 Melissa Burgess
Melissa Burgess Shannon Puhalla
Shannon Puhalla- Department of Medical Oncology, University of Pittsburgh Medical Center, University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA
No longer is histology solely predictive of cancer treatment and outcome. There is an increasing influence of tumor genomic characteristics on therapeutic options. Both breast and ovarian cancers are at higher risk of development in patients with BRCA 1/2-germline mutations. Recent data from The Cancer Genome Atlas and others have shown a number of genomic similarities between triple negative breast cancers (TNBCs) and ovarian cancers. Recently, poly (ADP-ribose) polymerase (PARP) inhibitors have shown promising activity in hereditary BRCA 1/2-mutated and sporadic breast and ovarian cancers. In this review, we will summarize the current literature regarding the genomic and phenotypic similarities between BRCA 1/2-mutation related cancers, sporadic TNBCs, and sporadic ovarian cancers. We will also review Phase I, II, and III data using PARP inhibitors for these malignancies and compare and contrast the results with respect to histology.
Introduction
BRCA1 and 2 proteins play integral functions in DNA homologous recombination repair (HRR). In normal cells, the HRR pathway is activated in response to DNA double-stranded breaks (1). In BRCA 1/2-deficient cells, HRR is faulty secondary to loss of BRCA function, and therefore, other more error-prone DNA repair pathways are activated. These less perfect mechanisms are felt to be accountable, in part, for carcinogenesis. Similarly, tumors with defective HRR mechanisms are more susceptible to the direct DNA damaging effects of chemotherapy.
Homologous recombination repair dysfunction can be exploited as a therapeutic strategy by the use of poly (ADP-ribose) polymerase (PARP) inhibitors, which inhibit PARP proteins, most commonly PARP1 and 2. As part of the base excision repair (BER) pathway, PARP1 attaches long polymers of ADP-ribose on itself, so that, XRCC1 and other repair proteins have the ability to rapidly locate single-stranded DNA breaks (2–4). Newer evidence reveals that the exact role of PARP1 in the BER pathway is perhaps more indirect and not yet clearly defined (5). Recent studies have also shown that PARP1 is more versatile, and has been implicated in other DNA repair pathways, such as the non-homologous end-joining (NHEJ) repair pathway (6, 7).
Several mechanisms by which PARP inhibition in HRR-deficient cells lead to cell death have been investigated. Most notably, the concept of synthetic lethality explains combinatory lethal effects of BER and HR repair dysfunction, whereas alone, HR or BER pathway disruptions are not lethal to the tumor cell (8). Additionally, other potential mechanisms have been explored including trapping of inhibited PARP1 at sites of DNA damage preventing other repair proteins access, failure to initiate HRR by PARP-dependent BRCA1 recruitment, and activation of the error-prone NHEJ repair pathway leading to genomic instability and subsequent cell death (9). Knowledge of PARP activity has led to effective treatment strategies for BRCA 1/2-germline mutation related tumors.
BRCA 1/2-Mutated Ovarian and Breast Cancer
BRCA 1/2-mutation related ovarian and breast cancers account for 5–10% of all female ovarian and breast cancers (10, 11). Ovarian cancers in the setting of BRCA 1/2-germline mutations can present with more aggressive, high-grade histologies, but are frequently responsive to chemotherapy, particularly platinum-based regimens, leading to an improved 5 years survival (12). The chemotherapy-sensitive mechanism is felt to be related to the intimate relationship between BRCA 1/2 proteins and defective HRR, as discussed above. Recent studies have demonstrated that women with BRCA-related ovarian cancers fare much better than sporadic ovarian cancers (13–16). A study, published by the National Israeli Study of Ovarian Cancer, showed women with BRCA mutations had a median survival of 55.7 months compared to 37.9 months in sporadic ovarian cancers (p = 0.002) (15). This may be in part explained by the standard use of carboplatin-based therapies for ovarian malignancies as the DNA damage induced by the platinum should be more efficacious in the DNA repair-deficient BRCA-related tumors.
Contrary to the more convincing outcomes in BRCA 1/2-related ovarian cancers, the outcomes of BRCA mutation-related breast cancers are less clear. Women with BRCA1 mutations typically develop breast cancer at an earlier age than BRCA2-related and sporadic breast cancers. BRCA1-related breast cancers tend to also be higher grade, hormone receptor-negative, and HER-2-negative, or “triple negative” (17), and also frequently express a basal phenotype (18–26). Patients with BRCA-mutated breast cancers generally respond to therapy as well as sporadic cancers; however, the risk of second ipsilateral or contralateral primaries may be as high as 3–5% per year, compared to 0.5–1% per year risk, seen in sporadic breast cancers (17). In contrast to ovarian cancer, platinum chemotherapy is not standardly administered to patients with breast cancer. The use of platinum agents has been evaluated in a small series which have demonstrated high efficacy in breast cancer in particular in the setting of a BRCA mutation. Silver et al. evaluated the use of neoadjuvant platinum-containing chemotherapy in patients with triple negative breast cancer (TNBC) (N = 28), and found those more likely to be platinum-sensitive were those with low BRCA1 gene expression (27). Likewise, in BRCA-mutated breast cancer patients who received cisplatin in the neoadjuvant setting showed a high rate of pathologic complete response (pCR) in a small series. Ten of 12 patients achieved pCR (83%). When non-platinum-containing regimens were used, the pCR rate was 14% (28). These studies highlight the rationale to further explore the use of platinum-containing regimens, specifically for patients with TNBC and BRCA mutations.
BRCAness: Sporadic Triple Negative Breast Cancers
Triple negative breast cancers account for ~20% of all breast cancers and are associated with an aggressive clinical picture (20, 25, 29). Due to lack of hormone receptor or HER-2 expression, and no other known target for tailored therapy, the only current treatment option is chemotherapy. Over 80% of hereditary BRCA1-mutated cancers are TNBCs. Several studies have investigated a potential role for BRCA1 inactivation in sporadic TNBC given the similar clinical outcomes and histological characteristics among these cancers and hereditary BRCA1-mutated breast cancers. Breast cancers developing in patients with BRCA1 mutations, in addition to frequently being triple negative, also often express basal markers (18–22, 25, 26). Gene microarray expression profiling has shown considerable similarities between BRCA1-mutated tumors and basal tumors (25). This shared phenotype has been termed “BRCAness” (26). What is unknown is whether the basal phenotype is a result of the BRCA loss or if the BRCA loss results in the basal phenotype (6).
Recently, Lehmann and colleagues delved further into the characterization of TNBC. They performed an analysis of gene expression profiles of 587 TNBC cases and identified six separate subtypes of TNBC. These six subtypes were: basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL), and luminal androgen receptor (LAR) subtype. Additional analysis of TNBC cell lines, representative of each of these identified subsets, revealed differential responses to various therapeutic agents. Both the BL1 and BL2 groups showed increased gene expression involved in DNA damage response, and showed higher response to cisplatin (30). In a follow-up study, Masuda et al. presented neoadjuvant chemotherapy response data in each of the aforementioned TNBC subtypes (31). In 130 TNBC patients, who received standard anthracycline- and taxane-based chemotherapy, the BL1 subtype achieved a pCR most frequently (52%). In contrast, the pCR in the BL2 subtype was 0%. The molecular differences in BL1 and BL2 may explain these differential responses. Specifically, the BL1 subtype involves the cell cycle, DNA replication reactome, and the BRCA pathway, among others, whereas the BL2 subtype involves growth factor, glycolysis, and gluconeogenesis pathways. This work demonstrates that even within “basal-like breast cancer (BLBC),” there may be a great deal of heterogeneity.
Telli and colleagues recently presented a study evaluating gemcitabine, carboplatin, and iniparib, a compound initially believed to have PARP inhibitory effects, in the neoadjuvant treatment of triple negative and BRCA-mutated breast cancer (32). This study demonstrated a pCR of 36% overall, with a pCR in BRCA 1/2-mutation carriers of 47%. Furthermore, patients who were both triple negative and had a BRCA 1/2-mutation, had a pCR of 56%. Although only 10 patients were classified as BL1 or BL2, there were an equal number of responders and non-responders to the neoadjuvant platinum regimen. It is also notable that only one patient classified as basal-like had a known BRCA mutation, whereas, there were BRCA-mutated tumors that were classified as IM, M, MSL, and unspecified (32). Although basal-like TNBC has become nearly synonymous with BRCAness, this study found that the basal-like subtype of TNBC was neither particularly responsive to the treatment combination, nor had a higher number of BRCA-germline mutations. In this study, the homologous recombination deficiency (HRD) score appeared to be more predictive of platinum response, as compared to TNBC intrinsic subtyping (30). The HRD assay has been developed to evaluate for loss of heterozygosity (LOH), which has been shown to be predictive of response to platinum in BRCA-related and sporadic cancers (33). While, this data is hypothesis-generating and thought-provoking, larger, prospective studies will be needed before any formal conclusions can be drawn.
