene (not shown). For the lack of ENV they are defective for cell to cell transmission. In addition to muERV-L, the mouse genome contains many thousands of ERV-L derived LTR elements termed MT2-Mm. LTRs are bound by transcription factors (TF) that drive transcription. ERV elements may also bind TF in the body of their genes, probably influencing chromatin dynamics or surveillance. After initial insertion (and transposition), ERVs over time may accumulate mutations and have contributed significantly to the generation of new transcription factor binding sites and hence impact on gene expression, genome function, and evolution. Transposon-derived promoters drive expression of alternative transcripts, including many of the LncRNAs detected by the ENCODE project, orchestrate stage, and tissue-dependent transcription, or serve as alternate promoters.
Based on the sequence of their RT genes and their relationship to described genera of exogenous retroviruses, ERVs are divided into three families or classes (31): ERVI/class I, ERV-K/class II (IAPs and MusD/ETn), and class III (ERV-L/MaLR). Class I ERVs comprise <1% of the mouse genome and this class is typified by the well-studied type-C murine leukemia viruses (MuLV), and also includes eMLVs, VL30, and MuRRS elements. Mouse mammary tumor virus (MMTV) is typical of the class II ERVs, which also includes IAP, musD, and deleted musD variants called ETn elements. MusD elements contain Gag-Pro-Pol genes, but lack an Env gene and vary from 5 to 7 kb in length. ETns are flanked by LTRs but mainly contain sequences of unknown function.
The Class III ERV in the mouse consists of two types of retrotransposon elements: murine ERV-L elements (muERV-L/MERVL) and the mammalian apparent LTR retrotransposons or MaLRs. After its initial identification in humans (32) the ERV-L family turned up in all placental mammals. The ERV-L family members are about 6.5 kbp in length and carry Gag and Pol but no Env genes, and carry an extra dUTPase gene (Figure 1). MERVL/MuERV-L (mouse ERV with a leucine tRNA primer binding site), is present in over 650 full-length copies in the C57BL/6 genome.
In addition, the C57BL/6 genome carries 37,000–38,000 copies of MERVL-derived LTRs called MT2-Mm. MT2 (MERVL) and MaLR LTRs share about 50% homology at the DNA level. MaLRs are very common, non-autonomous LTR elements that occupy 4.8% of the mouse genome. Among the MaLR retrotransposons three subfamilies are discerned, termed MT (about 2 kb), ORR1 (2.5 kb), and MLT. The non-autonomous but active MaLRs are all internally deleted, containing only non-coding repetitive DNA flanked by LTRs.
Elements of active TE/ERV families in the mouse (i.e., IAP and ETn in the mouse) display high levels of polymorphism, contributing to genetic variability within species. Elements detected in one strain (C57BL/6J) are absent in other three strains investigated (60 and 25% of IAP and of ETn/MusD elements, respectively), and differences in expression levels or the generation of alternative transcripts of ERV elements was also observed (33).
The activity of TEs in general and ERV in particular may contribute to pathogenic processes in different ways (1). Stress signals resulting from injury, infection, or inflammation may relieve repression of ERVs, provoking the expression and assembly of infectious virions. Independent of the pathogenicity of the resulting virions, reintegration may damage the genome (2). De-repressed ERVs may direct the production of retroviral proteins that either contribute to cell fusion (34) or display characteristics as superantigens causing inflammation (3, 35). Altered gene activity of ERV loci can affect the expression of neighboring cellular genes (25, 36).
To avoid deleterious effects of TEs/ERVs activation of any kind, mammalian cells have acquired a multitude of defense responses. They act at different stages of the TEs life cycle starting with proviral transcription and processing of resulting RNAs. Other mechanisms interfere with various steps required for productive infection and integration of retrovirus into the genome: release of particles, receptor binding, uncoating, deamination (APOBEC enzymes), and intracellular trafficking (31). In the mouse germline, transcriptional repression of retrotransposons is regulated through DNA methylation of their regulatory LTR regions (37). This process is dependent on co-operating small RNAs (piRNAs) and PIWI-proteins in a mechanism unique to the germline in mammals (38). This review will focus on the role of transposons as controlling elements of pre-implantation development and stem cell pluripotency at the level of transcriptional regulation. As there is no evidence that the PIWI pathway is relevant for these tissues, it will not be further discussed here.
The Contribution of TE to the Evolution of Regulatory DNA
Endogenous retroviral elements largely lack transcriptional activity in differentiated cells and tissues as a result of DNA methylation-dependent silencing (7). However, expression of ERV families in different species is elevated in the germ cells, in pre-implantation embryos and also in the placenta (39–42).
Retrotransposons modulate gene expression in these tissues in different ways [reviewed in Ref. (36), see also Figure 1]. Transcription of retrotransposons may serve as a source of small RNAs that interfere with gene transcription, or produce long non-coding RNAs (lncRNAs). A substantial portion of total lncRNA sequence across species (~30% in human) is TE-derived. Conversely, TEs contribute to a full two-thirds of mature lncRNA transcripts, as relative to their absence from protein-coding transcripts (43). TEs and particularly retrotransposon-derived LTRs profoundly shape expression of surrounding genomic loci through long distance enhancer or repressor activities, which may favor the use of alternative promoters, exons/splice sites, or polyadenylation sites (25, 36, 44).
It has been argued that the accumulation of TE in the genome has provided an abundant source of non-functional DNA with progressive mutations for evolution to act upon. Indeed (degenerated) LTR from ERV have been frequently recruited as binding sites for TFs (45). A well-studied example is the expansion of binding sites for the TF CTCF (CCCTC-binding factor), a DNA-binding protein that insulates transcriptional and chromatin domains (46). Mouse ES cells have thousands of CTCF-binding sites that are not conserved in the human genome, as they are found in B2 retrotransposons (47), a rodent-specific SINE, which originates from accidental retrotransposition of various polymerase III transcripts (31). Species-specific activation of retroelements has generated novel species-restricted CTCF-binding sites with active chromatin insulator function also in other species including rodents, dogs, and opossum (48).
Similarly, specific families of TE are enriched for TF binding sites, for example Class II elements for SOX2 and POU5F1 binding (47). Similar to the extension of CTCF-binding sites, regulatory elements carried on by transposons contribute to differences in gene expression patterns between different mammalian species (49). In line with remarkably restricted and regulated expression patterns, retrotransposon-directed transcription contributes to gene regulation in the endometrium (50), and during pre-implantation development (40). Transposon-derived promoters synchronize stage-specific gene expression in two-cell embryos (36, 51) and ERV-derived cis elements have been incorporated in regulatory networks in pluripotent stem cells (see below).
Pre-Implantation Development
After fertilization, the mammalian zygote develops into a multi-cellular blastocyst, which implants in the uterus for further development. Already at the first stage, genome-wide reorganization takes place to ensure that after protamine/histone exchange and demethylation of the male genome and the initiation of replication of haploid genomes, fusion of pronuclei takes place. Large-scale chromatin reorganization includes but is not limited to epigenetic modifications that are intertwined with zygotic gene activation (ZGA). Methylated cytosines are removed from both parental genomes in early cleavage embryos (23). While the maternal genome is passively and progressively demethylated during cleavage divisions (Figure 2), the paternal genome is actively demethylated in the zygote, at least in the mouse (52). During these early stages, a distinct chromatin structure and nuclear architecture are in place concomitant with the absence of structural restrictions to transcription.
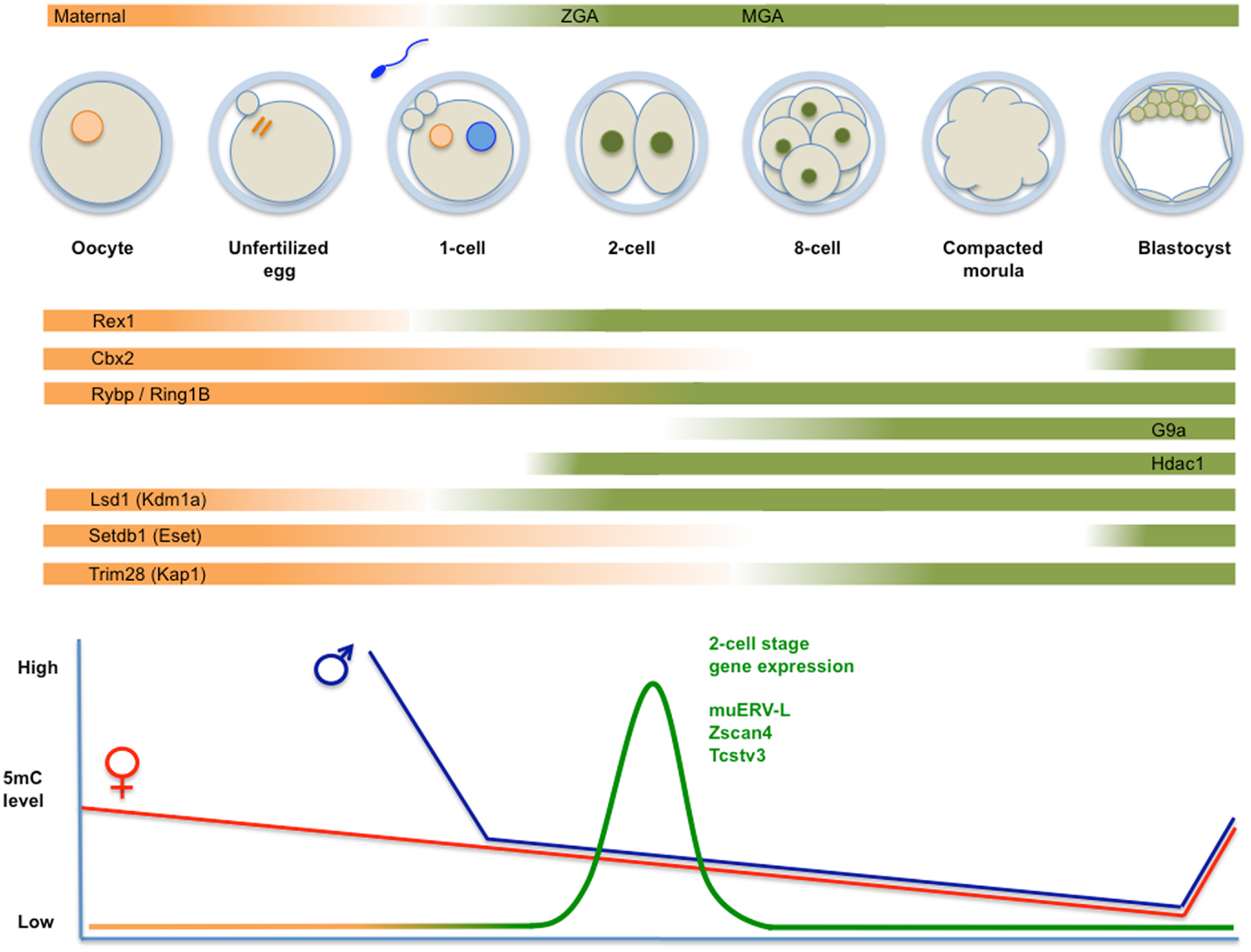
Figure 2. Top panel: schematic representation of fertilization, formation of the zygote, and cleavage divisions, followed by the successive generation of morula and blastocyst. Transcripts derive from maternal deposits, or correspond to the two transient waves of de novo transcription: zygotic gene activation (late zygote-two-cell) or ZGA, and mid pre-implantation gene activation (MGA). The temporal changes in expression levels of important chromatin modifiers are indicated. Bottom panel: time course of genome-wide (de)methylation. By the time of implantation, DNA methylation is re-established and TE are rapidly silenced. Demethylation is not uniform, i.e., certain TE (i.e., IAP) and imprinted loci are protected from demethylation. The diagram also shows a peak of unique expression of muERV-L, Zscan4, Tcstv3, and others in two-cell embryos.
Several excellent reviews detail the progress of cell division followed by cellular differentiation steps during pre-implantation development and the accompanying changes in chromatin states (15, 53, 54) (see also the legend of Figure 2). The diploid zygotic genome results from the gradual (55) fusion of paternal and maternal genomes into a totipotent embryo, referring to the ability to give rise to both embryonic and placental tissues. This property is maintained during cleavage divisions up to the eight-cell stage. Afterward, cell specification is initiated, concomitant with a loss of totipotency, which is definitive at the 32-cell stage.
