 Lakshmipathi Vadlakonda
Lakshmipathi Vadlakonda V. D. K. Reddy
V. D. K. Reddy Mukesh Pasupuleti
Mukesh Pasupuleti Pallu Reddanna
Pallu Reddanna- 1Cell Biology and Enzymology, Department of Zoology, Kakatiya University, Warangal, India
- 2Department of Animal Sciences, School of Life Sciences, University of Hyderabad, Hyderabad, India
- 3SRM Research Institute, Sri Ramaswamy Memorial University, Chennai, India
- 4National Institute of Animal Biotechnology, Hyderabad, India
In our previous article (1), we presented a unifying principle in the hypotheses of Pasteur, Warburg, and Crabtree that “an inverse relation exists between glucose uptake and respiration.” Warburg’s hypothesis laid emphasis on glucose metabolism and damaged respiration (mitochondria) for growth of cells in cancer (2). Pasteur recognized that yeast can grow only if ammonium tartrate (nitrogen source) is available, whether oxygen is available or not (3) and ammonium is transformed into a “complex albuminoid” (protein) material. Growth is faster in the presence of oxygen and for producing one unit of mass, yeast requires only 1/15th of glucose when compared to that in the absence of oxygen (4–10 parts as against 60–80 parts). Since the ratio of ATP produced per one glucose molecule in glycolysis/oxidative phosphorylation (OXPHOS) is 1:15, the energy consumed per unit of growth of yeast remains same (60X1 and 4X15 ATP). We call this: “the Pasteur’s dictum,” which is different from the “Pasteur effect” introduced by Warburg. Lagunas et al. (4) in early 80s reported that Pasteur effect is observed only in resting cells in the absence of nitrogen, but not in growing cells. Warburg’s hypothesis relies on energetics of glucose metabolism and damaged respiration but ignores Pasteur’s demonstration of nitrogen as the primary requirement for triggering growth. It also ignores the anabolic functions of mitochondria (5, 6) and the role of glutamine, another key nutrient avidly consumed by actively proliferating cells (7, 8). We present in this article, a hypothesis that activation of complexes of mechanistic target of rapamycin (mTORC1 and C2) by amino acids (nitrogen source) is the molecular explanation of the “Pasteur’s dictum.” Amino acids activate both mTORC1 and C2 independent of insulin or growth factor signaling (IIS). Amino acids are also required for activation of mTORC1 in IIS dependent pathway.
Mechanistic Target of Rapamycin as the HUB of Nutrient Sensing
Pasteur’s work demonstrates that the quality of nutrients is important for cell growth. PI3K–Akt–mTOR signaling is recognized as a key player in uptake of nutrients (9). mTOR is a multi-protein complex and exists in two complexes, mTORC1 and mTORC2. While mLST8 and raptor are key components of mTORC1, mLST8, rictor, mSIN1 are required for mTORC2 assembly and activity [reviewed in Ref. (10, 11)]. Nutrients, growth factors, and oxygen were shown to activate mTORC1 (12); the upstream regulators of mTORC2 are less understood. We have earlier shown that a reciprocal relation exists between mTORC1 and mTORC2, which is intertwined with Akt phosphorylations (13). While mTORC2 is an upstream kinase of Akt S473 phosphorylation (14), mTORC1 is the downstream effector of Akt T308 phosphorylated state. Activated mTORC1 inhibits mTORC2 assembly (see below).
Activation of mTOR Complexes by Amino Acids is the Modern Explanation of “Pasteur’s Dictum”
Amino acids were shown to activate mTORC1, independent of the growth factor signaling (15–17) and are also required for mTORC1 activity even under growth factor mediated conditions (18). Recent reports suggest that mTORC2 can also be activated by amino acids (19, 20). Tato et al. demonstrated that starvation and culture conditions influence activation of either mTORC1 or mTORC2 by amino acids (20). Rosario et al. demonstrated that in primary human placental cells, activation of amino acid transporters requires both mTORC1 and mTORC2 (19); localization of amino acid transporters is Tor2-independent in fission yeast (21). This indicates that mTORC1 could be the primary requirement for amino acid uptake. Activation of mTORC1 appears to precede mTORC2, which is in tune with its role in protein translation. But, Akt stability during translation requires mTORC2 (22); it later phosphorylates Akt at S473 (14).