In sporadic basal tumors, there are data that show reduced BRCA1 mRNA expression. It is felt that epigenetic modification of the BRCA gene, such as promoter hypermethylation, is responsible for this (34–36). Interestingly, no tumors showed both BRCA1 mutation and BRCA1 promoter methylation suggesting that these events are mutually exclusive in The Cancer Genome Atlas (TCGA) research network data (37). The association between BRCA1-mutated and BLBCs provides an important rationale to include this frequently encountered patient population in studies geared toward manipulation of the characteristic faulty DNA repair mechanisms in BRCA1-mutated tumors. As we move into an era where genomic analyses of tumors is becoming the norm, it will be important to link the genome, methylome, and proteome to clinical characteristics and outcomes.
BRCAness: Sporadic High-Grade Serous Ovarian Cancers
Similarly, there are many commonalities among BRCA 1/2-mutated cancers and sporadic epithelial ovarian cancers (EOCs). Although only 5–10% of ovarian cancers are directly attributable to a germline mutation in BRCA1 or 2, there is a growing body of evidence to suggest that additional mechanisms of BRCA dysfunction are involved in the pathogenesis of ovarian cancer (26, 38, 39). One study demonstrated alterations of BRCA1 and/or 2 in up to 82% of examined ovarian cancers (n = 92) (40). Methylation of the BRCA1 promoter has been demonstrated in up to 14% of sporadic breast and up to 30% of sporadic ovarian cancers (26, 35, 41–46). LOH has been described in ovarian tumors and may have multiple possible mechanisms leading to malignancy including co-existing LOH of BRCA1 and p53, and hypermethylation acting in a synergistic fashion (33, 47–51). In contrast, BRCA2 methylation has not been found to be a significant contributor (39, 52). Identifying and manipulating these BRCA-like deficiencies in DNA repair in sporadic ovarian cancers is of great importance and provides rationale for including these patients in clinical trials designed for BRCA-related malignancies.
Another important mechanism of BRCAness in ovarian cancers is the presence of somatic mutations in BRCA1 and 2 (53). Hennessy and colleagues performed BRCA1/2 sequencing on 235 unselected ovarian cancers and found that 19% of the sample had detectable mutations in BRCA1 (N = 31) or BRCA2 (N = 13). In the 28 samples, where germline DNA was also available, 42.9% of the BRCA1 mutations and 28.6% of the BRCA2 mutations were somatic. Of interest, somatic BRCA 1/2-mutations in breast cancer appear to be less frequent. In the TCGA BLBC cohort, about 20% had either germline (N = 12) or somatic (N = 8) BRCA 1/2-mutations. Another study evaluated 77 TNBC samples and only one harbored a somatic BRCA mutation (54). This potentially explains the seemingly higher activity of single agent PARP inhibitors, discussed later, in sporadic ovarian cancer as compared to sporadic TNBC.
Genomic Similarities: Basal-Like Breast Cancer and High-Grade Serous Ovarian Cancers
The Cancer Genome Atlas network recently published findings again demonstrating the four distinct molecular signatures in breast cancer from diverse genetic and epigenetic alterations: luminal A, luminal B, basal-like, and HER-2 enriched subtypes (55). Strikingly, BLBCs were notably different than the other three subtypes based on comprehensive analyses using multiple platforms. As expected, these cancers also often (80%) lacked expression of ER, PR, and HER-2 identifying as TNBCs. Specifically, most BLBCs showed a high frequency of TP53 deleterious mutations (80%), as well as, loss of RB1 and BRCA1. PIK3CA mutations (~9%) were also a common feature of BLBC. Analyses also highlighted increased MYC activation as a BLBC characteristic.
The BLBC mutation spectrum reported in the TCGA was similar to that identified in previously described serous ovarian cancers (56) and BLBC were more similar to serous ovarian carcinomas than to other subtypes of breast cancer. One gene, in particular, TP53, had a >10% mutation frequency in both basal-like breast and serous ovarian cancers. As well, both tumors when compared to luminal showed increased BRCA1 inactivation, RB1 loss, cyclin E1 amplification, high expression of AKT3, and MYC amplification. These molecular commonalities strongly suggest shared driving events in tumorigenesis, and similarly, show support for shared treatment strategies for TNBCs and high-grade serous ovarian cancers. Of note, p53 mutations have been described to have high frequency in BRCA mutation-related cancers as well (57, 58).
PARP Inhibitors: Preclinical Era
Bryant et al. and Farmer et al. demonstrated synthetic lethality in BRCA2-deficient cells with the use of two different PARP inhibitors (59, 60). PARP inhibitors have also shown efficacy preclinically in cells lacking other HRR proteins, such as RAD51, ATR, ATM, CHK1, and FANCA or FANCC (61). These studies have given basis for clinical trials in both BRCA-deficient cancer populations, as well as, those with malignancies sharing qualities of BRCAness or HRR-deficiency, such as basal-like or TNBC and serous ovarian cancer.
PARP Inhibitors in Clinical Trials
BRCA 1/2-Mutation Studies
The first published Phase I study evaluating PARP inhibitors in the clinic used olaparib (AZD2281) enrolling patients with varying malignancies (Tables 1 and 2) (62). An expansion cohort of BRCA-positive ovarian, breast, and prostate cancer patients was enrolled at the recommended Phase II dose of 400 mg twice daily. Nearly half of the evaluable patients had an objective response (19 patients, 47%). Results from this pivotal study showed olaparib was generally well tolerated. From here, two Phase II proof-of-concept trials (ICEBERG 1 and 2) (Tables 1 and 2) confirmed activity in both BRCA-mutated ovarian and breast cancers, with olaparib at 400 mg twice daily [ORR 11/33 (33%) and 11/27 (41%), respectively], with low overall toxicities (63, 64).
Olaparib was also evaluated in patients with sporadic cancers displaying a presumed BRCAness phenotype. Gelmon et al. performed a non-randomized Phase II trial using olaparib in heavily treated high-grade serous or undifferentiated ovarian carcinomas and TNBCs (65) (Tables 1–4). Stratified by BRCA mutation status, both BRCA-mutated and BRCA-wild type ovarian carcinoma patients showed response to olaparib. In contrast, neither BRCA-mutated nor sporadic breast cancer patients demonstrated significant response to olaparib. Potential explanations for these mixed results include that not all TNBCs have a BRCA-like phenotype, so there may have been some heterogeneity to this population (30).
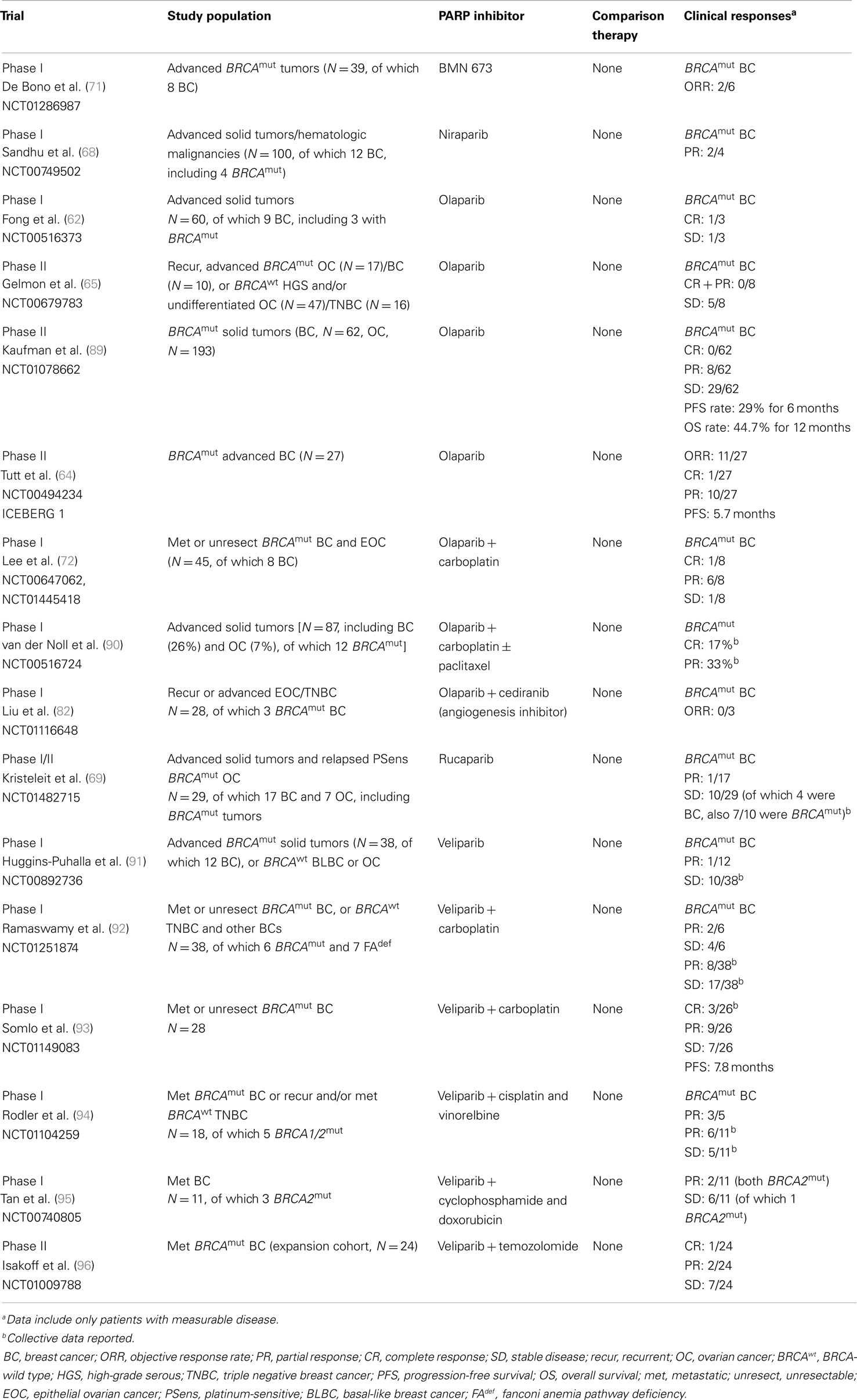
Table 1. Selected PARP inhibitor trials in BRCA 1/2-mutated (BRCAmut) breast cancers.