A next major event is compaction of the loosely connected cells into a more tightly associated structure, followed by the successive generation of morula and blastocyst. Simultaneous with compaction cell polarization takes place, which initiates a first cell differentiation, separating cells in the ICM from the surrounding Trophectoderm. Posteriorly, in the late blastocyst cells in the ICM separate into epiblast (or primitive ectoderm) and primitive endoderm (or hypoblast) in a stochastic process (56). As a result of these two successive differentiation steps, the late blastocyst contains three separated cell types or lineages: the trophectoderm, the primitive endoderm, and the epiblast. While the trophectoderm and the primitive endoderm contribute to extraembryonic tissues, the epiblast gives rise to all cell types of the developing embryo proper. Once these lineages have been specified in the early embryo, they are separated and become committed to a particular fate.
Formation of the zygote and posterior cleavage divisions are associated with specific mechanisms of regulation as a result of specific epigenetic features and the spatial organization of the newly formed diploid genome (53, 57). These include but are not limited to differential methylation of histones and DNA, as well as active DNA demethylation (see below and Figure 2). This period is associated with specific patterns of gene expression (40, 58, 59) that reconcile these mechanistic features with developmental requirements.
Among the earliest expressed genes are several MaLR and muERV-L family transposons (the latter collectively referred to as MERVL), and chimeric transcripts that initiate on them. In the mature oocyte/zygote, MaLR-derived sequences are overrepresented in transcripts (17, 40, 60). In two-cell embryos, muERV-L itself displays a transient increased expression (16) and MERVL sequences are incorporated into epsilon virus-like particles observed in the early mouse embryo (61). In addition to MERVL itself, over 50 transcripts have been identified that are expressed in two-cell stage embryos and are directly linked to MERVL elements (17). We will refer to these genes as LTR-linked 2C genes.
By contrast, class II IAP and musD/ETn elements are expressed from the blastocyst stage onward (62). ERV-driven gene expression is important at these stages, as at least in the case of LINE1 elements (L1Md_T class) (63) as well as muERV-L elements (64), cleavage division is impaired upon targeted depletion by antisense oligonucleotides.
Cellular Heterogeneity and Naive Pluripotency
Mouse ES cells can be derived from the ICM of blastocyst embryos (12), a stage posterior to ICM and trophectoderm lineage separation. ES cells share several characteristics with cells in the ICM of the blastocyst, and can differentiate in vitro into all the cell types of a fetus (including the germline). ES cells are considered pluripotent because in chimeric embryos they contribute to each of the three germ layers of the embryo proper (11). Only rarely do they contribute to the extraembryonic tissues in the placenta, providing a distinction with totipotent cells in the two-cell embryo. ES cells can be maintained in culture for an apparent indiscriminate number of cell divisions (a property called self-renewal).
Maintenance of ES cells in vitro depends on extracellular signaling by LIF and BMP4. Signals converge on the OCT4, NANOG, and SOX2 TFs, which mutually stimulate each other’s expression, and repress genes either promoting or associated with cell differentiation. Together these core pluripotency factors form a co-operating network of TFs, whose activity also relies on the recruitment of epigenetic regulators (13, 65, 66). Contrasting with the uniform expression of OCT4 and SOX2 in all pluripotent cells of mouse ES cell cultures, heterogeneous expression has been observed for several genes including Zfp42/Rex1 (67), Nanog (68, 69), Stella (Dppa3/Pgc7) (70), Esrrb, Pecam1 (71, 72), and Zscan4 (72, 73). The exact relationship between expression of these various markers is unknown. At least in the case of Stella, Pecam1, and Rex1, expression seems to mark the same cell population (70).
Interestingly, after selection for high expression of such factors, often a heterogeneous population is reconstituted spontaneously (67, 70) and cells negative for markers such as NANOG, REX1, or STELLA can regain expression for each of these factors. Therefore, cells shuttle between high and low expressing states and the resulting heterogeneity reflects the presence of subpopulations of cells, as opposed to an irreversible differentiation status. The differences in gene expression levels between subpopulations may also define distinct self-renewal and differentiation properties, which together define developmental potency. High expression of those genes correlates with self-renewal in vitro and enhanced contribution to chimeric embryos. Cells with low expression show increased (but reversible) expression of differentiation markers (67, 70, 74), exhibit enhanced sensitivity to differentiation-inducing conditions in vitro (70) and show altered contribution to chimeric embryos (67).
Different models have been proposed to describe this cellular heterogeneity. According to a first model, cells transition between metastable states referred to as “naive” and “primed.” The transition between different states is determined by differentiation-inducing ERK signaling, in such a way that inhibition of ERK signaling drives cells into a more “naive” homogeneous pluripotent state (75). In addition to compacted morphology, demethylation of the Oct4 promoter region, silencing of retroviral vectors, two active X-chromosomes in female cells, LIF dependency and competency for germline contributions (75), this “naïve” state or ground state is characterized by homogenous expression of Nanog and Rex1 (76).
MERVL-Driven Transcription is Specific for the Two-Cell Stage
A distinct level of heterogeneity within ES cell cultures is related to the sporadic and transient transition of mouse ES cells into more totipotent identities. A subset of cells in a pluripotent cell culture can be separated from the other cells based on the expression of a red fluorescent protein driven by the regulatory sequences from a MERVL-derived LTR element (17). As differences in regulation of MERVL-derived LTRs have already been described (77), it remains to be established whether expression of all MERVL elements is equally exclusive. This particular MERVL element directs gene expression exclusively in two-cell embryos and in a limited number of cells (in the order of 1%) within ES cell cultures. These cells (referred to as “2C-like”) express transcripts (see Figure 2) that can be ascribed to three (overlapping) groups: (1) the MERVL family of ERV (but not the vast majority of other retrotransposons) and the GAG protein it encodes; (2) their corresponding LTR promoters and chimeric transcripts with junctions to MERVL elements; (3) a group of genes previously shown to be restricted to the two to four-cell stage of development (see Figure 1), including Zscan4, Tcstv1/3, Eif1a, Gm4340/Thoc4, Tdpoz1–5, and Zfp352 (73, 78). Over 500 genes transcribed in 2C cells are also active at the two-cell stage in embryos, including 52 genes that generated chimeric transcripts linked to MERVL elements (17).
Surprisingly, similar to the absence of pluripotency-associated TFs OCT4, SOX2, and NANOG from two-cell mouse embryos, 2C-like cells do not express these markers at the protein level (17). In accordance with its gene expression pattern specific for two-cell stage embryos, the developmental potential of 2C-like cells is not restricted to embryonic cell types. In chimeric embryos, 2C-like cells contribute to the epiblast as expected, but also to extraembryonic tissues such as trophectoderm, the yolk sac, and placenta (17). Hence, muERV-L regulatory sequences are necessary and sufficient to drive expression in rare ES cells that express MERVL, and display enhanced developmental potential.
Chromatin Structure: DNA and Histone Modifications
As opposed to genetic alterations to the genome, epigenetic mechanisms are those mechanisms that control gene expression without changing the underlying DNA sequence. Such mechanisms involve remodeling of the chromatin structure as a result of the formation of functional or 3D domains, or posttranscriptional processing in the form of DNA (de)methylation and histone modifications. A score of non-coding RNAs including microRNA, lncRNA, and piRNA also contribute to epigenetic regulation. Modifying enzymes such as DNA (DNMT) and histone methyltransferases (HMTs) are recruited by DNA-binding factors or through affinity for relevant modifications in chromatin (79). Combined, these mechanisms impact on the activity of DNA polymerase and configure the transcriptional landscape of a given genetic locus in a cell type-specific context.
A first major epigenetic pathway is the presence of methylated cytosine (normally in CpG nucleotides) in DNA (80). Methylation at gene promoters usually correlates with repression. DNA methylation is reversible, either by deamination and base excision repair or by TET family proteins that catalyze the conversion of 5-methylcytosine to 5-hydroxymethylcytosine, which in subsequent steps can be removed from the DNA strand (81, 82). It follows that the default role of TET1 is related to transcriptional activity (83).
A second silencing mechanism alters chromatin structure through modifications of histone tails, which are laid down by specialized enzymes that form part of histone modification complexes such as HMTs, histone deacetylases (HDACs), or lysine-specific demethylases (KDMs). The modifier activities can either stimulate or repress gene expression, depending on the affected histone residue and the overall epigenetic landscape of the locus. Notwithstanding, specific modifications are generally associated with a defined activity status. The trimethylation of histone 3 at lysine 4 (H3K4) is associated with active loci. Well-known modifications associated with repression are the trimethylation of histone 3 at K27 (involved in Polycomb-mediated repression, see below) and methylation of histone 3 at lysine 9 (H3K9), imparted by SETDB1/ESET, G9a, and the KMT1A and KMT1B products of the Suv39h1/2 genes (84, 85).
HDAC1 belongs to the Class I HDAC, nuclear proteins with activity toward core histones (86). HDACs provide the catalytic entity for different multisubunit complexes (including SIN3A, NuRD, CoREST, and NODE). These complexes usually contribute to transcription silencing in cooperation with other chromatin modifiers, such as KDMs. A well-studied KDM is LSD1/KDM1A, which favorite substrate is mono- or di-methyl K4 in H3 (87). The activity of LSD1 can either stimulate or repress the locus affected. LSD1-mediated demethylation of H3K9 facilitates activation by androgen and estrogen receptors (88). By contrast, LSD1 represses as a subunit of HDAC1/2-containing CoREST complexes (89).
The interplay between the roles of DNA and histone modifying enzymes is an area of active research. Epigenetic marks and regulators are generally correlated with activity states, but do not determine them. As a generic model, loci are marked in a mutually exclusive way with H3K4 (active), as opposed to DNA methylation (inactive). Moreover, several instances have been described in which H3K9 trimethylation (H3K9me3)-associated silencing of Class II ERVs in ES cells, is required for posterior DNA methylation (90, 91).
Regulation of ERV by Methylation
DNA methylation is widely recognized as an important epigenetic mark in silencing of TE and ERV (7) in line with a heavy presence of methylated CpG within repeated DNA (38). In mice, deficient in genes encoding proteins involved directly or indirectly in DNA methylation, particular TE/ERVs are de-repressed, usually leading to reduced viability and fertility (92). It has become clear however, that this mechanism does not explain the dynamics of expression/silencing in undermethylated tissues such as germ cells, pre-implantation embryos, and stem cells. As an example, although differential methylation during pre-implantation development coincides with expression of LINE1 (93) and IAP elements (94), as well as ICR regions in imprinted genes [reviewed in Ref. (95)], transient expression of muERV-L in two-cell embryos is not related to DNA methylation levels.
During mammalian pre-implantation development, DNA methylation is highly dynamic (23, 83). Until recently, it was believed that 5-methylcytosine is removed from the paternal genome immediately upon fertilization, while this occurs more slowly and passively afterward on the maternal genome, reaching a minimum at the blastocyst stage [Ref. (23), see Figure 2]. A more recent genome-wide analysis (96) has revealed that general hypermethylation of specific families of LINE1 and ERV retroelements in the sperm is rapidly removed in the zygote. Generally, retrotransposons maintain this low level of 5-methylcytosine up to the ICM stage, and do not increase to somatic levels until later (E6.5/7.5). Thus, repeat elements are undermethylated during pre-implantation stages. As an exception to this rule, high methylation levels of IAPs are retained throughout cleavage divisions (94, 96–98).
While enzymatic activity of DNMT1 and DNMT3A/B is essential for mouse embryonic development, ES cells deficient for all three genes (TKO cells) mostly retain characteristics of undifferentiated ES cells (99). Surprisingly, expression of ERV is hardly affected in TKO cells (100). While a few Class I and II ERVs depend on DNMT-mediated repression, ERVIII is totally unresponsive to TKO (100, 101).