Activation of mTORC1 and creation of anabolic environment depend on ATP/ADP ratio (13). ATP production depends on glucose uptake. Studies on the relation between oxygen and glucose consumption (Crabtree effect) in the middle of twentieth Century [reviewed by Ibsen (23)] suggest that in response to glucose, cells consume initially high amounts of oxygen for about 20–120 s, which is followed by an inhibitory period but rises to stabilize around 30% of the original. Uncouplers of OXPHOS were shown to release the inhibitory effect on oxygen consumption, thus relating oxygen consumption and glucose uptake to ATP production. In terms of time frame, mitochondria devote only a short time (<2 min) for producing ATP in response to glucose availability and the buildup of ATP has regulatory effect on glucose uptake (24). ATP production, under OXPHOS, is 15-folds higher than in glycolysis and high ATP/ADP ratio is essential for functional stability of oncoprotein, Akt (25, 26) and to transform the intra cellular environment to ATP rich anabolic environment (Figure 1A).
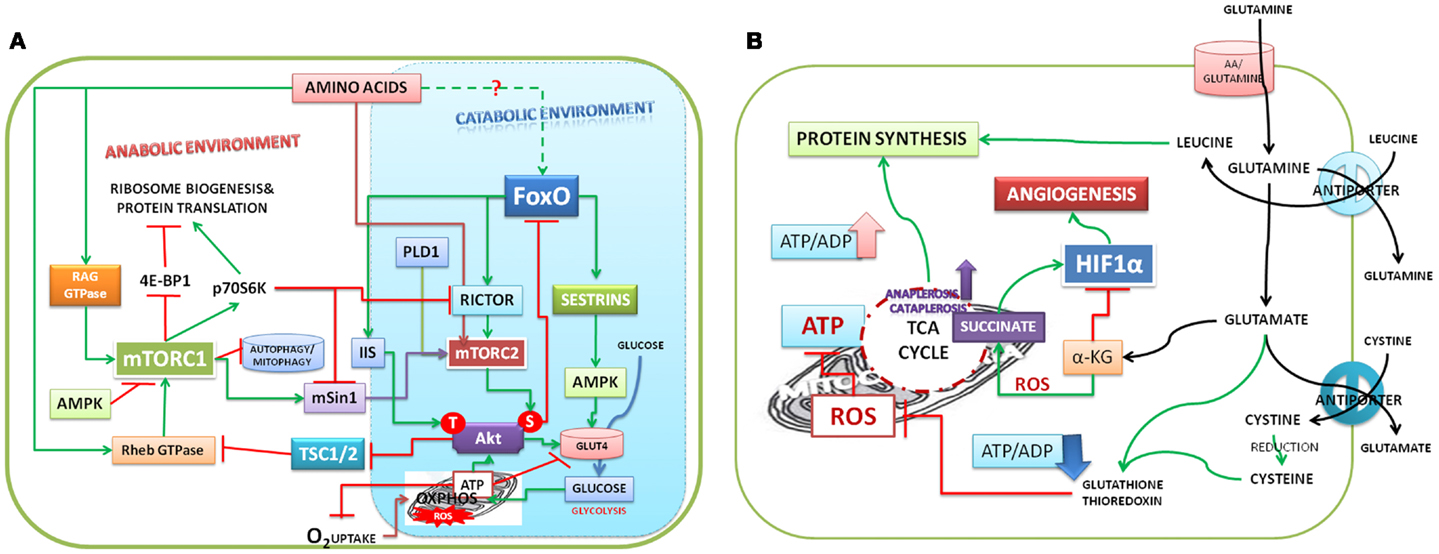
Figure 1. (A) Model depicting amino acids that trigger the downstream signals: amino acids activate mTORC1 and mTORC2 and protein biosynthesis. Both mTORC1 and mTORC2 cooperate with each other in stabilizing Akt translation. mTORC2 phosphorylates AktS473 and promotes glucose uptake. Akt S473 phosphorylates and causes nuclear exclusion of FoxO and promotes oxidative phosphorylation and ATP production (see Ref. 73). IIS activates PDPK1 (3-phosphoinositide dependent protein kinase-1), which phosphorylates the Akt at T308. Akt phosphorylations are stabilized by high ATP/ADP ratio. Akt under IIS activates mTORC1 to promote the anabolic environment. High ATP/ADP ratio inhibits uptake of glucose by phosphorylation of glucose transporter. S6K, one of the targets of mTORC1, inhibits mTORC2 by phosphorylating both rictor and mSin1. Inhibition of autophagy and mitophagy by mTORC1 shifts the mitochondrial function from ATP production to supply of amino acids, citrate for biosynthesis of proteins/lipids. (B) Model depicting the role of glutamine in mitochondrial function and anabolic environment in cells: glutamine entering into cells is hydrolyzed to generate glutamate by glutaminase. Glutamine – leucine antiporter promotes the uptake of leucine, which activates mTORC1 and protein synthesis. Glutamate has multiple functions; uptake of cystine in exchange for glutamate promotes glutathione biosynthesis, which regulates ROS. ROS accumulation inhibits ATP production. Glutamate replenishes α-ketoglutarate (α-KG) to Krebs cycle and promotes ATP production and inhibits HIF1-α. ROS promotes non-enzymatic decarboxylation of α-KG to succinate, which is an activator of HIF1-α, glycolysis, and angiogenesis.
