 Irina Häuselmann
Irina Häuselmann Lubor Borsig
Lubor Borsig- Zürich Center for Integrative Human Physiology, Institute of Physiology, University of Zürich, Zürich, Switzerland
Malignant transformation of cells is associated with aberrant glycosylation presented on the cell-surface. Commonly observed changes in glycan structures during malignancy encompass aberrant expression and glycosylation of mucins; abnormal branching of N-glycans; and increased presence of sialic acid on proteins and glycolipids. Accumulating evidence supports the notion that the presence of certain glycan structures correlates with cancer progression by affecting tumor-cell invasiveness, ability to disseminate through the blood circulation and to metastasize in distant organs. During metastasis tumor-cell-derived glycans enable binding to cells in their microenvironment including endothelium and blood constituents through glycan-binding receptors – lectins. In this review, we will discuss current concepts how tumor-cell-derived glycans contribute to metastasis with the focus on three types of lectins: siglecs, galectins, and selectins. Siglecs are present on virtually all hematopoietic cells and usually negatively regulate immune responses. Galectins are mostly expressed by tumor cells and support tumor-cell survival. Selectins are vascular adhesion receptors that promote tumor-cell dissemination. All lectins facilitate interactions within the tumor microenvironment and thereby promote cancer progression. The identification of mechanisms how tumor glycans contribute to metastasis may help to improve diagnosis, prognosis, and aid to develop clinical strategies to prevent metastasis.
Introduction
The majority of cancer deaths are attributed to the metastatic spread of cancer cells to vital organs rather than to the primary tumor outgrowth. During malignant transformation, the genetic alteration in the cells results in mutations of proto-oncogenes and tumor suppressor genes, which as a result give rise to tumor clones with different properties (1). Malignant cells thereby acquire characteristics enabling them dissociation from tumors, degradation of the extracellular matrix, invasion, adhesion, and metastasis to distant organs. Alteration of tumor-cell-surface glycosylation is one of the characteristic traits associated with enhanced malignancy (2–4). Glycans are oligosaccharide structures that are covalently bound to proteins, lipids, or present in a free form in tissues or tumors. Glycans are bound to the protein either through Asn (N-linked glycan) or through Ser or Thr (O-linked glycan). Lectins are a family of carbohydrate-binding proteins that specifically recognize glycans. Fundamental processes such as cell–cell recognition, cell adhesion, mobility, and pathogen–host interaction are facilitated by lectins in healthy organisms. The common expression of lectins on endothelial cells, immune cells, in the extracellular matrix or as soluble adhesion molecules enables them to bind to tumor-cell glycans and thereby affect tumor-cell progression (5). Subsequently, accumulating evidence supports the involvement of tumor-cell-surface glycans in tumor-cell migration, adhesion, and metastasis. This review addresses the role of cancer-associated glycans during metastasis with the focus on endogenous lectin interactions within the tumor microenvironment.
The Process of Metastasis
Hematogenous metastasis is a multistep process during which malignant cells detach from the primary tumors, degrade the extracellular matrix, invade the surrounding tissue, enter the blood or lymphatic vessels, and extravasate to form metastatic lesions. Tumor cells through the cell-surface glycans can engage with a variety of endogenous lectins both at the primary site of a tumor and in the circulation. Tumor cell upon reaching the blood circulation induces microthrombi, the formation of which is facilitated by platelet P-selectin binding to tumor-cell-surface glycans (6, 7). Tumor-cell emboli formation contributes to mechanical lodging in the microvasculature and/or adhesion to the endothelium thereby promoting tumor-cell extravasation and metastasis (8). There is accumulating evidence that vascular lectins–selectins facilitate tumor-cell interactions with all blood constituents, platelets, leukocytes, and endothelial cells, and thereby contribute to metastasis (3, 9, 10). In addition, recruitment of immune cells to the metastatic microenvironment is dependent on selectins (11–13).
Specific glycan structures on colonic epithelium provide immune-modulatory activity to tissue macrophages through sialic acid-binding lectins–siglecs (14, 15). In addition, galactose-binding lectins–galectins were shown to be involved in immune-suppression and metastasis (16). Consequently, altered glycosylation may both induce inflammatory reactions and promote immune-suppression, however; it is dependent on the cellular context within the tissue. Finally, glycan changes associated with cancer progression profoundly define the phenotype of cancer cells depending on interactions with endogenous lectins both in tumor and metastatic environments.
General Mechanisms for Altered Glycosylation in Cancer
Cancer progression requires a range of alterations in extracellular and intercellular signaling that promotes cell proliferation, emergence of invasive subsets, dissociation from the tumor, intravasation, and adhesive interactions within the circulation that finally facilitate metastasis. Within the tumor environment changes in glycosylation allow malignant cells to promote cell mobility, cell adhesion, and even receptor activation, and thereby contributing to the invasive phenotype (3–5). Malignant transformation leads to expression of oncofetal antigens, epitopes that are present on embryonic tissues and tumor cells, but are generally absent in healthy adult cells. Neo-synthesis and incomplete synthesis are the two major mechanisms for generation of cancer-specific glycans (2).
Altered glycosylation of N-linked glycans in cancer is typically associated with enhanced β1,6-branching (Table 1) that is facilitated by β1,6-N-acetylglucosaminyltransferase-5 (GnT5) (17, 18). Increased activity of GnT5 is associated with increased polylactosaminic sequences, and the inhibition of GnT5 resulted in attenuation of metastasis (19, 20). GnT5 deficiency (Mgat5-deficient mice) resulted in reduced tumor growth and metastasis (21). However, the functional role of branched N-glycosylation in cancer was later shown to be dependent on galectin binding and thereby altering the phenotype of the cell (22).
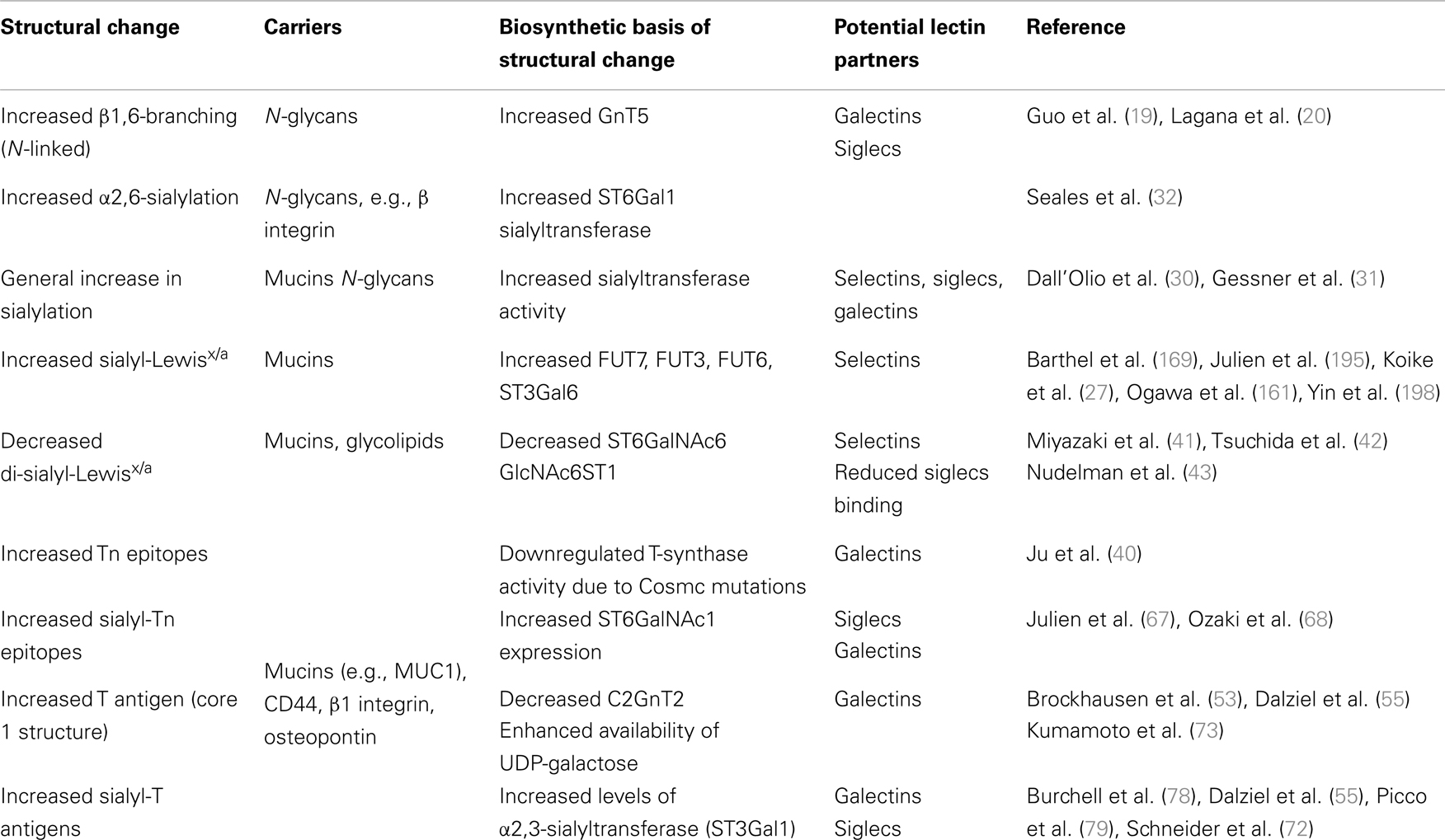
Table 1. Common glycan alterations on carcinoma cells and their effect on lectin recognition.
Virtually in every cancer type upregulation of glycosyltransferases has been detected, leading to expression of common tumor-cell epitopes such as sialyl-Lewisx and sialyl-Lewisa (sLex/sLea), Thomsen-nouvelle antigen (Tn), and sialyl-Tn (sTn) (3–5, 23, 24). Hypoxia has been identified as one of the factors leading to increased expression of glycosyltransferases (25, 26). For instance, increased expression of α1,3-fucosyltransferase-7 (FUT7) and α2,3-sialyltransferase ST3Gal1, enzymes involved in synthesis of sLex/a has been detected (27). The general increase in sialylation has been detected both in clinical settings and experimental models that is associated with a metastatic cell phenotype (25, 28, 29). An increase in α2,6-sialylation in tumors is usually attributed to the upregulation of ST6Gal1 sialyltransferase that is primarily active on N-linked glycans (30–32), or ST6GalNAc family of sialyltransferases, which are active on O-linked glycans or glycolipids (33). Accordingly, overexpression of Neu1 sialidase in colon cancer cells led to reduced liver metastasis in mice due to increased desialylation of β4 integrin whereas silencing of Neu1 sialidase increased cell migration, invasion, and adhesion in vitro (34).
Synthesis of shorter glycan structures like Thomsen–Friedenreich (TF or T), Tn, and sTn epitopes has been observed in a number of carcinomas (35–39). One of the factors affecting the synthesis of incomplete glycan structures is the frequent mutation of the Cosmc chaperone that is required for the galactosyltransferase activity that modifies O-linked glycans (40). Another example of shortened glycan synthesis is the reduced expression of disialyl-Lewisa (di-sLea) and sialyl 6-sulfo Lewisx structures in epithelial cancer. Disialyl-Lewisx (di-sLex) structure is synthesized with the α2,6-sialyltransferase ST6GalNAc6, and its expression is downregulated by epigenetic silencing in malignant epithelium (41, 42). Similarly, repressed expression of sulfotransferase responsible for 6-sulfo Lex was detected in cancer cells but not in normal epithelial cells (26).
Gangliosides are sialic acid-containing glycolipids, which expression is often dysregulated during malignant transformation (2). Apart from glycolipid specific glycan structures containing disialic acid in a α2,8-linkage (e.g., GD3), changes in glycosyltransferases promote expression of sLex epitopes (43). Overexpression of sialidase Neu2 led to reduced metastasis, while Neu2 was found to be downregulated in highly metastatic variants of colon carcinoma (44).
Despite many possibilities for the formation of glycans (linkage and sequence of monosaccharide units) there is a rather small number of structures commonly detected in cancer. Furthermore, terminal glycan structures exposed on the cell surfaces of tumor cells can be recognized through endogenous lectins and thereby modulate cancer progression.
Alterations of Cancer-Associated O-Linked Glycans
Mucins are high molecular weight glycoproteins exhibiting a rod like conformation due to heavy glycosylation with O-linked glycans (3, 45). O-linked glycosylation, which is based on GalNAc bound to the Ser/Thr of a protein, is further modified by galactose (core 1 structure) or GlcNAc (core 3 structure) in normal mucins (Figure 1). During malignant transformation mucins of intestine, colon, liver, and pancreas have reduced core 1 and core 3 structures that correlate with enhanced sialylation of Tn and T antigens (24, 46, 47). Core 3-derived glycans are a major type expressed by normal epithelial cells of the gastrointestinal tract, which are downregulated during malignancy due to loss of functional β3-N-acetylglucosaminyltransferase-6 (core 3 synthase) expression (48, 49). Consequently, overexpression of core 3 synthase in pancreatic cells was associated with decreased presence of Tn antigens and resulted in a reduced tumorigenicity and metastasis upon orthotopic injection. In addition, enhanced expression of the core 2 β1,6-N-acetylglucosaminyltransferase (C2GnT1) responsible for the core 2 synthesis was detected in colorectal and lung carcinomas, which correlated with high levels of sLex on O-glycans and therefore strong binding to E-selectin and metastasis compared to normal tissues (50–52). Mucins of normal mammary epithelial cells contain a mixture of O-glycans and the majority is core 2-based structures (53, 54). Reduced expression of C2GnT1 in mammary cancer is associated with enhanced presence of Tn and sTn (53–56). However, despite reduced core 2 structures on breast cancer cells, increased presence of sLex epitopes has been observed, which likely is a result of increased fucosylation (57).