Silencing of MERVL by RYBP and Associated Polycomb Complexes
In mouse ES cells retrotransposon silencing requires the activity of among others Eset or G9A-mediated histone modification machineries, and polycomb repressive complexes (PRC). Polycomb group (PcG) proteins are chromatin modifiers with important functions in cell proliferation, axial development, and X-chromosome inactivation (XCI) (102). In addition, PcG proteins contribute to pluripotency of mouse ES cells and coordinate genetic programs that orchaestrate differentiation of stem cells and cell fate decisions (103). PcG form two main multiprotein complexes termed PRC2 and PRC1 (104). These complexes bring about chromatin and histone modifications and catalyze the trimethylation of histone H3 at lysine 27 (H3K27me3) and the monoubiquitination of lysine 119 on histone H2A (H2AK119Ub1) for PRC2 and PRC1, respectively. A standard model postulates that chromatin-tethered PRC2 modifies histone H3, the resulting H3K27me3 is bound by the CBX component of PRC1, which in turn deposits H2AK119Ub1 (104). The concerted and sequential activities of PRC2 and PRC1 generate an inhibitory chromatin environment, limiting transcription of nearby genes (see Figure 3).
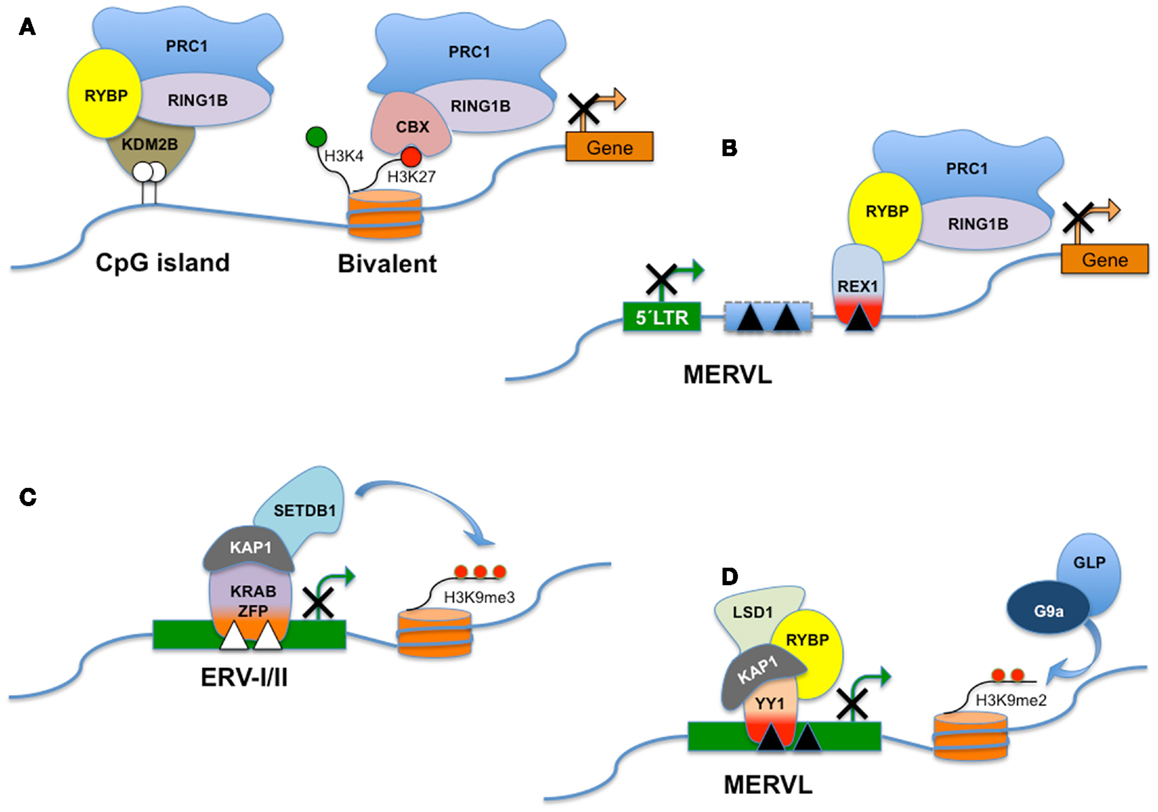
Figure 3. RYBP and distinct PRC1-like complexes. Polycomb Group genes (PcG) assemble into multiprotein complexes and, depending on the subunit composition, the complexes are recruited to different types of target sites. PRC1 (Polycomb repressive complex 1) is a canonical complex composed of either RING1A/or RING1B, MPH1/2/3, CBX2/7/8, and one of the Polycomb group RING fingers proteins (PCGF2–5) and is referred to here as CBX-PRC1 (A). CBX-PRC1 mediates H3K27me3-dependent silencing of bivalent promoters. PcG-regulated promoters devoid of H3K27me3 recruit a RYBP-containing variant complex termed RYBP-PRC1, which appears similar to a KDM2B-containing complex that recognizes non-methylated CpG islands (A). For clarity, only the subunits that differ between CBX-PRC1 and RYBP-PRC1 are shown, in addition to RING1B. RYBP silences the expression of transposable elements, including MERVL. RYBP can interact with both REX1 and/or RING1B. In this hypothetical model, REX1 might tether RYBP and the polycomb silencing complex to specific binding sites in MERVL or MERVL-derived control elements (B). Repression of class I and class II ERVs (C) is dependent on SETDB1 histone H3 lysine 9 trimethylase activity (H3K9me3). SETDB1 is believed to be recruited to its ERV targets via KRAB-containing zinc finger proteins and their co-repressor KAP1. MERVL repression is dependent on the presence of H3K9Me2 deposited through the combined action of G9a/GLP (D). Surprisingly, deletion of a variety of other chromatin proteins including LSD1, RYBP, and KAP1 also derepresses MERVL. The interplay between these pathways is presently unknown. LSD1/KAP1 and RYBP might all be recruited trough YY1-family DNA-binding factors as YY1 independently interacts with either KAP1 or RYBP (D).
RYBP was identified as a protein that interacts with both RING1 and Yin Yang 1 (YY1) (105). RYBP is a zinc finger protein, which in addition to its association with PcG complexes through RING1 is also a subunit of complexes containing the OCT4/POU5F1 TF essential for pluripotency in ES cells (106, 107). Despite its interaction with Ring1B, genome-wide chromatin binding of RYBP does not overlap well with PRC1 binding (108).
In addition to recruitment of PRC1 via interaction with PRC2 as outlined above, PcG complexes distinct from PRC1 are also recruited to a subset of CpG islands (CGIs) not marked by H3K27Me3 (109). CGIs lack DNA methylation and are associated with most mammalian gene promoters (110). Variant PRC1-related complexes that bind CGIs contain RYBP, and also KDM2B (FBXL10), which specifically recognizes non-methylated DNA in CGIs. KDM2B in turn recruits PRC1, leading to monoubiquitylation of H2AK119 and repression of associated genes. Yet another variant PRC1 complex also contains both RING1B and RYBP in addition to MEL18 (111) and differs from the canonical PRC1 in that the CBX7 subunit is excluded. The presence of CBX7 in these complexes appears to be mutually exclusive with association of the RYBP subunit (see also Figure 3).
Constitutive depletion of Rybp in a mouse model causes lethal gastrulation defects and obviates the generation of ES cell lines (112). By contrast, conditional inactivation of Rybp allows for the generation of ES cells with apparent normal expression of stem cell markers (108). Although RYBP associates with a subset of H3K27 and H2K119Ub-modified PRC1 target genes, it contributes only moderately to repression of these PRC1-regulated promoters and is dispensable for the association of PRC1 components MEL18 and RING1B to those targets (108). By contrast, in ES cells RYBP is necessary for repression of MERVL (but not musD and IAP) and two-cell stage-specific genes (see below). No direct evidence has yet implicated RING1B/PcG as a necessary cofactor in the observed regulation of MERVL levels by RYBP. However, in a limited analysis of mouse ES cells deficient in both RING1B and PRC2 (113), IAP, and RLTR33 (and to a lesser extent ORR1A1, RLTR4, and MMVL30) were upregulated, pointing toward a generic role for PcG in regulation of TE.
Regulation of MERVL by YY1-Family Members
Among the genes showing non-uniform expression in ES cell cultures is Rex1/Zfp42, a zinc finger protein with a unique expression pattern in ES cells (114, 115). Modest levels of expression are also detected during spermatogenesis and early trophectoderm derivatives and placenta in the mouse (114, 116). Rex1 is expressed throughout pre-implantation mouse development (117). Although deficiency of Rex1/Zfp42 affects ES cells only weakly (20, 115) expression is frequently used to identify pluripotent stem cells (118). REX1 has an essential role in the initiation of XCI in female ES cells (19), through regulation of the lncRNA Tsix (18). Although depletion of Rex1 does not affect expression levels of pluripotency markers (except for Tsix), it allows for upregulation of MERVL expression (20). REX1 has been shown to bind to MERVL loci in in vivo chromatin binding assays. Regulation of MERVL by REX1 is even more pronounced in vivo, as transient gain-and-loss of REX1 both influence MERVL levels during the late cleavage stages of the embryo, in a way compatible with a silencing function for REX1 (20). Thus, REX1 contributes to silencing of ERV expression in mouse ES cells and during pre-implantation development.
REX1 shares high homology in the DNA-binding zinc fingers with the well-studied, ubiquitously expressed TF YY1. YY1 is the prototype of a small family of DNA-binding TFs that also includes YY2 (119). Dependent on the epigenetic environment and numerous interactions with histone modifying complexes, YY1 can function as an activator, repressor, or initiator of gene transcription. YY1 contributes to the control of imprinted genes (120) and genes encoding regulators of the cell cycle (121), especially cytokinesis. Intriguingly, YY1 has been described already in the early 90s as a factor bound upstream of IAP and MMLV elements (122, 123). In addition, silencing of MMLV in embryonic cells is also dependent on YY1 (91). In line with the high homology in the DNA-binding zinc fingers between REX1, YY1, and YY2 (119), the latter two also bind both IAP and muERV-L elements, and weakly to musD in ES cells [Ref. (20); Raquel Pérez-Palacios, Pedro Muniesa, and Jon Schoorlemmer, manuscript in preparation]. In contrast to more efficient binding of REX1 in ES cells to muERV-L, YY1 and YY2 preferred IAP (20).
Consistent with in vivo chromatin binding, YY1-family proteins also play a role in regulation of a distinct subset of ERVs. While selected ERV class I and II elements are silenced by YY1 (in F9 EC cells), class III elements including muERV-L are not (91). Repression of IAP is also dependent on KAP1 in mouse ES cells and in early embryos (124). As KAP1 binding to IAP LTRs is lost in YY1 KD cells (91), and YY1 directly interacts with both KAP1 (91) and RYBP (105), YY1 may bridge RYBP/PRC1 and KAP1-dependent silencing (Figure 3). By contrast, REX1 (in mouse ES cells) contributes to silencing of muERV-L and musD (Class II) to a minor degree (20). Combined, these results are compatible with an important role for YY1-family members in ERV silencing.
Although RYBP interacts in vitro with YY1 (105) as well as with the related YY2 and REX1 proteins (125), neither of these proteins form part of RYBP-containing PcG complexes (111). As YY1-binding sites in ES do not significantly overlap with PRC1 nor with PRC2 binding (126), it was postulated that YY1 is unlikely to be relevant for PcG-dependent gene regulation (111, 127). However, this conclusion was reached ensuing the analysis of canonical protein-encoding target genes. In view of the data showing muERV-L deregulation in Rybp-deficient ES cells, we suggest that REX1 (and/or YY1 and YY2) may recruit RYBP and associated PcG/PRC1 complexes to binding sites in retroviral elements, ultimately contributing to silencing. This hypothetical model is depicted in Figure 3.
Different ERV Classes are Regulated by Distinct Epigenetic Mechanisms in Mouse ES Cells
Histone H3 lysine 9 methylation (H3K9me2/3) was initially observed as modification associated with centromeric repeat DNA and therefore presumed to be involved in repression (128, 129). It was subsequently observed that silencing of the promoter of the Oct4/Pou5f1 gene during differentiation is associated with the presence of H3K9me3 (130). In ES cells, different ERVs also carry this mark.