Glucose Uptake is Mediated by Akt S473 Which is Phosphorylated by mTORC2
Akt by phosphorylating its substrate AS160 was shown to mediate GLUT4 translocation to the membrane and promote glucose uptake. Interestingly, exercise/contractile activity also phosphorylates AS160 in muscle and AMP-activated protein kinase (AMPK) appears to have a role in this (27). Studies with rictor knock out cells had demonstrated that integrity of mTORC2 and phosphorylation of Akt S473 are essential for GLUT4 translocation and uptake of glucose (28, 29). Insulin resistance was shown to be the result of mTORC1 activation; one of its downstream targets, S6K, was shown to cause insulin resistance (30). Although phosphorylation of IRS1 by S6K was suggested to be the cause (31), recent studies demonstrated that S6k phosphorylation of rictor (32, 33) and mSin1 (34) results in mTORC2 inactivation. Further, inactivation of mTORC2 was shown to affect IIS mediated as well as PGDF and EGF mediated phosphorylation of Akt (34). This suggests that mTORC2 is upstream of Akt; it protects Akt during translation by phosphorylation of its turn motif site T450 and prevents its ubiquitination (22). Rapamycin inhibition of mTORC1 activates mTORC2 dependent AktS473. Rapamycin was also shown to up-regulate IGF-IR, Her2 expression, and reduced the phosphorylation of GSK-3β and NF-κB in an mTORC2 dependent way (35). These reports suggest that mTORC2 is up-regulated in metabolically starved or in mTORC1 inhibited cells and it is upstream of Akt activation.
FoxO is the Regulator of Both mTOR and IIS
FoxO, as the transcription factor of rictor, plays a key role in the assembly of mTORC2; FoxO also inhibits mTORC1, but the inhibition depends on expression of Sestrin3, rictor, and activation of AMPK (36). FoxO3a is mainly activated by reactive oxygen species (ROS) and inhibits the mitochondrial gene expression (37). FoxO is the transcriptional regulator of insulin receptor (38), its substrate IRS-2 (39), and a reciprocal relation between FoxO and IRS was demonstrated in β-cells (40). In addition, it can inhibit Wnt pathway and proliferation of cells (41). FoxO also inhibits Myc and controls cell metabolism (42, 43).
Aerobic Glycolysis, an Aberration on Mitochondrial Function
Warburg’s hypothesis of aerobic glycolysis centers on the respiratory damage (mitochondrial dysfunction) as the primary cause of malignancy. This, in our opinion, is an aberration of mitochondrial function. Traditionally mitochondria had been viewed as the ATP producing “power house” of the cell. It should be recognized that ATP production is only one of the short time functions of mitochondria. Substrate shuttles like, malate, aspartate, glutamate and citrate are critical for anaplerotic and cataplerotic reactions of Krebs cycle. They are the key sources of carbon and nitrogen requirements for protein and lipid biosynthesis of growing cells (5, 6). These reports suggest that mitochondria shift their function from ATP production to coordinate the biosynthetic function of proliferating cells and it has a time frame, which depends on ATP/ADP ratio and production of ROS in mitochondria. During active ATP production, ROS is kept under check and glutamine plays a critical role in redox homeostasis.
Glutamine in Mitochondrial Metabolic Reprograming
Glutamine, avidly consumed by actively proliferating cancer cells (7, 8), has pleiotropic functions; chief amongst which are activation of mTORC1 signaling (44), regulation of glutamate levels, uptake of cysteine and leucine through the amino acids antiporters [reviewed in Ref. (8)]. Deamination of glutamine by two step reactions, glutaminase (GLS) and glutamate dehydrogenase, replenishes α-ketoglutarate (α-KG), which is critical for sensing oxygen, ATP production in transformed cells (Figure 1B). It is also a substrate for prolyl hydroxylases (PHDs), which inhibit HIF1α signaling (45, 46). ROS decarboxylate α-KG non-enzymatically to succinate (47) and inactivate PHDs and activate HIF1α signaling.