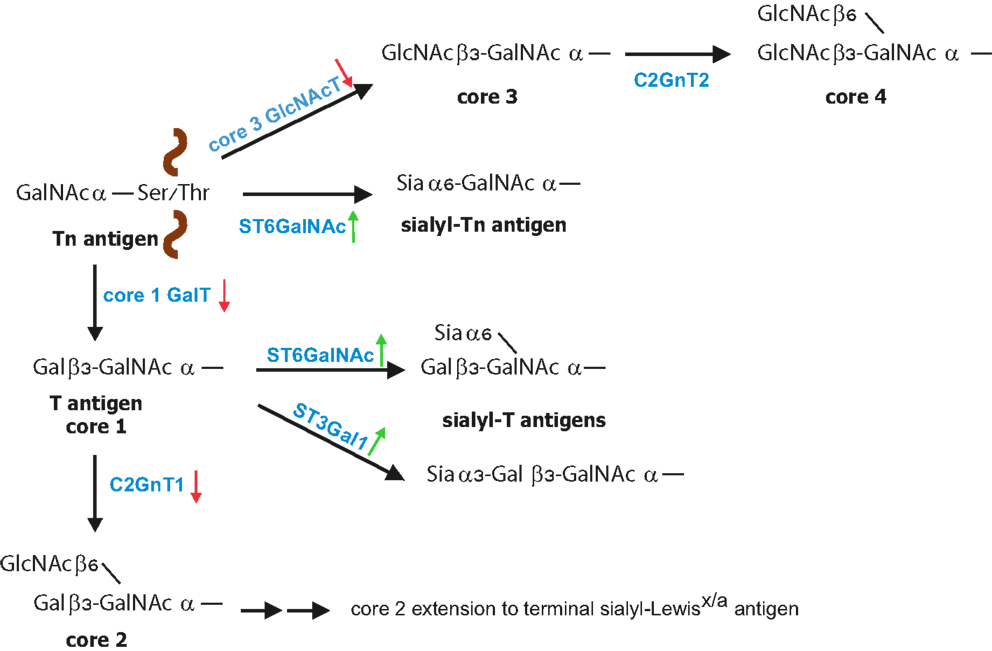
Figure 1. Biosynthesis of O-glycans. O-glycan synthesis is initiated by linking of GalNAc to the protein at Ser or Thr residue. The simplest O-glycan Tn antigen can be further converted to core 1 structure (T antigen) by β1,3 galactose extension; core 3 structure by addition of β1,3-GlcNAc. During cancer increased expression (green arrow) of sialyltransferases with concomitant reduced expression (red arrow) of core 1 GalT and core 3 GlcNAcT leads to increased formation of sialyl-Tn and sialyl-T antigens. Core 1 structure is further branched by C2GnT1 to form core 2 that can be further modified to poly-N-acetyllactosamine structures carrying sialyl-Lewisx/a [modified from Ref. (23)].
Formation of T, Tn, and sTn Antigens during Cancer Progression
In healthy tissues, core 1-based T and Tn epitopes are almost absent however; in about 90% of all human carcinomas these precursor structures are detected (36, 39). Unsubstituted Tn epitopes occur in human cancers of colon, breast, bladder, prostate, liver, ovary, and stomach; and their presence correlate with cancer progression and metastasis (35–37, 58–63). Similarly, sialylated T and Tn antigens correlate with progression of epithelial cancer and poor clinical prognosis of many carcinomas (25, 28, 39, 64–66). ST6GalNAc1-mediated α2,6-linked sialylation of GalNAc of the precursor Tn antigen results in formation of the sTn antigen (25, 67–69). The sialylation step prevents further glycan extension and therefore leads to truncation of O-linked glycans (47, 70).
Several mechanisms have been described to enable increased Tn, sTn, or T expression in cancer (Table 1) (33, 46). (1) Decreased activity of core 2 C2GnT1 enzyme leads to accumulation of T antigen (described above) that is further sialylated by ST6GalNAc1 and ST6GalNAc2 enzymes (71, 72). (2) Enhanced availability of the nucleotide sugar substrate UDP-galactose appears to promote increased T antigen biosynthesis through core 1 β1,3-galactosyltransferase (73). Colon cancer tissues expressed increased levels of the UDP-Galactose transporter, which brings the sugar donor into the Golgi apparatus compared to non-malignant mucosa. (3) Activity of β1,3-galactosyltransferase (T synthase) requires the presence of the molecular chaperon protein Cosmc, which is responsible for folding and stability of the enzyme (40, 74). The absence of Cosmc leads to β1,3-galactosyltransferase degradation. Mutation in Cosmc chaperone is associated with increased Tn expression in colon carcinoma and melanoma cell lines and also increased sTn expression (40, 75). Accordingly, down-regulation of T-synthase resulted in a marked increase of T, Tn, and particularly sTn in colon carcinoma cells (76). (4) Generation of sTn is facilitated by the sialyltransferase ST6GalNAc1 and ST6GalNAc2 (71, 72). Human gastric cancer cells with enhanced ST6GalNAc1 expression showed higher intraperitoneal metastasis compared to sTn-negative tumor cells. Similarly, overexpression of ST6GalNAc1, thereby sTn epitope, in human breast cancer cells led to increased tumor growth in immunodeficient mice (68, 77). In addition, enhanced sialylation of T antigen in breast cancer correlated with higher levels of α2,3-sialyltransferase (ST3Gal1) (72, 78). Overexpression of ST3Gal1 under the human MUC1 promoter in a spontaneous murine breast cancer model resulted in significantly decreased tumor latency compared to mice without ST3Gal1 overexpression (79). Furthermore, the sialyltransferase expression alone was responsible for enhanced tumorigenesis indicating that this enzyme per se acts as a tumor promoter (79).
Only few glycoproteins are known to present Tn, T, or sTn and sialyl-T (sT) antigens in malignant tissues (66). Mucin MUC1 and CD44v6 display sTn and sT antigens in colon, gastric, and breast cancers (80–83). MUC2 is a major carrier of shortened glycans in gastric cancer (84). Enhanced sTn expression in breast and gastric cancer is associated with overexpression of MUC1, CD44, and ST6GalNAc1 (68, 77). Although CD44v6 is expressed in some types of healthy epithelia, higher expression is observed in squamous cell carcinomas and adenocarcinomas including breast, lung, colon, and pancreatic carcinomas (85–87). Interestingly, serum levels of osteopontin, a CD44 ligand, that itself is a sTn carrier, have been detected in cancer patients and correlate with poor prognosis (87).
The enhanced expression of Tn, sTn, and T antigens on MUC1, osteopontin, and CD44 is associated with high metastatic potential and poor prognosis (84, 88, 89). However, there is little evidence for the functional consequence of this aberrant glycosylation during cancer progression. In human breast cancer cells, expression of sTn on MUC1 was associated with reduced cell adhesion and increased cell migration (77). In addition, β1 integrins carry aberrant forms of O-glycans that is associated with metastasis (90). Enhanced expression of ST6GalNAc1 in murine carcinoma cells led to an increase in sTn expression on β1 integrin subunit associated with morphological changes including loss of epithelial appearance, disorganization of actin stress fibers, and reduced ability to migrate on fibronectin. A recent study showed that high expression of the ppGalNAcT13, which initiates O-glycan synthesis by adding the first GalNAc to Ser/Thr, induced high metastatic potential of Lewis lung carcinoma by generating trimeric Tn antigens (GalNAc1-Ser/Thr)3 on syndecan 1 (91). The complex formation of trimeric Tn antigens on Syndecan 1 together with α5β1 integrin and MMP-9 resulted in enhanced invasion and metastasis. Recent findings provide evidence that cell-surface mucins are involved in signal transduction events [reviewed in Ref. (24, 45)]. Decreased sTn expression on neuroblastoma achieved by extension of core 1 structure with B3GNT3 expression reduced activation of focal adhesion kinase and thereby partially suppressed malignant phenotype (92). Aberrant glycosylation in cancer does not affect only the tumor-cell phenotype behavior (e.g., proliferation, differentiation, and adhesion), but also contribute to the control of the local microenvironment, immune responses, and metastasis. Therefore, these glycans serve as ligands for cells in the tumor microenvironment through endogenous lectins.
Siglecs
Sialic acid-binding immunoglobulin superfamily lectins (siglecs) are the largest family of sialic-acid-binding molecules (93–95). Siglecs are expressed on specific subpopulations of hematopoietic cells where they exert their immune-regulatory function. Many siglecs contain intracellular tyrosine motifs, which include one or more membrane-proximal immunoreceptor tyrosine-based inhibitory motif (ITIM) and a membrane-distal ITIM-like motif (93, 94). These motifs are involved in inhibitory signal transduction. Based on both sequence similarity and conservation between mammalian species siglecs are divided in two major subgroups. The first group comprises Siglec-1 (sialoadhesin, CD169), Siglec-2 (CD22), Siglec-4 (myeloid-associated glycoprotein), and Siglec-15. The second subfamily of CD33/Siglec-3 related siglecs consists of 10 human members (Siglec-3, -5, -6, -7, -8, -9, -10, -11, -14, and -16) and 5 rodent members (Siglec-3, -E, -F, -G, and -H) (93, 95). The first subgroup with its evolutionary conserved members has restricted expression patterns. For instance Siglec-1 is specifically expressed on macrophages, Siglec-2 on B-cells and Siglec-4 on oligodendrocytes and Schwann cells in the nervous system (96). On the other hand, CD33-related siglecs display a more divergent expression pattern dependent on developmental stage of immune cells (93, 95). The high sialic acid concentration on the cell-surface of siglec-expressing cells often leads to binding to the cell glycans (in cis) or adjacent cells (in trans). Siglecs can be affected by various stimuli including cytokines, toll-like receptor activation, and viral and bacterial infections, the biology of siglecs is therefore rather complex (96). The binding specificity of siglecs depends on the distinct types, linkages (α2,3, α2,6, and α2,8), arrangements of sialic acids, their way of presentation on different cells, organs, and organisms. Siglec binding to ligands modulates cell–cell interactions, cell proliferation, cell death, and endocytosis (96–99).
The Role of Siglecs in Cancer Progression
Accumulating evidence indicates that the interaction between tumor-specific glycans and lectins on immune cells are involved in modulation of the tumor microenvironment (100). The inhibitory nature of siglec upon binding of specific glycan may lead to dampening of immune responses and thereby escape of immune surveillance and clearance. Whether siglecs contribute to cancer progression through recognition of distinct cancer-specific glycan structures is currently under investigation. Non-malignant colon epithelial cells express di-sLea epitopes that serve as ligands for both Siglec-7 and -9 (15). The expression of siglec ligands was decreased upon malignant transformation, which was associated with enhanced expression of sLex and sLea epitopes (26). Expression of ST6GalNAc6, which synthesizes di-sLea in human colon cancer cells resulted in increased di-sLea, loss of sLea epitopes, and increased binding to Siglec-7 (41). Mainly resident macrophages were found to carry Siglec-7 and -9 in a colonic lamina propria and Siglec-7/9 ligation could suppress macrophage-mediated cyclooxygenase-2 (COX2) and prostaglandin E2 expression and thereby prevent inflammatory damage of the colonic mucosa (15). Siglec-15, which preferentially recognizes sTn antigen, is expressed in tumor-associated macrophages (TAMs) in various human carcinoma tissues including lung, liver, and rectum (101). Binding of myeloid cells through Siglec-15 to sTn on tumor cells resulted in increased TGF-β secretion into the tumor microenvironment that is associated with cancer progression. Interestingly, Siglec-15 expression was induced by M-CSF, which usually polarizes macrophages to M2 phenotype commonly detected in the tumor microenvironment.
Siglec-1 is expressed in a subset of macrophages that are involved in the pathophysiology of cancer (102). Clinical observation showed that increased Siglec-1 is present in splenic marginal cell lymphoma as well as in macrophage infiltrates of MUC1-positive breast cancers (103, 104). Siglec-1 positive macrophages were found to infiltrate into rat xenograft tumors in a CCL2-dependent manner (105). On contrary, recent study demonstrated that Siglec-1 positive macrophages in regional lymph nodes of colorectal carcinoma patients promote CD8+ T-cell mediated anti-tumor immunity and are associated with a better prognosis for these patients (106).
Siglec-9, a surface receptor on NK cells, B-cells, and monocytes, has been identified as a receptor for mucin MUC16 (14). Cell-surface bound as well as soluble MUC16 is overexpressed in human ovarian tumor cells and detected in peritoneal fluid of cancer patients (107). Engagement of Siglec-9 on monocytes also induced secretion of immunosuppressive cytokine IL-10 (108). Similar immune-suppression mediated by Siglec-7 on NK cells was observed in renal cell carcinoma expressing disialosyl globopentaosylceramide (DSGb5) as a major ganglioside (109). Recent study from C. Bertozzi group provided strong evidence that siglec-7-mediated cytotoxicity of NK cells can be modulated by the alteration of glycans on cell surfaces (110). Presentation of sialylated ligands on tumor cells recognized by siglec-7 resulted in enhanced phosphorylation of cytoplasmic tyrosine residues, causing dampening of cytolytic activity.
