 Carrie Danielle House
Carrie Danielle House Lidia Hernandez
Lidia Hernandez Christina Messineo Annunziata
Christina Messineo Annunziata- Women’s Malignancies Branch, National Cancer Institute, Bethesda, MD, USA
Serous epithelial ovarian cancer (SEOC) is the most lethal gynecological cancer in the United States with disease recurrence being the major cause of morbidity and mortality. Despite recent advances in our understanding of the molecular mechanisms responsible for the development of SEOC, the survival rate for women with this disease has remained relatively unchanged in the last two decades. Preclinical mouse models of ovarian cancer, including xenograft, syngeneic, and genetically engineered mice, have been developed to provide a mechanism for studying the development and progression of SEOC. Such models strive to increase our understanding of the etiology and dissemination of ovarian cancer in order to overcome barriers to early detection and resistance to standard chemotherapy. Although there is not a single model that is most suitable for studying ovarian cancer, improvements have led to current models that more closely mimic human disease in their genotype and phenotype. Other advances in the field, such as live animal imaging techniques, allow effective monitoring of the microenvironment and therapeutic efficacy. New and improved preclinical mouse models, combined with technological advances to study such models, will undoubtedly render success of future human clinical trials for patients with SEOC.
Introduction
Mouse models provide a wealth of information for understanding tumor biology. Through the validation of in vitro findings, cancer progression, regression, and relapse in the physiological setting is better understood. The continued high mortality associated with serous epithelial ovarian cancer (SEOC) underscores a need for tailored disease models and improved technology to study such models. Several recent advancements promise to facilitate the success of preclinical models in refining our understanding and treatment of SEOC. This mini-review will focus on the latest mouse models of ovarian cancer and improved technologies for using these models to study SEOC initiation, progression, relapse, and therapeutic response.
Ovarian cancer is the most common cause of gynecological mortality in the United States, accounting for more than 14,000 deaths annually (1). Most patients initially respond favorably to platinum-based therapy, however, there is a high incidence of recurrent, chemoresistant disease. Our knowledge of the clinical and molecular attributes of epithelial ovarian cancer has improved greatly over the last few decades, but survival rates for women with this disease remain low. This is partially explained by the inability of clinical trials to replicate the therapeutic response observed in animal models. So far, about one-third of highly cited animal studies saw success in human trials, however, improvements in study design and data interpretation should increase that figure (2, 3). Animal models in the personalized medicine era highlight the availability of affordable genomic sequencing and molecular profiling. As the pharmaceutical industry relies heavily on mouse models, such new refinements will be critical for producing reliable preclinical data on personalized ovarian cancer therapeutic approaches.
In order to generate accurate models, the biology of the disease must be understood. High grade SEOC is thought to arise in a rapid fashion de novo from the surface epithelium of the ovary or from the mucosa of the fallopian tube (4, 5). The remaining ovarian carcinomas, categorized as low grade, follow a stepwise adenoma-carcinoma sequence (4, 6). Whether high or low grade, SEOC usually does not reach clinical detection until late stage where it has spread well beyond the ovaries. This feature has hampered efforts to identify the site of origin and understand the pathophysiology of SEOC. Most existing mouse models of SEOC present a disseminated abdominal phenotype, which closely resembles late metastatic disease, and therefore may only provide a good model for therapeutic response in the “average” patient. Some recent mouse models provide a phenotype of early progressive disease coming from defined genetic abnormalities identified from patient subtypes.
Mouse Models of EOC
An extensive analysis of every mouse model is beyond the scope of this mini-review, however, a summary of recent advances in mouse models of ovarian cancer to place the technological advances in context is presented here. Several recent reviews are available detailing epithelial ovarian cancer experimental models (7–9). Mouse models of ovarian cancer generally fall into one of three categories (xenograft, syngeneic, genetically engineered), the most suitable being dependent on the information being sought.
Xenograft Models
A human tumor xenograft is the most widely used mouse model in which human tumor cells are transplanted under the skin (subcutaneous), into the abdominal cavity (intraperitoneal), or into the organ of origin (orthotopic) of an immune-compromised host. While intraperitoneal and orthotopic injections can mimic metastatic dissemination, subcutaneously injected cells are largely limited to tumor formation at the site of injection. The unique presence of a bursa, a sac encapsulating the ovaries and fallopian tubes, allows for intra-bursal injections in mice (10, 11). This technique permits the study of early, localized disease, tumor cell invasion, and dissemination in a more biologically relevant order of events (12).
Xenograft models are particularly useful for evaluating tumorigenesis in a timely fashion (13, 14). Within a few weeks, tumor formation can be measured in vivo with histology representative of the tumor of origin (12, 15, 16). Importantly, the pattern of spread to the ascites, liver, and spleen, typical in human disease, is replicated in many of these models and depending on the cell line used, tumors representing the different histological subtypes of epithelial ovarian cancer can be produced (8, 12). Xenografts are versatile and often used in parallel with in vitro studies to generate a majority of preclinical data.
Although quite valuable, xenografts carry important limitations. One major disadvantage is the lack of immune response inherent in these models. Nude mice are athymic and therefore have a limited T cell response, and severe combined immunodeficiency (SCID) strains lack both T and B cell responses. Because tumors can promote anti-tumor responses such as lymphocyte and macrophage infiltration, these models may not accurately represent disease progression and therapeutic response observed in otherwise immune-competent individuals (17–19). Furthermore, these models are not suitable for studying immunotherapy or mechanisms involving host–cell interactions. Cell line-derived xenografts have had little success in predicting therapeutic response in patients, thereby emphasizing a need for improvements to current models.