Glutaminase, which hydrolyzes glutamine to glutamate and ammonia, is up-regulated in several cancer cells (48, 49). Silencing of phosphate-activated mitochondrial GLS2 gene of HeLaR exposed to irradiation increased intracellular ROS and reduced the productions of antioxidants GSH, NADH, and NADPH (50). Glutamate exchange with cystine plays critical role in maintenance of the redox homeostasis mediated by cysteine thiol oxidation, thioredoxin system, and glutathione (GSH) peroxidases [reviewed in Ref. (51)].
Glutamine and glutamate are the substrates for leucine and cystine antiporters, while leucine is an activator of anabolism (52), cystine inside the cells is reduced to cysteine, which plays a key role in glutathione biosynthesis and redox homeostasis (53, 54). Interestingly, glutamine transport into cells is regulated by the inflammatory cytokine, the tumor necrosis factor α (TNF-α), which inhibits glutamine/Na+ co-transport (55) promotes the export of glutamate by activating cystine/glutamate transporters of microglia causes neurotoxicity in Japanese encephalitis (56). In contrast to cystine/glutamate transporter, activation of the leucine/glutamine antiporter SLC7A5/SLC3A2 and the amino acid sensor MAP4K3 improve leucine availability and were shown to activate mTORC1 and anabolic environment (57).
The Lactic Acid Puzzle
Production of lactic acid in cancer cells is one of the strong arguments in favor of aerobic glycolysis (58). Lactate is produced from pyruvate in glycolysis; but in actively proliferating cells, oncogenes and ROS inhibit pyruvate kinase (PK) and block pyruvate production (59). Accumulated phosphoenolpyruvate (PEP) acts as a feedback regulator of glycolysis (60); altered glycolytic pathway supplies building blocks for biosynthesis of lipids and nucleic acids (61–63). It has been suggested in support of lactate theory, that cancer tissue is a mixture of catabolic and anabolic cells with variable access to oxygen and exhibits metabolic symbiosis; lactate produced in non-proliferating cells could be a source of fuel for proliferating cells (64, 65).
Malate as a Source of Pyruvate
Mitochondrial substrate shuttles, like aspartate, glutamate, and succinate, which are key source of NADPH required for biosynthesis and the ATP, are also sources for malate [reviewed in Ref. (8, 45, 66)]. Malic enzyme (ME), which catalyzes the decarboxylation of malate to pyruvate exists in three isoforms, the cytosolic, mitochondrial NADP+ dependent forms, and the mitochondrial NAD+ dependent form (67, 68). ME plays a key role in lipogenesis and glutamine metabolism by generating the NADPH (69). Inhibition of MEs reciprocally activates p53, which regulates cell metabolism and proliferation (70). Anaplerotic flux of aspartate and glutamate in liver cells was shown to increase the lactate production (71); transamination of alanine to glutamate produces pyruvate, especially under glutamine deprived conditions.
In summary, we suggest that nitrogen source is critical to cell growth and oxygen’s role is to activate ATP production and limit glucose uptake. Nitrogen uptake and reduced dependency on glucose for growth in the presence of oxygen is the “Pasteur’s dictum.” Activation of mTORC1 by amino acids, independent of IIS, is the molecular recognition of “Pasteur’s dictum.” Activation of mTORC1 appears to precede mTORC2, which perhaps facilitates translation of Akt and its subsequent S473 phosphorylation and promotes glucose uptake and energy production as well as IIS activation. Stabilization of mTORC1 by IIS for anabolic activity of cells depends on ATP/ADP ratio in the absence of which, cells may recycle as stem cells. In anabolic environment, mitochondria reprogram their function for biosynthetic activity to replenish the carbon and amino acid resources for lipid and protein biosynthesis. Inhibition of mTORC1 in growing cells depends on ROS and AMPK. Higher levels of dietary amino acids result in longtime mTORC1 activation and inhibition of mTORC2 and insulin resistance, which appear to be the cause of metabolic pathologies and the secret of healthy life under dietary restriction (72).
