 Roberto S. Accolla
Roberto S. Accolla Letizia Lombardo
Letizia Lombardo Rawan Abdallah
Rawan Abdallah Giovanna Tosi
Giovanna Tosi- Department of Surgical and Morphological Sciences, University of Insubria, Varese, Italy
Although the existence of an immune response against tumor cells is well documented, the fact that tumors take off in cancer patients indicates that neoplastic cells can circumvent this response. Over the years many investigators have described strategies to rescue the anti-tumor immune response with the aim of creating specific and long-lasting protection against the disease. When exported to human clinical settings, these strategies have revealed in most cases a very limited, if any, positive outcome. We believe that the failure is mostly due to the inadequate triggering of the CD4+ T helper (TH) cell arm of the adaptive immunity, as TH cells are necessary to trigger all the immune effector mechanisms required to eliminate tumor cells. In this review, we focus on novel strategies that by stimulating MHC class II-restricted activation of TH cells generate a specific and persistent adaptive immunity against the tumor. This point is of critical importance for both preventive and therapeutic anti-tumor vaccination protocols, because adaptive immunity with its capacity to produce specific, long-lasting protection and memory responses is indeed the final goal of vaccination. We will discuss data from our as well as other laboratories which strongly suggest that triggering a specific and persistent anti-tumor CD4+ TH cell response stably modify not only the tumor microenvironment but also tumor-dependent extratumor microenvironments by eliminating and/or reducing the blood-derived tumor infiltrating cells that may have a pro-tumor growth function such as regulatory CD4+/CD25+ T cells and myeloid-derived-suppressor cells. Within this frame, therefore, we believe that the establishment of a pro-tumor environment is not the cause but simply the consequence of the tumor strategy to primarily counteract components of the adaptive cellular immunity, particularly TH lymphocytes.
Introduction
The onset, expansion, persistence, and spreading of tumors are under the control of a complex series of events that encompass both intrinsic modifications of cancer cells, such as genetic mutations in proto-oncogenes and in tumor suppressor genes, and alteration of the apoptotic process, which cumulatively impact on the homeostasis of the cell cycle, as well as extrinsic mechanisms related to the capacity of the host to counteract tumor growth (1). As far as host mechanisms counteracting the tumor, certainly the immune system plays a major role. The key functions of the immune system consist in the capacity to recognize and fight “foreign aggressors” (the non-self), while sparing self constituents (tolerance), and in conserving specific memory of this event, should foreign aggressors be encountered again (2). Under this view, the genetic modifications affecting tumor cells may lead to the generation of structurally altered or abundantly synthesized proteins that may be seen by the immune system as new antigens or “non-self” products against which the system has not learned to be tolerant during ontogenesis (3). Both innate and adaptive immunity have been shown to participate to this response (4). Nevertheless, the fact that the tumor process takes off in cancer patients demonstrates that the recognition of cancer cells and/or the equilibrium between tumor growth and anti-tumor immune response can be altered, favoring the progression of the malignancy (5, 6). Tumor development and response of the immune system have long been considered opposing actions in the cancer history. However, accumulating data strongly indicate that the two events can synergize, particularly for the tumor prosperity. Tumor cells may, for example, produce mediators that not only counteract the possible protective anti-tumor action of the immune system but also induce immune cells to produce factors or to polarize their functional phenotype in favor of the growth and the expansion of the tumor, thus completely deviating the goal of the immune response (7). Moreover, tumor infiltrating leukocytes, including macrophages (7, 8), regulatory T cells (Tregs) with CD4+/CD25+ phenotype and suppressive function on helper and effector T cells (9), as well as a heterogeneous group of similarly acting myeloid-derived suppressor cells (MDSCs) (10), can cooperate in antagonizing the protective immune response while favoring tumor growth. All together, these observations have created the diffuse belief that a pro-tumor polarization of the innate and adaptive immunity is a sort of inevitable event in the life history of the tumor and a major cause for cancer cells to survive, replicate, and spread.
We have recently discussed a different interpretation of this key issue (11, 12). Indeed, the establishment of a pro-tumor environment may not be the cause but the consequence of the initial inability of the immune system to be efficiently triggered against the tumor. Within this frame, one crucial aspect is the initial recognition of tumor-associated antigens (TAAs) by the adaptive immune system. Two fundamental events must take place in order to initiate the adaptive immune response: (a) antigen processing of complex antigens and presentation of appropriate peptides within the context of MHC class II molecules; (b) recognition of MHC class II-bound peptides displayed on the cell surface by MHC class II-restricted CD4+ T cells, designated T helper (TH) cells (13). TH cells are fundamental for optimal induction of both humoral and cellular effector mechanisms (14), particularly for the maturation of MHC class I-restricted CD8+ naïve T cells, their clonal expansions, and acquisition of cytolytic function (15). The latter function is of relevance in the context of anti-tumor immunity since CD8+ cytolytic T cells (CTLs) are believed to be the major lymphocyte effectors against cancer cells (16).
Initial priming and triggering of naïve antigen-specific TH cells are believed to be mediated by specialized MHC class II-positive cells, particularly dendritic cells (DC), which engulf exogenous antigenic material (pathogens, pathogen debris, cell debris), digest it in appropriate endosomal compartments, degrade the building blocks of life material, that is the proteins, into peptides, and allow the binding of antigenic peptides to MHC class II molecules. The MHC-II–peptide complexes can thus migrate to the cell surface and be displayed for TH cell scrutiny. This general mechanism would imply that endogenously synthesized protein, as tumor antigens are, may not be presented within the context of MHC class II unless they are captured as secreted products or as cell debris-associated components by DC surrounding or infiltrating tumor masses. However, previous elegant studies have shown that endogenous proteins could access the MHC class II pathway of antigen presentation (17, 18) and peptides of these proteins could be recognized and serve as immunogens for TH cell triggering (19, 20).