An alternative to traditional cell line-derived xenograft models involves the direct transfer of tumor fragments from individual patients. Minced fragments are delivered via orthotopic or intraperitoneal injection into immune-deficient mice to create “xenopatients” or tumor grafts (8, 20). Successful engraftment is higher in SCID mice compared to nude mice, likely due to the suppression of both cellular and humoral immunity (20–23). Several reports have demonstrated that tumor grafts stably maintain the histopathology, immunophenotype, and heterogeneity of the original tumor through multiple passages (21–26). Moreover, these models have the capacity to recapitulate the same therapeutic properties observed in patients (20, 25, 26). The better predictive response value makes these models superior to traditional cell line-based xenografts generated using a suspension of mostly homogenous cells. Engraftment of the native stromal extracellular matrix that would normally accompany a tumor graft may provide the most suitable microenvironment for replicating the biology of the original tumor. This feature renders tumor grafts more suitable for studying early metastasis, as it relies on dissemination of cells from a tumor fragment rather than dispersion of cells from a suspension (8). Thus, patient-derived tumor grafts provide a means to model inter-patient heterogeneity known to exist across high grade SEOC, and to study tumor evolution through exposure to therapy. Tumor grafts, although promising, are not without their own challenges. Generating a mouse model using a tumor graft is labor intensive and expensive and, as in traditional xenografts, the mice are immunocompromised; consequently immune responses cannot be studied. Although well suited for clinicians and personalized medicine, access to patient tumor samples can be challenging for many basic and translational investigators. Some research teams have generated banks of tumor grafts to make these models more accessible (20, 23, 24).
Syngeneic Models
Some challenges of xenograft models can be overcome using syngeneic mouse models, wherein tumors are established in immune-competent mice using cells from the same strain. In one of the most widely used syngeneic models, generated by Roby et al., ovarian surface epithelial cells isolated from immune-competent mice were repeatedly passaged in vitro until transformation occurred, and subsequently injected back into the same strain (27). Other syngeneic models have been created using genetically modified cells (28, 29) and highly metastatic cell lines stably expressing luciferase for monitoring disease (30). The histopathologic characteristics observed in the tumors of these models including the presence of papillary structures, nuclear atypia, and malignant ascites, closely resemble those seen in humans (29, 31).
The major advantage of this model is that the mice have an intact immune system; therefore the anti-tumor immune response can be examined and the risk of infection is minimized (19, 32). Syngeneic models provide the opportunity to study the tumor microenvironment, epithelial–stromal cell interactions, tumor-secreting factors, immune cell infiltration, and vasculature (28, 29, 31, 33). This model, however, is completely derived from the animal system and therefore may not mirror every element of human cancer. Although human and mouse tumors share similar features, the complexity of human disease coupled with the heterogeneity of cancer make it difficult to translate findings (34).
Genetically Engineered Models
Genetically engineered mouse models (GEMMs) are immune-competent mice with genetic defects introduced using RNA interference, inducible gene expression, viruses, or DNA recombination techniques. GEMMs provide a means for investigating the role of genetic alterations in cancer development. These models allow researchers to control and direct gene expression, which can be limited to the tissue of interest using a tissue-specific promoter to introduce the desired genetic alteration, or expressed throughout the organism using germ-line mutations (35). Furthermore, regulation of gene expression in the presence or absence of tetracycline and its receptor allows for inducible gene expression systems and provides the flexibility to turn on or off gene(s). For example, transgenic mice carrying both the tetracycline-regulated transcriptional transactivator and its respective binding site linked to a gene of interest permits amplified expression of that gene. If mice are provided with the tetracycline antibiotic in their drinking water, this expression is reversibly suppressed. Thus, GEMMs provide opportunities to identify which genes are necessary for disease progression, regression, and/or resistance to treatment.
Extensive analyses of human ovarian cancer specimens have identified several genetic alterations associated with malignancy including TP53, C-MYC, K-RAS, AKT, and BRCA1 and BRCA2 (36–38). Subsequently, several genetically modified ovarian cancer models, summarized in Table 1, have been developed to explore the contribution of these different aberrations to ovarian cancer development (39–44).