The association between Siglec-9 positive immune cells and MUC1-positive tumor cells has been detected in tissues of human colon, pancreas, and breast cancer. Interestingly, Siglec-9 binding to MUC1 expressing tumor cells was shown to induce recruitment of β-catenin in tumor cells resulting in promotion of cell growth in vitro (111). These findings suggest that Siglec-9 engagement of carcinoma mucin MUC1 may be involved in tumor growth, however; the nature of Siglec-9 ligands as well as the cellular context in vivo remains to be defined.
Taken together, the current evidence is largely based on clinical correlation of cancer–glycan expression and several experiments showing Siglec-cancer–glycan interaction in vitro. Whether these interactions indeed functionally modulate immune cell responses in the tumor microenvironment and thereby affect cancer progression in vivo requires experimental validation.
Siglecs as Target of Cancer Therapy
The identification of Siglec-2 and Siglec-3 as markers of acute myeloid leukemia (AML) and B-cell lymphomas raised interest in potential immunotherapy (112–114). Anti-Siglec-2 and siglec-3 specific antibodies were conjugated with variety of toxins and such immunotoxins have been targeted in several autoimmune diseases and hematological malignancies [reviewed in Ref. (93, 94, 115)]. In the majority of acute lymphoblastic leukemias (ALL) Siglec-2 (CD22) was identified as a useful target for cell-depletion therapy (116). Inotuzumab ozogamicin is an immunotoxin comprised of a humanized IgG4 monoclonal antibody covalently linked to calecheamicin (CMC-544). CMC-544 was active against B-cell tumors in preclinical models and has been evaluated in phase I study for patients with B-cell lineage ALL (117). Inotuzumab ozogamicin used as a single therapy in patients with refractory-relapsed ALL showed positive results.
The immunotoxin gemetuzumab ozogamicin (OG, Mylotarg; Wyeth, Madison, NJ, USA), which consists of a humanized anti-CD33 (siglec-3) murine antibody linked to calicheamicin, was approved by the FDA for treatment of CD33+ AML patients. Binding and endocytosis of the conjugate resulted in the intracellular release of the toxin causing cell death of CD33+ cells (94, 115). However the drug is off the market since 2010 because the key phase III trial (South West Oncology Group Study S0106) in which GO was combined with induction chemotherapy failed to improve disease-free survival and caused higher fatal induction toxicity rate compared to chemotherapy alone (118). Recent studies using lower or fractionated dose of GO suggest that GO may still improve survival of distinct subsets of AML patients, particularly patients with favorable cytogenetics (119). New approaches with humanized CD33 antibody conjugated to synthetic DNA cross-linking pyrrolobenzodiazepine (SGN-CD33A) have been developed and revealed promising effectiveness in animal models (120). SGN-CD33A is now currently being tested in a phase I trial (ClinicalTrials.gov: NCT01902329).
Galectins
In contrast to siglecs and selectins, which are mostly cell-surface-bound receptors, galectins are soluble immunomodulatory lectins (121). Galectins bind to galactose that is either β1,3- or β1,4-linked to N-acetylglucosamine, a common disaccharide found both on N- and O-linked glycans and glycolipids. Galectins act both intracellularly by modulating signaling pathways and extracellularly as regulatory receptors (100). Up to date the galectin family consists of 15 members, which are classified into three groups based on structural differences: prototype galectins (Galectin-1, -2, -5, -7, -10, -11, -13, -14, and -15) having one carbohydrate recognition domain (CRD), tandem repeat-type galectins (Galectin-4, -6, -8, -9, and -12) having two CRDs, and the single member Galectin-3, which has one CRD connected to a non-lectin N-terminal region responsible for oligomerization (100). Galectins are expressed by various cell types including epithelial and immune cells, but their expression is altered during progression of colon, breast, lung, pancreatic, head and neck, and cervical cancers (16, 122). Many studies indicate that cancer-associated galectins could regulate cancer cell proliferation, signaling, adhesion, invasion, and metastasis (122–124). Galectin-1 and Galectin-3 were most intensively studied in context of cancer.
Galectin-1
Accumulating evidence indicate that tumor-derived Galectin-1 contributes to immunosuppressive activity in different tumors, including lung and pancreatic carcinoma, melanoma, and neuroblastoma (16, 125–127). It has been shown that Galectin-1 binding to T-cells through N- and O-linked glycans on CD43 or CD45 mucins induces apoptosis of activated T-cells (128, 129). Galectin-1 expression by melanoma cells induced apoptosis of tumor-specific effector T-cells, and Galectin-1 inhibition allowed generation of a tumor-specific T1 response (126). Modification of cell-surface glycosylation affects glycan pattern on T-cells and thereby changes Galectin-1 binding. Enhanced expression of α2,6-sialyltransferase-1 (ST6Gal1) selectively modified N-glycans on CD45 and thereby inhibited Galectin-1 binding (130). How Galectin-1 contributes to immune-suppression in tumors has been delineated in lung cancer (131). High expression of Galectin-1 in lung cancer cell lines, as well as in human tumor tissues, alters the phenotype of monocyte-derived dendritic cells and impairs T-cell response, concomitant with increased presence of regulatory T-cells (Tregs). The regulatory effect of Galectin-1 is mediated by increased expression of IL-10 in monocytes thereby inducing a Th2-dominant cytokine profile. The enhanced infiltration of CD11c+ dendritic cells in human lung cancer samples has been recapitulated in a mouse model, which was completely omitted after transplantation of Galectin-1 silenced tumor cells. In another study, the amount of Galectin-1 positive cells correlated with the tumor grade in human breast cancer (132). Silencing of Galectin-1 in a metastatic murine mammary tumor led to a reduction of tumor growth and lung metastasis with a concomitant reduction in infiltrating regulatory T-cells.
Experimental evidence also suggests that Galectin-1 expressed on various tumor-cell types including hepatocellular carcinoma, melanoma, ovarian, and prostate cancer cells mediates tumor-cell adhesion to the extracellular matrix (133, 134). In addition, Galectin-1 mediated attachment of cancer cells to the extracellular matrix and endothelial cells through binding to CD44 and CD326 on murine breast and colon cancer cells (16). Galectin-1 might also be involved in formation of platelet-cancer cell complexes since it was shown to activate platelets (135). Murine breast, colon, and Lewis lung cancer cells with silenced Galectin-1 showed decreased lung metastasis, which was associated with increased T-cell numbers and reduced angiogenesis (16, 125). Taken together, tumor-derived Galectin-1 exerts its immunosuppressive function through binding to endogenous (non-tumor-derived) glycans and thereby contributes to cancer progression.
Galectin-3
There is accumulating evidence that the cancer-associated T, Tn, and sTn structures promote metastasis through binding to Galectin-3. Galectin-3 expression is also increased in patient sera of several cancer types and associated with increased risk of metastasis (136, 137). For instance, T antigen expression by breast and prostate cancer cells facilitated interactions with cancer-associated Galectin-3 or with endothelial associated Galectin-3 (66, 138–140). These interactions lead to homotypic aggregation of cancer cells, which protects cancer cells from apoptosis induced by the lack of adhesion to the extracellular matrix (139). In addition, cancer cell-associated T antigens can induce Galectin-3 expression on the endothelium, which enabled cancer-endothelium adhesion (140). Another study has shown that lysosomal-associated membrane protein-1 (LAMP-1) on highly metastatic melanoma cells carries N-acetyllactosaminyl structures, which are recognized by Galectin-3 on lung endothelial cells suggesting that lung endothelial galectin-3 can serve as anchor for LAMP-1 expressing tumor cells in the circulation (141).
A characteristic feature of galectins is the induction of complex formation by cross-linking glycoproteins, which can form multimers “lattice” microdomain (121). Complex N-glycans are formed by GnT5 modification of N-glycans that are the ligands for Galectin-3 (142). Expression of GnT5 has long been implicated in tumor progression and metastasis (17). In particular, the absence of GnT5 delayed tumor formation and suppressed metastasis (21). Accordingly, up-regulated GnT5 expression has been observed in various human cancers (18, 143); and the ectopic expression of the GnT-V in multiple epithelial cells resulted in increased cell motility, tumor formation, and enhanced metastasis (144, 145). Furthermore, GnT5-dependent modifications of tyrosine kinase receptors such as EGF, TGF-β, IGFR, and PDGF enhanced affinity to galectin-3 and thereby prolonged their cell-surface expression (22, 146). Galectin-3-induced lattice formation prevented the surface clearance of receptors by clathrin-dependent endocytosis and enabled interaction with inhibitory caveolin-1 domains.
Branched O-glycans with poly-N-acetyllactosamine structures are recognized by Galectin-3 (147). In C2GnT1-expressing bladder tumor cells core 2 O-glycans present on MHC class I-related chain A are bound to Galectin-3 that reduced the affinity for the activating NK cell receptors NKG2D, thereby impairing NK cell function and anti-tumor activity.
Recent findings suggest that Galectin-3 also regulates dynamics of N-cadherin and the lipid raft marker ganglioside GM1 (148). Accumulation of N-cadherin and GM1 at cell–cell junctions destabilized cell–cell junctions and thereby promoted tumor-cell migration. N-glycans on α5β1 integrin are important for their proper binding to fibronectin (149, 150). Increased GnT5 mediated β1,6-branching reduces cell-surface clustering of α5β1 integrin, specifically of the β1 subunit, resulting in a less adhesive phenotype due to reduced adhesion to fibronectin and modulates fibronectin matrix remodeling in tumors (20, 151). Thus, Galectin-3 lattice formation provides another mechanism how altered glycosylation contributes to the malignant and invasive phenotype of tumor cells (148).
Selectins
Selectins are vascular cell adhesion molecules that belong to a family of C-type lectins, which facilitate the initial attachment of leukocytes to the endothelium during the process of leukocyte extravasation. The selectin family consists of L-, E-, and P-selectin, which share around 50% sequence homology in their C-type lectin domain (152). L-selectin (LECAM-1 and CD62L) is constitutively expressed on almost all hematopoietic cell types including myeloid cells, naïve, and some activated memory T-cells (152) and enables adhesion of leukocytes to the activated endothelium or in high endothelial venules of the peripheral lymph nodes (153, 154). E-selectin (ELAM-1 and CD62E) is exclusively displayed on endothelial cells, which requires de novo expression in response to inflammatory stimuli such as TNF-α and Il-1β. However, skin and parts of the bone marrow microvasculature have been shown to constitutively express certain E-selectin levels (155). On contrary, P-selectin (PADGEM and CD62P) is stored in alpha-granules of platelets as well as in Weibel–Pallade bodies of endothelial cells and can be rapidly mobilized to the cell-surface upon activation of platelets or the endothelia. E- and P-selectin bind to ligands on myeloid cells (156), certain types of lymphocytes (152) but also to several types of tumor cells (157–159). Selectins are the most-studied lectins in cancer biology, which promote cell–cell interaction with tumor cells and their microenvironment (9). All three selectins have been shown to contribute to tumor dissemination and specifically facilitate processes when the tumor cells are in the circulation.
Selectin Ligand Expression Correlates with Cancer Progression
There is compelling clinical and experimental evidence that overexpression of tetrasaccharides sLex and sLea correlates with poor prognosis due to enhanced metastatic phenotype in a number of cancer types, including colon, gastric, prostate, renal, pancreatic, and lung cancer (89, 160–165). Enhanced expression of sLex/a on cancer cells correlated with increased ability to adhere to E-selectin or to the activated endothelial cells and stromal cells in vitro (157, 166–168). Furthermore, high cell-surface expression levels of sLex were linked to enhanced metastatic activity in various experimental metastasis models using human carcinoma cells compared to lower or minimal sLex expression (169–171).
The minimal recognition motif for all three selectins are tetrasaccharides sLex/a (Figure 2) (172). SLex are terminal structures of N-or O-linked glycans attached to glycoproteins and glycolipids displayed by most circulating leukocytes and endothelial cells whereas sLea is detected on some epithelial cells but mostly on various tumor cells (3, 4, 173). The four glycosyltransferases N-acetylglucosaminyltransferase, β1,4-galactosyltransferase, α2,3-sialyltransferase, and α1,3-fucosyltransferase-7 are responsible for synthesis of sialyl-Lewisa/x structures on cells of the hematopoietic system (172, 174). Efficient selectin binding to carbohydrates usually requires a glycoprotein scaffold that facilitates the presentation of selectin ligands in clusters (175). One of the best characterized ligands for all three selectins is the P-selectin glycoprotein ligand-1 (PSGL-1), which is concentrated on the tips of microvilli on leukocyte surface (176). To the most common mucins carrying selectin ligands that are associated with cancer progression belong MUC1, MUC2, MUC4, and MUC16 (35, 45, 177, 178). Apart from mucins, several other selectin ligand carriers on tumor cells have been identified that includes CD24, CD44, death-receptor 3, E-selectin ligand-1, PSGL-1, and podocalyxin-like protein and this list is by far not complete (179–183). Several of these ligands are also expressed on tumor cells and are associated with cancer progression. For instance, CD44 glycoproteins exist in several isoforms and are expressed on epithelial and endothelial cells as well as on multiple cancer cell types such as gastric, colorectal, pancreatic, and lung cancer (184–186). The aberrant expression of CD44 in colorectal carcinoma cells correlated with increased metastatic potential in vivo (187, 188). Based on flow-based adhesion assays in vitro, CD44v on human colon carcinoma cells binds to P-, E-, and L-selectin (189, 190). The majority of selectin ligands are presented on mucins, but they can be found equally functional also on N-linked glycans or glycolipids. Finally, P- and L-selectins also bind to heparin, heparan sulfate, and sulfated glycolipids, which also indicates certain flexibility in ligand recognition (9, 175). In addition, chondroitin sulfate glycosaminoglycans (CS-GAGs) on breast cancer cells were identified to serve as a P-selectin ligand that is associated with breast cancer metastasis (191). Despite the large variety of glycans, tumor cells express sialylated and fucosylated molecules, mostly on mucins which are also recognized by selectins (158, 159, 167, 192, 193).