The majority of tumors, particularly carcinomas, usually do not express MHC class II molecules that are instead expressed by professional antigen presenting cells (APCs) such as DC, macrophages, and B cells (13). Furthermore, although in some circumstances tumor cells may express MHC class II molecules, these cells are believed to act at most as “peptide antigen presenters” to antigen-specific primed TH cells but not as “antigen processors and presenters” for naïve TH cell priming. This is only partially true, since we have demonstrated that, at least in vitro, human or murine engineered-tumor cells, stably expressing MHC class II molecules, can process protein antigens and present relevant peptides to MHC-II-restricted antigen-specific T cell clones (21, 22). These results have shown that the intracellular machinery responsible for digesting and exposing MHC-II–peptide complexes on the cell surface for TH cell recognition may work properly also in cells which are not classical APC, and notably in MHC-II-positive tumor cells.
Within this frame, the fact that most tumor cells do not express MHC class II molecules prevents even the possibility that these cells may act as potential APCs for their TAAs and by consequence that they may trigger tumor-specific TH cells.
In this review, we will discuss data from our as well as from other laboratories that strongly suggest how the initial insurgence of the tumor process and the efficient response of the adaptive immune system to the tumor are strongly conditioned by the fact that tumor cells may or may not express MHC class II molecules in the appropriate “physiological” condition, and by the fact that insufficient amount of tumor antigens is offered in a MHC class II-restricted fashion to TH cells.
These two events dramatically influence the formation and the functional role of the tumor microenvironment, conceived not only as tumor tissue but also as tumor-dependent microenvironments at distant sites, particularly in lymphoid tissues of tumor-bearing hosts. We will stress the concept that the generation of a pro-tumor microenvironment is not the cause but the consequence of the failure of the adaptive immune system to recognize MHC-II–tumor peptide complexes by tumor-specific TH cells.
Induction of Canonical MHC Class II Expression in Tumor Cells may Trigger a Protective Anti-Tumor Immune Response in vivo
The idea to promote anti-tumor adaptive immune responses in vivo by providing adequate antigen availability (AAA), that means not only sufficient amount of antigen but also access of this antigen to MHC-II binding for optimal triggering of TH cells (12), has been approached from different standpoints. For example, irradiated or genetically modified tumor cells have been used even in clinical trials with the goal to provide host APC with sufficient amount of TAA or to generate, within the tissue injected with tumor cells, a suitable milieu for optimal APC uptake and presentation of tumor antigens by APC via their MHC class II molecules (23). DC loaded with TAAs have been also used with the aim of providing a direct source of ready-to-use MHC-II–tumor peptide complexes for optimal priming and triggering of TH cells (24, 25) and recent clinical results in melanoma patients give further hope in improving clinical responses by this approach (26). Several groups, including ours, have instead investigated the possibility to render tumor cells themselves MHC class II-positive and thus used them as potential surrogate APC for triggering tumor-specific TH cells (27–29). Within this frame, two distinct approaches have been described. The group of Ostrand-Rosenberg induced MHC class II expression in tumor cells by transfecting isolated MHC alpha- and beta chain-encoding genes (28, 30), whereas our group has privileged the transfection of tumor cells with the MHC class II transcriptional activator (CIITA), which is the physiological regulator of expression of all MHC class II genes (31–33). CIITA regulates also the expression of other fundamental genes necessary for MHC-II transport to endosomal compartments and loading of peptides, including the invariant chain (In chain) and DM (34–37). In the first approach, by expressing only isolated MHC class II molecules without expression of In chain, both the district of interaction and the quality of interacting peptides, including tumor-associated peptides, are totally different as compared to the site and the peptides interacting with physiologically expressed MHC class II molecules. The rationale underlying this approach was to allow peptides from TAAs, which are endogenous proteins, to associate with MHC class II molecules in the ER, similarly to what happens for MHC class I-peptide binding, and thus allow better recognition of putative tumor antigens by MHC class II-restricted TH cells. However, although with the SaI sarcoma model (mostly used by the Ostrand-Rosenberg group) protective immunity could be generated in vivo by vaccinating mice with MHC-II (alpha-beta)-transfected cells, the cellular correlates of protection remained not completely clarified, because no other tumor models were studied intensively. In the SaI tumor model, it has been suggested that tumor cells may not act directly as surrogate APCs but as donors of peptide–MHC class II complexes for professional APC, such as DC, that in turn stimulate TH cells (30). However, it must be kept in mind that in the absence of In chain hardly any MHC class II molecules are in a stable peptide-loaded form. Cells from In chain knock-out mice show a dramatic reduction in cell surface MHC class II molecules, resulting from both defective association of class II alpha- and beta-chains and markedly decreased post-Golgi transport. The few class II alpha/beta heterodimers reaching the cell surface behave as empty molecules or as molecules occupied by an easily displaced peptide, and display a distinct structure. Moreover spleen cells from these mice are defective in their ability to present intact protein antigens (36, 38).
Our approach based on CIITA-mediated expression of MHC class II molecules as well as upregulation of In chain, seemed to us more prone to mimic the physiological condition by which TAA-derived peptides may be charged onto MHC-II in a more stable way and exposed to the cell surface for TH cell scrutiny. Indeed, it was possible to show that CIITA-transduced tumor cells of at least four distinct histological origin can be efficiently rejected or strongly prevented in their growth when injected into immunocompetent syngeneic mice (22, 27, 39), demonstrating the general applicability of our model system. Furthermore, it was shown that mice vaccinated with CIITA-transfected tumor cells develop an anamnestic response not only against the CIITA-expressing tumor but also, most importantly, against the CIITA-negative parental tumor cells leading to a very efficient rejection of the parental tumor (22, 27, 39). CIITA-dependent MHC class II expression in tumor cells was instrumental to trigger the anamnestic protective immune response against the parental tumor, as also demonstrated by vaccination experiments with non-replicating CIITA-transfected tumor cells (40). The search for cellular correlates of protection highlighted three fundamental points. Firstly, long-lasting immunity was generated in CIITA-vaccinated mice since these animals were able to reject parental tumors after 6 months from initial vaccination. Second, rejection and/or reduced tumor growth were mediated by tumor-specific CD4+ TH and CD8+ CTL, because ablation of these subpopulations in vivo by injection of specific anti-CD4 or anti-CD8, but not by anti-B cell or anti-NK cell monoclonal antibodies, prevented the acquisition of protective immunity after injection of CIITA-expressing tumor cells. Third, and of particular relevance, transfer of purified CD4+ T cells from vaccinated mice into naïve recipients was sufficient to confer protection against parental tumors (22, 27, 39, 40).