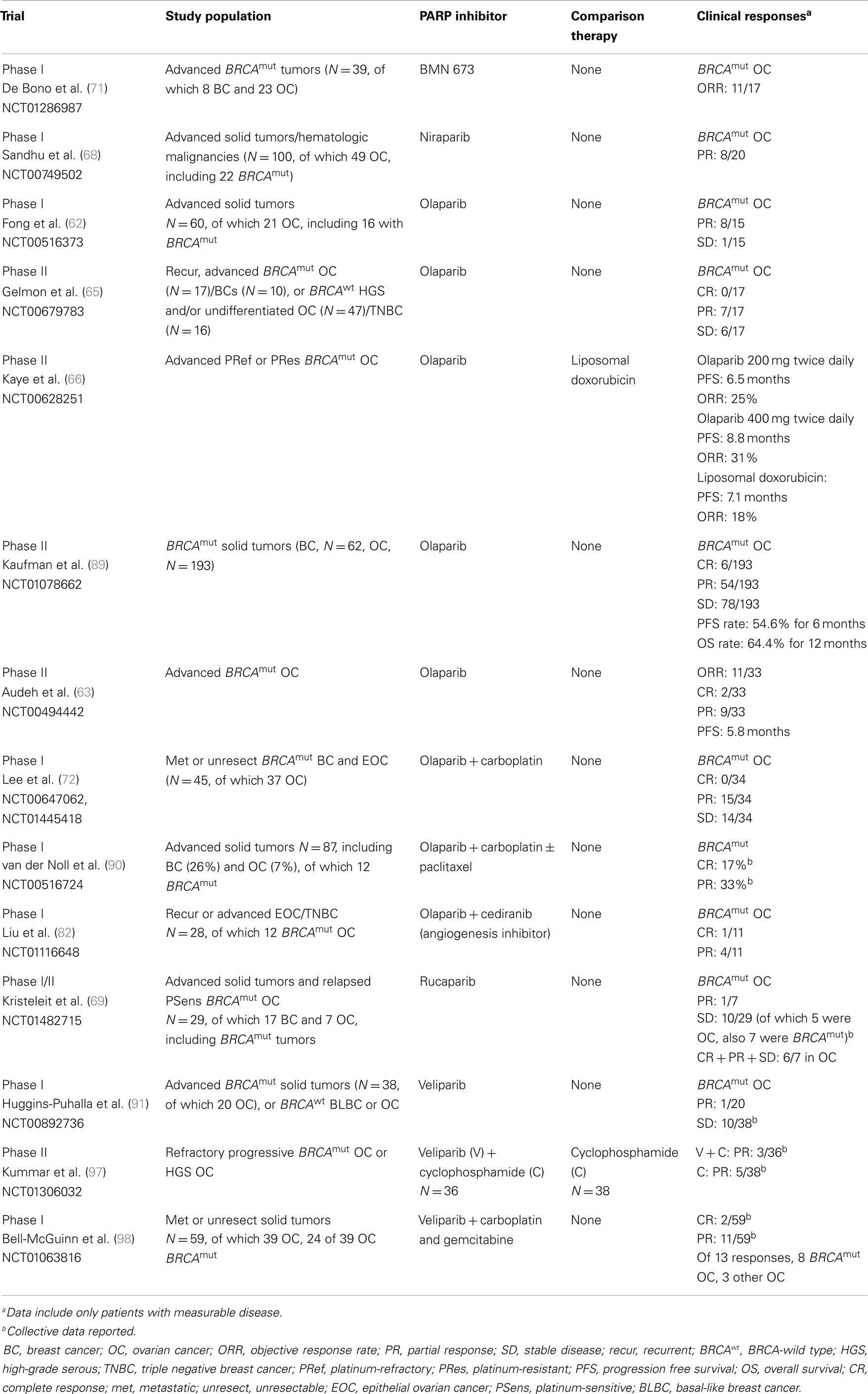
Table 2. Selected PARP inhibitor trials in BRCA 1/2-mutated (BRCAmut) ovarian cancers.
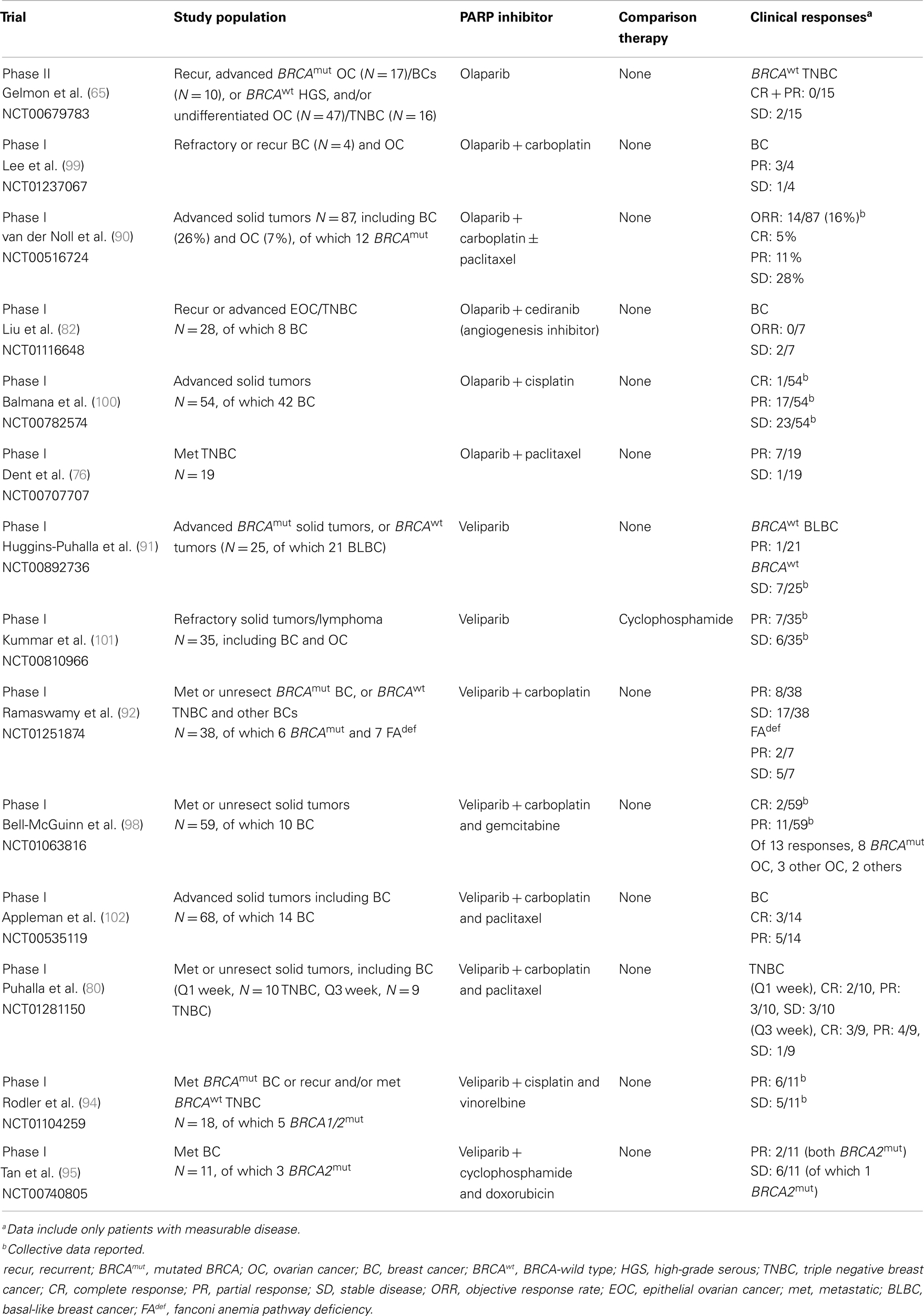
Table 3. Selected PARP inhibitor trials in sporadic breast cancers.