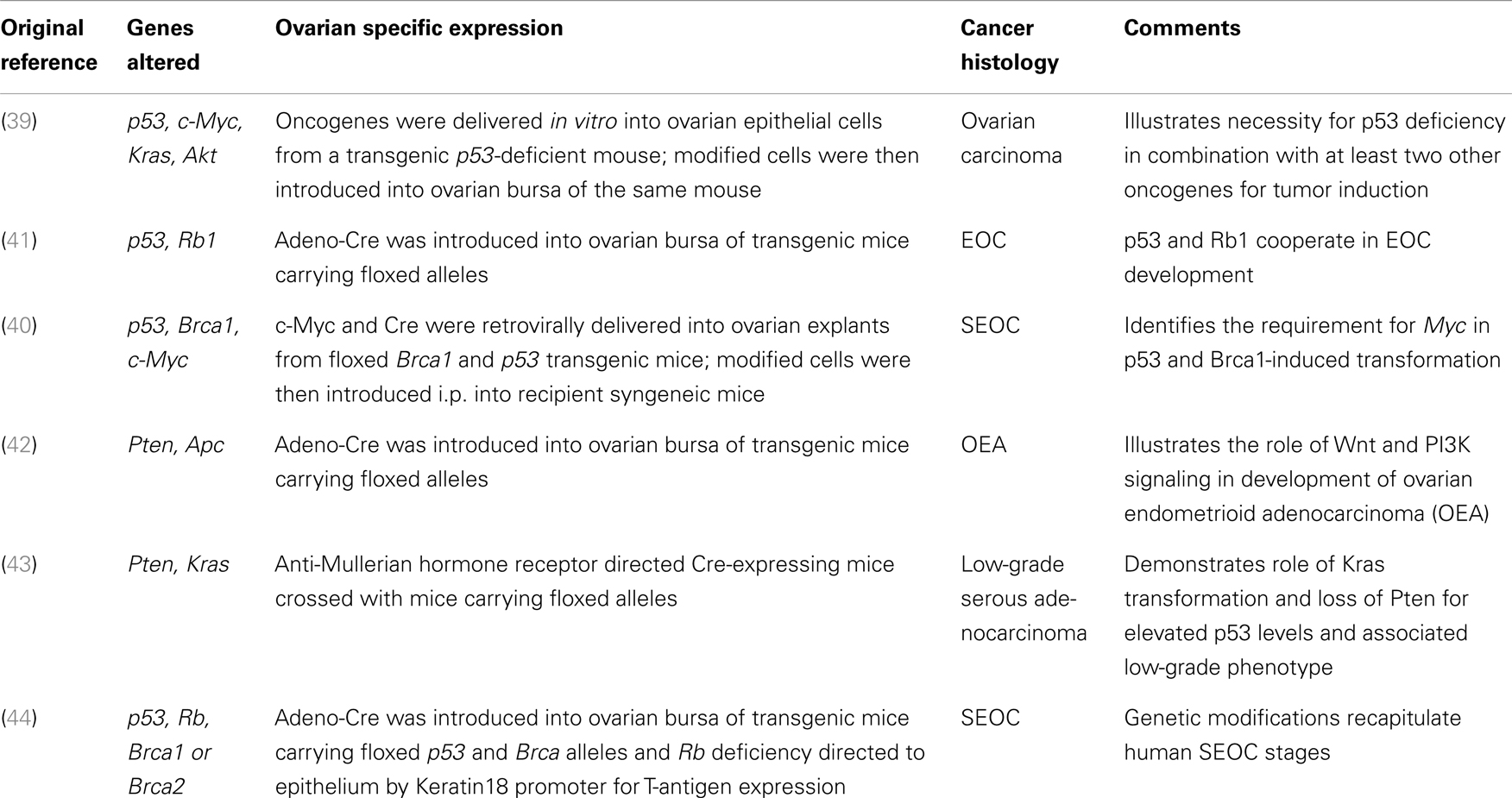
Table 1. GEMMs for ovarian cancer.
Although GEMMs are labor-, time-, and resource-intensive, they provide information that cannot be attained in xenograft or syngeneic models. Early tumorigenesis and genetic events leading to tumor initiation, maintenance, and relapse can be analyzed. The flexibility provided by genetic manipulation permits the study of different mutation combinations. These models are ideal for target validation, treatment response, and chemoprevention (45). The major challenge with this model is the scarcity of tissue-specific promoters in ovarian surface epithelium or distal fallopian tube. It is also challenging to accurately replicate the contribution of genetic elements given that genes over-expressed in mice are often at non-physiological levels or deleted throughout the organism (46). GEMMs may fail to recapitulate the genetic complexity of human SEOC, and the varied genetic background of different mouse strains can influence findings and conclusions (8).
Technological Advances in Using Animal Models
Reporters
Most ovarian cancer cell lines can be stably transfected with a fluorescent and/or bioluminescent reporter for monitoring tumor cell growth and dissemination, pathway activity, and receptor interactions.
This technology has been adapted to xenograft and syngeneic mouse models of ovarian cancer. For example, NF-κB activity was tracked in a syngeneic model of SEOC to confirm that activation correlated with progression and influenced immune cells of the microenvironment (47). Similarly, reporter-tagged tumor cells can be used to monitor tumor response in real-time using digital imaging following systemic targeted therapy (48–50). Using reporters in live animals to track tumor cell dissemination allows for studying cancer progression and therapeutic response, especially in syngeneic models where the immune response is integral.
Luciferase complementation-based assays measure receptor activation and protein interactions using monomeric enzyme components that have enzymatic activity only when complementation is induced by the interaction of binding partners or small molecules (51). Activation is proportional to the production of light that occurs upon complementation. The flexibility of this technology allows detailed quantitative measurements of complexes, assessment of nuclear translocation, and identification of pathway modulators (52). For example, this assay was successfully implemented for live imaging of the chemokine, CXCL12, interacting with its receptor, CXCR4, in animal models of ovarian cancer (53, 54).
Imaging
Quantitative measurements of late-stage disease in ovarian cancer models are challenging due to the presence of varying levels of ascites and the poor correlation between total body weight gain and tumor burden. Diagnostic imaging is a reproducible means to quantify tumor mass, monitor tumor progression, and interrogate the tumor microenvironment. Imaging techniques used in the clinic [e.g., magnetic resonance imaging (MRI), computed tomography (CT), positron emission tomography (PET), ultrasound] have been adapted for use in animals (55, 56). These modalities are especially informative as they can be performed in intact living animals. Interval imaging reduces the number of animals needed for experiments as measurements are taken without sacrificing the animal. The major challenge to imaging ovarian cancer in animal models, as in humans, is the difficulty in detecting early disease; by the time mice begin to show signs of morbidity the cancer has often spread beyond the ovaries and throughout the peritoneum.