These results demonstrated that expression of CIITA-mediated MHC class II molecules in tumor cells was instrumental in triggering a protective adaptive immune response in immunocompetent mice. One possible conclusion from these findings was that tumor cells expressing CIITA-dependent MHC-II molecules could serve themselves as APC of their own TAA for initial triggering and priming of tumor-specific TH cells. Alternatively, preformed MHC-II–TAA peptide complexes derived from CIITA-transfected tumor cells could be shed from the cells, captured by professional APC and used by these cells to reach the AAA necessary to trigger an efficient priming of tumor-specific CD4+ TH cells.
Characterization of the Tumor and Lympoid Organs Microenvironment in CIITA-Tumor Vaccinated Mice and in Parental Tumor-Bearing Mice
To further investigate the crucial issue of the possible direct APC function of CIITA-transfected tumor cells, a series of experiments were undertaken in our laboratory focusing first on the histological characterization of the tumor microenvironment in CIITA-tumor and in parental tumor-injected mice (22). While tumors derived by injecting parental, MHC-II-negative cells showed very poor, if any, infiltration of lymphocytes and scarce presence of macrophage and neutrophils in several tumor models, CIITA-transfected MHC-II positive tumors were rapidly and firstly infiltrated by CD4+ T cells, followed by CD8+ T cells, and later by DC, macrophage, and neutrophils. Already in the early phase of CD4+ and CD8+ T cell infiltration, extensive areas of tumor cell necrosis surrounded by the above T cells and progressively infiltrated by large numbers of neutrophils could be observed. Indeed, the histological picture observed in the tumor microenvironment generated by CIITA-transfected tumor cells was the classical picture of an inflammatory lesion. Most importantly, parental tumor rechallenge of CIITA-vaccinated mice showed a histological aspect of the tumor tissue virtually superimposable to the one observed in naïve mice injected with CIITA-tumor cells. Indeed, in CIITA-tumor vaccinated and parental tumor challenged mice, the lymphocyte infiltration and the tumor necrotic areas were even more massive than in naïve mice injected with CIITA-tumor cells (39).
This finding, along with the capacity of CIITA-expressing tumor cells to process and present antigenic peptides to CD4+ T cells in vitro (21, 22), supports the hypothesis that much of the tumor-specific TH cell triggering takes place in the tumor tissue as consequence of the recognition of MHC-II–TAA peptide complexes expressed by CIITA-transfected tumor cells.
If confirmed, this will provide further evidence that tumor cells can act as APC for MHC-II-restricted TH cells in the absence of DC. This in turn will show that alternative mechanisms of antigen presentation for TH cell priming in vivo can not only occur but they can occur in milieus other than lymphoid tissues. A further, and in our opinion, extremely important conclusion of these findings is that MHC-II molecules of CIITA-transfected tumor cells are indeed loaded with the relevant immunogenic tumor-derived peptides; these peptides might be eluted out and sequenced to assess the tumor-associated peptidome, a fundamental step for constructing more promising anti-tumor vaccines aimed at optimally stimulating TH cells. Extending the knowledge of the repertoire of MHC-bound tumor-derived peptides by purifying and characterizing the tumor-associated peptidome and choosing the most appropriate cocktail of peptides to prepare anti-tumor vaccines has been shown recently to be a very promising approach for the construction of suitable multi-peptide vaccine in human renal cancer (41).
The increased immunogenicity of the tumor generated by the CIITA-mediated MHC-II expression and the consequent adequate tumor antigen availability at the right time and at the right place affects dramatically the tumor microenvironment, moving the balance from a pro-tumor to an anti-tumor microenvironment. The subversion of the tumor microenvironment affected also the number of Tregs observed in tumor draining lymph nodes. Tregs are believed to play an important role in down-modulating the function of CD4+ TH cells in adaptive immunity, and their number and function were found to increase in tumor-bearing hosts (9). In human colorectal cancer, very recently, the presence of Tregs and the consequent reduced response of anti-tumor CD4+ TH cells has been associated also to the progression of the diseases (42). Accordingly, we found that parental tumor-bearing mice had a substantial increase in the number of Tregs in tumor draining lymph nodes (40), although the relative suppressive capacity of these cells on TH function in vitro and on tumor growth in vivo was not increased (43). On the other hand, the number of Tregs in CIITA-tumor injected and protected mice was found to be much lower and virtually superimposable to that of naïve mice (40). Thus, initial optimal triggering of the adaptive immune response against the tumor prevented the increase of a crucial cellular component with suppressive function on CD4+ TH cells and with pro-tumor behavior on tumor microenvironment. Active investigation is also focused, at present, on the possibility that CIITA-tumor vaccination may also affect the function and/or the number of other cells, like MDSC, with suppressive function on adaptive anti-tumor immunity, particularly in light of recent findings suggesting that MDSC can act as tolerizing cells toward tumor antigen recognition in specialized sites of the splenic lymphoid tissue (44).
Anti-Tumor Immunity Elicited after Chemotherapy, Radiotherapy, and Cytokine Therapy: The Concept of Therapy-Induced Anti-Tumor Vaccination
In recent years, it has become apparent that the beneficial effects of various conventional therapeutic modalities, including chemotherapy and radiation therapy, are associated with the rescue of an immune response against the tumor.