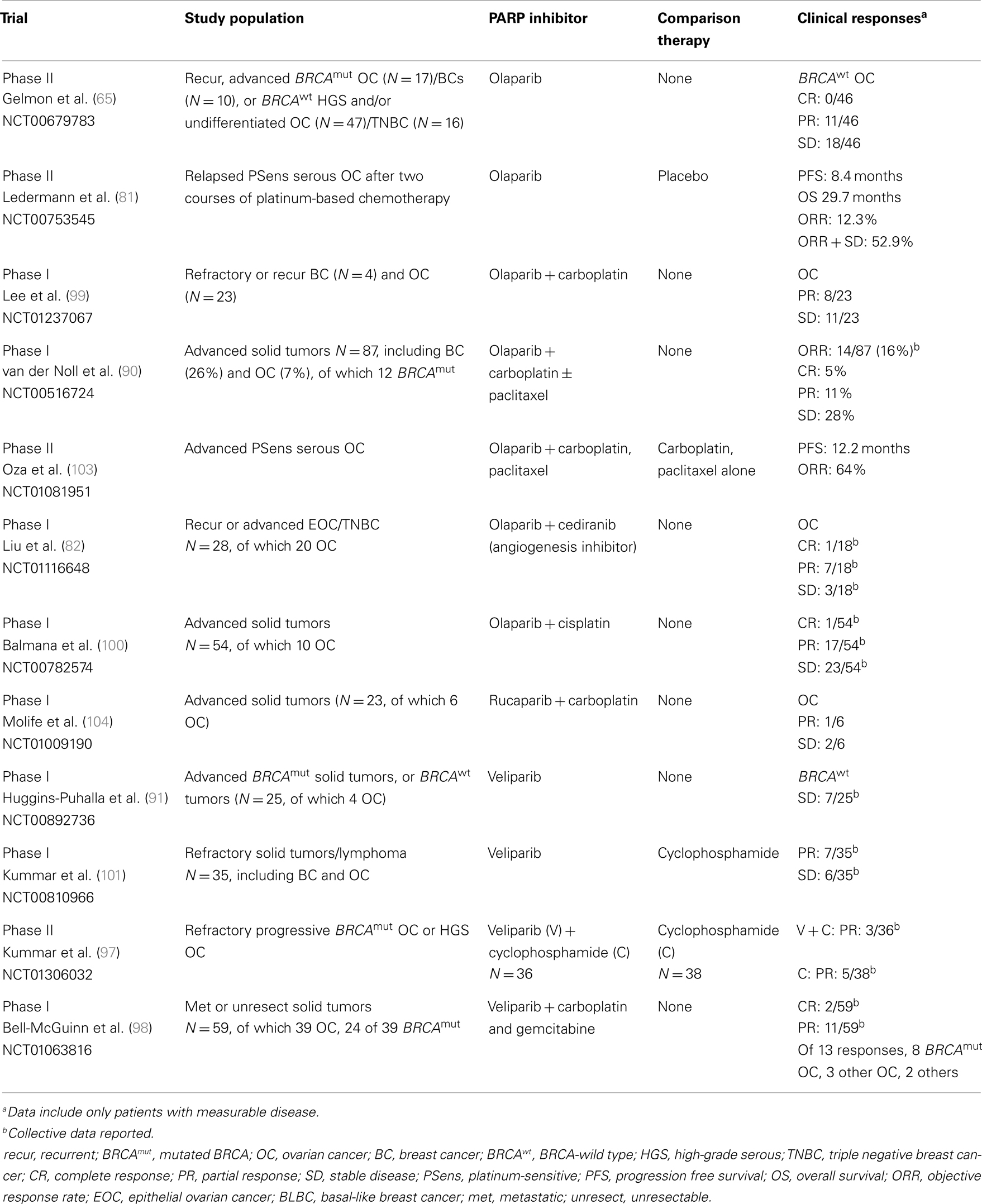
Table 4. Selected PARP inhibitor trials in sporadic ovarian cancers.
In a population of BRCA-positive recurrent ovarian cancer patients with a platinum-free interval of ≤12 months, olaparib was compared to pegylated liposomal doxorubicin (PLD) in a randomized Phase II trial (N = 97) (66) (Table 2). Progression free survival (PFS) was not statistically significantly different for olaparib 200 or 400 mg twice daily (combined or individually) versus PLD (PFS 6.5 versus 8.8 versus 7.1 months, respectively). Where the PFS and ORR were consistent with prior studies for olaparib at 400 mg twice daily, the efficacy of PLD was higher than expected when compared with previous trials. Toxicity profiles were distinct between olaparib (nausea, vomiting, and fatigue) and PLD (stomatitis and palmar-plantar erythrodysesthesia), and overall, the drugs were well tolerated. Although olaparib did not show an improvement in PFS over chemotherapy, these results show that targeted therapy with a PARP inhibitor is as effective as chemotherapy, with potential for improved tolerability.
Other PARP inhibitors have also been studied in clinical trials including niraparib (MK4827) in both BRCA-positive and sporadic tumors. This compound’s mechanism of action includes PARP inhibition via a novel PARP trapping mechanism (67). A Phase I study utilizing niraparib monotherapy was recently published that established a maximum tolerated dose of 300 mg/day (N = 100) (68) (Table 1). Dose-limiting toxicities (DLTs) were reported in the first cycle including grade 4 thrombocytopenia at a dose of 400 mg/day. Non-hematologic DLTs included grade 3 fatigue and grade 3 pneumonitis at lower doses (30 and 60 mg/day, respectively). Common treatment-related effects were anemia, nausea, fatigue, thrombocytopenia, anorexia, neutropenia, constipation, and vomiting, but were predominantly grade 1 or 2. There were anti-tumor responses seen in the BRCA-mutated breast and ovarian cancer population, and these were recorded at doses >60 mg/day. Results from this study show promise for this newer PARP inhibitor and currently there are multiple Phase III trials recruiting in BRCA-positive breast and ovarian, and sporadic ovarian cancer populations (NCT01905592, NCT01847274) (Tables 5 and 6).
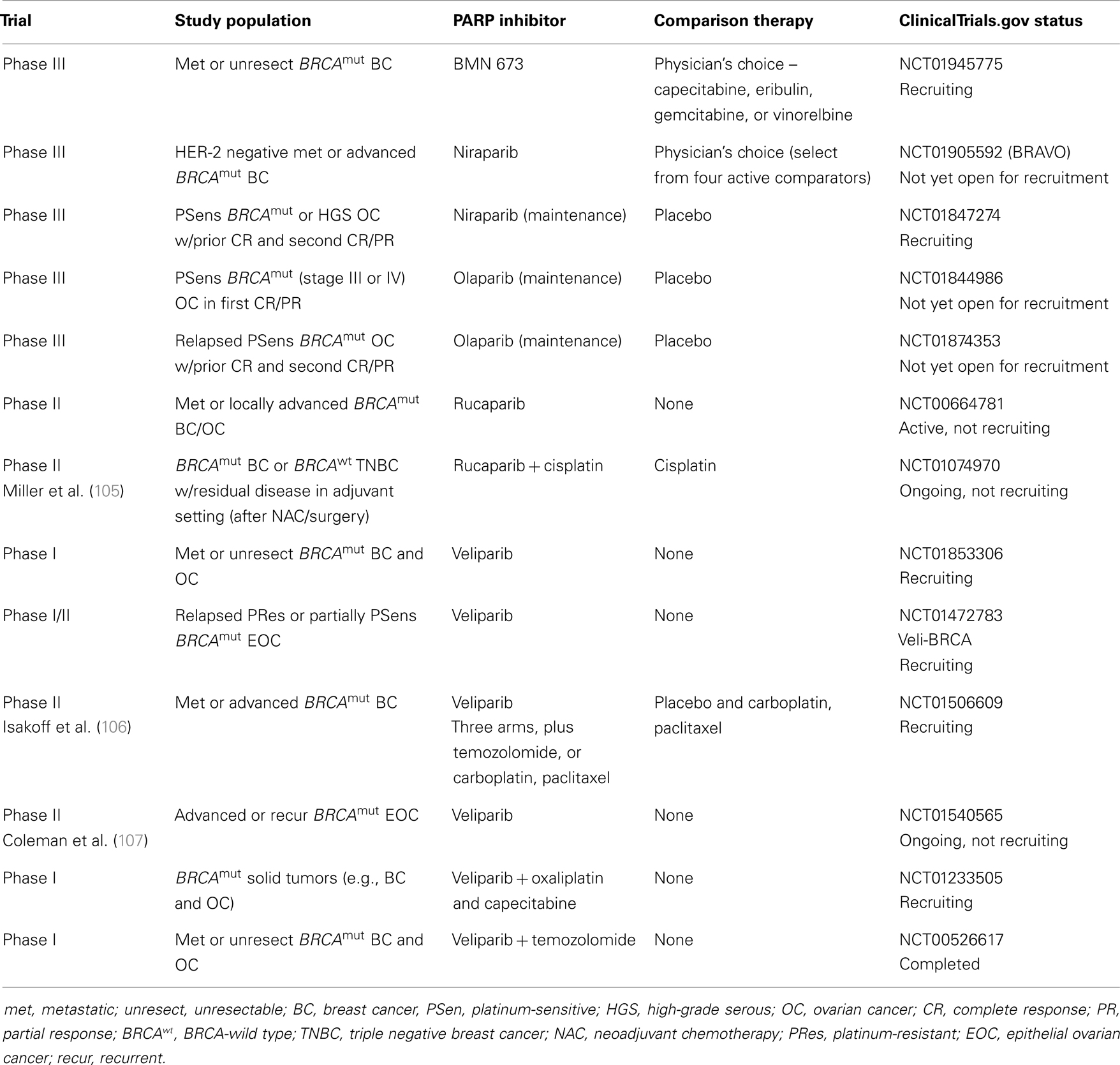
Table 5. Ongoing or future PARP inhibitor trials in BRCA 1/2-mutated (BRCAmut) breast and ovarian cancers.