In accordance with H3K9me3 and H4K20me3 marking of ERVs specifically in ES cells (131), a role for SETDB1 in regulation was postulated. Indeed, a wide variety of Class I and II ERVs including ERVK10C and other IAP elements, musD, and ETn elements have lost the H3K9me3 mark and are ectopically expressed in Setbd1-deficient cells (100, 101). SETDB1 (also known as ESET) is recruited to chromatin through interactions with the KAP1 co-repressor (132). KRAB-associated protein 1 (KAP1) is a co-repressor of transcriptional repressors of the Zinc Finger Protein with Krüppel-Associated Box (KRAB-ZFP) family (133).
KRAB-associated protein 1 (also known as TRIM28 or TIF1β) recruits HDAC and histone methyltransferase machinery to its chromatin targets. Kap1 deficiency causes developmental arrest shortly after implantation and gastrulation failure (134). Kap1 cooperates with Cnot3, c-Myc, and Zfx in functions essential for the maintenance of pluripotency and self-renewal of ES cells (107, 135). Following up on initial reports that KAP1 mediates silencing of an exogenous virus in ES cells (136, 137), it was subsequently discovered that KAP1 is essential for repression of a large set of endogenous retroelements (100, 124). KAP1/SETDB1 complexes are tethered in turn to TE targets through interactions with KRAB-ZFP.
The genomes of higher vertebrates (including mice and humans) encode a large group of over 300 TFs named KRAB-containing zinc finger proteins (138). They contain a varying number of C2H2 zinc fingers (ZF) that bind DNA in a sequence-specific way and an N-terminal Krüppel-associated box (KRAB) domain with transcriptional repression activity (133). As the KRAB domain associates with KAP1, it was proposed that the sequence-specific docking of KRAB-ZFPs at given genomic loci can induce transcriptional repression mediated by KAP1 and associated proteins (139). This silencing mechanism is important in ES cells and operates during preimplantation development (90), in accordance with high expression of Kap1 at this stage (Figure 2).
More recent results indicate that different classes of ERV are regulated by distinct chromatin-modification pathways. Class III muERV-L/MERVL and MaLR (i.e., ORR1A3) families are generally devoid of the H3K9me3 mark in ES cells (101, 131). By contrast, MERVL elements carry the H3K9me2 mark, and de-repression is associated with loss of H3K9me2 (77). Moreover, while repression of both ERV I and ERV II elements in ES cells is dependent on SETDB1, this is hardly the case for ERV III (60, 100, 101). MERVL silencing is dependent on chromatin association and HMTase activity of both G9a (EHMT2) and GLP (EuHMTase1), two closely related HKMT that form a heteromeric complex in vivo (60). The complex represents the main HKMTs to catalyze H3K9me1 and H3K9me2 in euchromatin. Deletion of either G9a or GLP in ES cells dramatically reduces overall H3K9me1 and H3K9me2 levels (140, 141). The mechanisms underlying G9a recruitment to genomic targets including MERVL are not understood. G9A/GLP does not interact with either KAP1 or KRAB-ZFP (142). This observation is consistent with the notion that a wave of MERVL expansion has taken place in the mouse relatively recently (32), potentially precluding KRAB-ZFP adaptation to these novel sites. Recruitment mechanisms may rely on other chromatin proteins that reportedly interact with G9A/GLP, including Blimp1, ZNF200, HP1, and DNMT1 (142).
However, class III including MERVL ERVs are also upregulated in ES cells following Kap1 depletion (124). Silencing relies on targeting through the 5′UTR/LTR (124). MERVL expression is also de-repressed in ES cells that lack LSD1/KDM1 (87), a mono- and di-methyl lysine-specific histone demethylase 1 (77). Consistent with the detection of histone modification changes in Lsd1-mutant ES cells that are incompatible with LSD1 specificity, additional modifiers copurify with LSD1 complexes including KAP1-associated HDAC. HDAC acts in concert with LSD1 to properly repress muERV-L/MERVL in pluripotent stem cells (77, 143).
MERVL repression is dependent on the presence of H3K9Me2 deposited through the combined action of G9a/GLP (60) and LSD1/HDAC1 activity, although the interplay between these two pathways is presently unknown. Furthermore, although RYBP is not a component of LSD1 complexes (77), this PcG-associated protein also plays a role (see Figures 3B,D). As outlined above, RYBP may contribute through association with either REX1 or YY1. The former may attract demethylating activity through its interaction with LSD1 (20), while YY1 was shown in turn to selectively bind KAP1 in undifferentiated stem cells (91), providing a potential docking site for KAP1 and its associated activities.
The variety of potentially independent mechanisms contributing to MERVL silencing could be explained by two different but not mutually exclusive arguments. First of all, different copies of MERVL may be regulated by distinct mechanisms. The mouse genome contains 500–600 full-length and nearly 350 protein-coding MERVL elements (60, 61). Except the LSD1-deficiency, which affects many different copies (77), in most studies describing de-repression of MERVL the difference between induction of multiple copies versus several fold induction of a few copies has not been addressed. Alternatively, different mechanisms may be initiated at different time-points of the life cycle, which is discussed below.
The Contribution of Retrotransposons to Gene Expression Specific for Totipotency and Pluripotency
In contrast to silencing of retrotransposons in differentiated cells and tissues, activity of retrotransposons in the germline and/or in pluripotent cells destined for the next generation is a crucial prerequisite to ensure genetic propagation. In line with the resulting restricted and regulated expression patterns, retrotransposon-derived control elements underlie transcriptional networks in pluripotent stem cells. The binding regions for several core pluripotency factors including SOX2, NANOG, and POU5F1 show meaningful overlap with degenerated TEs, i.e., ERV-derived LTRs. In particular, remnants of class II elements (IAP and ETn) overlap binding sites for SOX2/NANOG/OCT4 in the mouse (49, 144). Despite evolutionary innovation and diversification at the level of individual binding sites, overlap with retrotransposons is evident in both mouse and human (49). Hence, ERV-contributed cis-binding sites may underlie the coordination of gene expression in pluripotent cells in a species-specific manner. The prevalence of MERVL-derived control elements and LTRs (Class III ERV elements) in the vicinity of two-cell stage gene promoters (17, 60), suggests that they orchestrate coordinated regulation of gene expression at this stage. The contribution of ERV-derived sequences to cis-acting regulatory elements is not restricted to mammals (47, 48, 144). A chicken retroviral (Ens-1) LTR contributes to both the transcriptional networks in ES cells and to extraembryonic transcription (145).
In placental mammals (Eutherians), female cells silence one of their X-chromosomes in a process called XCI to accomplish equal expression levels of X-encoded genes compared with males (146, 147). In the mouse, the activity of both X chromosomes is re-established in pluripotent cells in the ICM. This process calls for the expression of the lncRNA Tsix. The activity of both X-chromosomes in female ES cell lines is similarly dependent on Tsix, which counteracts painting of one of the X-chromosomes by Xist. The level of Tsix RNA depends, in addition to the promoter, on a cis-acting element called DxPas34 (148). In rodents DxPas34 contains binding motifs for several TFs including CTCF and REX1 [Ref. (18); Guallar et al. submitted] and DxPas34 activity is dependent on the presence of REX1 (in addition to c-MYC and KLF4) (18). Surprisingly enough, the DxPas34 element has the structure of a repeat element very similar at the sequence level to ERVs (149). In fact, it is most similar to, and can be considered a fossilized version of muERV-L/MERVL. The activity of this element seems unrelated to LTR-originated promoter activity, as the sequence similarity is in the internal portion of the retroviral element, and not in the LTRs. Although XCI is a female-associated process (while most commonly used mouse ES lines are male), re-activation of both X-chromosomes is a hallmark of pluripotency. Naive female ES cells invariably express Tsix resulting in the presence of two active X chromosomes. This observation suggests that MERVL-related sequence elements direct gene expression required for naive pluripotency (75).
Several genes identified initially as transcripts expressed exclusively in two-cell stage embryos represent transcripts that initiate in MERVL/MT2 LTRs. These include Zscan4, Tcstv1/3, Eif1a (Gm2022), similar to Tho4, Tdpoz1–5, and Zfp352 (16, 61, 64, 78, 150). We will refer to these genes as LTR-linked 2C genes. ES cells devoid of individual MERVL regulators discussed above all suffer de-repression of both MERVL and (a subset of) LTR-linked 2C genes. This holds in addition to LSD1 for G9A (60), KAP1 (101), HDAC1 (77), and RYBP (108). Although de-repression of muERV-L might be a consequence of ectopic expression of LTR-linked 2C genes, no direct evidence for such a mechanism is available. Therefore, we favor the hypothesis that aberrant transcription of 2C genes results from sharing the silencing machinery with MERVL. In line with this hypothesis, MERVL-positive ES cells are believed to represent a transient two-cell state accompanied by induction of LTR-linked 2C genes.
Developmental Potency and Developmental Timing
In contrast to the well-established exclusive contribution of ES cells to the ICM of the blastocyst of chimeras (and embryonic tissues during later development), 2C-like cells also contribute to the trophectoderm and to the yolk sac and placenta in later embryos. The proportion of 2C-like cells within an ES cell culture is controlled by LSD1, and to a lesser extent by LSD1-associated HDAC1 (17). Without LSD1/HDAC1 activity, more cells reside in the 2C-like state. Not surprisingly then, Lsd1-deficient ES cells also show enhanced lineage potential in mouse chimera assays and contribute to embryonic tissues and primordial germ cells, the amnion, yolk sac, and placental tissues, including giant trophoblast cells. Therefore, the separation between trophectodermal and embryonic lineages established in post cleavage-stage embryos is overturned in 2C-like cells, which transiently exhibit an expanded cell fate relative to pluripotent cells. In ES cells RYBP is necessary for repression of germline-specific genes, as well as for repression of MERVL class ERVs (not musD norIAP) and two-cell stage-specific genes (including Tcstv3, Zscan4, Zfp352, and Ubtfl1). In addition, the collection of genes controlled by RYBP overlapped very well with LSD1 target genes (77, 108), including a set of extraembryonic endoderm markers. This coincidence reinforces the hypothetical model presented (in Figure 3) suggesting that RYBP and LSD1 act in concert.
Neither self-renewal properties, nor pluripotency (measured in vitro or by contribution to chimeric embryos) have been rigorously established so far for the majority of ES cells devoid of individual MERVL regulators discussed above (except for Lsd1 and Rybp). However, available data are compatible with the notion that deficiency in these MERVL regulators causes defects during differentiation (100, 151), which can be interpreted as altered pluripotency. It is not clear at present whether the shared de-repression of MERVL in these cells drives a higher percentage of cells into a 2C-like state which in turn allows differentiation into extraembryonic lineages. Alternatively, de-repression of MERVL and 2C-stage-specific transcripts initiating on ERV may be indicative of transcriptional deregulation, allowing for enhanced plasticity of cell fate and increased extraembryonic differentiation.
The absence of REX1 in mouse ES cells is accompanied by a (relatively minor) de-repression of MERVL, suggesting that REX1 may control only few copies among the MERVL elements (20). It is unclear at present whether this induction reflects increased MERVL expression in a constant number of expressing cells, or results from an increased subpopulation of 2C-like cells. In the latter case, REX1 may negatively influence transition into the 2C-like state, or stimulate reversal into pluripotent cells (Figure 4). The latter would be in accordance with its repressing function on MERVL after its peak expression in two-cell embryos (20). REX1 may regulate this transition indirectly through its interaction with LSD1 (20), or directly as a transcriptional repressor in concert with RYBP (125). A final alternative resides in a distinct function of REX1 in protecting a regulatory ERV locus from generic TE silencing mechanisms. The above considerations imply that REX1 is present in 2C-like ES cells based on the comparison with two-cell embryos. In support of this view, Rex1 mRNA levels were not identified as differentially regulated between 2C-like and pluripotent ES cells (17). REX1 is subject to RNF12-mediated ubiquitination and subsequent degradation in pluripotent cells (19). It remains possible that a similar mechanism lowers REX1 protein levels in 2C-like cells.