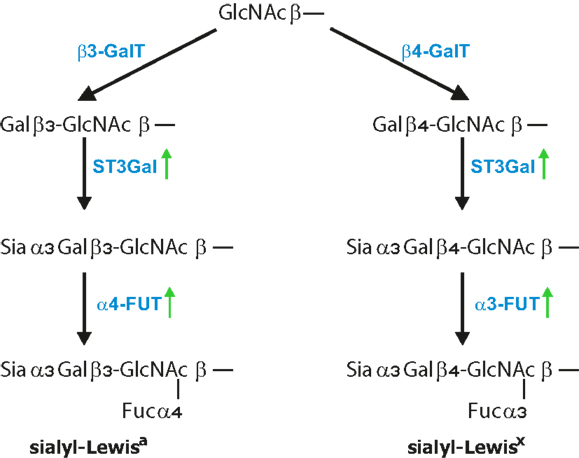
Figure 2. Formation of Lewis antigens. Terminal GlcNAc residues, particularly on core 2 structures, are further extended by addition of β1,4 galactose, for Lewisx epitope, and β1,3 galactose, for Lewisa epitope. This is further followed by the addition of α2,3-linked sialic acid to Gal by ST3Gal enzymes and finalized by the addition of α1,3-linked fucose for sLex and α1,4-linked fucose for the sLea antigen. FUT3 finalized the synthesis of Lea antigen, while FUT6 and FUT7 were shown to finalize Lex epitopes.
Increased expression of sLex/a in tumor cells has been attributed to elevated levels of α1,3-fucosyltransferase-7 (FUT7), which has also been shown to correspond with increased malignancy in lung cancer patients (161). In addition, overexpression of α1,3-fucosyltransferase-3 and -6 in metastatic prostate cancer cells correlated with higher sLex levels and more metastasis that was dependent on E-selectin-mediated recruitment to distant sites (169, 194). Genes encoding for FUT3, FUT4, and ST3GAL6 enzymes that are involved in sLex synthesis were significantly increased in breast cancers and correlated with metastasis to the bone where sLex receptor E-selectin is constitutively expressed (195). Inflammatory cytokines might also be involved in sLex production. TNF-α enhanced motility and invasion properties of prostatic cancer cells were associated with selective upregulation of genes related to sLex synthesis (196). Studies analyzing prostate and pancreatic cancer cell homing into bone showed that E-selectin-mediated adhesion is dependent on enhanced α1,3-fucosyltransferase, FUT3, FUT6, and FUT7 activity (197, 198). Consequently, down-regulation of α1,3-fucosyltransferase activity dramatically reduced prostate cancer incidence. However, there is also the possibility that selectin-mediated activation of either tumor cells or the tumor microenvironment further promote inflammation that is a hallmark of cancer progression.
P-Selectin
The association between circulating cancer cells, platelets, and formation of tumor microemboli is widely accepted (199–202). Many studies showed that platelets enhance hematogenous dissemination, intravascular tumor-cell survival, and metastasis (203–206). However, the major mechanism of platelet-adhesion to tumor cells has been found to be mediated by platelet P-selectin (6). Platelet-tumor cell interactions were significantly reduced in P-selectin deficient mice, and consequently attenuation of metastasis was observed. Enzymatic removal of carcinoma mucins carrying selectin ligands from tumor cells prior to tail vein injection resulted in attenuated metastasis comparable to the absence of P-selectin (158, 203). In addition, endothelial P-selectin-mediated interactions also contributed to metastasis indicating that both platelet and endothelial P-selectin promote early events during tissues colonization (11, 207). Another study shows that platelets promote lung metastasis of B16F1 melanoma and 4T1.2 breast cancer cells (208). Platelet depletion resulted in a significant reduction of lung metastasis when compared to NK cell depleted animals, indicating an additional pro-metastatic function of platelets. These findings are in agreement with a direct effect of platelet–tumor-cell interactions that promotes the metastatic behavior of tumor cells (209). Taken together, P-selectin-mediated interactions significantly contribute to the early steps of metastasis when tumor cells are in circulation.
L-Selectin
L-selectin binds to a variety of tumor cells and contributes to metastasis (167, 210). Intravenous injection of human and murine tumor cells in L-selectin deficient mice resulted in reduced recruitment of leukocytes and subsequently attenuated metastasis that confirmed the active role of L-selectin-mediated interaction in this process (11, 13). Metastasis was further attenuated in P- and L-selectin double deficient mice providing evidence that both selectins synergistically contribute to metastasis (11). In addition, the enhanced expression of selectin ligands around the metastatic tumor cells was detected with L-selectin chimera, which correlated with the recruitment of leukocytes (13). These findings indicated that L-selectin is either responsible for recruitment of leukocytes or their interactions within the metastatic microenvironment. Enhanced presence of inflammatory cells, primarily myeloid-derived cells, in the tumor microenvironment is usually associated with tumor growth and metastatic dissemination (211, 212). Thus, L-selectin represents a potential facilitator of myeloid cell recruitment to metastatic sites and thereby promotes early steps of metastasis, e.g., tumor-cell extravasation (13, 213). During inflammation, leukocyte interaction with the endothelium results in induced vascular permeability. However, whether L-selectin promotes metastasis through a direct engagement with selectin ligands on tumor cells or rather mimics inflammatory-like reaction accompanying the process of tumor-cell seeding in distant organs remains to be determined.
E-Selectin
E-selectin has been the first selectin intensively studied in context of metastasis (9, 10). The original hypothesis was that E-selectin mediates metastatic dissemination to distant organs through binding to ligands on tumor cells, similarly to leukocyte adhesion during inflammation (3). Numerous studies provided evidence that tumor cells expressing selectin ligands adhere to activated endothelium under flow condition in vitro (157, 168, 181). While different E-selectin ligands were linked to enhanced metastasis, the majority of them belong to the mucin type molecules. Despite the observation of increased primary tumor growth in selectin deficient mice, which seems to be linked to reduced anti-tumorigenic infiltration of immune cells (214), there is accumulating evidence that E-selectin promotes cancer metastasis in animal models. Enhanced E-selectin expression was observed in the liver during metastatic colonization and the down-regulation of E-selectin resulted in attenuation of metastasis (215, 216). Metastasis was redirected to the E-selectin overexpressing liver using experimental lung metastasis model, which provided direct evidence for involvement of E-selectin in facilitation of tumor-cell seeding (217). Accordingly, experimental liver metastasis of human colon carcinoma cells was also E-selectin-dependent (218). However, experimental lung metastasis of human colon adenocarcinoma cells remained unchanged in E-selectin deficient mice (219). On contrary, spontaneous metastasis of human breast cancer cells to the lungs was significantly attenuated in E-selectin-deficient mice (220). Interestingly, Hiratsuka et al. showed that factors secreted from primary tumors can activate endothelial focal adhesion kinase and E-selectin expression in the lung vasculature and thereby induce the formation of permissible sites for metastasis (221). Enhanced homing of metastatic tumor cells to these sites was observed and was associated with metastasis. These observations indicate that primary tumors can actively form a distant metastatic niche and upregulate expression of cell adhesion molecules involved in tumor cell-endothelial interactions. In conclusion, there is convincing evidence that endothelial E-selectin facilitates metastasis by enabling tumor-cell adhesion to vasculature. Nevertheless, the exact mechanism of E-selectin facilitation of metastasis remains to be defined.
Carcinoma Mucins as Initiators of Cancer-Related Prothrombotic Activity
Altered cancer glycosylation is not reflected only on cell-surface molecules, but aberrantly glycosylated proteins are detected in the circulation (26). Antibodies raised against tumor cells, were shown to specifically recognize glycan structures, e.g., sLea, which are currently used for cancer diagnostics (45). The presence of carcinoma mucins (e.g., CA-125, CA19-9), which are shedded from tumors, are routinely used as serum tumor markers in diagnosis of cancer. Besides, efficient binding of recombinant soluble selectin to carcinoma mucins has been observed (158, 222). Increased thromboembolism is a recognized complication in various carcinomas, particularly mucinous carcinomas, however; there are several pathologic mechanisms likely to be involved (7). Idiopathic thromboembolism, which is frequently associated with occult carcinomas, belongs to the Trousseau syndrome. Recent studies provided evidence that intravenous injection of carcinoma mucins carrying selectin ligands into mice resulted in generation of platelet-rich microthrombi (222). This pathology was markedly diminished in P-selectin or L-selectin deficient mice. Interestingly, carcinoma mucins could not activate platelets and thereby could not generate microthrombi in mice lacking PSGL-1 (223). Carcinoma mucins initiated thrombosis only in the presence of platelets that induced release of cathepsin G from neutrophils through a selectin-dependent, reciprocal activation of neutrophils and platelets. Taken together, carcinoma mucins carrying selectin ligands in blood circulation may serve as initiators of thrombi formation observed in cancer patients.
Selectins Shape the Metastatic Microenvironment
There is accumulating evidence that selectins facilitate heterotypic interactions between tumor cells and blood components, including the endothelium and thereby promote tumor-cell seeding, survival and extravasation (8, 9, 224). When circulating tumor cells arrest in the microvasculature of distant organs, early on markers of endothelial cell activation and inflammation, including E-selectin, were upregulated in experimental lung and liver metastasis models (219, 225–228). Enhanced E-selectin expression was detected also in the metastatic lungs using a spontaneous metastatic model with Lewis lung carcinoma (219, 221). Consequently, inhibition of endothelial activation and/or E-selectin function attenuated metastasis (227, 229). Endothelial activation caused by factors derived from primary tumor or from arrested tumors in the vasculature promoted selectin-mediated interactions and formation of a permissive microenvironment within the vasculature prior to tumor-cell extravasation (11, 13, 213). Tumor-cell glycan-induced and P-selectin-dependent endothelial activation resulted in enhanced expression of E-selectin and vascular cell adhesion molecule 1 (VCAM-1) and promoted lung colonization and metastasis (213). In addition, elevated production of chemokine CCL5 contributed to the recruitment of monocytes. Accordingly, endothelial VCAM-1 expression was induced by tumor-cell embolus that resulted in increased recruitment of myeloid cells supporting metastasis (225). Recruitment of inflammatory cells, especially myeloid-derived cells, is strongly associated with enhanced metastatic colonization that is at least partially dependent on L-selectin (12, 13, 213, 230–232). Taken together, the selectin-mediated interactions play a critical role during the establishment of metastasis that is co-initiated by aberrant glycans on tumor cells in circulation. Whether tumor glycans only initiate the inflammatory-like cascade leading to metastasis or have further function in shaping this process remains to be defined.
Conclusion and Perspectives
Cancer-associated aberrant glycosylation has been identified in virtually every type of cancer. Expression of cancer-specific glycan epitopes represents a great opportunity to explore them for diagnostics and potentially specific targeting of tumors. Considering that genes only indirectly regulate glycan formation, it is still puzzling that glycan epitopes have been consistently validated as cancer markers. Based on the broad expression and high specificity for cancer tissues, T antigen is currently explored as a potential target for the development of cancer diagnostics and immunotherapeutics (16, 233). Since the expression of sTn antigens on the majority of tumors correlated with poor prognosis, the sTn antigen has become a target for cancer vaccine (58, 61). Administration of sTn disaccharide conjugate to highly immunogenic protein induced antibodies against sTn and showed protective effects in a mouse model of breast cancer (234). Although a randomized phase III clinical trial using the same sTn vaccine did not improve overall survival, patients with high titer against the sTn had significantly prolonged overall survival (235).
The accumulating knowledge about the function of lectin–tumor-cell glycan interactions in cancer will open ways for new approaches to interfere with cancer progression. However, the exploitation of such therapeutic opportunities requires a comprehensive knowledge about the underlying mechanisms of lectin-mediated interactions. Nevertheless, the role of selectins in cancer progression has been extensively investigated in number of preclinical models and the mechanism at least partially characterized (9). Clearly, further studies in the exact mechanism of action are still required, but selectin inhibition in cancer has been inadvertently clinically tested in cancer patients treated with antithrombotic therapies (236). Unfractionated heparin as well as low molecular weight heparin has a strong P- and L-selectin inhibitory activity at clinically relevant concentrations. Retrospective analysis of clinical studies revealed that apart from antithrombotic activity, heparin improved survival of cancer patients especially in patients with early stage disease. Still, prospective and well-designed clinical study remains to be performed. Similarly, development of highly specific ligand probes for siglecs (e.g., Siglec-2) revealed the ability to target siglec-expressing cells (94). Further investigations are required for deciding whether glycan-specific targeting of lectins involved in cancer modulation (e.g., siglec, selectins, or galectins) or rather development of glycan-specific targeting of tumor cells represents the right approach for the treatment of cancer. The cell-surface presentation of unique glycan epitopes makes them an “ideal” candidate for targeting since they are both specific and therapeutically accessible. Future studies need to validate the therapeutic potential in clinically relevant experimental models prior to clinical evaluation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from Swiss National Foundation #31003A-133025 to Lubor Borsig.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Hakomori S. Aberrant glycosylation in cancer cell membranes as focused on glycolipids: overview and perspectives. Cancer Res (1985) 45:2405–14.