Most chemotherapeutic drugs exert their action through the induction of apoptosis, a mechanism that has long been interpreted as generating tolerogenic signals for the immune system. This was generally attributed to the scarce or absent inflammation generated by the apoptotic process and thus by the impossibility to trigger leukocyte migration and lymphocyte activation. However, after the important work of the Novak and colleagues, it appeared that drug-induced apoptotic tumor cells behave as potent “providers” of immunogenic tumor antigens for APC-dependent cross-stimulation of tumor-specific CTL (45). After this observation, many studies have confirmed that not only gemcitabine (the drug initially used by Novak et al.) but also cyclophosphamide, oxaliplatin, cisplatin, etoposide, topotecan, paclitaxel, and vinblastine may increase tumor recognition by the immune system via several mechanism not only related to apoptosis induction but also to the capacity of these drugs to upregulate the expression of tumor antigens and to increase MHC class I expression on tumor cells, thus favoring recognition and triggering of tumor-specific CTL (46, 47). Within this frame, it has been shown that chemotherapy may activate also innate immune effectors such as NK cells (48). The detailed study of the effect of chemotherapy on the anti-tumor immune response has provided new information on the complexity of actions and targets that the treatment brings about. Indeed, chemotherapy may be beneficial not only because it activates immune effectors but also because it counteracts negative regulators of anti-tumor immunity in the tumor microenvironment and at distant sites. In fact, chemotherapy may reduce the infiltration of Tregs (49–51) and may reduce the action and/or decrease the number of MDSC (52, 53).
Similarly to chemotherapy, radiotherapy can be associated to the elicitation of an immune response against the tumor (54). Radiotherapy very often induces an inflammatory reaction. It is widely believed that radiation-mediated inflammation promotes tissue necrosis (54, 55) and necrotic tissue may constitute a major source of tumor antigen for triggering adaptive immune responses. Indeed, radiation can induce antigen release in the tumor microenvironment, consequent activation of APCs and triggering of CTL responses (56, 57). Interestingly, radiotherapy may also influence the activation of tumor-associated DC, disclosing the possibility that potentially tolerized DC may re-acquire strong tumor antigen APC function for immune lymphocyte effectors (58) Moreover, radiation can increase the expression of tumor antigens (59) and the cell surface expression of MHC class I molecules by various modalities. Together, these events increase the potential to present TAA peptides to CTL (60–62). In recent years, it has become apparent that radiation can also induce apoptosis and apoptotic cell death can efficiently support stimulation of adaptive immunity (63, 64) in a similar way to the observed effect of chemotherapy.
Not only classical chemotherapy and radiotherapy may induce triggering of the adaptive immunity, biological therapies such as the ones that tag the tumor tissue with certain anti-tumor cytokines may also be responsible of a strong anti-tumor immunity that in certain cases is indeed the mechanism through which the therapy generates tumor cure and permanent tumor clearance (11, 12, 40, 65). Together with the group of Luciano Zardi and Laura Borsi, we have investigated the role of TNF-alpha and Il-2 cytokines conjugated with an scFv antibody (L19) specific for the extra domain B of fibronectin, selectively expressed in the neovasculature (66). Injection of L19-mouse TNFα conjugate (L19mTNFα) induces a dramatic cell death of established tumors in several murine tumor models including the WEHI-164 fibrosarcoma, C51 colon carcinoma (67), and N2A neuroblastoma (65), because it allows therapeutically active doses of TNFα to be reached at the tumor site in which the cytokine increases intravasal coagulation and trombosis around the tumor, further favoring tumor necrosis. In this approach of targeted tumor therapy, three observations lead us to envisage a crucial participation of the adaptive immune system to the tumor cure: (a) the high rate of complete and long-lasting tumor eradication, (b) the fact that tumor-bearing immunodeficient SCID mice were partially resistant to the L19mTNFα treatment even when combined with the cytostatic drug melphalan, and (c) most importantly, the fact that cured mice were resistant to challenge with parental tumor (67). Indeed, immunological studies demonstrated that cured mice developed both anti-tumor CD8+ and CD4+ T cell immunity. Importantly, adoptive cell transfer of CD4+ T cells from cured mice into naïve recipients was sufficient to protect the animals from tumor take (43, 65, 67). These studies lead us to formulate the concept of therapy-induced anti-tumor vaccination because definitive cure of the tumor was achieved after elicitation of a potent adaptive immune response and the treatment was instrumental in reaching protective immunity. The study of the tumor microenvironment in this tumor model was of particular relevance for refining the concept of therapy-induced anti-tumor vaccination. In fact the tumor microenvironment after L19mTNFα alone or in combination with L19mIL-2 showed a marked cellular remodeling with respect to the one observed before therapy. Extensive areas of necrosis infiltrated by granulocytes and macrophages and particularly by CD4+ and CD8+ T cells were observed. The generation of an abundant tumor necrotic material, together with the known effect of TNFα in recruiting DC in inflammatory sites (68) could be a crucial element to generate AAA for fueling professional APC, thus reaching optimal saturation of MHC class II molecule-tumor peptide complexes on DC. These APC could then stimulate specific anti-tumor TH cells and effector CTL, leading to the complete rejection of the tumor and to the establishment of a critical reservoir of memory effector cells responsible for the accelerated rejection of the tumor upon challenge. Again, the therapeutic approach was instrumental in creating the conditions for a protective anti-tumor adaptive immune response responsible of the tumor elimination. The immune-related modification of the tumor microenvironment following therapeutic treatment was also strongly appreciated in the tumor draining lymphonodes and in the spleen. The predominant presence of CD4+ TH2-type cells observed in tumor-bearing hosts before therapy was reconverted in a predominant, although not exclusive, presence of TH1-type cells (43, 65). A further element of remodeling of the tumor microenvironment consisted in the modification of the number, but not the function, of regulatory Tregs with suppressive characteristics on CD4+ TH cells (43). Thus, similarly to the preventive vaccination approach with CIITA-expressing tumor cells, the protective anti-tumor immune response generated by the treatment with L19mTNFα was associated to a rapid appearance and conversion toward a TH1 immune phenotype. This preliminary in vivo experimental investigation has paved the way to initiate clinical trials with L19mTNFα and melphalan in patients with selected tumors (69, 70).