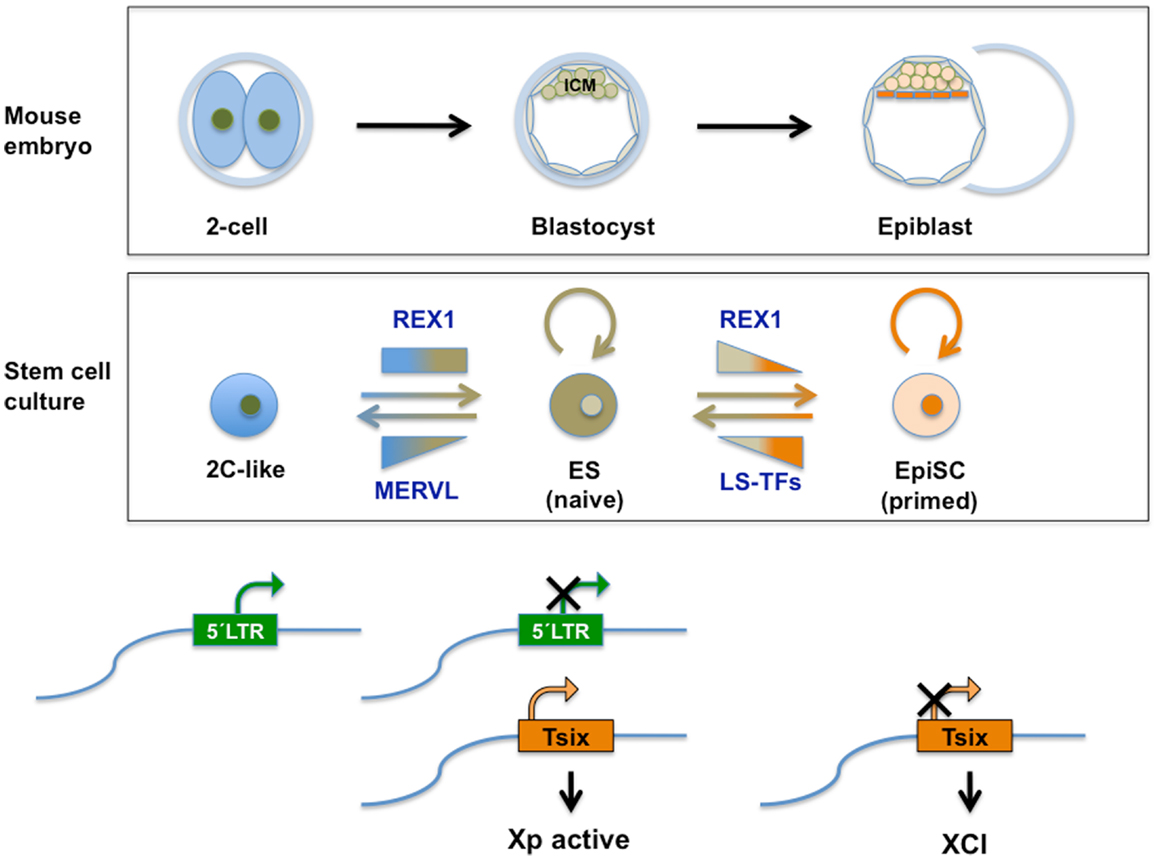
Figure 4. Top panel: schematic representation of relevant stages during mouse pre-implantation development. A specific set of genes transiently expressed at the two-cell mouse embryo (see also Figure 2), contributes to the totipotent status distinctive of those early stages. See text for further information. Middle panel: transient subpopulations of mouse ES cells. MERVL-derived elements drive the expression of genes specific to two-cell embryos in a transient subset of ES cells, discriminating pluripotent ES cells with ICM characteristics from totipotent 2C-like cells with expanded developmental potential. Independently, self-renewing (OCT4/SOX2 positive) ES cells shuttle between a so-called naive pluripotent status of gene expression and a primed state, characterized by the ability of the former to contribute to germline chimeras. These populations can be separated based on the expression of various heterogeneously expressed genes including Rex1. Among other characteristics of naivety, figures high expression of Rex1 in all cells, as opposed to heterogeneous or low expression. Primed cells exhibit a lower potency of development and no expression of Rex1. These populations bear similarity to ICM cells in the blastocyst and Epiblast Stem cells (EpiSC), respectively. REX1 contributes to regulation of MERVL-derived transcriptional control elements relevant to these cellular transitions. REX1 is present throughout pre-implantation development (Figure 2) and regulates MERVL and Zscan4 expression at these stages. Therefore a function of REX1 in the transition between totipotent 2C-like and pluripotent ES cells is speculated on (see text). Levels of Zfp42 are not significantly affected in 2C-like cells (17). On the other hand, REX1 positively affects re-activation of the inactive X-chromosome typical of the ICM and naive ES cells, by promoting the transcription of Tsix. Bottom panel: LTR regulation. The LTR drives expression of MERVL and totipotency-specific gene expression in 2C-like cells with totipotent characteristics, which co-exist in culture with pluripotent REX1-expressing ES cells. REX1 silences MERVL in ES cells, and is necessary for full expression of Tsix. Tsix expression in turn is instrumental in both the re-activation of the paternal X-chromosome in the blastocyst, which is silenced at earlier stages and maintenance of re-activation compatible with naive pluripotency. In primed ES cells and in EpiSC Tsix is silenced and XCI is installed. LS-TFs, lineage specific transcription factors expressed in primed ES cells and EpiSC; Xp, paternally derived X-chromosome XCI; X-chromosome inactivation; Tsix, lncRNA counteracting XCI, whose transcription is driven by a MERVL-related repeat sequence.
A potential enhanced contribution of Rex1-deficient cells to extraembryonic tissues in chimeras has not been rigorously tested [see Ref. (115)]. The influence of Rex1 on developmental potency in vitro concerns some extraembryonic endoderm markers, but is much more restricted than reported for Lsd1 and Rybp. A role for Rex1 in MERVL-driven totipotency is not easily reconciled with the reported differentiation characteristics of Rex1-deficient ES cells in vitro (115), potentially due to the separate role of Rex1 in naive pluripotency (Figure 4).
Based on the different requirements for RYBP during the generation of ES cells from blastocyst versus depletion after lines have been established, it has been suggested that Rybp is required for the generation, but not for the maintenance of the ES cells state (108). This notion coincides with the magnitude of MERVL regulation by Rex1 during these stages. It is therefore a possibility that REX1 attracts a repressing complex to MERVL during pre-implantation development via its interaction with RYBP, while repression at later stages (including in the ICM/ES cells) is exerted in conjunction with parallel pathways.
Compared to two-cell embryos, an increase in H3K9me2 methylation at heterochromatin has been reported at the four-cell stage (152), with the increased level maintained up to the blastocyst stage. A similar increase associated with MERVL elements may underlie G9A-mediated repression of MERVL after transient expression in two-cell embryos.
Zscan4, Pluripotency, and Two-Cell Embryos
An alternative pathway conferring characteristics of two-cell embryos on ES cells implicates the Zscan4 locus. Zscan4 is the common name for several transcripts generated from a cluster of six paralogous genes with high homology (73). ZSCAN4 is a nuclear protein with DNA-binding Zinc Fingers and a SCAN domain for protein–protein interactions. In a pattern reminiscent of MERVL, Zscan4 is expressed exclusively in two-cell embryos and transiently and reversibly in only a small percentage of ES cells in culture (73, 153). Zscan4-expressing cells selected to near homogeneity (using a reporter gene inserted in the locus) do exhibit standard pluripotency characteristics such as expression of OCT4/POU5F1, in vitro differentiation into three germ layers and contribution to chimeric embryos. Despite the relatively small percentage of Zscan4-expressing cells in an ES culture at a given time point, all cells cycle through a Zscan+ state over a span of 9–10 passages. Failure to do so results in cell death associated with telomerase-independent telomere shortening, loss of normal karyotype, and genomic instability. These observations are consistent with a role for ZSCAN4 in the lengthening of telomeres through a recombination process unique to one- and two-cell embryos called sister-chromatid exchange (154).
At this moment, there is no direct proof that implicates ZSCAN4 in regulation of either MERVL-driven transcription or MERVL-directed passage to the 2C-like state (17). They seem to be coregulated as Zscan4 is manifold induced in 2C-like cells, depletion of 2C-like cells also depleted Zscan4 levels (17), and both Zscan4 and MERVL are upregulated in KAP1- and LSD1-deficient ES cells, and to a minor extent in G9A deficient cells. Hence, ZSCAN4 might directly regulate MERVL-driven conversion to 2C, and/or MERVL may somehow coordinate induction of genes required for SCE. Integration of both processes might rely on REX1, which has been shown to regulate expression of MERVL in both early mouse embryos and ES cells (20), and Zscan4 in embryos (117). A difference between regulation of Zscan4 and MERVL expression is the dependence of Zscan4 levels on the presence of REX1 in two-cell stage embryos (117). By contrast, MERVL expression is indifferent to REX1 levels at this stage (20), suggesting a potential mechanism underlying the differences between Zscan4- and MERVL-expressing ES populations.
Human ERV and Embryonic Stem Cells
The contributions of MERVL to gene expression restricted to different states of developmental potency, and its epigenetic control mechanisms (Figures 3 and 4) have been described in mouse stem cells. Studies to determine the role of similar HERV -driven processes for HESCs are ongoing. We will briefly discuss present knowledge and issues being addressed in this respect. Definitely, many differences exist between HESC and mouse ES cells (155). Although pluripotent, the differentiation properties and growth factor dependency of HESC are more similar to mouse Epiblast Stem Cells [reviewed in Ref. (14)]. If remnants of MERVL-related sequences are present in human they are much more degenerated (156). Whether human MERVL-related sequences that influence TSIX expression and XCI in HESC exist, is an unresolved issue at present (157, 158). Surprisingly, few data are available regarding the expression of HERV in human tissue in general. In HESC, HERV-K expression is very high (159). As opposed to a wide variety of differentiated cells, this element is marked in HESC with the activity associated H3K4Me3 modification. Moreover, binding sites for the core pluripotency factors NANOG, OCT4, and SOX2 are located within HERV-K LTRs. HERV-K(HML-2) RNA and protein are both expressed in undifferentiated HESCs (160). As exemplified by rapid silencing upon differentiation, HERV-K expression is representative of the pluripotent state of HESC (159, 160), potentially displaying regulation similar to MERVL in the mouse. HERV hardly displays retrotransposition activity, as opposed to rodent ERVs. However, present inactivity does not rule out its previous recruitment as cis-control element. Considering the dearth of information on gene expression in human embryos and placenta (161, 162), HERV expression may be more prominent than presently known. By contrast, as a result of expanded life span, human cells are equipped with a more extended set of anti-tumor mechanisms compared to rodents. It cannot be excluded that expanded life span also has required a tighter control of TEs, which has evolved differently from rodents.
Moreover, the muERV-L/MERVL regulator REX1 is a well-defined marker for both mouse and HESCs (163, 164). REX1 expression also defines heterogeneity within OCT4 and TRA-1-60 positive populations of HESCs (164). Low or absent REX1 expression identifies an OCT4 positive cell type that does not revert back to REX1 positivity under normal conditions and is prone to differentiation into definitive and extraembryonic endoderm. It is an outstanding question whether pluripotency in HESC is fine-tuned by REX1-mediated regulation of HERV-K similar to the situation in the mouse.
In human pluripotent cells, cellular heterogeneity has been studied mainly in the context of standardization of human induced pluripotent stem (HiPS) cells. HiPS are pluripotent cells with characteristics identical to or very similar to HESC, which can be generated by reprograming of adult cells (14, 165). Different lines of HiPS cells display a higher level of variability at the level of gene expression and epigenetic signatures compared to ES from either mouse or human. This can be attributed to differences in derivation methods including donor cell types, and to the differences in age, genetic background, and pathology states of the donors (14). However, overcoming longstanding impediments, HESC with properties similar to ground state pluripotency described in the mouse have recently been derived (166). Conditions to generate such ground state HESC/HiPS cells are expected to diminish the cellular heterogeneity observed at the gene expression level. As HERV-K expression is very high in HESC (159), it will be of interest to see if ERV-driven reporters could now be used to classify HESC according to potency levels similar to mouse ES cells.