3. Kannagi R. Carbohydrate-mediated cell adhesion involved in hematogenous metastasis of cancer. Glycoconj J (1997) 14:577–84. doi:10.1023/A:1018532409041
4. Kim YJ, Varki A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J (1997) 14:569–76. doi:10.1023/A:1018580324971
5. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer (2005) 5:526–42. doi:10.1038/nrc1649
6. Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci U S A (1998) 95:9325–30. doi:10.1073/pnas.95.16.9325
7. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood (2007) 110:1723–9. doi:10.1182/blood-2006-10-053736
8. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov (2012) 2:1091–9. doi:10.1158/2159-8290.CD-12-0329
9. Läubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol (2010) 20:169–77. doi:10.1016/j.semcancer.2010.04.005
10. Witz IP. The selectin-selectin ligand axis in tumor progression. Cancer Metastasis Rev (2008) 27:19–30. doi:10.1007/s10555-007-9101-z
11. Borsig L, Wong R, Hynes RO, Varki NM, Varki A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc Natl Acad Sci U S A (2002) 99:2193–8. doi:10.1073/pnas.261704098
12. Hoos A, Protsyuk D, Borsig L. Metastatic growth progression caused by PSGL-1-mediated recruitment of monocytes to metastatic sites. Cancer Res (2014) 74:695–704. doi:10.1158/0008-5472.CAN-13-0946
13. Läubli H, Stevenson JL, Varki A, Varki NM, Borsig L. L-selectin facilitation of metastasis involves temporal induction of fut7-dependent ligands at sites of tumor cell arrest. Cancer Res (2006) 66:1536–42. doi:10.1158/0008-5472.CAN-05-3121
14. Belisle JA, Horibata S, Jennifer GA, Petrie S, Kapur A, Andre S, et al. Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Mol Cancer (2010) 9:118. doi:10.1186/1476-4598-9-118
15. Miyazaki K, Sakuma K, Kawamura YI, Izawa M, Ohmori K, Mitsuki M, et al. Colonic epithelial cells express specific ligands for mucosal macrophage immunosuppressive receptors siglec-7 and -9. J Immunol (2012) 188:4690–700. doi:10.4049/jimmunol.1100605
16. Ito K, Stannard K, Gabutero E, Clark AM, Neo SY, Onturk S, et al. Galectin-1 as a potent target for cancer therapy: role in the tumor microenvironment. Cancer Metastasis Rev (2012) 31:763–78. doi:10.1007/s10555-012-9388-2
17. Dennis JW, Laferte S, Waghorne C, Breitman ML, Kerbel RS. Beta 1-6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science (1987) 236:582–5. doi:10.1126/science.2953071
18. Lau KS, Dennis JW. N-Glycans in cancer progression. Glycobiology (2008) 18:750–60. doi:10.1093/glycob/cwn071
19. Guo HB, Randolph M, Pierce M. Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J Biol Chem (2007) 282:22150–62. doi:10.1074/jbc.M611518200
20. Lagana A, Goetz JG, Cheung P, Raz A, Dennis JW, Nabi IR. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol Cell Biol (2006) 26:3181–93. doi:10.1128/MCB.26.8.3181-3193.2006
21. Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med (2000) 6:306–12. doi:10.1038/73163
22. Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science (2004) 306:120–4. doi:10.1126/science.1102109
23. Borsig L. Glycans in cancer. In: Pavao MS, editor. Biology of Extracellular Matrix. Glycans in Disease and Therapeutics. Berlin: Springer Verlag (2011). p. 63–81.
24. Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol (2013) 10:607–20. doi:10.1038/nrgastro.2013.120
25. Dall’Olio F, Malagolini N, Trinchera M, Chiricolo M. Mechanisms of cancer-associated glycosylation changes. Front Biosci (Landmark Ed) (2012) 17:670–99. doi:10.2741/3951
26. Kannagi R, Sakuma K, Miyazaki K, Lim KT, Yusa A, Yin J, et al. Altered expression of glycan genes in cancers induced by epigenetic silencing and tumor hypoxia: clues in the ongoing search for new tumor markers. Cancer Sci (2010) 101:586–93. doi:10.1111/j.1349-7006.2009.01455.x
27. Koike T, Kimura N, Miyazaki K, Yabuta T, Kumamoto K, Takenoshita S, et al. Hypoxia induces adhesion molecules on cancer cells: a missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc Natl Acad Sci U S A (2004) 101:8132–7. doi:10.1073/pnas.0402088101
28. Schultz MJ, Swindall AF, Bellis SL. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev (2012) 31:501–18. doi:10.1007/s10555-012-9359-7
29. Varki NM, Varki A. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab Invest (2007) 87:851–7. doi:10.1038/labinvest.3700656
30. Dall’Olio F, Malagolini N, di Stefano G, Minni F, Marrano D, Serafini-Cessi F. Increased CMP-NeuAc:Gal beta 1,4GlcNAc-R alpha 2,6 sialyltransferase activity in human colorectal cancer tissues. Int J Cancer (1989) 44:434–9. doi:10.1002/ijc.2910440309
31. Gessner P, Riedl S, Quentmaier A, Kemmner W. Enhanced activity of CMP-neuAc:Gal beta 1-4GlcNAc:alpha 2,6-sialyltransferase in metastasizing human colorectal tumor tissue and serum of tumor patients. Cancer Lett (1993) 75:143–9. doi:10.1016/0304-3835(93)90056-F
32. Seales EC, Jurado GA, Brunson BA, Wakefield JK, Frost AR, Bellis SL. Hypersialylation of beta1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res (2005) 65:4645–52. doi:10.1158/0008-5472.CAN-04-3117
33. Marcos NT, Bennett EP, Gomes J, Magalhaes A, Gomes C, David L, et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front Biosci (Elite Ed) (2011) 3:1443–55.
34. Uemura T, Shiozaki K, Yamaguchi K, Miyazaki S, Satomi S, Kato K, et al. Contribution of sialidase NEU1 to suppression of metastasis of human colon cancer cells through desialylation of integrin beta4. Oncogene (2009) 28:1218–29. doi:10.1038/onc.2008.471
35. Baldus SE, Monig SP, Hanisch FG, Zirbes TK, Flucke U, Oelert S, et al. Comparative evaluation of the prognostic value of MUC1, MUC2, sialyl-Lewis(a) and sialyl-Lewis(x) antigens in colorectal adenocarcinoma. Histopathology (2002) 40:440–9. doi:10.1046/j.1365-2559.2002.01389.x
36. Campbell BJ, Finnie IA, Hounsell EF, Rhodes JM. Direct demonstration of increased expression of Thomsen-Friedenreich (TF) antigen in colonic adenocarcinoma and ulcerative colitis mucin and its concealment in normal mucin. J Clin Invest (1995) 95:571–6. doi:10.1172/JCI117700
37. Kumar SR, Sauter ER, Quinn TP, Deutscher SL. Thomsen-Friedenreich and Tn antigens in nipple fluid: carbohydrate biomarkers for breast cancer detection. Clin Cancer Res (2005) 11:6868–71. doi:10.1158/1078-0432.CCR-05-0146
38. Sotozono MA, Okada Y, Tsuji T. The Thomsen-Friedenreich antigen-related carbohydrate antigens in human gastric intestinal metaplasia and cancer. J Histochem Cytochem (1994) 42:1575–84. doi:10.1177/42.12.7527063
39. Springer GF. T and Tn, general carcinoma autoantigens. Science (1984) 224:1198–206. doi:10.1126/science.6729450
40. Ju T, Lanneau GS, Gautam T, Wang Y, Xia B, Stowell SR, et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res (2008) 68:1636–46. doi:10.1158/0008-5472.CAN-07-2345
41. Miyazaki K, Ohmori K, Izawa M, Koike T, Kumamoto K, Furukawa K, et al. Loss of disialyl Lewis(a), the ligand for lymphocyte inhibitory receptor sialic acid-binding immunoglobulin-like lectin-7 (Siglec-7) associated with increased sialyl Lewis(a) expression on human colon cancers. Cancer Res (2004) 64:4498–505. doi:10.1158/0008-5472.CAN-03-3614
42. Tsuchida A, Okajima T, Furukawa K, Ando T, Ishida H, Yoshida A, et al. Synthesis of disialyl Lewis a (Le(a)) structure in colon cancer cell lines by a sialyltransferase, ST6GalNAc VI, responsible for the synthesis of alpha-series gangliosides. J Biol Chem (2003) 278:22787–94. doi:10.1074/jbc.M211034200
43. Nudelman E, Fukushi Y, Levery SB, Higuchi T, Hakomori S. Novel fucolipids of human adenocarcinoma: disialosyl Lea antigen (III4FucIII6NeuAcI V3NeuAcLc4) of human colonic adenocarcinoma and the monoclonal antibody (FH7) defining this structure. J Biol Chem (1986) 261:5487–95.
44. Sawada M, Moriya S, Saito S, Shineha R, Satomi S, Yamori T, et al. Reduced sialidase expression in highly metastatic variants of mouse colon adenocarcinoma 26 and retardation of their metastatic ability by sialidase overexpression. Int J Cancer (2002) 97:180–5. doi:10.1002/ijc.1598
45. Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer (2004) 4:45–60. doi:10.1038/nrc1251
46. Brockhausen I. Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep (2006) 7:599–604. doi:10.1038/sj.embor.7400705
47. Taylor-Papadimitriou J, Burchell J, Miles DW, Dalziel M. MUC1 and cancer. Biochim Biophys Acta (1999) 1455:301–13. doi:10.1016/S0925-4439(99)00055-1
48. Iwai T, Kudo T, Kawamoto R, Kubota T, Togayachi A, Hiruma T, et al. Core 3 synthase is down-regulated in colon carcinoma and profoundly suppresses the metastatic potential of carcinoma cells. Proc Natl Acad Sci U S A (2005) 102:4572–7. doi:10.1073/pnas.0407983102
49. Radhakrishnan P, Grandgenett PM, Mohr AM, Bunt SK, Yu F, Chowdhury S, et al. Expression of core 3 synthase in human pancreatic cancer cells suppresses tumor growth and metastasis. Int J Cancer (2013) 133:2824–33. doi:10.1002/ijc.28322
50. Machida E, Nakayama J, Amano J, Fukuda M. Clinicopathological significance of core 2 beta1,6-N-acetylglucosaminyltransferase messenger RNA expressed in the pulmonary adenocarcinoma determined by in situ hybridization. Cancer Res (2001) 61:2226–31.
51. Shimodaira K, Nakayama J, Nakamura N, Hasebe O, Katsuyama T, Fukuda M. Carcinoma-associated expression of core 2 beta-1,6-N-acetylglucosaminyltransferase gene in human colorectal cancer: role of O-glycans in tumor progression. Cancer Res (1997) 57:5201–6.
52. St Hill CA, Farooqui M, Mitcheltree G, Gulbahce HE, Jessurun J, Cao Q, et al. The high affinity selectin glycan ligand C2-O-sLex and mRNA transcripts of the core 2 beta-1,6-N-acetylglucosaminyltransferase (C2GnT1) gene are highly expressed in human colorectal adenocarcinomas. BMC Cancer (2009) 9:79. doi:10.1186/1471-2407-9-79
53. Brockhausen I, Yang JM, Burchell J, Whitehouse C, Taylor-Papadimitriou J. Mechanisms underlying aberrant glycosylation of MUC1 mucin in breast cancer cells. Eur J Biochem (1995) 233:607–17. doi:10.1111/j.1432-1033.1995.607_2.x
54. Burchell JM, Mungul A, Taylor-Papadimitriou J. O-linked glycosylation in the mammary gland: changes that occur during malignancy. J Mammary Gland Biol Neoplasia (2001) 6:355–64. doi:10.1023/A:1011331809881
55. Dalziel M, Whitehouse C, McFarlane I, Brockhausen I, Gschmeissner S, Schwientek T, et al. The relative activities of the C2GnT1 and ST3Gal-I glycosyltransferases determine O-glycan structure and expression of a tumor-associated epitope on MUC1. J Biol Chem (2001) 276:11007–15. doi:10.1074/jbc.M006523200
56. Solatycka A, Owczarek T, Piller F, Piller V, Pula B, Wojciech L, et al. MUC1 in human and murine mammary carcinoma cells decreases the expression of core 2 beta1,6-N-acetylglucosaminyltransferase and beta-galactoside alpha2,3-sialyltransferase. Glycobiology (2012) 22:1042–54. doi:10.1093/glycob/cws075
57. Matsuura N, Narita T, Hiraiwa N, Hiraiwa M, Murai H, Iwase T, et al. Gene expression of fucosyl- and sialyl-transferases which synthesize sialyl Lewisx, the carbohydrate ligands for E-selectin, in human breast cancer. Int J Oncol (1998) 12:1157–64.
58. Cao Y, Stosiek P, Springer GF, Karsten U. Thomsen-Friedenreich-related carbohydrate antigens in normal adult human tissues: a systematic and comparative study. Histochem Cell Biol (1996) 106:197–207. doi:10.1007/BF02484401
59. Coon JS, Weinstein RS, Summers JL. Blood group precursor T-antigen expression in human urinary bladder carcinoma. Am J Clin Pathol (1982) 77:692–9.