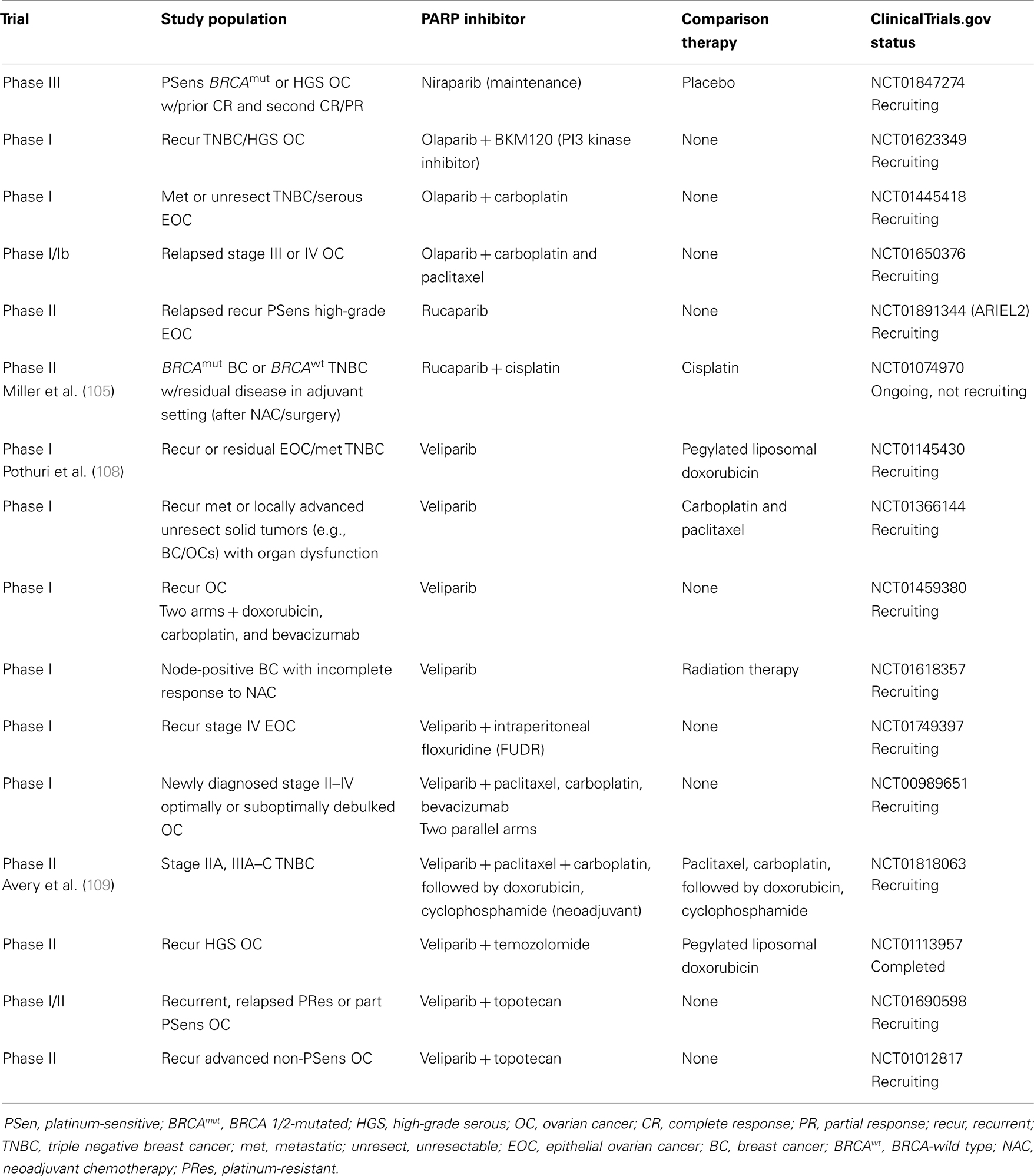
Table 6. Ongoing or future PARP inhibitor trials in sporadic breast and ovarian cancers.
Rucaparib (CO-338/AG-014699, also previously PF-01367338) was recently evaluated in Phase I and II studies in advanced solid tumors, including BRCA-positive breast and ovarian cancers. The PARP inhibitor as monotherapy and in combinations with cytotoxic chemotherapy is under investigation. In a standard dose-escalation fashion, a Phase I/II study (Tables 1 and 2) is currently evaluating rucaparib monotherapy in advanced solid tumors (N = 29) including ovarian/primary peritoneal (N = 7) and breast (N = 17) cancer patients (69). Thus far, no DLTs at 360 mg twice daily (study not yet complete) have been reported. To date, two PRs were seen in one BRCA-positive ovarian cancer, and one BRCA-positive breast cancer patient at 300 mg daily dosing during the sixth week of therapy. Ten additional patients (ovarian N = 5, breast N = 4, and colorectal N = 1) have experienced stable disease (SD) at >12 weeks so far; seven of which are BRCA-positive. Overall the disease control rate (PR + SD > 12 weeks) for ovarian cancer patients is 86% (6/7). Further results are anticipated from this study. These promising results to date have supported ARIEL2, a Phase II study of rucaparib in platinum-sensitive, relapsed, high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer patients, which is currently recruiting patients (Table 6).
BMN 673, a novel, highly potent PARP 1/2 inhibitor, demonstrated high efficacy in preclinical studies (70). BMN 673 elicits DNA repair biomarkers at much lower concentrations [PARP1 half maximal inhibitory concentration (IC50) <1 nmol/L] than earlier generation PARP inhibitors, i.e., olaparib, veliparib, and rucaparib. Its anti-tumor activity has been tested in vitro and in xenograft cancer models, as monotherapy and in combination. Anti-tumor activity was seen in BRCA1, BRCA2, and PTEN deficient cells with a 20 to more than 200-fold greater potency than existing PARP 1/2 inhibitors. Synergism was also seen when BMN 673 was combined with temozolomide, SN38, or platinum drugs. Thus far, BMN 673 has been the most specific PARP inhibitor in its class.
The first in-human Phase I, clinical trial using BMN 673 in solid tumor patients was recently presented at ASCO 2013 (71) (Tables 1 and 2). Patients with advanced solid tumors defective in DNA repair, including BRCA-mutated breast (N = 6), and ovarian (N = 17) cancer patients, were eligible for the stage II expansion phase at the maximum tolerated dose of 1000 mcg daily. In total, 39 patients with advanced solid tumors were enrolled, including those tumors with deleterious BRCA mutations. Thrombocytopenia was dose-limiting and occurred in three patients at doses 900 or 1100 mcg daily. Most potential treatment-related adverse events (AEs) were grade 1/2 and included fatigue, nausea, flatulence, anemia, neutropenia, thrombocytopenia, and alopecia. Objective responses were seen in 11/17 BRCA-mutated ovarian/primary peritoneal cancer patients and 2/6 BRCA-mutated breast cancer patients. Based on these encouraging results, the recommended dose, 1000 mcg daily, will be studied in a Phase III trial in BRCA-carrier metastatic or locally advanced breast cancer patients (NCT01945775) (Table 5).
In addition to the single agent studies described above, PARP inhibitors have been combined with chemotherapy in BRCA mutation-related malignancies. Lee et al. in a Phase I/Ib study, utilized olaparib, in combination with carboplatin, in a standard dose-escalation study design in BRCA 1/2-mutated breast and ovarian cancers (N = 45) (72) (Tables 1 and 2). The recommended Phase II dose was 400 mg twice daily for 14 days with carboplatin AUC 5. As noted in several other trials utilizing olaparib, and other PARP inhibitors, myelosuppression was frequently present with grade 3/4 AEs (neutropenia 42%), as well as, thrombocytopenia (20%), anemia (13%), carboplatin-hypersensitivity (9%), and fatigue (7%). Responses included one CR in a breast cancer patient that was durable (duration of 17 months), and a PR in 15/34 (44%) ovarian cancer (duration 3–28+ months) and 6/8 breast cancer (duration 5–24+ months) patients. Prolonged SD was seen in 14/34 (41%) ovarian cancer patients for as long as 25 months and for 11 months in a breast cancer patient. Remarkably, the overall clinical benefit rate was 100% in breast cancer patients and 85% in ovarian cancer patients. A summary of Phase I–III studies utilizing PARP inhibitors in BRCA 1/2-mutated breast and ovarian cancers can be found in Tables 1 and 2.