An important difference between mouse and HESCs also is the latter’s capacity for trophectodermal differentiation. Placentation is a relatively recent event on the scale of evolution, and the diversity of placental morphology among mammalian species suggests that it has appeared various times independently (167). Moreover, generic difference in genome management may already underlie different timing of ZGA between mouse and human (161), and posterior separation of lineages. Such species-specific differences may also have altered cis-acting functions and regulation of ERV-L in different species.
Concluding Remarks: Relationships with Cancer
We finally comment on the relevance of HERV -directed transcription and the mechanisms that control it for human cancer biology. Cancerous cells often display genome-wide loss of DNA methylation resulting from a loss of methylation on TE (24, 168), and progressive loss of silencing. Similar to DNA hypomethylation, dramatic changes in histone modification patterns are rampant (169). Relaxation of alternative silencing mechanisms described here may re-activate TE/ERVs elements in tumors, as initially demonstrated in mice (170).
Mechanisms that explain how the activity of HERVs may contribute to tumor formation have been reviewed elsewhere [i.e., Ref. (8, 171)]. Both de novo transposition events and deregulation of HERV-derived control elements may affect the expression of tumor suppressor genes or (proto-)oncogenes nearby. Occasionally, proto-oncogenes may be incorporated in variant HERV. As these genes control cell growth and proliferation, their deregulation may give rise to a malignant or transformed cell with enhanced growth characteristics. In addition, HERV-encoded ENV proteins may provide cell fusion properties that help the transition toward metastasis or help tumor cells evade an anti-tumoral immune response by virtue of its immunosuppressive properties (172). Alternatively, uncontrolled expression of TE has been related to genome instability caused by DNA breaks and chromosome translocations. Deregulation of retroviral elements in tumors might be indicative of improper activity of genome stability mechanisms, potentially preceding insult resulting from de-stabilization. RT activity (encoded in HERV) might be used as a measure of the extent of ERV/TE-deregulation (173), and be instrumental in tumor classification. Moreover, as pluripotency markers are overexpressed in tumor cell lines coinciding with increased RT activity (173), the level of RT activity might even indicate the presence of tumor stem cells.
The potential contribution of HERV to tumor formation should also be taken into account in the context of gene therapy, as insertion of new genes in a location that relieves HERV silencing may trigger these activation processes. Moreover, as modified viruses are commonly used as vectors for gene therapy, the relief of HERV silencing commonly associated with viral infections and inflammation (174) constitutes a risk factor for gene therapy by itself and HERV-encoded proteins might provide essential proteins that allow the mobilization of replication-defective provirus.
YY1 overexpression has been reported in tumors, and overexpression levels are related to clinical progression (175). Differences (usually overexpression) of YY1 transcript levels in tumor tissue relative to normal counterparts have also been extracted by computational analysis from gene expression omnibus (GEO) datasets (176). It seems obvious to presume that YY1’s well documented function in the regulation of gene networks that control cell proliferation underlies its role in oncogenesis. However, YY1 participates in distinct mechanisms potentially related to oncogenesis, including increased mutagenesis, regulation of chromosome dynamics (such as imprinting and X-inactivation), and DNA repair and chromosome segregation (177). We suggest that the contribution of YY1-family proteins to oncogenesis may as well reside in silencing of TE (and ERV in particular).
The judge is still out on the question whether HERV activation is a triggering event in tumor development, or whether they are upregulated as a consequence of previous alterations associated with tumor development, i.e., genomic instability or demethylation. So far, the expression of HERVs has been linked to germ cell tumors, breast cancer, ovarian cancer, and melanoma [reviewed in Ref. (8, 10)]. Enhanced knowledge of the mechanisms that control HERV expression will aid the understanding of how heterogeneous HERV activity levels may contribute to differences among individuals in susceptibility to cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the authors’ laboratory has been supported by grant PI07119 from FIS/CarlosIII, Spanish Ministry of Health; grant PI110/09 from the Departamento de Ciencia, Tecnología y Universidad, Gobierno de Aragón and redes DGA (B77), DGA, Spain; PAMER grants, Aragon Health Sciences Institute, Spain.
References
1. McClintock B. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci U S A (1950) 36(6):344–55. doi:10.1073/pnas.36.6.344
2. Fedoroff N. How jumping genes were discovered. Nat Struct Biol (2001) 8(4):300–1. doi:10.1038/86161
3. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature (2002) 420(6915):520–62. doi:10.1038/nature01262
4. de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet (2011) 7(12):e1002384. doi:10.1371/journal.pgen.1002384
5. Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet (2006) 2(1):e2. doi:10.1371/journal.pgen.0020002
6. Löwer R, Löwer J, Kurth R. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci U S A (1996) 93(11):5177–84. doi:10.1073/pnas.93.11.5177
7. Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet (1997) 13(8):335–40. doi:10.1016/S0168-9525(97)01181-5
8. Kurth R, Bannert N. Beneficial and detrimental effects of human endogenous retroviruses. Int J Cancer (2010) 126(2):306–14. doi:10.1002/ijc.24902
9. Flockerzi A, Ruggieri A, Frank O, Sauter M, Maldener E, Kopper B, et al. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV transcriptome project. BMC Genomics (2008) 9:354. doi:10.1186/1471-2164-9-354
10. McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta (2008) 1782(3):127–50. doi:10.1016/j.bbadis.2007.12.005
11. Beddington RS, Robertson EJ. An assessment of the developmental potential of embryonic stem cells in the midgestation mouse embryo. Development (1989) 105(4):733–7.
12. Rossant J. Stem cells and early lineage development. Cell (2008) 132(4):527–31. doi:10.1016/j.cell.2008.01.039
13. Ng HH, Surani MA. The transcriptional and signalling networks of pluripotency. Nat Cell Biol (2011) 13(5):490–6. doi:10.1038/ncb0511-490
14. Cahan P, Daley GQ. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat Rev Mol Cell Biol (2013) 14(6):357–68. doi:10.1038/nrm3584
15. Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat Rev Mol Cell Biol (2009) 10(8):526–37. doi:10.1038/nrm2727
16. Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev Biol (2004) 269(1):276–85. doi:10.1016/j.ydbio.2004.01.028
17. Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature (2012) 487(7405):57–63. doi:10.1038/nature11244
18. Navarro P, Oldfield A, Legoupi J, Festuccia N, Dubois A, Attia M, et al. Molecular coupling of Tsix regulation and pluripotency. Nature (2010) 468(7322):457–60. doi:10.1038/nature09496
19. Gontan C, Achame EM, Demmers J, Barakat TS, Rentmeester E, van Ijcken W, et al. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature (2012) 485(7398):386–90. doi:10.1038/nature11070
20. Guallar D, Pérez-Palacios R, Climent M, Martínez-Abadía I, Larraga A, Fernández-Juan M, et al. Expression of endogenous retroviruses is negatively regulated by the pluripotency marker Rex1/Zfp42. Nucleic Acids Res (2012) 40(18):8993–9007. doi:10.1093/nar/gks686
21. Spencer TE, Palmarini M. Application of next generation sequencing in mammalian embryogenomics: lessons learned from endogenous betaretroviruses of sheep. Anim Reprod Sci (2012) 134(1–2):95–103. doi:10.1016/j.anireprosci.2012.08.016
22. Taruscio D, Mantovani A. Factors regulating endogenous retroviral sequences in human and mouse. Cytogenet Genome Res (2004) 105(2–4):351–62. doi:10.1159/000078208
23. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature (2007) 447(7143):425–32. doi:10.1038/nature05918
24. Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol (2006) 310:211–50.
25. Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu Rev Genet (2008) 42:709–32. doi:10.1146/annurev.genet.42.110807.091501
26. Harris JR. The evolution of placental mammals. FEBS Lett (1991) 295(1–3):3–4. doi:10.1016/0014-5793(91)81370-N
27. Benveniste RE, Lieber MM, Livingston DM, Sherr CJ, Todaro GJ, Kalter SS. Infectious C-type virus isolated from a baboon placenta. Nature (1974) 248(443):17–20. doi:10.1038/248017a0
28. Kalter SS, Heberling RL, Helmke RJ, Panigel M, Smith GC, Kraemer DC, et al. A comparative study on the presence of C-type viral particles in placentas from primates and other animals. Bibl Haematol (1975) 40:391–401.
29. Weiss RA. The discovery of endogenous retroviruses. Retrovirology (2006) 3:67. doi:10.1186/1742-4690-3-67
30. Martin MA, Bryan T, Rasheed S, Khan AS. Identification and cloning of endogenous retroviral sequences present in human DNA. Proc Natl Acad Sci U S A (1981) 78(8):4892–6. doi:10.1073/pnas.78.8.4892
31. Stocking C, Kozak CA. Murine endogenous retroviruses. Cell Mol Life Sci (2008) 65(21):3383–98. doi:10.1007/s00018-008-8497-0
32. Bénit L, Lallemand JB, Casella JF, Philippe H, Heidmann T. ERV-L elements: a family of endogenous retrovirus-like elements active throughout the evolution of mammals. J Virol (1999) 73(4):3301–8.
33. Zhang Y, Maksakova IA, Gagnier L, van de Lagemaat LN, Mager DL. Genome-wide assessments reveal extremely high levels of polymorphism of two active families of mouse endogenous retroviral elements. PLoS Genet (2008) 4(2):e1000007. doi:10.1371/journal.pgen.1000007
34. Frendo JL, Olivier D, Cheynet V, Blond JL, Bouton O, Vidaud M, et al. Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Mol Cell Biol (2003) 23(10):3566–74. doi:10.1128/MCB.23.10.3566-3574.2003
35. Conrad B, Weissmahr RN, Böni J, Arcari R, Schüpbach J, Mach B. A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell (1997) 90(2):303–13. doi:10.1016/S0092-8674(00)80338-4
36. Cowley M, Oakey RJ. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet (2013) 9(1):e1003234. doi:10.1371/journal.pgen.1003234
37. Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev (2008) 22(7):908–17. doi:10.1101/gad.1640708
38. Zamudio N, Bourc’His D. Transposable elements in the mammalian germline: a comfortable niche or a deadly trap? Heredity (Edinb) (2010) 105(1):92–104. doi:10.1038/hdy.2010.53
39. Dupressoir A, Heidmann T. Germ line-specific expression of intracisternal A-particle retrotransposons in transgenic mice. Mol Cell Biol (1996) 16(8):4495–503.
40. Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, et al. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell (2004) 7(4):597–606. doi:10.1016/j.devcel.2004.09.004
41. Blond JL, Besème F, Duret L, Bouton O, Bedin F, Perron H, et al. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J Virol (1999) 73(2):1175–85.