60. Ghazizadeh M, Oguro T, Sasaki Y, Aihara K, Araki T, Springer GF. Immunohistochemical and ultrastructural localization of T antigen in ovarian tumors. Am J Clin Pathol (1990) 93:315–21.
61. Itzkowitz SH, Yuan M, Montgomery CK, Kjeldsen T, Takahashi HK, Bigbee WL, et al. Expression of Tn, sialosyl-Tn, and T antigens in human colon cancer. Cancer Res (1989) 49:197–204.
62. Limas C, Lange P. T-antigen in normal and neoplastic urothelium. Cancer (1986) 58:1236–45. doi:10.1002/1097-0142(19860915)58:6<1236::AID-CNCR2820580611>3.0.CO;2-I
63. Zhang S, Zhang HS, Cordon-Cardo C, Reuter VE, Singhal AK, Lloyd KO, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: II. Blood group-related antigens. Int J Cancer (1997) 73:50–6. doi:10.1002/(SICI)1097-0215(19970926)73:1<42::AID-IJC8>3.0.CO;2-1
64. Davidson B, Gotlieb WH, Ben-Baruch G, Kopolovic J, Goldberg I, Nesland JM, et al. Expression of carbohydrate antigens in advanced-stage ovarian carcinomas and their metastases-A clinicopathologic study. Gynecol Oncol (2000) 77:35–43. doi:10.1006/gyno.1999.5708
65. Devine PL, McKenzie IF. Mucins: structure, function, and associations with malignancy. Bioessays (1992) 14:619–25. doi:10.1002/bies.950140909
66. Yu LG. The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj J (2007) 24:411–20. doi:10.1007/s10719-007-9034-3
67. Julien S, Krzewinski-Recchi MA, Harduin-Lepers A, Gouyer V, Huet G, Le Bourhis X, et al. Expression of sialyl-Tn antigen in breast cancer cells transfected with the human CMP-Neu5Ac: GalNAc alpha2,6-sialyltransferase (ST6GalNac I) cDNA. Glycoconj J (2001) 18:883–93. doi:10.1023/A:1022200525695
68. Ozaki H, Matsuzaki H, Ando H, Kaji H, Nakanishi H, Ikehara Y, et al. Enhancement of metastatic ability by ectopic expression of ST6GalNAcI on a gastric cancer cell line in a mouse model. Clin Exp Metastasis (2012) 29:229–38. doi:10.1007/s10585-011-9445-1
69. Yang JM, Byrd JC, Siddiki BB, Chung YS, Okuno M, Sowa M, et al. Alterations of O-glycan biosynthesis in human colon cancer tissues. Glycobiology (1994) 4:873–84. doi:10.1093/glycob/4.6.873
70. Brockhausen I. Pathways of O-glycan biosynthesis in cancer cells. Biochim Biophys Acta (1999) 1473:67–95. doi:10.1016/S0304-4165(99)00170-1
71. Marcos NT, Pinho S, Grandela C, Cruz A, Samyn-Petit B, Harduin-Lepers A, et al. Role of the human ST6GalNAc-I and ST6GalNAc-II in the synthesis of the cancer-associated sialyl-Tn antigen. Cancer Res (2004) 64:7050–7. doi:10.1158/0008-5472.CAN-04-1921
72. Schneider F, Kemmner W, Haensch W, Franke G, Gretschel S, Karsten U, et al. Overexpression of sialyltransferase CMP-sialic acid:Galbeta1,3GalNAc-R alpha6-Sialyltransferase is related to poor patient survival in human colorectal carcinomas. Cancer Res (2001) 61:4605–11.
73. Kumamoto K, Goto Y, Sekikawa K, Takenoshita S, Ishida N, Kawakita M, et al. Increased expression of UDP-galactose transporter messenger RNA in human colon cancer tissues and its implication in synthesis of Thomsen-Friedenreich antigen and Sialyl Lewis A/X determinants. Cancer Res (2001) 61:4620–7.
74. Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A (2002) 99:16613–8. doi:10.1073/pnas.262438199
75. Schietinger A, Philip M, Yoshida BA, Azadi P, Liu H, Meredith SC, et al. A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science (2006) 314:304–8. doi:10.1126/science.1129200
76. Barrow H, Tam B, Duckworth CA, Rhodes JM, Yu LG. Suppression of core 1 Gal-transferase is associated with reduction of TF and reciprocal increase of Tn, sialyl-Tn and Core 3 glycans in human colon cancer cells. PLoS One (2013) 8:e59792. doi:10.1371/journal.pone.0059792
77. Julien S, Adriaenssens E, Ottenberg K, Furlan A, Courtand G, Vercoutter-Edouart AS, et al. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology (2006) 16:54–64. doi:10.1093/glycob/cwj033
78. Burchell J, Poulsom R, Hanby A, Whitehouse C, Cooper L, Clausen H, et al. An alpha2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology (1999) 9:1307–11. doi:10.1093/glycob/9.12.1307
79. Picco G, Julien S, Brockhausen I, Beatson R, Antonopoulos A, Haslam S, et al. Over-expression of ST3Gal-I promotes mammary tumorigenesis. Glycobiology (2010) 20:1241–50. doi:10.1093/glycob/cwq085
80. Baldus SE, Hanisch FG, Kotlarek GM, Zirbes TK, Thiele J, Isenberg J, et al. Coexpression of MUC1 mucin peptide core and the Thomsen-Friedenreich antigen in colorectal neoplasms. Cancer (1998) 82:1019–27. doi:10.1002/(SICI)1097-0142(19980315)82:6<1019::AID-CNCR3>3.0.CO;2-9
81. Burdick MD, Harris A, Reid CJ, Iwamura T, Hollingsworth MA. Oligosaccharides expressed on MUC1 produced by pancreatic and colon tumor cell lines. J Biol Chem (1997) 272:24198–202. doi:10.1074/jbc.272.39.24198
82. Singh R, Campbell BJ, Yu LG, Fernig DG, Milton JD, Goodlad RA, et al. Cell surface-expressed Thomsen-Friedenreich antigen in colon cancer is predominantly carried on high molecular weight splice variants of CD44. Glycobiology (2001) 11:587–92. doi:10.1093/glycob/11.7.587
83. Storr SJ, Royle L, Chapman CJ, Hamid UM, Robertson JF, Murray A, et al. The O-linked glycosylation of secretory/shed MUC1 from an advanced breast cancer patient’s serum. Glycobiology (2008) 18:456–62. doi:10.1093/glycob/cwn022
84. Conze T, Carvalho AS, Landegren U, Almeida R, Reis CA, David L, et al. MUC2 mucin is a major carrier of the cancer-associated sialyl-Tn antigen in intestinal metaplasia and gastric carcinomas. Glycobiology (2010) 20:199–206. doi:10.1093/glycob/cwp161
85. Hofmann M, Rudy W, Zoller M, Tolg C, Ponta H, Herrlich P, et al. CD44 splice variants confer metastatic behavior in rats: homologous sequences are expressed in human tumor cell lines. Cancer Res (1991) 51:5292–7.
86. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol (2003) 4:33–45. doi:10.1038/nrm1004
87. Wai PY, Kuo PC. The role of Osteopontin in tumor metastasis. J Surg Res (2004) 121:228–41. doi:10.1016/j.jss.2004.03.028
88. Bresalier RS, Niv Y, Byrd JC, Duh QY, Toribara NW, Rockwell RW, et al. Mucin production by human colonic carcinoma cells correlates with their metastatic potential in animal models of colon cancer metastasis. J Clin Invest (1991) 87:1037–45. doi:10.1172/JCI115063
89. Nakamori S, Kameyama M, Imaoka S, Furukawa H, Ishikawa O, Sasaki Y, et al. Increased expression of sialyl Lewisx antigen correlates with poor survival in patients with colorectal carcinoma: clinicopathological and immunohistochemical study. Cancer Res (1993) 53:3632–7.
90. Clement M, Rocher J, Loirand G, Le Pendu J. Expression of sialyl-Tn epitopes on beta1 integrin alters epithelial cell phenotype, proliferation and haptotaxis. J Cell Sci (2004) 117:5059–69. doi:10.1242/jcs.01350
91. Matsumoto Y, Zhang Q, Akita K, Nakada H, Hamamura K, Tokuda N, et al. pp-GalNAc-T13 induces high metastatic potential of murine Lewis lung cancer by generating trimeric Tn antigen. Biochem Biophys Res Commun (2012) 419:7–13. doi:10.1016/j.bbrc.2012.01.086
92. Ho WL, Che MI, Chou CH, Chang HH, Jeng YM, Hsu WM, et al. B3GNT3 expression suppresses cell migration and invasion and predicts favorable outcomes in neuroblastoma. Cancer Sci (2013) 104:1600–8. doi:10.1111/cas.12294
93. Crocker PR, McMillan SJ, Richards HE. CD33-related siglecs as potential modulators of inflammatory responses. Ann N Y Acad Sci (2012) 1253:102–11. doi:10.1111/j.1749-6632.2011.06449.x
94. O’Reilly MK, Paulson JC. Siglecs as targets for therapy in immune-cell-mediated disease. Trends Pharmacol Sci (2009) 30:240–8. doi:10.1016/j.tips.2009.02.005
95. Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci (2012) 1253:16–36. doi:10.1111/j.1749-6632.2012.06517.x
96. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol (2007) 7:255–66. doi:10.1038/nri2056
97. Avril T, Attrill H, Zhang J, Raper A, Crocker PR. Negative regulation of leucocyte functions by CD33-related siglecs. Biochem Soc Trans (2006) 34:1024–7. doi:10.1042/BST0341024
98. Lock K, Zhang J, Lu J, Lee SH, Crocker PR. Expression of CD33-related siglecs on human mononuclear phagocytes, monocyte-derived dendritic cells and plasmacytoid dendritic cells. Immunobiology (2004) 209:199–207. doi:10.1016/j.imbio.2004.04.007
99. Nutku E, Aizawa H, Hudson SA, Bochner BS. Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood (2003) 101:5014–20. doi:10.1182/blood-2002-10-3058
100. Rabinovich GA, van Kooyk Y, Cobb BA. Glycobiology of immune responses. Ann N Y Acad Sci (2012) 1253:1–15. doi:10.1111/j.1749-6632.2012.06492.x
101. Takamiya R, Ohtsubo K, Takamatsu S, Taniguchi N, Angata T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-beta secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology (2013) 23:178–87. doi:10.1093/glycob/cws139
102. Crocker PR, Gordon S. Properties and distribution of a lectin-like hemagglutinin differentially expressed by murine stromal tissue macrophages. J Exp Med (1986) 164:1862–75. doi:10.1084/jem.164.6.1862
103. Marmey B, Boix C, Barbaroux JB, Dieu-Nosjean MC, Diebold J, Audouin J, et al. CD14 and CD169 expression in human lymph nodes and spleen: specific expansion of CD14+CD169− monocyte-derived cells in diffuse large B-cell lymphomas. Hum Pathol (2006) 37:68–77. doi:10.1016/j.humpath.2005.09.016
104. Nath D, Hartnell A, Happerfield L, Miles DW, Burchell J, Taylor-Papadimitriou J, et al. Macrophage-tumour cell interactions: identification of MUC1 on breast cancer cells as a potential counter-receptor for the macrophage-restricted receptor, sialoadhesin. Immunology (1999) 98:213–9. doi:10.1046/j.1365-2567.1999.00827.x
105. Yamashiro S, Takeya M, Nishi T, Kuratsu J, Yoshimura T, Ushio Y, et al. Tumor-derived monocyte chemoattractant protein-1 induces intratumoral infiltration of monocyte-derived macrophage subpopulation in transplanted rat tumors. Am J Pathol (1994) 145:856–67.
106. Ohnishi K, Komohara Y, Saito Y, Miyamoto Y, Watanabe M, Baba H, et al. CD169-positive macrophages in regional lymph nodes are associated with a favorable prognosis in patients with colorectal carcinoma. Cancer Sci (2013) 104:1237–44. doi:10.1111/cas.12212
107. Buller RE, Berman ML, Bloss JD, Manetta A, DiSaia PJ. Serum CA125 regression in epithelial ovarian cancer: correlation with reassessment findings and survival. Gynecol Oncol (1992) 47:87–92. doi:10.1016/0090-8258(92)90082-T
108. Ando M, Tu W, Nishijima K, Iijima S. Siglec-9 enhances IL-10 production in macrophages via tyrosine-based motifs. Biochem Biophys Res Commun (2008) 369:878–83. doi:10.1016/j.bbrc.2008.02.111
109. Kawasaki Y, Ito A, Withers DA, Taima T, Kakoi N, Saito S, et al. Ganglioside DSGb5, preferred ligand for Siglec-7, inhibits NK cell cytotoxicity against renal cell carcinoma cells. Glycobiology (2010) 20:1373–9. doi:10.1093/glycob/cwq116
110. Hudak JE, Canham SM, Bertozzi CR. Glygocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol (2014) 10:69–75. doi:10.1038/nchembio.1388
111. Tanida S, Akita K, Ishida A, Mori Y, Toda M, Inoue M, et al. Binding of the sialic acid-binding lectin. J Biol Chem (2013) 288:31842–52. doi:10.1074/jbc.M113.471318
112. Ball ED. In vitro purging of bone marrow for autologous marrow transplantation in acute myelogenous leukemia using myeloid-specific monoclonal antibodies. Bone Marrow Transplant (1988) 3:387–92.