A Hierarchic Model for the Protective Anti-Tumor Immunity
We have seen how either preventive or therapeutic anti-tumor modalities may generate an anti-tumor immune response that in many cases strongly contribute to the modification of the tumor microenvironment and to the eradication of the neoplastic lesion. These results give strong support to the idea that a correct stimulation of the immune system, particularly of the adaptive arm of it, constitutes a primary strategy to be pursued for combating and defeating cancer (see Figure 1). With the exception of preventive vaccination approaches whose final goal is indeed to primarily trigger the adaptive immune system and generate long-lasting immunity against cancer, other classical therapeutic approaches have unveiled the participation of different components of the immune system to a possible successful outcome upon therapy. Recent excellent reviews have summarized the mechanisms and the distinct components of the immune systems that are involved in such therapeutic approaches (46, 54, 71). However, the many possible players that the immune system may use for either triggering a positive response or counteracting a suppressive response against the tumor are not, in our opinion, equally important for the elicitation of a protective immune response against cancer. Based on our experience and on the results of many groups described in this review, we believe that a crucial and hierarchically predominant step is constituted by the efficacy of MHC class II-restricted tumor antigen presentation to CD4+ TH cells (see Figure 2). We have defined this step as AAA to indicate the optimal tumor antigen dose and related antigen processing and MHC-II-restricted presentation necessary to efficiently trigger tumor-specific TH cells (12). We have shown that AAA can be obtained in several ways either by inducing MHC class II expression in tumor cells by transfection with the AIR-1 gene-encoded MHC CIITA, thus providing functionally sufficient MHC-II–tumor peptide complexes for TH cell scrutiny, or by increasing the availability of tumor antigens by specific treatment of established tumors with L19mTNFα. The fundamental finding of both approaches was the elicitation of long-lived anti-tumor specific TH cells capable to eradicate the tumor and to protect the host against further tumor challenge. The primary importance of AAA in the hierarchic scale of anti-tumor immunity stems also from the fact that the correct triggering of TH cells was sufficient to unleash the chain of events leading to a strong effector CTL response and to the abrogation or, at least, the attenuation of suppressor mechanisms operating on the anti-tumor immune response (Figure 1). Moreover, and importantly, the optimal initiation of the adaptive immune response dictated by AAA was sufficient to reorient the tumor microenvironment from a pro-tumor to an anti-tumor microenvironment. Thus, in a sort of re-edition of the immunological homunculus theory originally proposed by Cohen to explain autoimmunity (72), we propose that most of the protective control exerted by the adaptive immune system on cancer derives from AAA (APC and MHC-II, the hand in Figure 2) and consequent activation of tumor specific TH cells (the face in Figure 2).
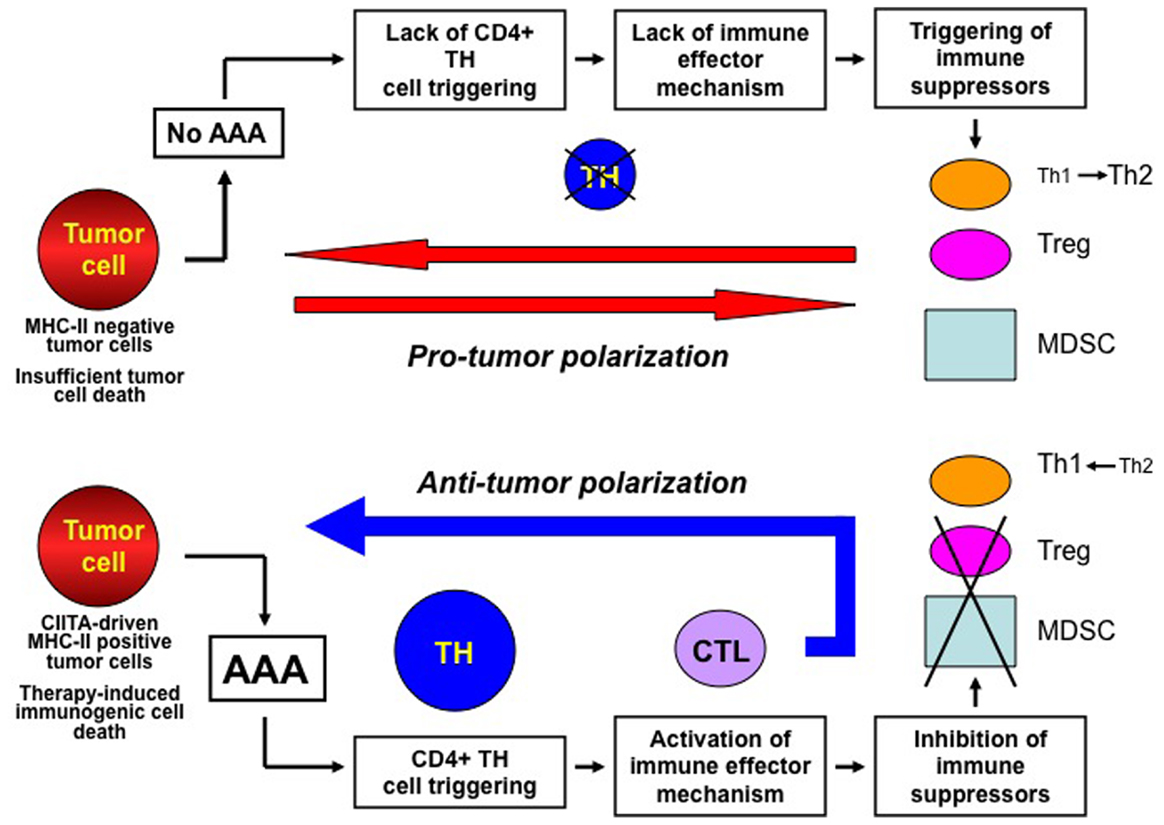
Figure 1. The key actions for establishing a protective anti-tumor immunity against cancer. The immune response against cancer is put on break by several mechanisms among which tumor antigen availability and stimulation of MHC class II-restricted CD4+ TH cells are key features (upper part of the Figure). Insufficient tumor antigen availability and/or insufficient MHC-II–tumor peptide complexes (cumulatively defined as adequate antigen availability or AAA) lead to insufficient stimulation of TH cells. This results in lack of immune effector responses which may favor the establishment of immune suppressor mechanism on anti-tumor responses, such as polarization of TH responses toward a TH2 phenotype, activation and increase number of regulatory T cells (Treg) and myeloid-derived-suppressor cells (MDSC), which cumulatively create a pro-tumor polarization of the tumor microenvironment. Presence of AAA (lower part of the Figure) generated, for example, by MHC class II expression in tumor cells or by therapy-induced immunogenic cell death efficiently triggers tumor specific TH cells and this is instrumental to both activate immune effector mechanisms such as CTL and repress and/or prevent suppressor mechanisms on protective immunity. This results in the generation of an anti-tumor microenvironment and in a strong adaptive immune response against the tumor.