Positron emission tomography imaging is a standard diagnostic radiological technique commonly used to monitor drug action in cancer patients. This modality allows the measurement of metabolic activity in cancer cells and is especially useful in quantitative monitoring of tumor response to anti-cancer therapies (56). PET imaging can assess targeted therapies in both transgenic (57) and xenograft (58) models of ovarian cancer.
Ultrasound imaging is another common tool used in small animal models and is often combined with other imaging techniques for a more comprehensive analysis (57). Ultrasound is cost-effective and convenient for measuring individual tumors in live animals (59). Doppler ultrasonography can measure changes in blood flow and angiogenesis associated with disease progression or response to anti-angiogenic therapy (59, 60).
Magnetic resonance imaging with gadolinium-based contrast agent permits high-resolution serial imaging with minimum scanning duration, allowing quantification of tumor volume over time. MRI data are comparable to caliper-based measurements taken at necropsy. This longitudinal imaging protocol is well suited for monitoring therapeutic response (61). MRI can also be combined with fluorescence molecular tomography (FMT) to monitor tumor-specific biology, such as protease and integrin activity (62). When coupled with a reporter gene such as ferritin heavy chain (FHC), MRI can evaluate recruitment of other cell types, such as fibroblasts, to the tumor site (63). Alternatively, MRI combined with magnetic resonance spectroscopy (MRS) can characterize tumor physiology and metabolic profiles over time (64).
Metabolic Profiling
Measurement of metabolites and their intermediates can illustrate the response of an organism to a genetic manipulation or therapy. Metabolites are small, low molecular weight analytes and include amino acids, oligopeptides, sugars, fatty acids, and various intermediates of biochemical pathways, in contrast to large proteins and nucleotides that are assessed using proteomics and genomics, respectively (65). Nuclear magnetic resonance (NMR) spectroscopy, liquid and gas chromatography, and mass spectrometry (MS) are generally used to analyze serum, urine, or tissue extracts. Such measurements provide insight into drug mechanisms and toxicities. Metabolic profiles represent a snapshot of the biochemical reactions occurring at a point functionally downstream of genome, transcriptome, and proteome (65).
Commonly used in human studies, (66, 67) this technology was adopted in a GEMM of SEOC. The metabolic profile overlapped with human SEOC and showed a temporal correlation with disease progression (44, 68), highlighting the feasibility of metabolic profiling for identifying biomarkers and monitoring treatment response in animal models (44).
Tumor-Initiating Cells
The cancer stem cell (CSC) or tumor-initiating cell (TIC) hypothesis suggests that a small population of chemoresistant cells reside in the tumor, capable of reconstituting the tumor. These cells share properties of normal stem cells, such as self-renewal and multi-potency. Given the high recurrence of ovarian cancer, the TIC hypothesis is an attractive model for explaining ovarian cancer relapse.
Mouse models have been especially useful in evaluating TICs. When injected into mice, these cells must recapitulate the heterogeneity of the original tumor. Animal models are essential for defining TICs and for evaluating drugs and pathways important for eradicating these cells. Patient-derived xenografts might allow further characterization of the frequency of TICs in human tumors, and their relevant biomarkers.
A number of markers have been used to identify and isolate ovarian cancer TICs including CD133, CD44, CD117, and ALDH activity; however it is unlikely that a single marker defines ovarian TICs (69, 70). Several studies have demonstrated heterogeneous tumor formation in xenograft mice after subcutaneous injection of sorted ovarian cancer cells from primary tumors, cell lines, or ascites (71–74). TICs have also been propagated in vitro using low attachment culture plates and specialized serum-free media to enhance the formation of multicellular spheroids with stem-like features (69, 73).
Although much research has focused on characterizing tumorigenesis of human TICs in xenograft models, recent studies evaluated endogenous TICs in mice (75–77). Syngeneic or GEMMs offer the possibility of studying the role of the immune system in TIC biology. Furthermore, with direct or indirect labeling of the TICs, each of these models can facilitate tracking of the cells to monitor tumor initiation and dissemination.
Immune Therapies
The role of the immune system in ovarian cancer is studied extensively using animal models (19). Representing a robust predictor of outcome, tumor-infiltrating lymphocytes are associated with better survival for ovarian cancer patients (78, 79). Immune therapies involving vaccines, dendritic cell therapy, engineered T cells, and immune modulators thus hold promise for ovarian cancer treatment (80–86).
Current goals aim to enhance the anti-tumor immune response through increased immune activation and decreased immune suppression. Programed death-1 (PD-1) and CTL antigen-4 (CTLA-4) signals silence the immune response in tumors. A syngeneic mouse model of ovarian cancer showed that simultaneously blocking these pathways enhanced T cell infiltration into the tumor and increased long-term survival (81). A related model found that the therapeutic effect of gemcitabine is limited because of the immunosuppressive network of CTLA-4 (83). Gemcitabine plus anti-CTLA-4 antibody exhibited synergy in a strong anti-tumor immune response. Likewise, anti PD-1 therapy shows synergism with a variety of immunotherapies or vaccines (86). These findings have translated well and are currently under evaluation in the clinic.
Genetically modified T cells engineered to over-express receptors for tumor-associated antigens have shown great success in mouse models of ovarian cancer (87, 88). This emerging technology is a logical avenue for ovarian cancer, an apparently immunogenic disease where T cell infiltration is associated with improved survival (19, 88).