42. Dunlap KA, Palmarini M, Varela M, Burghardt RC, Hayashi K, Farmer JL, et al. Endogenous retroviruses regulate periimplantation placental growth and differentiation. Proc Natl Acad Sci U S A (2006) 103(39):14390–5. doi:10.1073/pnas.0603836103
43. Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet (2013) 9(4):e1003470. doi:10.1371/journal.pgen.1003470
44. Rebollo R, Miceli-Royer K, Zhang Y, Farivar S, Gagnier L, Mager DL. Epigenetic interplay between mouse endogenous retroviruses and host genes. Genome Biol (2012) 13(10):R89. doi:10.1186/gb-2012-13-10-r89
45. Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet (2003) 19(2):68–72. doi:10.1016/S0168-9525(02)00006-9
46. Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell (2009) 137(7):1194–211. doi:10.1016/j.cell.2009.06.001
47. Bourque G, Leong B, Vega VB, Chen X, Lee YL, Srinivasan KG, et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res (2008) 18(11):1752–62. doi:10.1101/gr.080663.108
48. Schmidt D, Schwalie PC, Wilson MD, Ballester B, Gonçalves A, Kutter C, et al. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell (2012) 148(1–2):335–48. doi:10.1016/j.cell.2011.11.058
49. Xie D, Chen CC, Ptaszek LM, Xiao S, Cao X, Fang F, et al. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res (2010) 20(6):804–15. doi:10.1101/gr.100594.109
50. Lynch VJ, Leclerc RD, May G, Wagner GP. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat Genet (2011) 43(11):1154–9. doi:10.1038/ng.917
51. Rebollo R, Romanish MT, Mager DL. Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet (2012) 46:21–42. doi:10.1146/annurev-genet-110711-155621
52. Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature (2000) 403(6769):501–2. doi:10.1038/35000654
53. Albert M, Peters AHFM. Genetic and epigenetic control of early mouse development. Curr Opin Genet Dev (2009) 19(2):113–21. doi:10.1016/j.gde.2009.03.004
54. Leeb M, Wutz A. Establishment of epigenetic patterns in development. Chromosoma (2012) 121(3):251–62. doi:10.1007/s00412-012-0365-x
55. Mayer W, Smith A, Fundele R, Haaf T. Spatial separation of parental genomes in preimplantation mouse embryos. J Cell Biol (2000) 148(4):629–34. doi:10.1083/jcb.148.4.629
56. Plusa B, Piliszek A, Frankenberg S, Artus J, Hadjantonakis AK. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development (2008) 135(18):3081–91. doi:10.1242/dev.021519
57. Probst AV, Santos F, Reik W, Almouzni G, Dean W. Structural differences in centromeric heterochromatin are spatially reconciled on fertilisation in the mouse zygote. Chromosoma (2007) 116(4):403–15. doi:10.1007/s00412-007-0106-8
58. Ko MS, Kitchen JR, Wang X, Threat TA, Hasegawa A, Sun T, et al. Large-scale cDNA analysis reveals phased gene expression patterns during preimplantation mouse development. Development (2000) 127(8):1737–49.
59. Hamatani T, Carter M, Sharov A, Ko M. Dynamics of global gene expression changes during mouse preimplantation development. Dev Cell (2004) 6(1):117–31. doi:10.1016/S1534-5807(03)00373-3
60. Maksakova IA, Thompson PJ, Goyal P, Jones SJ, Singh PB, Karimi MM, et al. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin (2013) 6(1):15. doi:10.1186/1756-8935-6-15
61. Ribet D, Louvet-Vallée S, Harper F, de Parseval N, Dewannieux M, Heidmann O, et al. Murine endogenous retrovirus MuERV-L is the progenitor of the “orphan” epsilon virus like particles of the early mouse embryo. J Virol (2008) 82(3):1622–5. doi:10.1128/JVI.02097-07
62. Brulet P, Condamine H, Jacob F. Spatial distribution of transcripts of the long repeated ETn sequence during early mouse embryogenesis. Proc Natl Acad Sci U S A (1985) 82(7):2054–8. doi:10.1073/pnas.82.7.2054
63. Beraldi R, Pittoggi C, Sciamanna I, Mattei E, Spadafora C. Expression of LINE-1 retroposons is essential for murine preimplantation development. Mol Reprod Dev (2006) 73(3):279–87. doi:10.1002/mrd.20423
64. Kigami D, Minami N, Takayama H, Imai H. MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol Reprod (2003) 68(2):651–4. doi:10.1095/biolreprod.102.007906
65. Surface L, Thornton S, Boyer L. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell (2010) 7(3):288–98. doi:10.1016/j.stem.2010.08.004
66. Boyer L, Mathur D, Jaenisch R. Molecular control of pluripotency. Curr Opin Genet Dev (2006) 16(5):455–62. doi:10.1016/j.gde.2006.08.009
67. Toyooka Y, Shimosato D, Murakami K, Takahashi K, Niwa H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development (2008) 135(5):909–18. doi:10.1242/dev.017400
68. Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, et al. Nanog safeguards pluripotency and mediates germline development. Nature (2007) 450(7173):1230–4. doi:10.1038/nature06403
69. Suzuki A, Raya A, Kawakami Y, Morita M, Matsui T, Nakashima K, et al. Maintenance of embryonic stem cell pluripotency by Nanog-mediated reversal of mesoderm specification. Nat Clin Pract Cardiovasc Med (2006) 3(Suppl 1):S114–22. doi:10.1038/ncpcardio0442
70. Hayashi K, Lopes SM, Tang F, Surani MA. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell (2008) 3(4):391–401. doi:10.1016/j.stem.2008.07.027
71. Furusawa T, Ohkoshi K, Honda C, Takahashi S, Tokunaga T. Embryonic stem cells expressing both platelet endothelial cell adhesion molecule-1 and stage-specific embryonic antigen-1 differentiate predominantly into epiblast cells in a chimeric embryo. Biol Reprod (2004) 70(5):1452–7. doi:10.1095/biolreprod.103.024190
72. Carter MG, Stagg CA, Falco G, Yoshikawa T, Bassey UC, Aiba K, et al. An in situ hybridization-based screen for heterogeneously expressed genes in mouse ES cells. Gene Expr Patterns (2008) 8(3):181–98. doi:10.1016/j.gep.2007.10.009
73. Falco G, Lee SL, Stanghellini I, Bassey UC, Hamatani T, Ko MS. Zscan4: a novel gene expressed exclusively in late 2-cell embryos and embryonic stem cells. Dev Biol (2007) 307(2):539–50. doi:10.1016/j.ydbio.2007.05.003
74. Kalmar T, Lim C, Hayward P, Muñoz-Descalzo S, Nichols J, Garcia-Ojalvo J, et al. Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. PLoS Biol (2009) 7(7):e1000149. doi:10.1371/journal.pbio.1000149
75. Nichols J, Smith A. Naive and primed pluripotent states. Cell Stem Cell (2009) 4(6):487–92. doi:10.1016/j.stem.2009.05.015
76. Wray J, Kalkan T, Smith AG. The ground state of pluripotency. Biochem Soc Trans (2010) 38(4):1027–32. doi:10.1042/BST0381027
77. Macfarlan TS, Gifford WD, Agarwal S, Driscoll S, Lettieri K, Wang J, et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev (2011) 25(6):594–607. doi:10.1101/gad.2008511
78. Zhang W, Walker E, Tamplin OJ, Rossant J, Stanford WL, Hughes TR. Zfp206 regulates ES cell gene expression and differentiation. Nucleic Acids Res (2006) 34(17):4780–90. doi:10.1093/nar/gkl631
79. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell (2007) 128(4):635–8. doi:10.1016/j.cell.2007.02.006
80. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev (2002) 16(1):6–21. doi:10.1101/gad.947102
81. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333(6047):1303–7. doi:10.1126/science.1210944
82. Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev (2011) 25(7):679–84. doi:10.1101/gad.2036011
83. Sui X, Price C, Li Z, Chen J. Crosstalk between DNA and histones: tet’s new role in embryonic stem cells. Curr Genomics (2012) 13(8):603–8. doi:10.2174/138920212803759730
84. Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci (2003) 116(Pt 11):2117–24. doi:10.1242/jcs.00493
85. Ringrose L, Paro R. Epigenetic regulation of cellular memory by the polycomb and trithorax group proteins. Annu Rev Genet (2004) 38:413–43. doi:10.1146/annurev.genet.38.072902.091907
86. Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol (2008) 9(3):206–18. doi:10.1038/nrm2346
87. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell (2004) 119(7):941–53. doi:10.1016/j.cell.2004.12.012
88. Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature (2005) 437(7057):436–9. doi:10.1038/nature04020
89. Andrés ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, et al. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci U S A (1999) 96(17):9873–8. doi:10.1073/pnas.96.17.9873
90. Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, et al. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development (2013) 140(3):519–29. doi:10.1242/dev.087585
91. Schlesinger S, Lee AH, Wang GZ, Green L, Goff SP. Proviral silencing in embryonic cells is regulated by Yin Yang 1. Cell Rep (2013) 4(1):50–8. doi:10.1016/j.celrep.2013.06.003
92. Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet (1998) 20(2):116–7. doi:10.1038/2413
93. Vitullo P, Sciamanna I, Baiocchi M, Sinibaldi-Vallebona P, Spadafora C. LINE-1 retrotransposon copies are amplified during murine early embryo development. Mol Reprod Dev (2012) 79(2):118–27. doi:10.1002/mrd.22003
94. Gaudet F, Rideout WM, Meissner A, Dausman J, Leonhardt H, Jaenisch R. Dnmt1 expression in pre- and postimplantation embryogenesis and the maintenance of IAP silencing. Mol Cell Biol (2004) 24(4):1640–8. doi:10.1128/MCB.24.4.1640-1648.2004
95. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet (2013) 14(3):204–20. doi:10.1038/nrg3354
96. Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature (2012) 484(7394):339–44. doi:10.1038/nature10960
97. Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis (2003) 35(2):88–93. doi:10.1002/gene.10168
98. Kim SH, Kang YK, Koo DB, Kang MJ, Moon SJ, Lee KK, et al. Differential DNA methylation reprogramming of various repetitive sequences in mouse preimplantation embryos. Biochem Biophys Res Commun (2004) 324(1):58–63. doi:10.1016/j.bbrc.2004.09.023
99. Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells (2006) 11(7):805–14. doi:10.1111/j.1365-2443.2006.00984.x
100. Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature (2010) 464(7290):927–31. doi:10.1038/nature08858
101. Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell (2011) 8(6):676–87. doi:10.1016/j.stem.2011.04.004
102. Vidal M. Role of polycomb proteins Ring1A and Ring1B in the epigenetic regulation of gene expression. Int J Dev Biol (2009) 53(2–3):355–70. doi:10.1387/ijdb.082690mv
103. Christophersen NS, Helin K. Epigenetic control of embryonic stem cell fate. J Exp Med (2010) 207(11):2287–95. doi:10.1084/jem.20101438
104. Müller J, Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev (2009) 19(2):150–8. doi:10.1016/j.gde.2009.03.001
105. García E, Marcos-Gutiérrez C, del Mar Lorente M, Moreno J, Vidal M. RYBP, a new repressor protein that interacts with components of the mammalian polycomb complex, and with the transcription factor YY1. EMBO J (1999) 18(12):3404–18. doi:10.1093/emboj/18.12.3404
106. van den Berg D, Snoek T, Mullin N, Yates A, Bezstarosti K, Demmers J, et al. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell (2010) 6(4):369–81. doi:10.1016/j.stem.2010.02.014
107. Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature (2006) 444(7117):364–8. doi:10.1038/nature05284
108. Hisada K, Sánchez C, Endo TA, Endoh M, Román-Trufero M, Sharif J, et al. RYBP represses endogenous retroviruses and preimplantation- and germ line-specific genes in mouse embryonic stem cells. Mol Cell Biol (2012) 32(6):1139–49. doi:10.1128/MCB.06441-11
109. Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, et al. KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. Elife (2012) 1:e00205. doi:10.7554/eLife.00205
110. Illingworth RS, Bird AP. CpG islands – ‘a rough guide’. FEBS Lett (2009) 583(11):1713–20. doi:10.1016/j.febslet.2009.04.012
111. Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell (2012) 148(4):664–78. doi:10.1016/j.cell.2011.12.029
112. Pirity M, Locker J, Schreiber-Agus N. Rybp/DEDAF is required for early postimplantation and for central nervous system development. Mol Cell Biol (2005) 25(16):7193–202. doi:10.1128/MCB.25.16.7193-7202.2005
113. Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev (2010) 24(3):265–76. doi:10.1101/gad.544410
114. Rogers M, Hosler B, Gudas L. Specific expression of a retinoic acid-regulated, zinc-finger gene, Rex-1, in preimplantation embryos, trophoblast and spermatocytes. Development (1991) 113(3):815–24.