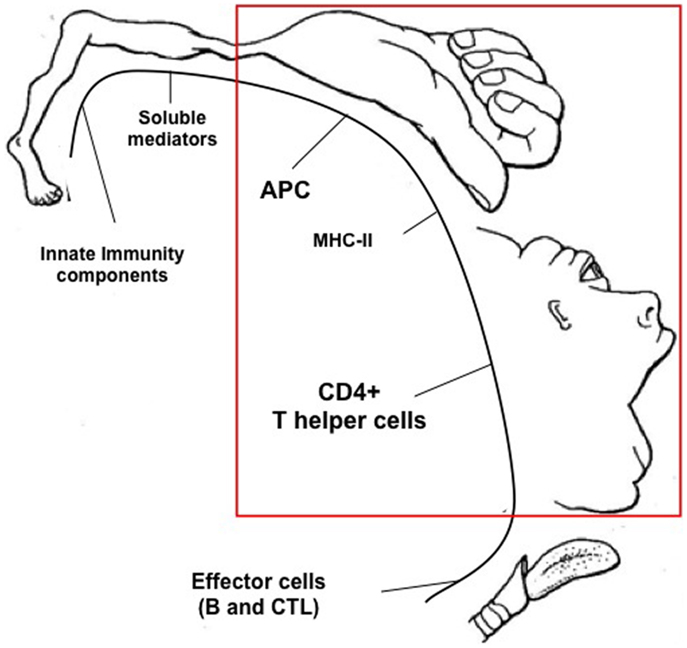
Figure 2. Proposed hierarchic position of components of the immune system involved in the productive response against cancer. By similarity with the pictorial representation of the “Cortical Homunculus” of W. Penfield, a hypothetical “Immunological Homunculus” is depicted identifying specific components of the immune system as the hierarchically most important actors against the tumor. These are, in primis, the tumor antigen presenting function exerted, for example, by dendritic cells or by tumor cells expressing MHC class II molecules, and the MHC class II-restricted CD4+ T helper cells. Other important components are defined as effector cells, innate immunity components, and soluble mediators.
By similarity, we believe that classical anti-tumor chemotherapeutic or radiotherapeutic approaches that generate or increase an underlying anti-tumor adaptive immune response are most successful when they can generate AAA and indeed many examples supporting this concept have been described (56, 60, 61, 73).
Concluding Remarks and Perspective for Vaccination Against Human Tumors
A large number of studies both in animal experimental tumor models and in human tumor-bearing hosts have strongly suggested that anti-tumor immunity can be triggered and/or implemented by several preventive and therapeutical modalities. Can we translate this information to provide in the very near future better anti-tumor treatments and, hopefully, more reliable anti-tumor cure? Spontaneous or experimentally induced HLA class II expression by exogenously transferred CIITA in tumor cells may be instrumental to purify and sequence more immunogenically relevant HLA class II-bound TAA peptides. In conjunction with similarly derived HLA class I-restricted TAA peptides, they will be the basis for the construction of a multi-peptide, multi-epitope vaccine that can target both CD4+ TH and CD8+ CTL anti-tumor responses.
In relation to chemotherapy, radiotherapy, and cytokine-mediated therapy and their effects on anti-tumor immunity, we believe it will be of particular relevance to select new drugs, to apply radiotherapeutic modalities, and to construct cytokine-based compounds, respectively, that may offer the best effects in stimulating AAA and consequent triggering of anti-tumor specific TH cells. This, in turn, will set the ground for the generation of what we have defined as therapy-induced anti-tumor vaccination.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the following grants to RSA: European Community FP7 Grant no. 602893 “Cancer Vaccine Development for Hepatocellular Carcinoma-HepaVAC”; Associazione Italiana Ricerca sul Cancro “New strategies of tumor vaccination and immunotherapy based on optimized triggering of anti-tumor CD4+ T cells” (AIRC IG 8862); Italian Ministry of University and Research project PRIN “New strategies of immunointervention against tumors” (2008-WXF7KK); Fondazione Cariplo “Cellular and molecular basis of human retroviral-dependent pathology”(2008-2230); University of Insubria “FAR 2011 and 2012.” “FAR 2009” and “FAR 2010” to Giovanna Tosi. Letizia Lombardo, Rawan Abdallah, and Goutham Raval are students in the PhD Program of Experimental Medicine and Oncology, University of Insubria; For Rawan Abdallah the PhD program is granted by Fondazione Cariplo: Post-graduate Program of Excellence in Innovative Biomedical Technologies 2009–2957. For Goutham Raval the PhD Program is granted by Fondazione Anna Villa e Felice Rusconi. Greta Forlani was supported by University of Insubria and Regione Lombardia Consortium Project DOTE-UNIRE: TumVac, New Strategies of Anti-Tumor Vaccination.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
2. Zinkernagel RM. On ‘reactivity’ versus ‘tolerance’. Immunol Cell Biol (2004) 82:343–52. doi:10.1111/j.0818-9641.2004.01255.x
3. Van Der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev (2002) 188:51–64. doi:10.1034/j.1600-065X.2002.18806.x
4. Dunn JP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
5. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10:909–15. doi:10.1038/nm1100
6. Vesely M, Kershaw M, Schreiber R, Smyth M. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi:10.1146/annurev-immunol-031210-101324
7. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6:24–37. doi:10.1038/nrc1782
8. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18:349–55. doi:10.1016/j.semcancer.2008.03.004
9. Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of the immune response. Annu Rev Immunol (2004) 22:531–62. doi:10.1146/annurev.immunol.21.120601.141122
10. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
11. Accolla RS, Tosi G. Optimal MHC-II-restricted tumor antigen presentation to CD4+ T helper cells: the key issue for development of anti-tumor vaccines. J Transl Med (2012) 9:106. doi:10.1186/1479-5876-9-106
12. Accolla RS, Tosi G. Adequate antigen availability: a key issue for novel approaches to tumor vaccination and tumor immunotherapy. J Neuroimmune Pharmacol (2013) 8:28–36. doi:10.1007/s11481-012-9423-7
13. Germain RN, Margulies DH. The biochemistry and cellular biology of antigen processing and presentation. Annu Rev Immunol (1993) 11:403–50. doi:10.1146/annurev.iy.11.040193.002155
14. Pardoll DM, Topalian SL. The role of CD4+ T cell responses in anti-tumor immunity. Curr Opin Immunol (1998) 10:588–94. doi:10.1016/S0952-7915(98)80228-8
15. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll DM, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med (1998) 188:2357–68. doi:10.1084/jem.188.12.2357
16. Boon T, Cerottini JC, Van den Eynde B, Van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol (1994) 12:337–65. doi:10.1146/annurev.iy.12.040194.002005
17. Rudensky A, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CAJ. Sequence analysis of peptides bound to MHC class II molecules. Nature (1991) 353:622–7. doi:10.1038/353622a0
18. Schimd D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility class II molecules continuously receive input from autophagosomes. Immunity (2007) 26:79–92. doi:10.1016/j.immuni.2006.10.018
19. Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med (1993) 178:27–47. doi:10.1084/jem.178.1.27
20. Jaraquemada D, Marti M, Long EO. An endogenous processing pathway in vaccinia virus-infected cells for presentation of cytosolic antigens to MHC class II-restricted T cells. J Exp Med (1990) 172:947–54. doi:10.1084/jem.172.3.947
21. Sartoris S, Valle MT, De Lerma Barbaro A, Tosi G, Cestari T, D’Agostino A, et al. HLA class II expression in uninducible hepatocarcinoma cells after transfection of the AIR-1 gene product CIITA. Acquisition of antigen processing and presentation capacity. J Immunol (1998) 161:814–20.
22. Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, et al. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res (2006) 12:3435–43. doi:10.1158/1078-0432.CCR-06-0165
23. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer (2004) 4:11–22. doi:10.1038/nrc1252
24. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature (2007) 449:419–26. doi:10.1038/nature06175
25. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity (2013) 39:38–48. doi:10.1016/j.immuni.2013.07.004
26. Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, et al. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res (2013) 73:19–29. doi:10.1158/0008-5472.CAN-12-1127
27. Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol (2003) 33:1183–92. doi:10.1002/eji.200323712
28. Armstrong TD, Clements VK, Martin BK, Ting JPY, Ostrand-Rosenberg S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc Natl Acad Sci U S A (1997) 94:6886–91. doi:10.1073/pnas.94.13.6886
29. Jabrane-Ferrat N, Campbell MJ, Esserman LJ, Peterlin BM. Challenge with mammary tumor cells expressing MHC class II and CD80 prevents the development of spontaneously arising tumors in MMTV-neu transgenic mice. Cancer Gene Ther (2006) 13:1002–10. doi:10.1038/sj.cgt.7700974
30. Dolan BP, Gibbs KDJ, Ostrand-Rosenberg S. Tumor-specific CD4+ T cells are activated by “cross-dressed” dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J Immunol (2006) 176:1447–55.
31. Accolla RS, Scarpellino L, Carra G, Guardiola J. Trans-acting element(s) operating across species barriers positively regulate the expression of major histocompatibility complex class II genes. J Exp Med (1985) 162:1117–33. doi:10.1084/jem.162.4.1117
32. Accolla RS, Jotterand-Bellomo M, Scarpellino L, Maffei A, Carra G, Guardiola J. Air-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. J Exp Med (1986) 164:369–74. doi:10.1084/jem.164.1.369
33. Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell (1993) 75:135–46. doi:10.1016/0092-8674(93)90685-J
34. Accolla RS, Carra G, Buchegger F, Carrel S, Mach J-P. The human Ia-associated invariant chain is synthesized in Ia-negative variants and is not expressed on the cell surface of both Ia-negative and Ia-positive parental cells. J Immunol (1985) 134:3265–71.
35. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11:823–36. doi:10.1038/nri3084
36. Viville S, Neefjes J, Lotteau V, Dierich A, Lemeur M, Ploegh H, et al. Mice lacking the MHC class II-associated invariant chain. Cell (1993) 72:635–48. doi:10.1016/0092-8674(93)90081-Z
37. Harton JA, Ting JPY. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol (2000) 20:6185–94. doi:10.1128/MCB.20.17.6185-6194.2000
38. Bikoff EK, Huang LY, Episkopou V, van Meerwijk J, Germain RN, Robertson EJ. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+ T cell selection in mice lacking invariant chain expression. J Exp Med (1993) 177:1699–712. doi:10.1084/jem.177.6.1699
39. Frangione V, Mortara L, Castellani P, De Lerma Barbaro A, Accolla RS. CIITA-driven MHC-II positive tumor cells: preventive vaccines and superior generators of anti-tumor CD4+ T lymphocytes for immunotherapy. Int J Cancer (2010) 127:1614–24. doi:10.1002/ijc.25183
40. Mortara L, Frangione V, Castellani P, De Lerma Barbaro A, Accolla RS. Irradiated CIITA-positive mammary adenocarcinoma cells act as a potent anti-tumor-preventive vaccine by inducing tumor-specific CD4+ T cell priming and CD8+ T cell effector functions. Int Immunol (2009) 21:655–65. doi:10.1093/intimm/dxp034
41. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med (2012) 18:1254–61. doi:10.1038/nm.2883
42. Scurr M, Bloom A, Pembroke T, Srinivasan R, Brown C, Smart K, et al. Escalating regulation of 5T4-specific IFN-γ+ CD4+ T cells distinguishes colorectal cancer patients from healthy controls and provides a target for in vivo therapy. Cancer Immunol Res (2013) 1(6):416–25. doi:10.1158/2326-6066
43. Mortara L, Balza E, Sassi F, Castellani P, Carnemolla B, De Lerma Barbaro A, et al. Therapy-induced antitumor vaccination by targeting tumor necrosis factor alpha to tumor vessels in combination with melphalan. Eur J Immunol (2007) 37:3381–92. doi:10.1002/eji.200737450
44. Ugel S, Peranzoni E, Desantis G, Chioda M, Walter S, Weinschenk T, et al. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep (2012) 2:628–39. doi:10.1016/j.celrep.2012.08.006
45. Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol (2003) 170:4905–13.
46. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39:74–88. doi:10.1016/j.immuni.2013.06.014
47. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One (2012) 7(3):e32542. doi:10.1371/journal.pone.0032542
48. Ménard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C, et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res (2009) 69:3563–9. doi:10.1158/0008-5472.CAN-08-3807
49. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24:5373–80. doi:10.1200/JCO.2006.05.9584
50. Ladoire S, Mignot G, Dabakuyo S, Arnould L, Apetoh L, Rébé C, et al. In situ immune response after neoadjuvant chemotherapy for breast cancer predicts survival. J Pathol (2011) 224:389–400. doi:10.1002/path.2866
51. Maeda K, Hazama S, Tokuno K, Kan S, Maeda Y, Watanabe Y, et al. Impact of chemotherapy for colorectal cancer on regulatory T-cells and tumor immunity. Anticancer Res (2011) 31:4569–74.
52. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11:6713–21. doi:10.1158/1078-0432.CCR-05-0883
53. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16:4583–94. doi:10.1158/1078-0432.CCR-10-0733
54. Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol (2013) 4:273–80. doi:10.1016/j.semradonc.2013.05.009
55. Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity (2011) 35:467–77. doi:10.1016/j.immuni.2011.09.006
56. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood (2009) 114:589–95. doi:10.1182/blood-2009-02-206870
57. Wu CY, Yang LH, Yang HY, Knoff J, Peng S, Lin YH, et al. Enhanced cancer radiotherapy through immunosuppressive stromal cell destruction in tumors. Clin Cancer Res (2014) 20(3):644–57. doi:10.1158/1078-0432.CCR-13-1334
58. Gupta A, Sharma A, von Boehmer L, Surace L, Knuth A, van den Broek M. Radiotherapy supports protective tumor-specific immunity. Oncoimmunology (2012) 1:1610–1. doi:10.4161/onci.21478
59. Sharma A, Bode B, Wenger RH, Lehmann K, Sartori AA, Moch H, et al. γ-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS One (2011) 6:e28217. doi:10.1371/journal.pone.0028217
60. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol (2005) 174:7516–23.
61. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med (2006) 203:1259–71. doi:10.1084/jem.20052494
62. Takeshima T, Chamoto K, Wakita D, Ohkuri T, Togashi Y, Shirato H, et al. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res (2010) 70:2697–706. doi:10.1158/0008-5472.CAN-09-2982
63. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 9:1050–9. doi:10.1038/nm1622
64. Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell (2010) 140:798–804. doi:10.1016/j.cell.2010.02.015
65. Balza E, Carnemolla B, Mortara L, Castellani P, Soncini D, Accolla RS, et al. Therapy-induced antitumor vaccination in neuroblastomas by the combined targeting of IL-2 and TNFalpha. Int J Cancer (2010) 127:101–10. doi:10.1002/ijc.25018
66. Borsi L, Balza E, Carnemolla B, Sassi F, Castellani P, Berndt A, et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood (2003) 102:4384–92. doi:10.1182/blood-2003-04-1039
67. Balza E, Mortara L, Sassi F, Monteghirfo S, Carnemolla B, Castellani P, et al. Targeted delivery of tumor necrosis factor alpha to tumor vessels induces a therapeutic T cell-mediated immune response that protects the host against syngeneic tumors of different histologic origin. Clin Cancer Res (2006) 12:2575–82. doi:10.1158/1078-0432.CCR-05-2448
68. Roake JA, Rao AS, Morris PJ, Larsen CP, Hankins DF, Austyn JM. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J Exp Med (1995) 181:2237–47. doi:10.1084/jem.181.6.2237
69. Papadia F, Basso V, Patuzzo R, Maurichi A, Di Florio A, Zardi L, et al. Isolated limb perfusion with the tumor-targeting human monoclonal antibody-cytokine fusion protein L19-TNF plus melphalan and mild hyperthermia in patients with locally advanced extremity melanoma. J Surg Oncol (2013) 107:173–9. doi:10.1002/jso.23168
70. Hemmerle T, Probst P, Giovannoni L, Green AJ, Meyer T, Neri D. The antibody-based targeted delivery of TNF in combination with doxorubicin eradicates sarcomas in mice and confers protective immunity. Br J Cancer (2013) 103:1206–13. doi:10.1038/bjc.2013.421
71. Lake RA, Robinson BWS. Immunotherapy and chemotherapy – a practical partnership. Nat Rev Cancer (2005) 5:397–404. doi:10.1038/nrc1613
72. Cohen IR. The cognitive paradigm and the immunological homunculus. Immunol Today (1992) 13:490–4. doi:10.1016/0167-5699(92)90024-2
Keywords: CIITA, MHC class II, T helper cells, tumor vaccines, anti-tumor immunity
Citation: Accolla RS, Lombardo L, Abdallah R, Raval G, Forlani G and Tosi G (2014) Boosting the MHC class II-restricted tumor antigen presentation to CD4+ T helper cells: a critical issue for triggering protective immunity and re-orienting the tumor microenvironment toward an anti-tumor state. Front. Oncol. 4:32. doi: 10.3389/fonc.2014.00032
Received: 20 December 2013; Paper pending published: 18 January 2014;
Accepted: 04 February 2014; Published online: 18 February 2014.
Edited by:
Jozsef Dudas, Medical University Innsbruck, AustriaReviewed by:
Jozsef Dudas, Medical University Innsbruck, AustriaSieghart Sopper, Medical University Innsbruck, Austria
Copyright: © 2014 Accolla, Lombardo, Abdallah, Raval, Forlani and Tosi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto S. Accolla, Laboratory of General Pathology and Immunology, Department of Surgical and Morphological Sciences, University of Insubria, Via Ottorino Rossi 9, Varese 21100, Italy e-mail:cm9iZXJ0by5hY2NvbGxhQHVuaW5zdWJyaWEuaXQ=;YWNjb2xsYS5yb2JlcnRvQGdtYWlsLmNvbQ==