Conclusion
Despite our progress in understanding ovarian cancer biology, there remains a high mortality associated with this disease. Exciting advances in reporter assays, live imaging, metabolomics, TICs, and immune therapies, provide new information about the tumor microenvironment and further our understanding of SEOC development, progression, and recurrence (Figure 1). Further refinement of mouse models of ovarian cancer, an awareness of the limitations each model presents, and taking advantage of the technologies available to study these models will undoubtedly expedite the success of new treatments.
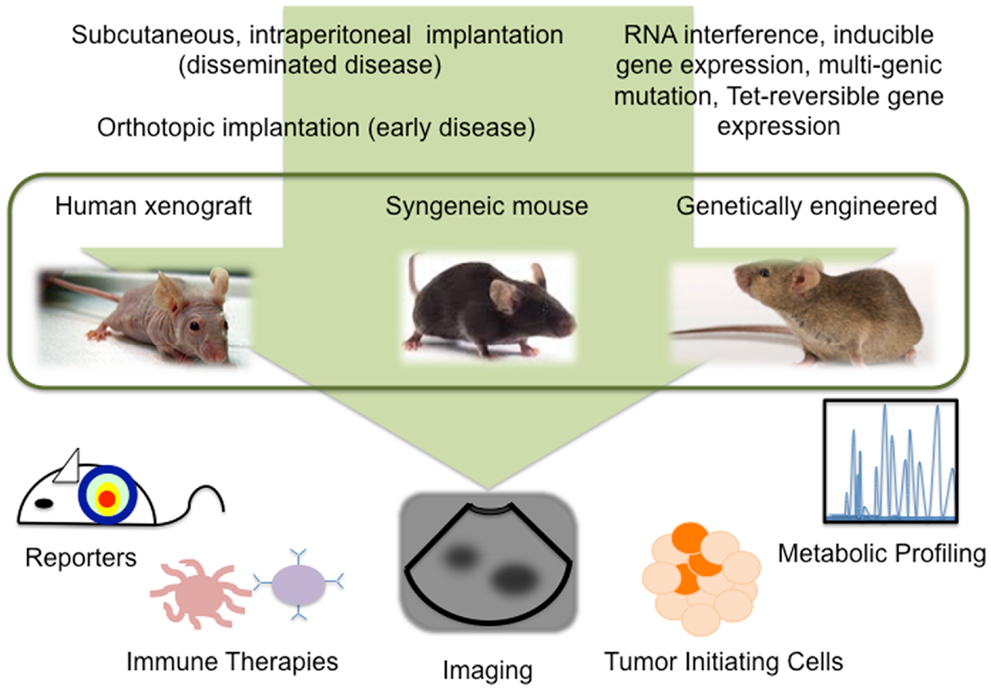
Figure 1. Technological advances in mouse models allow detailed study of ovarian cancer biology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin (2012) 62(1):10–29. doi:10.3322/caac.20138
2. Hackam DG, Redelmeier DA. Translation of research evidence from animals to humans. JAMA (2006) 296(14):1731–2. doi:10.1001/jama.296.14.1731
3. van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, et al. Can animal models of disease reliably inform human studies? PLoS Med (2010) 7(3):e1000245. doi:10.1371/journal.pmed.1000245
4. Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol (2004) 164(5):1511–8. doi:10.1016/S0002-9440(10)63708-X
5. Smith Sehdev AE, Sehdev PS, Kurman RJ. Noninvasive and invasive micropapillary (low-grade) serous carcinoma of the ovary: a clinicopathologic analysis of 135 cases. Am J Surg Pathol (2003) 27(6):725–36. doi:10.1097/00000478-200306000-00003
6. Kobel M, Kalloger SE, Huntsman DG, Santos JL, Swenerton KD, Seidman JD, et al. Differences in tumor type in low-stage versus high-stage ovarian carcinomas. Int J Gynecol Pathol (2010) 29(3):203–11. doi:10.1097/PGP.0b013e3181c042b6
7. Jones PM, Drapkin R. Modeling high-grade serous carcinoma: how converging insights into pathogenesis and genetics are driving better experimental platforms. Front Oncol (2013) 3:217. doi:10.3389/fonc.2013.00217
8. Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, et al. Epithelial ovarian cancer experimental models. Oncogene (2013). doi:10.1038/onc.2013.321
9. Ricci F, Broggini M, Damia G. Revisiting ovarian cancer preclinical models: implications for a better management of the disease. Cancer Treat Rev (2013) 39(6):561–8. doi:10.1016/j.ctrv.2013.01.005
10. Fu X, Hoffman RM. Human ovarian carcinoma metastatic models constructed in nude mice by orthotopic transplantation of histologically-intact patient specimens. Anticancer Res (1993) 13(2):283–6.
11. Vanderhyden BC, Shaw TJ, Ethier JF. Animal models of ovarian cancer. Reprod Biol Endocrinol (2003) 1:67. doi:10.1186/1477-7827-1-67
12. Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther (2004) 10(6):1032–42. doi:10.1016/j.ymthe.2004.08.013
13. Fiebig HH, Berger DP, Winterhalter BR, Plowman J. In vitro and in vivo evaluation of US-NCI compounds in human tumor xenografts. Cancer Treat Rev (1990) 17(2–3):109–17. doi:10.1016/0305-7372(90)90034-D
14. Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res (2003) 9(11):4227–39.
15. Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst (1977) 59(1):221–6.