115. Masui S, Ohtsuka S, Yagi R, Takahashi K, Ko M, Niwa H. Rex1/Zfp42 is dispensable for pluripotency in mouse ES cells. BMC Dev Biol (2008) 8:45. doi:10.1186/1471-213X-8-45
116. Kim JD, Kim H, Ekram MB, Yu S, Faulk C, Kim J. Rex1/Zfp42 as an epigenetic regulator for genomic imprinting. Hum Mol Genet (2011) 20(7):1353–62. doi:10.1093/hmg/ddr017
117. Climent M, Alonso-Martin S, Pérez-Palacios R, Guallar D, Benito AA, Larraga A, et al. Functional analysis of Rex1 during preimplantation development. Stem Cells Dev (2013) 22(3):459–72. doi:10.1089/scd.2012.0211
118. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature (2007) 448(7151):313–7. doi:10.1038/nature05934
119. Kim J, Faulk C, Kim J. Retroposition and evolution of the DNA-binding motifs of YY1, YY2 and REX1. Nucleic Acids Res (2007) 35(10):3442–52. doi:10.1093/nar/gkm235
120. Kim J, Hinz A, Bergmann A, Huang J, Ovcharenko I, Stubbs L, et al. Identification of clustered YY1 binding sites in imprinting control regions. Genome Res (2006) 16(7):901–11. doi:10.1101/gr.5091406
121. Affar EB, Gay F, Shi Y, Liu H, Huarte M, Wu S, et al. Essential dosage-dependent functions of the transcription factor Yin Yang 1 in late embryonic development and cell cycle progression. Mol Cell Biol (2006) 26(9):3565–81. doi:10.1128/MCB.26.9.3565-3581
122. Flanagan JR, Becker KG, Ennist DL, Gleason SL, Driggers PH, Levi BZ, et al. Cloning of a negative transcription factor that binds to the upstream conserved region of Moloney murine leukemia virus. Mol Cell Biol (1992) 12(1):38–44.
123. Satyamoorthy K, Park K, Atchison ML, Howe CC. The intracisternal A-particle upstream element interacts with transcription factor YY1 to activate transcription: pleiotropic effects of YY1 on distinct DNA promoter elements. Mol Cell Biol (1993) 13(11):6621–8.
124. Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature (2010) 463(7278):237–40. doi:10.1038/nature08674
125. Garcia-Tuñon I, Guallar D, Alonso-Martin S, Benito AA, Benítez-Lázaro A, Pérez-Palacios R, et al. Association of Rex-1 to target genes supports its interaction with polycomb function. Stem Cell Res (2011) 7(1):1–16. doi:10.1016/j.scr.2011.02.005
126. Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, et al. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet (2010) 6(12):e1001244. doi:10.1371/journal.pgen.1001244
127. Kim J, Kim H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J (2012) 53(3–4):232–9. doi:10.1093/ilar.53.3-4.232
128. Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell (2003) 12(6):1591–8. doi:10.1016/S1097-2765(03)00479-9
129. Martens JH, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, et al. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J (2005) 24(4):800–12. doi:10.1038/sj.emboj.7600545
130. Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol (2006) 8(2):188–94. doi:10.1038/ncb1353
131. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature (2007) 448(7153):553–60. doi:10.1038/nature06008
132. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev (2002) 16(8):919–32. doi:10.1101/gad.973302
133. Thomas JH, Emerson RO. Evolution of C2H2-zinc finger genes revisited. BMC Evol Biol (2009) 9:51. doi:10.1186/1471-2148-9-51
134. Cammas F, Mark M, Dollé P, Dierich A, Chambon P, Losson R. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development (2000) 127(13):2955–63.
135. Hu G, Kim J, Xu Q, Leng Y, Orkin S, Elledge S. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev (2009) 23(7):837–48. doi:10.1101/gad.1769609
136. Wolf D, Goff SP. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell (2007) 131(1):46–57. doi:10.1016/j.cell.2007.07.026
137. Wolf D, Goff SP. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature (2009) 458(7242):1201–4. doi:10.1038/nature07844
138. Corsinotti A, Kapopoulou A, Gubelmann C, Imbeault M, Santoni de Sio FR, Rowe HM, et al. Global and stage specific patterns of Krüppel-associated-box zinc finger protein gene expression in murine early embryonic cells. PLoS One (2013) 8(2):e56721. doi:10.1371/journal.pone.0056721
139. Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem (2011) 286(30):26267–76. doi:10.1074/jbc.R111.252569
140. Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev (2002) 16(14):1779–91. doi:10.1101/gad.989402
141. Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev (2005) 19(7):815–26. doi:10.1101/gad.1284005
142. Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev (2011) 25(8):781–8. doi:10.1101/gad.2027411
143. Reichmann J, Crichton JH, Madej MJ, Taggart M, Gautier P, Garcia-Perez JL, et al. Microarray analysis of LTR retrotransposon silencing identifies Hdac1 as a regulator of retrotransposon expression in mouse embryonic stem cells. PLoS Comput Biol (2012) 8(4):e1002486. doi:10.1371/journal.pcbi.1002486
144. Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet (2010) 42(7):631–4. doi:10.1038/ng.600
145. Mey A, Acloque H, Lerat E, Gounel S, Tribollet V, Blanc S, et al. The endogenous retrovirus ENS-1 provides active binding sites for transcription factors in embryonic stem cells that specify extra embryonic tissue. Retrovirology (2012) 9:21. doi:10.1186/1742-4690-9-21
146. Navarro P, Avner P. When X-inactivation meets pluripotency: an intimate rendezvous. FEBS Lett (2009) 583(11):1721–7. doi:10.1016/j.febslet.2009.03.043
147. Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev (2009) 23(16):1831–42. doi:10.1101/gad.1811209
148. Courtier B, Heard E, Avner P. Xce haplotypes show modified methylation in a region of the active X chromosome lying 3’ to Xist. Proc Natl Acad Sci U S A (1995) 92(8):3531–5. doi:10.1073/pnas.92.8.3531
149. Cohen DE, Davidow LS, Erwin JA, Xu N, Warshawsky D, Lee JT. The DXPas34 repeat regulates random and imprinted X inactivation. Dev Cell (2007) 12(1):57–71. doi:10.1016/j.devcel.2006.11.014
150. Liu TY, Chen HH, Lee KH, Choo KB. Display of different modes of transcription by the promoters of an early embryonic gene, Zfp352, in preimplantation embryos and in somatic cells. Mol Reprod Dev (2003) 64(1):52–60. doi:10.1002/mrd.10218
151. Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A (2010) 107(18):8242–7. doi:10.1073/pnas.1000478107
152. Yeo S, Lee KK, Han YM, Kang YK. Methylation changes of lysine 9 of histone H3 during preimplantation mouse development. Mol Cells (2005) 20(3):423–8.
153. Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee SL, et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature (2010) 464(7290):858–63. doi:10.1038/nature08882
154. Liu L, Bailey SM, Okuka M, Muñoz P, Li C, Zhou L, et al. Telomere lengthening early in development. Nat Cell Biol (2007) 9(12):1436–41. doi:10.1038/ncb1664
155. De Los Angeles A, Loh YH, Tesar PJ, Daley GQ. Accessing naïve human pluripotency. Curr Opin Genet Dev (2012) 22(3):272–82. doi:10.1016/j.gde.2012.03.001
156. Kolesnikov NN, Elisafenko EA. [Comparative organization and the origin of noncoding regulatory RNA genes from X-chromosome inactivation center of human and mouse]. Genetika (2010) 46(10):1386–91.
157. van den Berg IM, Galjaard RJ, Laven JS, van Doorninck JH. XCI in preimplantation mouse and human embryos: first there is remodelling…. Hum Genet (2011) 130(2):203–15. doi:10.1007/s00439-011-1014-9
158. Escamilla-Del-Arenal M, da Rocha ST, Heard E. Evolutionary diversity and developmental regulation of X-chromosome inactivation. Hum Genet (2011) 130(2):307–27. doi:10.1007/s00439-011-1029-2
159. Santoni FA, Guerra J, Luban J. HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology (2012) 9:111. doi:10.1186/1742-4690-9-111
160. Fuchs NV, Loewer S, Daley GQ, Izsvák Z, Löwer J, Löwer R. Human endogenous retrovirus K (HML-2) RNA and protein expression is a marker for human embryonic and induced pluripotent stem cells. Retrovirology (2013) 10(1):115. doi:10.1186/1742-4690-10-115
161. Vassena R, Boué S, González-Roca E, Aran B, Auer H, Veiga A, et al. Waves of early transcriptional activation and pluripotency program initiation during human preimplantation development. Development (2011) 138(17):3699–709. doi:10.1242/dev.064741
162. Niakan KK, Eggan K. Analysis of human embryos from zygote to blastocyst reveals distinct gene expression patterns relative to the mouse. Dev Biol (2013) 375(1):54–64. doi:10.1016/j.ydbio.2012.12.008
163. Brivanlou A, Gage F, Jaenisch R, Jessell T, Melton D, Rossant J. Stem cells. Setting standards for human embryonic stem cells. Science (2003) 300(5621):913–6. doi:10.1126/science.1082940
164. Bhatia S, Pilquil C, Roth-Albin I, Draper JS. Demarcation of stable subpopulations within the pluripotent hESC compartment. PLoS One (2013) 8(2):e57276. doi:10.1371/journal.pone.0057276
165. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131(5):861–72. doi:10.1016/j.cell.2007.11.019
166. Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, et al. Derivation of novel human ground state naive pluripotent stem cells. Nature (2013) 504(7479):282–6. doi:10.1038/nature12745
167. Dupressoir A, Lavialle C, Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta (2012) 33(9):663–71. doi:10.1016/j.placenta.2012.05.005
168. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature (1983) 301(5895):89–92. doi:10.1038/301089a0
169. Ballestar E. Epigenetic contributions in autoimmune disease. Preface. Adv Exp Med Biol (2011) 711:v–vi.
170. Howard G, Eiges R, Gaudet F, Jaenisch R, Eden A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene (2008) 27(3):404–8. doi:10.1038/sj.onc.1210631
171. Kozeretska IA, Demydov SV, Ostapchenko LI. Mobile genetic elements and cancer. From mutations to gene therapy. Exp Oncol (2011) 33(4):198–205.
172. Mangeney M, Heidmann T. Tumor cells expressing a retroviral envelope escape immune rejection in vivo. Proc Natl Acad Sci U S A (1998) 95(25):14920–5. doi:10.1073/pnas.95.25.14920
173. Jeon BG, Kumar BM, Kang EJ, Ock SA, Lee SL, Kwack DO, et al. Characterization and comparison of telomere length, telomerase and reverse transcriptase activity and gene expression in human mesenchymal stem cells and cancer cells of various origins. Cell Tissue Res (2011) 345(1):149–61. doi:10.1007/s00441-011-1191-9
174. Cho K, Lee YK, Greenhalgh DG. Endogenous retroviruses in systemic response to stress signals. Shock (2008) 30(2):105–16. doi:10.1097/SHK.0b013e31816a363f
175. Castellano G, Torrisi E, Ligresti G, Malaponte G, Militello L, Russo AE, et al. The involvement of the transcription factor Yin Yang 1 in cancer development and progression. Cell Cycle (2009) 8(9):1367–72. doi:10.4161/cc.8.9.8314
176. Zaravinos A, Spandidos DA. Yin Yang 1 expression in human tumors. Cell Cycle (2010) 9(3):512–22. doi:10.4161/cc.9.3.10588
Keywords: MERVL, REX1/ZFP42, embryonic stem cells, two-cell state, developmental potential
Citation: Schoorlemmer J, Pérez-Palacios R, Climent M, Guallar D and Muniesa P (2014) Regulation of mouse retroelement MuERV-L/MERVL expression by REX1 and epigenetic control of stem cell potency. Front. Oncol. 4:14. doi: 10.3389/fonc.2014.00014
Received: 30 September 2013; Accepted: 21 January 2014;
Published online: 06 February 2014.
Edited by:
Iyoko Katoh, University of Yamanashi, JapanReviewed by:
Sathish Kumar Mungamuri, Mount Sinai School of Medicine, USAXiaoping Zhang, Mount Sinai School of Medicine, USA
Copyright: © 2014 Schoorlemmer, Pérez-Palacios, Climent, Guallar and Muniesa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jon Schoorlemmer, Centro de Investigación Biomédica de Aragón (CIBA), Avda. San Juan Bosco 13, Planta A, Zaragoza 50009, Spain e-mail:anNjaG9vcmxlbW1lci5pYWNzQGFyYWdvbi5lcw==
†Present address: María Climent, Stem Cell Center, Lund, Sweden; Diana Guallar, Black Family Stem Cell Institute, Icahn School of Medicine at Mount Sinai, New York, USA