113. Drexler HG. Classification of acute myeloid leukemias – a comparison of FAB and immunophenotyping. Leukemia (1987) 1:697–705.
114. Ziegler-Heitbrock HW, Munker R, Dorken B, Gaedicke G, Thiel E. Induction of features characteristic of hairy cell leukemia in chronic lymphocytic leukemia and prolymphocytic leukemia cells. Cancer Res (1986) 46:2172–8.
115. Jandus C, Simon HU, von Gunten S. Targeting siglecs – a novel pharmacological strategy for immuno- and glycotherapy. Biochem Pharmacol (2011) 82:323–32. doi:10.1016/j.bcp.2011.05.018
116. Jain N, O Brien S, Thomas D, Kantarjian H. Inotuzumab ozogamicin in the treatment of acute lymphoblastic leukemia. Front Biosci (Elite Ed) (2014) 6:40–5. doi:10.2741/E688
117. Kantarjian H, Thomas D, Jorgensen J, Kebriaei P, Jabbour E, Rytting M, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer (2013) 119:2728–36. doi:10.1002/cncr.28136
118. Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood (2013) 121:4854–60. doi:10.1182/blood-2013-01-466706
119. Gasiorowski RE, Clark GJ, Bradstock K, Hart DN. Antibody therapy for acute myeloid leukaemia. Br J Haematol (2013) 164:481–75. doi:10.1111/bjh.12691
120. Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood (2013) 122:1455–63. doi:10.1182/blood-2013-03-491506
121. Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol (2009) 9:338–52. doi:10.1038/nri2536
122. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer (2005) 5:29–41. doi:10.1038/nrc1527
123. Califice S, Castronovo V, Van Den Brule F. Galectin-3 and cancer (Review). Int J Oncol (2004) 25:983–92.
124. Takenaka Y, Fukumori T, Raz A. Galectin-3 and metastasis. Glycoconj J (2004) 19:543–9. doi:10.1023/B:GLYC.0000014084.01324.15
125. Banh A, Zhang J, Cao H, Bouley DM, Kwok S, Kong C, et al. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res (2011) 71:4423–31. doi:10.1158/0008-5472.CAN-10-4157
126. Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; a potential mechanism of tumor-immune privilege. Cancer Cell (2004) 5:241–51. doi:10.1016/S1535-6108(04)00024-8
127. Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer (2012) 130:2337–48. doi:10.1002/ijc.26290
128. Hernandez JD, Nguyen JT, He J, Wang W, Ardman B, Green JM, et al. Galectin-1 binds different CD43 glycoforms to cluster CD43 and regulate T cell death. J Immunol (2006) 177:5328–36.
129. Nguyen JT, Evans DP, Galvan M, Pace KE, Leitenberg D, Bui TN, et al. CD45 modulates galectin-1-induced T cell death: regulation by expression of core 2 O-glycans. J Immunol (2001) 167:5697–707.
130. Amano M, Galvan M, He J, Baum LG. The ST6Gal I sialyltransferase selectively modifies N-glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J Biol Chem (2003) 278:7469–75. doi:10.1074/jbc.M209595200
131. Kuo PL, Hung JY, Huang SK, Chou SH, Cheng DE, Jong YJ, et al. Lung cancer-derived galectin-1 mediates dendritic cell anergy through inhibitor of DNA binding 3/IL-10 signaling pathway. J Immunol (2011) 186:1521–30. doi:10.4049/jimmunol.1002940
132. Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Mendez-Huergo SP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res (2013) 73:1107–17. doi:10.1158/0008-5472.CAN-12-2418
133. Perillo NL, Marcus ME, Baum LG. Galectins: versatile modulators of cell adhesion, cell proliferation, and cell death. J Mol Med (Berl) (1998) 76:402–12. doi:10.1007/s001090050232
134. van den Brule F, Califice S, Garnier F, Fernandez PL, Berchuck A, Castronovo V. Galectin-1 accumulation in the ovary carcinoma peritumoral stroma is induced by ovary carcinoma cells and affects both cancer cell proliferation and adhesion to laminin-1 and fibronectin. Lab Invest (2003) 83:377–86. doi:10.1097/01.LAB.0000059949.01480.40
135. Pacienza N, Pozner RG, Bianco GA, D’Atri LP, Croci DO, Negrotto S, et al. The immunoregulatory glycan-binding protein galectin-1 triggers human platelet activation. FASEB J (2008) 22:1113–23. doi:10.1096/fj.07-9524com
136. Iurisci I, Tinari N, Natoli C, Angelucci D, Cianchetti E, Iacobelli S. Concentrations of galectin-3 in the sera of normal controls and cancer patients. Clin Cancer Res (2000) 6:1389–93.
137. Vereecken P, Zouaoui Boudjeltia K, Debray C, Awada A, Legssyer I, Sales F, et al. High serum galectin-3 in advanced melanoma: preliminary results. Clin Exp Dermatol (2006) 31:105–9. doi:10.1111/j.1365-2230.2005.01992.x
138. Khaldoyanidi SK, Glinsky VV, Sikora L, Glinskii AB, Mossine VV, Quinn TP, et al. MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J Biol Chem (2003) 278:4127–34.
139. Zhao Q, Barclay M, Hilkens J, Guo X, Barrow H, Rhodes JM, et al. Interaction between circulating galectin-3 and cancer-associated MUC1 enhances tumour cell homotypic aggregation and prevents anoikis. Mol Cancer (2010) 9:154. doi:10.1186/1476-4598-9-154
140. Glinsky VV, Glinsky GV, Rittenhouse-Olson K, Huflejt ME, Glinskii OV, Deutscher SL, et al. The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res (2001) 61:4851–7.
141. Krishnan V, Bane SM, Kawle PD, Naresh KN, Kalraiya RD. Altered melanoma cell surface glycosylation mediates organ specific adhesion and metastasis via lectin receptors on the lung vascular endothelium. Clin Exp Metastasis (2005) 22:11–24. doi:10.1007/s10585-005-2036-2
142. Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell (2007) 129:123–34. doi:10.1016/j.cell.2007.01.049
143. Kobata A, Amano J. Altered glycosylation of proteins produced by malignant cells, and application for the diagnosis and immunotherapy of tumours. Immunol Cell Biol (2005) 83:429–39. doi:10.1111/j.1440-1711.2005.01351.x
144. Chen L, Zhang W, Fregien N, Pierce M. The her-2/neu oncogene stimulates the transcription of N-acetylglucosaminyltransferase V and expression of its cell surface oligosaccharide products. Oncogene (1998) 17:2087–93. doi:10.1038/sj.onc.1202124
145. Demetriou M, Nabi IR, Coppolino M, Dedhar S, Dennis JW. Reduced contact-inhibition and substratum adhesion in epithelial cells expressing GlcNAc-transferase V. J Cell Biol (1995) 130:383–92. doi:10.1083/jcb.130.2.383
146. Lajoie P, Partridge EA, Guay G, Goetz JG, Pawling J, Lagana A, et al. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol (2007) 179:341–56. doi:10.1083/jcb.200611106
147. Tsuboi S, Sutoh M, Hatakeyama S, Hiraoka N, Habuchi T, Horikawa Y, et al. A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. EMBO J (2011) 30:3173–85. doi:10.1038/emboj.2011.215
148. Boscher C, Dennis JW, Nabi IR. Glycosylation, galectins and cellular signaling. Curr Opin Cell Biol (2011) 23:383–92. doi:10.1016/j.ceb.2011.05.001
149. Veiga SS, Chammas R, Cella N, Brentani RR. Glycosylation of beta-1 integrins in B16-F10 mouse melanoma cells as determinant of differential binding and acquisition of biological activity. Int J Cancer (1995) 61:420–4. doi:10.1002/ijc.2910610324
150. Zheng M, Fang H, Hakomori S. Functional role of N-glycosylation in alpha 5 beta 1 integrin receptor. De-N-glycosylation induces dissociation or altered association of alpha 5 and beta 1 subunits and concomitant loss of fibronectin binding activity. J Biol Chem (1994) 269:12325–31.
151. Guo HB, Lee I, Kamar M, Akiyama SK, Pierce M. Aberrant N-glycosylation of beta1 integrin causes reduced alpha5beta1 integrin clustering and stimulates cell migration. Cancer Res (2002) 62:6837–45.
152. Kansas GS. Selectins and their ligands: current concepts and controversies. Blood (1996) 88:3259–87.
153. Guyer DA, Moore KL, Lynam EB, Schammel CM, Rogelj S, McEver RP, et al. P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood (1996) 88:2415–21.
154. Sperandio M, Smith ML, Forlow SB, Olson TS, Xia L, McEver RP, et al. P-selectin glycoprotein ligand-1 mediates L-selectin-dependent leukocyte rolling in venules. J Exp Med (2003) 197:1355–63. doi:10.1084/jem.20021854
155. Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature (2005) 435:969–73. doi:10.1038/nature03703
156. Lenter M, Levinovitz A, Isenmann S, Vestweber D. Monospecific and common glycoprotein ligands for E- and P-selectin on myeloid cells. J Cell Biol (1994) 125:471–81. doi:10.1083/jcb.125.2.471
157. Burdick MM, McCaffery JM, Kim YS, Bochner BS, Konstantopoulos K. Colon carcinoma cell glycolipids, integrins, and other glycoproteins mediate adhesion to HUVECs under flow. Am J Physiol Cell Physiol (2003) 284:C977–87. doi:10.1152/ajpcell.00423.2002
158. Kim YJ, Borsig L, Han HL, Varki NM, Varki A. Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am J Pathol (1999) 155:461–72. doi:10.1016/S0002-9440(10)65142-5
159. McCarty OJ, Mousa SA, Bray PF, Konstantopoulos K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood (2000) 96:1789–97.
160. Jorgensen T, Berner A, Kaalhus O, Tveter KJ, Danielsen HE, Bryne M. Up-regulation of the oligosaccharide sialyl LewisX: a new prognostic parameter in metastatic prostate cancer. Cancer Res (1995) 55:1817–9.
161. Ogawa J, Inoue H, Koide S. Expression of alpha-1,3-fucosyltransferase type IV and VII genes is related to poor prognosis in lung cancer. Cancer Res (1996) 56:325–9.
162. Renkonen J, Paavonen T, Renkonen R. Endothelial and epithelial expression of sialyl Lewis(x) and sialyl Lewis(a) in lesions of breast carcinoma. Int J Cancer (1997) 74:296–300. doi:10.1002/(SICI)1097-0215(19970620)74:3<296::AID-IJC11>3.0.CO;2-A
163. Takahashi S, Oda T, Hasebe T, Sasaki S, Kinoshita T, Konishi M, et al. Overexpression of sialyl Lewis x antigen is associated with formation of extratumoral venous invasion and predicts postoperative development of massive hepatic metastasis in cases with pancreatic ductal adenocarcinoma. Pathobiology (2001) 69:127–35. doi:10.1159/000048767
164. Tatsumi M, Watanabe A, Sawada H, Yamada Y, Shino Y, Nakano H. Immunohistochemical expression of the sialyl Lewis x antigen on gastric cancer cells correlates with the presence of liver metastasis. Clin Exp Metastasis (1998) 16:743–50. doi:10.1023/A:1006584829246
165. Tozawa K, Okamoto T, Kawai N, Hashimoto Y, Hayashi Y, Kohri K. Positive correlation between sialyl Lewis X expression and pathologic findings in renal cell carcinoma. Kidney Int (2005) 67:1391–6. doi:10.1111/j.1523-1755.2005.00216.x
166. Inaba Y, Ohyama C, Kato T, Satoh M, Saito H, Hagisawa S, et al. Gene transfer of alpha1,3-fucosyltransferase increases tumor growth of the PC-3 human prostate cancer cell line through enhanced adhesion to prostatic stromal cells. Int J Cancer (2003) 107:949–57. doi:10.1002/ijc.11513
167. Mannori G, Crottet P, Cecconi O, Hanasaki K, Aruffo A, Nelson RM, et al. Differential colon cancer cell adhesion to E-, P-, and L-selectin: role of mucin-type glycoproteins. Cancer Res (1995) 55:4425–31.
168. St Hill CA, Bullard KM, Walcheck B. Expression of the high-affinity selectin glycan ligand C2-O-sLeX by colon carcinoma cells. Cancer Lett (2005) 217:105–13. doi:10.1016/j.canlet.2004.06.038
169. Barthel SR, Wiese GK, Cho J, Opperman MJ, Hays DL, Siddiqui J, et al. Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc Natl Acad Sci U S A (2009) 106:19491–6. doi:10.1073/pnas.0906074106
170. Izumi Y, Taniuchi Y, Tsuji T, Smith CW, Nakamori S, Fidler IJ, et al. Characterization of human colon carcinoma variant cells selected for sialyl Lex carbohydrate antigen: liver colonization and adhesion to vascular endothelial cells. Exp Cell Res (1995) 216:215–21. doi:10.1006/excr.1995.1027
171. Weston BW, Hiller KM, Mayben JP, Manousos GA, Bendt KM, Liu R, et al. Expression of human alpha(1,3)fucosyltransferase antisense sequences inhibits selectin-mediated adhesion and liver metastasis of colon carcinoma cells. Cancer Res (1999) 59:2127–35.