16. Molthoff CF, Calame JJ, Pinedo HM, Boven E. Human ovarian cancer xenografts in nude mice: characterization and analysis of antigen expression. Int J Cancer (1991) 47(1):72–9. doi:10.1002/ijc.2910470114
17. Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J Immunol (2007) 178(3):1268–76.
18. Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol (2000) 1(6):515–20. doi:10.1038/82771
19. Gavalas NG, Karadimou A, Dimopoulos MA, Bamias A. Immune response in ovarian cancer: how is the immune system involved in prognosis and therapy: potential for treatment utilization. Clin Dev Immunol (2010) 2010:791603. doi:10.1155/2010/791603
20. Garber K. From human to mouse and back: ‘tumorgraft’ models surge in popularity. J Natl Cancer Inst (2009) 101(1):6–8. doi:10.1093/jnci/djn481
21. Elkas JC, Baldwin RL, Pegram M, Tseng Y, Slamon D, Karlan BY. A human ovarian carcinoma murine xenograft model useful for preclinical trials. Gynecol Oncol (2002) 87(2):200–6. doi:10.1006/gyno.2002.6819
22. Xu Y, Silver DF, Yang NP, Oflazoglu E, Hempling RE, Piver MS, et al. Characterization of human ovarian carcinomas in a SCID mouse model. Gynecol Oncol (1999) 72(2):161–70. doi:10.1006/gyno.1998.5238
23. Lee CH, Xue H, Sutcliffe M, Gout PW, Huntsman DG, Miller DM, et al. Establishment of subrenal capsule xenografts of primary human ovarian tumors in SCID mice: potential models. Gynecol Oncol (2005) 96(1):48–55. doi:10.1016/j.ygyno.2004.09.025
24. DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med (2011) 17(11):1514–20. doi:10.1038/nm.2454
25. Khabele D, Fadare O, Liu AY, Wilson AJ, Wass E, Osteen K, et al. An orthotopic model of platinum-sensitive high grade serous fallopian tube carcinoma. Int J Clin Exp Pathol (2012) 5(1):37–45.
26. Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res (2006) 12(15):4652–61. doi:10.1158/1078-0432.CCR-06-0113
27. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis (2000) 21(4):585–91. doi:10.1093/carcin/21.4.585
28. Quinn BA, Xiao F, Bickel L, Martin L, Hua X, Klein-Szanto A, et al. Development of a syngeneic mouse model of epithelial ovarian cancer. J Ovarian Res (2010) 3:24. doi:10.1186/1757-2215-3-24
29. Zhang L, Yang N, Garcia JR, Mohamed A, Benencia F, Rubin SC, et al. Generation of a syngeneic mouse model to study the effects of vascular endothelial growth factor in ovarian carcinoma. Am J Pathol (2002) 161(6):2295–309. doi:10.1016/S0002-9440(10)64505-1
30. Toyoshima M, Tanaka Y, Matumoto M, Yamazaki M, Nagase S, Sugamura K, et al. Generation of a syngeneic mouse model to study the intraperitoneal dissemination of ovarian cancer with in vivo luciferase imaging. Luminescence (2009) 24(5):324–31. doi:10.1002/bio.1112
31. Greenaway J, Moorehead R, Shaw P, Petrik J. Epithelial-stromal interaction increases cell proliferation, survival and tumorigenicity in a mouse model of human epithelial ovarian cancer. Gynecol Oncol (2008) 108(2):385–94. doi:10.1016/j.ygyno.2007.10.035
32. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410(6832):1107–11. doi:10.1038/35074122
33. Hagemann T, Robinson SC, Thompson RG, Charles K, Kulbe H, Balkwill FR. Ovarian cancer cell-derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol Cancer Ther (2007) 6(7):1993–2002. doi:10.1158/1535-7163.MCT-07-0118
34. Bhatia S, Frangioni JV, Hoffman RM, Iafrate AJ, Polyak K. The challenges posed by cancer heterogeneity. Nat Biotechnol (2012) 30(7):604–10. doi:10.1038/nbt.2294
35. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov (2006) 5(9):741–54. doi:10.1038/nrd2110
36. Aunoble B, Sanches R, Didier E, Bignon YJ. Major oncogenes and tumor suppressor genes involved in epithelial ovarian cancer (review). Int J Oncol (2000) 16(3):567–76.
37. Lynch HT, Casey MJ, Lynch J, White TE, Godwin AK. Genetics and ovarian carcinoma. Semin Oncol (1998) 25(3):265–80.
38. Lynch HT, Casey MJ, Snyder CL, Bewtra C, Lynch JF, Butts M, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Mol Oncol (2009) 3(2):97–137. doi:10.1016/j.molonc.2009.02.004
39. Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell (2002) 1(1):53–62. doi:10.1016/S1535-6108(01)00002-2
40. Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res (2006) 66(18):8949–53. doi:10.1158/0008-5472.CAN-06-1495
41. Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res (2003) 63(13):3459–63.
42. Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell (2007) 11(4):321–33. doi:10.1016/j.ccr.2007.02.016
43. Mullany LK, Fan HY, Liu Z, White LD, Marshall A, Gunaratne P, et al. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene (2011) 30(32):3522–36. doi:10.1038/onc.2011.70
44. Szabova L, Yin C, Bupp S, Guerin TM, Schlomer JJ, Householder DB, et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res (2012) 72(16):4141–53. doi:10.1158/0008-5472.CAN-11-3834
45. Teicher BA. Tumor Models in Cancer Research. 2nd ed. (Vol. xiv). New York, NY: Humana Press (2011). 693 p.
46. Lum DH, Matsen C, Welm AL, Welm BE. Overview of human primary tumorgraft models: comparisons with traditional oncology preclinical models and the clinical relevance and utility of primary tumorgrafts in basic and translational oncology research. Curr Protoc Pharmacol (2012) Chapter 14:Unit1422. doi:10.1002/0471141755.ph1422s59
47. Wilson AJ, Barham W, Saskowski J, Tikhomirov O, Chen L, Lee HJ, et al. Tracking NF-kappaB activity in tumor cells during ovarian cancer progression in a syngeneic mouse model. J Ovarian Res (2013) 6(1):63. doi:10.1186/1757-2215-6-63
48. Skubitz AP, Taras EP, Boylan KL, Waldron NN, Oh S, Panoskaltsis-Mortari A, et al. Targeting CD133 in an in vivo ovarian cancer model reduces ovarian cancer progression. Gynecol Oncol (2013) 130(3):579–87. doi:10.1016/j.ygyno.2013.05.027
49. Sprowl JA, van Doorn L, Hu S, van Gerven L, de Bruijn P, Li L, et al. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: influence on antitumor efficacy and systemic clearance. Clin Pharmacol Ther (2013) 94(5):585–92. doi:10.1038/clpt.2013.145
50. Gaikwad SM, Gunjal L, Junutula AR, Astanehe A, Gambhir SS, Ray P. Non-invasive imaging of phosphoinositide-3-kinase-catalytic-subunit-alpha (PIK3CA) promoter modulation in small animal models. PLoS One (2013) 8(2):e55971. doi:10.1371/journal.pone.0055971
51. Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA (2004) 101(33):12288–93. doi:10.1073/pnas.0404041101
52. Ilagan MX, Lim S, Fulbright M, Piwnica-Worms D, Kopan R. Real-time imaging of notch activation with a luciferase complementation-based reporter. Sci Signal (2011) 4(181):rs7. doi:10.1126/scisignal.2001656
53. Salomonnson E, Stacer AC, Ehrlich A, Luker KE, Luker GD. Imaging CXCL12-CXCR4 signaling in ovarian cancer therapy. PLoS One (2013) 8(1):e51500. doi:10.1371/journal.pone.0051500
54. Ray P, Lewin SA, Mihalko LA, Schmidt BT, Luker KE, Luker GD. Noninvasive imaging reveals inhibition of ovarian cancer by targeting CXCL12-CXCR4. Neoplasia (2011) 13(12):1152–61. doi:10.1593/neo.111076
55. Olive KP, Tuveson DA. The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res (2006) 12(18):5277–87. doi:10.1158/1078-0432.CCR-06-0436
56. Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov (2008) 7(7):591–607. doi:10.1038/nrd2290
57. Kinross KM, Brown DV, Kleinschmidt M, Jackson S, Christensen J, Cullinane C, et al. In vivo activity of combined PI3K/mTOR and MEK inhibition in a Kras(G12D);Pten deletion mouse model of ovarian cancer. Mol Cancer Ther (2011) 10(8):1440–9. doi:10.1158/1535-7163.MCT-11-0240
58. Munk Jensen M, Erichsen KD, Bjorkling F, Madsen J, Jensen PB, Sehested M, et al. [18F]FLT PET for non-invasive assessment of tumor sensitivity to chemotherapy: studies with experimental chemotherapy TP202377 in human cancer xenografts in mice. PLoS One (2012) 7(11):e50618. doi:10.1371/journal.pone.0050618
59. Cho S, Sun Y, Soisson AP, Dodson MK, Peterson CM, Jarboe EA, et al. Characterization and evaluation of pre-clinical suitability of a syngeneic orthotopic mouse ovarian cancer model. Anticancer Res (2013) 33(4):1317–24.
60. Barua A, Bitterman P, Bahr JM, Bradaric MJ, Hales DB, Luborsky JL, et al. Detection of tumor-associated neoangiogenesis by Doppler ultrasonography during early-stage ovarian cancer in laying hens: a preclinical model of human spontaneous ovarian cancer. J Ultrasound Med (2010) 29(2):173–82.