Sporadic Breast and Ovarian Cancer Trials
The earliest trials reported for sporadic TNBCs evaluated iniparib (BSI-201) in combination with gemcitabine and carboplatin. The Phase II trials showed promising anti-tumor activity, prolonged median progression-free survival, and median overall survival (OS) with minimal overall toxicity (73). Disappointingly, the results were not significant in the Phase III trial (74). There are a number of potential explanations for the lack of efficacy seen in the Phase III study, including the heterogeneity within the subtypes of TNBC. Importantly, it was discovered that iniparib was actually not a PARP inhibitor, at physiologic concentrations. Rather, iniparib was shown to cause telomere-centric DNA damage (75).
There are also a number of reported and ongoing studies with “true” PARP inhibitors in sporadic TNBCs, although, only a few studies that have been published in final format. A Phase I/II study of mention explored the use of olaparib in combination with paclitaxel in the first or second-line setting for metastatic TNBC patients (N = 19) (76) (Table 3). Notably, patients were treated with olaparib 200 mg daily with paclitaxel 90 mg/m2 weekly for 3 of 4 weeks and 15 of the patients had had previous taxane-based therapy. Thirty-seven percent of patients had a PR, although, there were significant dose modifications due to the greater than expected rate of neutropenia, even despite use of growth factor support. While taxanes are proven agents in TNBC (77–79), this class is not typically thought to be a potentiating agent for PARP inhibitors. Most studies have used a platinum agent for potentiation, exploiting the DNA damage/dysfunctional DNA repair pathways concept. Perhaps utilizing two agents that are active in different parts of the cell cycle would potentially target more tumor cells, overall, including those in different phases of growth. Additionally, the utility of PARP inhibitor/taxane-based combination may have potentially overcome taxane resistance. There are ongoing studies with platinum and taxane combinations with a PARP inhibitor. Early looks at efficacy are promising (80).
Similarly in ovarian cancer, there have been a number of studies evaluating PARP inhibitors with chemotherapy, including in the maintenance setting. Ledermann et al. studied olaparib in the maintenance setting after second CR in platinum-sensitive recurrent serous ovarian cancer patients. This was a Phase II, randomized, double-blinded, placebo-controlled trial (N = 265) (81) (Table 4). Median PFS was statistically significant between the groups, 8.4 versus 4.8 months, in the olaparib and placebo arms, respectively (p < 0.001). OS was not significantly different (29.7 versus 29.9 months in the olaparib and placebo groups, respectively). Further studies are needed to identify a population of patients that may experience greater clinical benefit, such as those with BRCA 1/2-mutations or those with a BRCAness phenotype.
Combination therapies with PARP inhibitors have also been investigated in sporadic ovarian and breast cancers, specifically with other novel targeted agents. Cediranib, an anti-angiogenesis agent, was studied with olaparib in recurrent epithelial ovarian or TNBCs (N = 28, 20 ovarian and 8 breast) (82) (Tables 1–4). Patients were enrolled to four dose levels and the recommended Phase II dose was cediranib 30 mg daily and olaparib 200 mg twice daily was based on one occurrence of grade 4 neutropenia (≥4 days) and one of grade 4 thrombocytopenia with dosages of cediranib 30 mg daily and olaparib 400 mg twice daily. Seventy-five percent of patients experienced grade 3 or higher toxicities with grade 3 hypertension and fatigue, occurring in 25 and 18% of subjects, respectively. Despite the frequent hematologic and non-hematologic toxicities, the ORR was 44% in the evaluable ovarian cancer population (N = 18). Sixty-one percent of ovarian patients had clinical benefit (including those with SD). None of the breast cancer patients experienced clinical response, but two patients had SD for >24 weeks. A summary of Phase I–III studies utilizing PARP inhibitors in sporadic breast and ovarian cancers can be found in Tables 3 and 4.
Platinum and PARP Inhibitor Resistance
BRCA 1/2-deficient cancers are known to be hypersensitive to platinum agents which are thought account for, in part, better overall prognosis for those patients with BRCA 1/2-germline mutation-related breast and ovarian cancer. Not all patients respond to platinum, however, and indeed, it is likely that the majority of tumors will eventually become platinum-resistant. Additionally, not all patients with BRCA 1/2-germline mutations or those with an expected BRCAness phenotype respond to PARP inhibition. Several mechanisms of resistance to both agents have been hypothesized and are likely to be multifactorial in etiology. Current evidence suggests that secondary mutations occur in the BRCA1 or BRCA2 gene restoring the wild type BRCA 1/2 open reading frame which may provide return of DNA repair through a functional HR pathway. These reversion mutations are thought to lead to platinum resistance, as well as PARP inhibitor resistance (83–87). It is imperative that these secondary mutations are identified to help modulate therapeutic management of these populations. Of interest, PARP inhibitor resistance may, in fact, not affect subsequent therapy response, including subsequent platinum regimens (88).
Conclusion
Poly (ADP-ribose) polymerase inhibitors have shown promising activity as both monotherapy and in combination with cytotoxic chemotherapy in BRCA 1/2-mutated cancers. More recently, this concept has been implicated in sporadic high-grade serous ovarian cancers and TNBCs. Like platinum agents, PARP inhibitors have been efficacious in this population. Published data from the TCGA network further support this therapeutic strategy by showcasing the genomic similarities between high-grade serous ovarian cancers and TNBCs. It may be worthwhile in the future to study new drug therapies in tandem in these two populations. New strategies are needed to combat tumor resistance mechanisms, such as secondary mutations that revert BRCA genes to wild type, to both platinum agents and PARP inhibitors. Future directions for PARP inhibition include when best to use these agents, in what combinations, and precisely, how to define the optimal populations that will get the most benefit.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer (2003) 3:23–34. doi: 10.1038/nrc970
2. El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res (2003) 31(19):5526–33. doi:10.1093/nar/gkg761
3. Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature (1992) 356:356–8. doi:10.1038/356356a0
4. Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol (1998) 18(6):3563–71.
5. Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res (2011) 39(8):3166–75. doi:10.1093/nar/gkq1241
6. Ashworth A. A synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol (2008) 26:3785–90. doi:10.1200/JCO.2008.16.0812
7. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature (2001) 411:366–74. doi:10.1038/35077232
8. Dobzhansky T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics (1946) 31:269–90.
9. De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The elephant and the blind men: making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front Oncol (2013) 3:228. doi:10.3389/fonc.2013.00228
11. Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol (2012) 30(21):2654–63. doi:10.1200/JCO.2011.39.8545
12. Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer (2003) 97(9):2187–95. doi:10.1002/cncr.11310
13. Ben David Y, Chetrit A, Hirsh-Yechezkel G, Friedman E, Beck BD, Beller U, et al. Effect of BRCA mutations on the length of survival in epithelial ovarian tumors. J Clin Oncol (2002) 20(2):463–6. doi:10.1200/JCO.20.2.463
14. Boyd J, Sonoda Y, Federici MG, Bogomolniy F, Rhei E, Maresco DL, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA (2000) 283(17):2260–5. doi:10.1001/jama.283.17.2260
15. Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol (2008) 26(1):20–5. doi:10.1200/JCO.2007.11.6905
16. Rubin SC, Benjamin I, Behbakht K, Takahashi H, Morgan MA, LiVolsi VA, et al. Clinical and pathological features of ovarian cancer in women with germ-line mutations of BRCA1. N Engl J Med (1996) 335(19):1413–6. doi:10.1056/NEJM199611073351901
17. Brekelmans CT, Seynaeve C, Menke-Pluymers M, Brüggenwirth HT, Tilanus-Linthorst MM, Bartels CC, et al. Survival and prognostic factors in BRCA1-associated breast cancer. Ann Oncol (2006) 17(3):391–400. doi:10.1093/annonc/mdj095
18. Arnes JB, Brunet JS, Stefansson I, Bégin LR, Wong N, Chappuis PO, et al. Placental cadherin and the basal epithelial phenotype of BRCA1-related breast cancer. Clin Cancer Res (2005) 11(11):4003–11. doi:10.1158/1078-0432.CCR-04-2064
19. Foulkes WD, Brunet JS, Stefansson IM, Straume O, Chappuis PO, Bégin LR, et al. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res (2004) 64(3):830–5. doi:10.1158/0008-5472.CAN-03-2970
20. Foulkes WD, Stefansson IM, Chappuis PO, Bégin LR, Goffin JR, Wong N, et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst (2003) 95(19):1482–5. doi:10.1093/jnci/djg050
21. Laakso M, Loman N, Borg A, Isola J. Cytokeratin 5/14-positive breast cancer: true basal phenotype confined to BRCA1 tumors. Mod Pathol (2005) 18(10):1321–8. doi:10.1038/modpathol.3800456
22. Lakhani SR, Van De Vijver MJ, Jacquemier J, Anderson TJ, Osin PP, McGuffog L, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol (2002) 20(9):2310–8. doi:10.1200/JCO.2002.09.023
23. Palacios J, Honrado E, Osorio A, Cazorla A, Sarrió D, Barroso A, et al. Immunohistochemical characteristics defined by tissue microarray of hereditary breast cancer not attributable to BRCA1 or BRCA2 mutations: differences from breast carcinomas arising in BRCA1 and BRCA2 mutation carriers. Clin Cancer Res (2003) 9(10 Pt 1):3606–14.
24. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol (2008) 26(15):2568–81. doi:10.1200/JCO.2007.13.1748
25. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A (2003) 100(14):8418–23. doi:10.1073/pnas.0932692100
26. Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer (2004) 4(10):814–9. doi:10.1038/nrc1457
27. Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol (2010) 28(7):1145–53. doi:10.1200/JCO.2009.22.4725
28. Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol (2010) 28(3):375–9. doi:10.1200/JCO.2008.20.7019
29. Park Y, Moriyama A, Kitahara T, Yoshida Y, Urita T, Kato R. Triple-negative breast cancer and poly(ADP-ribose) polymerase inhibitors. Anticancer Agents Med Chem (2012) 12(6):672–7. doi:10.2174/187152012800617759
30. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest (2011) 121(7):2750–67. doi:10.1172/JCI45014
31. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, et al. Differential pathologic complete response rates after neoadjuvant chemotherapy among molecular subtypes of triple-negative breast cancer. J Clin Oncol (2013) 31(Suppl):abstr 1005.
32. Telli ML, Jensen KC, Kurian AW, Vinayak S, Lipson JA, Schackmann EA, et al. PrECOG 0105: final efficacy results from a phase II study of gemcitabine (G) and carboplatin (C) plus iniparib (BSI-201) as neoadjuvant therapy for triple-negative (TN) and BRCA 1/2 mutation-associated breast cancer. J Clin Oncol (2013) 31(Suppl):abstr 1003.
33. Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer (2012) 107(10):1776–82. doi:10.1038/bjc.2012.451
34. Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene (2006) 25(43):5846–53. doi:10.1038/sj.onc.1209876
35. Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene (2007) 26(14):2126–32. doi:10.1038/sj.onc.1210014
36. Yang Q, Sakurai T, Mori I, Yoshimura G, Nakamura M, Nakamura Y, et al. Prognostic significance of BRCA1 expression in Japanese sporadic breast carcinomas. Cancer (2001) 92(1):54–60. doi:10.1002/1097-0142(20010701)92:1<54::AID-CNCR1291>3.0.CO;2-8
37. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer (2013) 108(10):2172–7. doi:10.1038/bjc.2013.144
38. Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol (2008) 26(19):3259–67. doi:10.1200/JCO.2007.11.3902
39. George J, Alsop K, Etemadmoghadam D, Hondow H, Mikeska T, Dobrovic A, et al. Nonequivalent gene expression and copy number alterations in high-grade serous ovarian cancers with BRCA1 and BRCA2 mutations. Clin Cancer Res (2013) 19(13):3474–84. doi:10.1158/1078-0432.CCR-13-0066
40. Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst (2002) 94(18):1396–406. doi:10.1093/jnci/94.18.1396
41. Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res (2000) 60(19):5329–33.
42. Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene (1999) 18(11):1957–65. doi:10.1038/sj.onc.1202509
43. Chan KY, Ozcelik H, Cheung AN, Ngan HY, Khoo US. Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer. Cancer Res (2002) 62(14):4151–6.
44. Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst (2000) 92(7):564–9. doi:10.1093/jnci/92.7.564
45. Geisler JP, Hatterman-Zogg MA, Rathe JA, Buller RE. Frequency of BRCA1 dysfunction in ovarian cancer. J Natl Cancer Inst (2002) 94(1):61–7. doi:10.1093/jnci/94.1.61
46. Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis (2000) 21(9):1761–5. doi:10.1093/carcin/21.9.1761
47. Berchuck A, Heron KA, Carney ME, Lancaster JM, Fraser EG, Vinson VL, et al. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res (1998) 4(10):2433–7.
48. Merajver SD, Frank TS, Xu J, Pham TM, Calzone KA, Bennett-Baker P, et al. Germline BRCA1 mutations and loss of the wild-type allele in tumors from families with early onset breast and ovarian cancer. Clin Cancer Res (1995) 1(5):539–44.
49. Smith SA, Easton DF, Evans DG, Ponder BA. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat Genet (1992) 2(2):128–31. doi:10.1038/ng1092-128
50. Tong D, Kucera E, Schuster E, Schmutzler RK, Swoboda H, Reinthaller A, et al. Loss of heterozygosity (LOH) at p53 is correlated with LOH at BRCA1 and BRCA2 in various human malignant tumors. Int J Cancer (2000) 88(2):319–22. doi:10.1002/1097-0215(20001015)88:2<319::AID-IJC27>3.0.CO;2-X
51. Werness BA, Parvatiyar P, Ramus SJ, Whittemore AS, Garlinghouse-Jones K, Oakley-Girvan I, et al. Ovarian carcinoma in situ with germline BRCA1 mutation and loss of heterozygosity at BRCA1 and TP53. J Natl Cancer Inst (2000) 92(13):1088–91. doi:10.1093/jnci/92.13.1088
52. Collins N, Wooster R, Stratton MR. Absence of methylation of CpG dinucleotides within the promoter of the breast cancer susceptibility gene BRCA2 in normal tissues and in breast and ovarian cancers. Br J Cancer (1997) 76(9):1150–6. doi:10.1038/bjc.1997.526
53. Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD II, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol (2010) 28(22):3570–6. doi:10.1200/JCO.2009.27.2997
54. Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res (2011) 17(5):1082–9. doi:10.1158/1078-0432.CCR-10-2560
55. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490(7418):61–70. doi:10.1038/nature11412
56. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474(7353):609–15. doi:10.1038/nature10166
57. Crook T, Brooks LA, Crossland S, Osin P, Barker KT, Waller J, et al. p53 Mutation with frequent novel condons but not a mutator phenotype in BRCA1- and BRCA2-associated breast tumours. Oncogene (1998) 17(13):1681–9. doi:10.1038/sj.onc.1202106
58. Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res (2001) 61(10):4092–7.
59. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434(7035):913–7. doi:10.1038/nature03443
60. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi:10.1038/nature03445
61. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res (2006) 66(16):8109–15. doi:10.1158/0008-5472.CAN-06-0140
62. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361(2):123–34. doi:10.1056/NEJMoa0900212
63. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet (2010) 376(9737):245–51. doi:10.1016/S0140-6736(10)60893-8
64. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet (2010) 376(9737):235–44. doi:10.1016/S0140-6736(10)60892-6
65. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol (2011) 12(9):852–61. doi:10.1016/S1470-2045(11)70214-5
66. Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol (2012) 30:372–9. doi:10.1200/JCO.2011.36.9215
67. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res (2012) 72(21):5588–99. doi:10.1158/0008-5472.CAN-12-2753
68. Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol (2013) 14(9):882–92. doi:10.1016/S1470-2045(13)70240-7
69. Kristeleit RS, Shapiro G. A phase I dose-escalation and PK study of continuous oral rucaparib in patients with advanced solid tumors. J Clin Oncol (2013) 31(Suppl):abstr 2585.
70. Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res (2013) 19(18):5003–15. doi:10.1158/1078-0432.CCR-13-1391
71. De Bono JS, Mina LA, Gonzalez M, Curtin NJ, Wang E, Henshaw JW, et al. First in-human trial of novel oral PARP inhibitor BMN 673 in patients with solid tumors. J Clin Oncol (2013) 31(Suppl):abstr 2580.
72. Lee J, Annunziata CM, Hays JL, Noonan AM, Minasian LM, Zujewski J, et al. Phase I/Ib study of the PARP inhibitor olaparib (O) with carboplatin (C) in BRCA1/2 mutation carriers with breast or ovarian cancer (Br/OvCa) (NCT00647062). J Clin Oncol (2013) 31(Suppl):abstr 2514.
73. O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med (2011) 364(3):205–14. doi:10.1056/NEJMoa1011418
74. O’Shaughnessy J, Schwartzberg LS, Danso MA, Rugo HS, Miller K, Yardley DA, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC). J Clin Oncol (2011) 29(15s):abstr 1007.
75. Ji J, Lee MP, Kadota M, Zhang Y, Parchment RE, Tomaszewski JE, et al. Pharmacodynamic and pathway analysis of three presumed inhibitors of poly (ADP-ribose) polymerase: ABT-888, AZD2281, and BSI201. In: Proceedings of the AACR 102nd Annual Meeting. Vol. 52. Orlando, FL (2011). 1080 p.
76. Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res (2013) 15(5):R88. doi:10.1186/bcr3484
77. Hayes DF, Thor AD, Dressler LG, Weaver D, Edgerton S, Cowan D, et al. HER2 and response to paclitaxel in node-positive breast cancer. N Engl J Med (2007) 357:1496–506. doi:10.1056/NEJMoa071167
78. Martín M, Rodríguez-Lescure A, Ruiz A, Alba E, Calvo L, Ruiz-Borrego M, et al. Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Res Treat (2010) 123:149–57. doi:10.1007/s10549-009-0663-z
79. Sparano JA, Wang M, Martino S, Jones V, Perez EA, Saphner T, et al. Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med (2008) 358:1663–71. doi:10.1056/NEJMoa0707056
80. Puhalla SL, Appleman LJ, Beumer JH, Tawbi H, Stoller RG, Owonikoko TK, et al. Two phase I trials exploring different dosing schedules of carboplatin (C), paclitaxel (P), and the poly-adp-ribose polymerase (PARP) inhibitor, veliparib (ABT-888) (V) with activity in triple negative breast cancer (TNBC). Cancer Res (2012) 72(24 Suppl):abstr PD09-6. doi:10.1158/0008-5472.SABCS12-PD09-06
81. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med (2012) 366:1382–92. doi:10.1056/NEJMoa1105535
82. Liu JF, Tolaney SM, Birrer M, Fleming GF, Buss MK, Dahlberg SE, et al. A phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer (2013) 49(14):2972–8. doi:10.1016/j.ejca.2013.05.020
83. Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol (2010) 28(22):3555–61. doi:10.1200/JCO.2009.27.5719
84. Dhillon KK, Swisher EM, Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci (2011) 102(4):663–9. doi:10.1111/j.1349-7006.2010.01840.x
85. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res (2008) 68(8):2581–6. doi:10.1158/0008-5472.CAN-08-0088
86. Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol (2013) 229(3):422–9. doi:10.1002/path.4140
87. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol (2011) 29(22):3008–15. doi:10.1200/JCO.2010.34.2980
88. Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V, et al. Efficacy of chemotherapy in BRCA 1/2 mutation carrier ovarian cancer in the setting of parp inhibitor resistance: a multi-institutional study. Clin Cancer Res (2013) 19(19):5485–93. doi:10.1158/1078-0432.CCR-13-1262
89. Kaufman B, Shapira-Frommer R, Schmutzler RK. Olaparib monotherapy in patients with advanced cancer and a germ-line BRCA1/2 mutation: an open-label phase II study. J Clin Oncol (2013) 31(Suppl):abstr 11024.
90. van der Noll R, Ang JE, Jager A, Marchetti S, Mergui-Roelvink M, DeBono JS, et al. Phase I study of olaparib in combination with carboplatin and/or paclitaxel in patients with advanced solid tumors. J Clin Oncol (2013) 31(Suppl):abstr 2579.
91. Huggins-Puhalla SL, Beumer JH, Appleman LJ, Tawbi HA, Stoller RG, Lin Y, et al. A phase I study of chronically dosed, single-agent veliparib (ABT-888) in patients (pts) with either BRCA 1/2-mutated cancer (BRCA+), platinum-refractory ovarian cancer, or basal-like breast cancer (BRCA-wt). J Clin Oncol (2012) 30(Suppl):abstr 3054.
92. Ramaswamy B, Zhang J, Hall N, Schregel K, Lustberg MB, Weslowski R, et al. NCI 8609: interim fluoro-3’-deoxythymidine (FLT) PET imaging findings from the phase I trial of PARP inhibitor veliparib (V) and carboplatin (C) in advanced breast cancer. J Clin Oncol (2013) 31(Suppl):abstr 1023.
93. Clark CC, Weitzel JN, O’Connor TR. Efficacy of the combination of ABT-888 (veliparib) and carboplatin in patients with BRCA-associated breast cancer. J Clin Oncol (2013) 31(Suppl):abstr 1024.
94. Rodler ET, Specht JM, Gadi VK, Kurland BF, Griffin MJ, Hammon JJ, et al. Phase I study of PARP inhibitor ABT-888 (veliparib) in combination with cisplatin and vinorelbine for patients with advanced triple negative breast cancer and/or BRCA-mutation associated breast cancer. Cancer Res (2011) 71(24 Suppl):abstr P1-17-04. doi:10.1158/0008-5472.SABCS11-P1-17-04
95. Tan A, Toppmeyer D, Tsan Lai Wong S, Lin H, Gounder M, Moss RA, et al. Assessment of γH2AX levels in circulating tumor cells in patients treated with veliparib in combination with doxorubicin and cyclophosphamide in metastatic breast cancer. J Clin Oncol (2013) 31(Suppl):abstr 2582.
96. Isakoff SJ, Overmoyer B, Tung NM, Gelman RS, Giranda VL, Bernhard KM, et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol (2010) 28(Suppl):abstr 1019.
97. Kummar S, Oza AM, Fleming GF, Sullivan D, Gandara DR, Erlichman C, et al. Randomized trial of oral cyclophosphamide (C) with or without veliparib (V), an oral poly (ADP-ribose) polymerase (PARP) inhibitor, in patients with recurrent BRCA-positive ovarian, or primary peritoneal or high-grade serous ovarian carcinoma. J Clin Oncol (2012) 30(Suppl):abstr 5020.
98. Bell-McGuinn K, Gray HJ, Fleming GF, Cristea MC, Medina DM, Xiong H, et al. Phase I study of ABT-888 in combination with carboplatin and gemcitabine in subjects with advanced solid tumors. J Clin Oncol (2013) 31(Suppl):abstr 2584.
99. Lee J, Annunziata CM, Minasian LM, Zujewski J, Prindiville SA, Kotz HL, et al. Phase I study of the PARP inhibitor olaparib (O) in combination with carboplatin (C) in BRCA1/2 mutation carriers with breast (Br) or ovarian (Ov) cancer (Ca). J Clin Oncol (2011) 29(Suppl):abstr 2520.
100. Balmana J, Tung NM, Isakoff SJ. Phase I, open-label study of olaparib plus cisplatin in patients with advanced solid tumors. J Clin Oncol (2012) 30(Suppl):abstr 1009.
101. Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res (2012) 18(6):1726–34. doi:10.1158/1078-0432.CCR-11-2821
102. Appleman LJ, Beumer JH, Jiang Y, Puhalla S, Lin Y, Owonikoko TK, et al. A phase I study of veliparib (ABT-888) in combination with carboplatin and paclitaxel in advanced solid malignancies. J Clin Oncol (2012) 30(Suppl):abstr 3049.
103. Oza AM, Cibula D, Oaknin A, Poole CJ, Mathijssen RHJ, Sonke GS, et al. Olaparib plus paclitaxel plus carboplatin (P/C) followed by olaparib maintenance treatment in patients (pts) with platinum-sensitive recurrent serous ovarian cancer (PSR SOC): a randomized, open-label phase II study. J Clin Oncol (2012) 30(Suppl):abstr 5001.
104. Molife LR, Roxburgh R, Wilson RH, Gupta A, Middleton MR, Evans TRJ, et al. A phase I study of oral rucaparib in combination with carboplatin. J Clin Oncol (2013) 31(Suppl):abstr 2586.
105. Miller KD, Perkins SM, Badve SS, Sledge GW, Schneider BP. PARP inhibition after preoperative chemotherapy in patients with triple-negative breast cancer (TNBC) or known BRCA 1/2 mutations: Hoosier Oncology Group BRE09-146. Cancer Res (2011) 71(24 Suppl):abstr OT3-01-5. doi:10.1158/0008-5472.SABCS11-OT3-01-05
106. Isakoff SJ, Puhalla S, Shepherd SP. A randomized, phase 2 study of the poly (ADP-ribose) polymerase (PARP) inhibitor veliparib (ABT-888) in combination with temozolomide (TMZ) or in combination with carboplatin (C) and paclitaxel (P) versus placebo plus C/P in subjects with BRCA1 or BRCA2 mutation and metastatic breast cancer. Cancer Res (2012) 72(24 Suppl):abstr OT2-3-07. doi:10.1158/0008-5472.SABCS12-OT2-3-07
107. Coleman R, Sill M, Aghajanian C. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation – a Gynecologic Oncology Group study. Gynecol Oncol (2013) 130(1):e168. doi:10.1016/j.ygyno.2013.04.465
108. Pothuri B, Muggia F, Sparano JA, Blank SV, Curtin JP, Chuang E, et al. Dose-dependent pharmacokinetic (PK) interaction of pegylated liposomal doxorubicin (PLD) with escalating doses of veliparib in a phase I study. J Clin Oncol (2013) 31(Suppl):abstr 2591.
109. Avery T, Berger AC, Kovatich AJ, Rui H, Hyslop T, Mitchell EP, et al. An adaptive randomized phase II trial to determine pathologic complete response with the addition of carboplatin with and without ABT-888 to standard chemotherapy in the neoadjuvant treatment of triple-negative breast cancer. J Clin Oncol (2013) 31(Suppl):abstr TS1135.
Keywords: BRCA 1/2-mutations, breast cancer, ovarian cancer, BRCAness, PARP inhibitor, reversion mutations
Citation: Burgess M and Puhalla S (2014) BRCA 1/2-mutation related and sporadic breast and ovarian cancers: more alike than different. Front. Oncol. 4:19. doi: 10.3389/fonc.2014.00019
Received: 14 October 2013; Accepted: 24 January 2014;
Published online: 27 February 2014.
Edited by:
Kristin Zorn, Magee-Womens Hospital of UPMC, USAReviewed by:
Jian Lu, Johns Hopkins University, USAChristine Walsh, Cedars-Sinai Medical Center, USA
Bhavana Pothuri, New York University School of Medicine, USA
Copyright: © 2014 Burgess and Puhalla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shannon Puhalla, University of Pittsburgh Medical Center, University of Pittsburgh Cancer Institute, Magee-Women’s Hospital, 300 Halket Street, Pittsburgh, PA 15213, USA e-mail:cHVoYWxsYXNsQG1haWwubWFnZWUuZWR1