172. Rosen SD, Bertozzi CR. The selectins and their ligands. Curr Opin Cell Biol (1994) 6:663–73. doi:10.1016/0955-0674(94)90092-2
173. Kim YS, Gum J Jr, Brockhausen I. Mucin glycoproteins in neoplasia. Glycoconj J (1996) 13:693–707. doi:10.1007/BF00702333
174. Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunol Rev (2009) 230:97–113. doi:10.1111/j.1600-065X.2009.00795.x
175. Varki A. Selectin ligands: will the real ones please stand up? J Clin Invest (1997) 99:158–62. doi:10.1172/JCI119142
176. McEver RP. Selectins: lectins that initiate cell adhesion under flow. Curr Opin Cell Biol (2002) 14:581–6. doi:10.1016/S0955-0674(02)00367-8
177. Chaturvedi P, Singh AP, Batra SK. Structure, evolution, and biology of the MUC4 mucin. FASEB J (2008) 22:966–81. doi:10.1096/fj.07-9673rev
178. Chen SH, Dallas MR, Balzer EM, Konstantopoulos K. Mucin 16 is a functional selectin ligand on pancreatic cancer cells. FASEB J (2012) 26:1349–59. doi:10.1096/fj.11-195669
179. Aigner S, Sthoeger ZM, Fogel M, Weber E, Zarn J, Ruppert M, et al. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood (1997) 89:3385–95.
180. Burdick MM, Chu JT, Godar S, Sackstein R. HCELL is the major E- and L-selectin ligand expressed on LS174T colon carcinoma cells. J Biol Chem (2006) 281:13899–905. doi:10.1074/jbc.M513617200
181. Dimitroff CJ, Descheny L, Trujillo N, Kim R, Nguyen V, Huang W, et al. Identification of leukocyte E-selectin ligands, P-selectin glycoprotein ligand-1 and E-selectin ligand-1, on human metastatic prostate tumor cells. Cancer Res (2005) 65:5750–60. doi:10.1158/0008-5472.CAN-04-4653
182. Gout S, Morin C, Houle F, Huot J. Death receptor-3, a new E-Selectin counter-receptor that confers migration and survival advantages to colon carcinoma cells by triggering p38 and ERK MAPK activation. Cancer Res (2006) 66:9117–24. doi:10.1158/0008-5472.CAN-05-4605
183. Thomas SN, Schnaar RL, Konstantopoulos K. Podocalyxin-like protein is an E-/L-selectin ligand on colon carcinoma cells: comparative biochemical properties of selectin ligands in host and tumor cells. Am J Physiol Cell Physiol (2009) 296:C505–13. doi:10.1152/ajpcell.00472.2008
184. Heider KH, Hofmann M, Hors E, van den Berg F, Ponta H, Herrlich P, et al. A human homologue of the rat metastasis-associated variant of CD44 is expressed in colorectal carcinomas and adenomatous polyps. J Cell Biol (1993) 120:227–33. doi:10.1083/jcb.120.1.227
185. Penno MB, August JT, Baylin SB, Mabry M, Linnoila RI, Lee VS, et al. Expression of CD44 in human lung tumors. Cancer Res (1994) 54:1381–7.
186. Rall CJ, Rustgi AK. CD44 isoform expression in primary and metastatic pancreatic adenocarcinoma. Cancer Res (1995) 55:1831–5.
187. Harada N, Mizoi T, Kinouchi M, Hoshi K, Ishii S, Shiiba K, et al. Introduction of antisense CD44S CDNA down-regulates expression of overall CD44 isoforms and inhibits tumor growth and metastasis in highly metastatic colon carcinoma cells. Int J Cancer (2001) 91:67–75. doi:10.1002/1097-0215(20010101)91:1<67::AID-IJC1011>3.0.CO;2-D
188. Reeder JA, Gotley DC, Walsh MD, Fawcett J, Antalis TM. Expression of antisense CD44 variant 6 inhibits colorectal tumor metastasis and tumor growth in a wound environment. Cancer Res (1998) 58:3719–26.
189. Hanley WD, Burdick MM, Konstantopoulos K, Sackstein R. CD44 on LS174T colon carcinoma cells possesses E-selectin ligand activity. Cancer Res (2005) 65:5812–7. doi:10.1158/0008-5472.CAN-04-4557
190. Hanley WD, Napier SL, Burdick MM, Schnaar RL, Sackstein R, Konstantopoulos K. Variant isoforms of CD44 are P- and L-selectin ligands on colon carcinoma cells. FASEB J (2006) 20:337–9.
191. Cooney CA, Jousheghany F, Yao-Borengasser A, Phanavanh B, Gomes T, Kieber-Emmons AM, et al. Chondroitin sulfates play a major role in breast cancer metastasis: a role for CSPG4 and CHST11 gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res (2011) 13:R58. doi:10.1186/bcr2895
192. Kaytes PS, Geng JG. P-selectin mediates adhesion of the human melanoma cell line NKI-4: identification of glycoprotein ligands. Biochemistry (1998) 37:10514–21. doi:10.1021/bi9730846
193. Stone JP, Wagner DD. P-selectin mediates adhesion of platelets to neuroblastoma and small cell lung cancer. J Clin Invest (1993) 92:804–13. doi:10.1172/JCI116654
194. Li J, Guillebon AD, Hsu JW, Barthel SR, Dimitroff CJ, Lee YF, et al. Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br J Cancer (2013) 109:3014–22. doi:10.1038/bjc.2013.690
195. Julien S, Ivetic A, Grigoriadis A, Qize D, Burford B, Sproviero D, et al. Selectin ligand Sialyl-Lewis x antigen drives metastasis of hormone-dependent breast cancers. Cancer Res (2011) 71:7683–93. doi:10.1158/0008-5472.CAN-11-1139
196. Radhakrishnan P, Chachadi V, Lin MF, Singh R, Kannagi R, Cheng PW. TNFalpha enhances the motility and invasiveness of prostatic cancer cells by stimulating the expression of selective glycosyl- and sulfotransferase genes involved in the synthesis of selectin ligands. Biochem Biophys Res Commun (2011) 409:436–41. doi:10.1016/j.bbrc.2011.05.019
197. Barthel SR, Hays DL, Yazawa EM, Opperman M, Walley KC, Nimrichter L, et al. Definition of molecular determinants of prostate cancer cell bone extravasation. Cancer Res (2013) 73:942–52. doi:10.1158/0008-5472.CAN-12-3264
198. Yin X, Rana K, Ponmudi V, King MR. Knockdown of fucosyltransferase III disrupts the adhesion of circulating cancer cells to E-selectin without affecting hematopoietic cell adhesion. Carbohydr Res (2010) 345:2334–42. doi:10.1016/j.carres.2010.07.028
199. Borsig L. The role of platelet activation in tumor metastasis. Expert Rev Anticancer Ther (2008) 8:1247–55. doi:10.1586/14737140.8.8.1247
200. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer (2011) 11:123–34.
201. Honn KV, Tang DG, Crissman JD. Platelets and cancer metastasis: a causal relationship? Cancer Metastasis Rev (1992) 11:325–51. doi:10.1007/BF01307186
202. Karpatkin S, Pearlstein E. Role of platelets in tumor cell metastases. Ann Intern Med (1981) 95:636–41. doi:10.7326/0003-4819-95-5-636
203. Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci U S A (2001) 98:3352–7. doi:10.1073/pnas.061615598
204. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood (2004) 104:397–401. doi:10.1182/blood-2004-02-0434
205. Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res (1999) 59:1295–300.
206. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood (2005) 105:178–85. doi:10.1182/blood-2004-06-2272
207. Ludwig RJ, Boehme B, Podda M, Henschler R, Jager E, Tandi C, et al. Endothelial P-selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res (2004) 64:2743–50. doi:10.1158/0008-5472.CAN-03-1054
208. Coupland LA, Chong BH, Parish CR. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res (2012) 72:4662–71. doi:10.1158/0008-5472.CAN-11-4010
209. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell (2011) 20:576–90. doi:10.1016/j.ccr.2011.09.009
210. Jadhav S, Bochner BS, Konstantopoulos K. Hydrodynamic shear regulates the kinetics and receptor specificity of polymorphonuclear leukocyte-colon carcinoma cell adhesive interactions. J Immunol (2001) 167:5986–93.
211. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer (2009) 9:239–52. doi:10.1038/nrc2618
212. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
213. Läubli H, Spanaus KS, Borsig L. Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood (2009) 114:4583–91. doi:10.1182/blood-2008-10-186585
214. Taverna D, Moher H, Crowley D, Borsig L, Varki A, Hynes RO. Increased primary tumor growth in mice null for beta3- or beta3/beta5-integrins or selectins. Proc Natl Acad Sci U S A (2004) 101:763–8. doi:10.1073/pnas.0307289101
215. Laferriere J, Houle F, Taher MM, Valerie K, Huot J. Transendothelial migration of colon carcinoma cells requires expression of E-selectin by endothelial cells and activation of stress-activated protein kinase-2 (SAPK2/p38) in the tumor cells. J Biol Chem (2001) 276:33762–72. doi:10.1074/jbc.M008564200
216. Tremblay PL, Auger FA, Huot J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene (2006) 25:6563–73. doi:10.1038/sj.onc.1209664
217. Biancone L, Araki M, Araki K, Vassalli P, Stamenkovic I. Redirection of tumor metastasis by expression of E-selectin in vivo. J Exp Med (1996) 183:581–7. doi:10.1084/jem.183.2.581
218. Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer (1997) 71:612–9. doi:10.1002/(SICI)1097-0215(19970516)71:4<612::AID-IJC17>3.3.CO;2-1
219. Läubli H, Borsig L. Selectins as mediators of lung metastasis. Cancer Microenviron (2010) 3:97–105. doi:10.1007/s12307-010-0043-6
220. Stubke K, Wicklein D, Herich L, Schumacher U, Nehmann N. Selectin-deficiency reduces the number of spontaneous metastases in a xenograft model of human breast cancer. Cancer Lett (2012) 321:89–99. doi:10.1016/j.canlet.2012.02.019
221. Hiratsuka S, Goel S, Kamoun WS, Maru Y, Fukumura D, Duda DG, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U S A (2011) 108:3725–30. doi:10.1073/pnas.1100446108
222. Wahrenbrock M, Borsig L, Le D, Varki N, Varki A. Selectin-mucin interactions as a probable molecular explanation for the association of Trousseau syndrome with mucinous adenocarcinomas. J Clin Invest (2003) 112:853–62. doi:10.1172/JCI200318882
223. Shao B, Wahrenbrock MG, Yao L, David T, Coughlin SR, Xia L, et al. Carcinoma mucins trigger reciprocal activation of platelets and neutrophils in a murine model of Trousseau syndrome. Blood (2011) 118:4015–23. doi:10.1182/blood-2011-07-368514
224. Gil-Bernabe AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood (2012) 119:3164–75. doi:10.1182/blood-2011-08-376426
225. Ferjancic S, Gil-Bernabe AM, Hill SA, Allen PD, Richardson P, Sparey T, et al. VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood (2013) 121:3289–97. doi:10.1182/blood-2012-08-449819
226. Khatib AM, Kontogiannea M, Fallavollita L, Jamison B, Meterissian S, Brodt P. Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res (1999) 59:1356–61.
227. Matsuo Y, Amano S, Furuya M, Namiki K, Sakurai K, Nishiyama M, et al. Involvement of p38alpha mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem (2006) 281:36767–75. doi:10.1074/jbc.M604371200
228. Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes AM, Anasagasti MJ, Martin J, et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci U S A (2000) 97:734–9. doi:10.1073/pnas.97.2.734
229. Kobayashi K, Matsumoto S, Morishima T, Kawabe T, Okamoto T. Cimetidine inhibits cancer cell adhesion to endothelial cells and prevents metastasis by blocking E-selectin expression. Cancer Res (2000) 60:3978–84.
230. Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem (2009) 284:29087–96. doi:10.1074/jbc.M109.035899
231. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One (2009) 4:e6562. doi:10.1371/journal.pone.0006562
232. Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell (2012) 22:91–105. doi:10.1016/j.ccr.2012.05.023
233. Fujita-Yamaguchi Y. Renewed interest in basic and applied research involving monoclonal antibodies against an oncofetal Tn-antigen. J Biochem (2013) 154:103–5. doi:10.1093/jb/mvt052
234. Julien S, Picco G, Sewell R, Vercoutter-Edouart AS, Tarp M, Miles D, et al. Sialyl-Tn vaccine induces antibody-mediated tumour protection in a relevant murine model. Br J Cancer (2009) 100:1746–54. doi:10.1038/sj.bjc.6605083
Keywords: glycosylation, cancer, metastasis, glycan ligands, mucins, siglecs, galectins, selectins
Citation: Häuselmann I and Borsig L (2014) Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 4:28. doi: 10.3389/fonc.2014.00028
Received: 13 December 2013; Accepted: 29 January 2014;
Published online: 13 February 2014.
Edited by:
Roger Chammas, Universidade de São Paulo, BrazilReviewed by:
Stephan Von Gunten, University of Bern, SwitzerlandShoukat Dedhar, University of British Columbia, Canada
Copyright: © 2014 Häuselmann and Borsig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lubor Borsig, Zürich Center for Integrative Human Physiology, Institute of Physiology, University of Zurich, Winterthurerstrasse 190, Zurich CH-8057, Switzerland e-mail:bGJvcnNpZ0BhY2Nlc3MudXpoLmNo