61. Hensley H, Quinn BA, Wolf RL, Litwin SL, Mabuchi S, Williams SJ, et al. Magnetic resonance imaging for detection and determination of tumor volume in a genetically engineered mouse model of ovarian cancer. Cancer Biol Ther (2007) 6(11):1717–25. doi:10.4161/cbt.6.11.4830
62. Hensley HH, Roder NA, O’Brien SW, Bickel LE, Xiao F, Litwin S, et al. Combined in vivo molecular and anatomic imaging for detection of ovarian carcinoma-associated protease activity and integrin expression in mice. Neoplasia (2012) 14(6):451–62. doi:10.1596/neo.12480
63. Vandsburger MH, Radoul M, Addadi Y, Mpofu S, Cohen B, Eilam R, et al. Ovarian carcinoma: quantitative biexponential MR imaging relaxometry reveals the dynamic recruitment of ferritin-expressing fibroblasts to the angiogenic rim of tumors. Radiology (2013) 268(3):790–801. doi:10.1148/radiol.13122053
64. Canese R, Pisanu ME, Mezzanzanica D, Ricci A, Paris L, Bagnoli M, et al. Characterisation of in vivo ovarian cancer models by quantitative 1H magnetic resonance spectroscopy and diffusion-weighted imaging. NMR Biomed (2012) 25(4):632–42. doi:10.1002/nbm.1779
65. Clarke CJ, Haselden JN. Metabolic profiling as a tool for understanding mechanisms of toxicity. Toxicol Pathol (2008) 36(1):140–7. doi:10.1177/0192623307310947
66. Zhang T, Wu X, Ke C, Yin M, Li Z, Fan L, et al. Identification of potential biomarkers for ovarian cancer by urinary metabolomic profiling. J Proteome Res (2013) 12(1):505–12. doi:10.1021/pr3009572
67. Denkert C, Budczies J, Kind T, Weichert W, Tablack P, Sehouli J, et al. Mass spectrometry-based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer Res (2006) 66(22):10795–804. doi:10.1158/0008-5472.CAN-06-0755
68. Fong MY, McDunn J, Kakar SS. Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer. PLoS One (2011) 6(5):e19963. doi:10.1371/journal.pone.0019963
69. Curley MD, Garrett LA, Schorge JO, Foster R, Rueda BR. Evidence for cancer stem cells contributing to the pathogenesis of ovarian cancer. Front Biosci (Landmark Ed) (2011) 16:368–92. doi:10.2741/3693
70. Foster R, Buckanovich RJ, Rueda BR. Ovarian cancer stem cells: working towards the root of stemness. Cancer Lett (2013) 338(1):147–57. doi:10.1016/j.canlet.2012.10.023
71. Alvero AB, Chen R, Fu HH, Montagna M, Schwartz PE, Rutherford T, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle (2009) 8(1):158–66. doi:10.4161/cc.8.1.7533
72. Chen J, Wang J, Chen D, Yang J, Yang C, Zhang Y, et al. Evaluation of characteristics of CD44+CD117+ ovarian cancer stem cells in three dimensional basement membrane extract scaffold versus two dimensional monocultures. BMC Cell Biol (2013) 14:7. doi:10.1186/1471-2121-14-7
73. Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S, et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer (2012) 130(1):29–39. doi:10.1002/ijc.25967
74. Shank JJ, Yang K, Ghannam J, Cabrera L, Johnston CJ, Reynolds RK, et al. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol Oncol (2012) 127(2):390–7. doi:10.1016/j.ygyno.2012.07.115
75. Chen L, Kasai T, Li Y, Sugii Y, Jin G, Okada M, et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS One (2012) 7(4):e33544. doi:10.1371/journal.pone.0033544
76. Ma J, Lanza DG, Guest I, Uk-Lim C, Glinskii A, Glinsky G, et al. Characterization of mammary cancer stem cells in the MMTV-PyMT mouse model. Tumour Biol (2012) 33(6):1983–96. doi:10.1007/s13277-012-0458-4
77. Flesken-Nikitin A, Hwang CI, Cheng CY, Michurina TV, Enikolopov G, Nikitin AY. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature (2013) 495(7440):241–5. doi:10.1038/nature11979
78. Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol (2012) 124(2):192–8. doi:10.1016/j.ygyno.2011.09.039
79. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA (2005) 102(51):18538–43. doi:10.1073/pnas.0509182102
80. Dai MS, Vassaux G, Xu M, You RI, Hsieh YF, Ouisse LH, et al. Early Treg suppression by a listeriolysin-O-expressing E. coli vaccine in heterologous prime-boost vaccination against cancer. Vaccine (2012) 30(48):6903–11. doi:10.1016/j.vaccine.2012.09.001
81. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res (2013) 73(12):3591–603. doi:10.1158/0008-5472.CAN-12-4100
82. Fujisawa T, Joshi BH, Puri RK. IL-13 regulates cancer invasion and metastasis through IL-13Ralpha2 via ERK/AP-1 pathway in mouse model of human ovarian cancer. Int J Cancer (2012) 131(2):344–56. doi:10.1002/ijc.26366
83. Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One (2013) 8(4):e61895. doi:10.1371/journal.pone.0061895
84. Tanyi JL, Chu CS. Dendritic cell-based tumor vaccinations in epithelial ovarian cancer: a systematic review. Immunotherapy (2012) 4(10):995–1009. doi:10.2217/imt.12.100
85. Zhang Z, Chen X, Chang X, Ye X, Li Y, Cui H. Vaccination with embryonic stem cells generates effective antitumor immunity against ovarian cancer. Int J Mol Med (2013) 31(1):147–53. doi:10.3892/ijmm.2012.1195
86. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi:10.1158/0008-5472.CAN-13-1550
87. Chekmasova AA, Rao TD, Nikhamin Y, Park KJ, Levine DA, Spriggs DR, et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res (2010) 16(14):3594–606. doi:10.1158/1078-0432.CCR-10-0192
Keywords: mouse models, serous epithelial ovarian cancer, imaging techniques, immune therapy, cancer stem cells, reporter, metabolite profiling
Citation: House CD, Hernandez L and Annunziata CM (2014) Recent technological advances in using mouse models to study ovarian cancer. Front. Oncol. 4:26. doi: 10.3389/fonc.2014.00026
Received: 18 October 2013; Accepted: 28 January 2014;
Published online: 13 February 2014.
Edited by:
Viive Maarika Howell, University of Sydney, AustraliaReviewed by:
Tamara Louise Kalir, Mount Sinai School of Medicine, USAJudy Luborsky, Rush University Medical Center, USA
Copyright: © 2014 House, Hernandez and Annunziata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina Messineo Annunziata, Women’s Malignancies Branch, National Cancer Institute, 10 Center Drive, Room 12N226, Bethesda, MD 20892-1906, USA e-mail:YW5udW56aWNAbWFpbC5uaWguZ292