Acknowledgments
Lakshmipathi Vadlakonda recollects his brief stay during 2008 at “Cell Works, Bangalore,” which enabled him to have deeper insights into cell signaling. Thanks are due to Dr. Shireen Vali, Chief Scientific Officer and Dr. Taher Abbasi, the CEO for their invitation to be part of their team.
Abbreviations
Akt, protein kinase B (T308, S473 – phosphorylated sites threonine 308 and serine 473); AMPK, AMP-activated protein kinase; FoxO, fork head transcription factors of O group; GLUT, glucose transporter; GLS, Glutaminase; Her2, human epidermal growth factor receptor-2; IGF, insulin growth factor; IRS, insulin receptor substrate; IGF-IR, insulin-like growth factor-I receptor; mTORC1, 2, mechanistic target of rapamycin complex 1 and 2 (mTOR, formerly known as mammalian target of rapamycin); mLST8, MTOR associated protein, LST8 homolog (S. cerevisiae); mSIN1, mitogen-activated protein kinase associated protein 1; Myc, v-myc avian myelocytomatosis viral oncogene homolog (c-Myc); OXPHOS, oxidative phosphorylation; PEP, phosphoenolpyruvate; PA, phosphatidic acid; PDGF, platelet-derived growth factor; PI3FK, phosphatidylinositol 3-kinases; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PLD1, phospholipase D1, phosphatidylcholine-specific ROS, reaction oxygen species; S6K, the p70 ribosomal, Sestrins, stress response proteins.
References
1. Vadlakonda L, Dash A, Pasupuleti M, Anil Kumar K, Reddanna P. Did we get Pasteur, Warburg, and Crabtree on a right note? Front Oncol (2013) 3:186. doi: 10.3389/fonc.2013.00186
2. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi:10.1126/science.123.3191.309
3. Barnett JA. A history of research on yeasts 2: Louis Pasteur and his contemporaries, 1850–1880. Yeast (2000) 16:755–71. doi:10.1002/1097-0061(20000615)16:8<755::AID-YEA587>3.0.CO;2-4
4. Lagunas R, Dominguez C, Busturia A, Saez MJ. Mechanisms of appearance of the Pasteur effect in Saccharomyces cerevisiae: inactivation of sugar transport systems. J Bacteriol (1982) 152:19–25.
5. Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res (2012) 25:375–83. doi:10.1111/j.1755-148X.2012.00989.x
6. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem (2002) 277:30409–12. doi:10.1074/jbc.R200006200
7. Souba WW. Glutamine: a key substrate for the splanchnic bed. Annu Rev Nutr (1991) 11:285–308. doi:10.1146/annurev.nu.11.070191.001441
8. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123:3678–84. doi:10.1172/JCI69600
9. Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell (2002) 13:2276–88. doi:10.1091/mbc.01-12-0584
10. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2010) 12:21–35. doi:10.1038/nrm3025
11. Laplante M, Sabatini DM. mTOR signaling. Cold Spring Harb Perspect Biol (2012) 4. doi:10.1101/cshperspect.a011593
12. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol (2013) 15:555–64. doi:10.1038/ncb2763
13. Vadlakonda L, Dash A, Pasupuleti M, Anil Kumar K, Reddanna P. The paradox of Akt-mTOR interactions. Front Oncol (2013) 3:165. doi:10.3389/fonc.2013.00165
14. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi:10.1126/science.1106148
15. Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem (1998) 273:14484–94. doi:10.1074/jbc.273.23.14484
16. Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab (2009) 296:E592–602. doi:10.1152/ajpendo.90645.2008
17. Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med (2012) 18:524–33. doi:10.1016/j.molmed.2012.05.007
18. Groenewoud MJ, Zwartkruis FJ. Rheb and Rags come together at the lysosome to activate mTORC1. Biochem Soc Trans (2013) 41:951–5. doi:10.1042/BST20130037
19. Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol (2013) 591:609–25. doi:10.1113/jphysiol.2012.238014
20. Tato I, Bartrons R, Ventura F, Rosa JL. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem (2013) 286:6128–42. doi:10.1074/jbc.M110.166991
21. Nakase Y, Nakase M, Kashiwazaki J, Murai T, Otsubo Y, Mabuchi I, et al. The fission yeast beta-arrestin-like protein Any1 is involved in TSC-Rheb signaling and the regulation of amino acid transporters. J Cell Sci (2013) 126:3972–81. doi:10.1242/jcs.128355
22. Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J (2010) 29:3939–51. doi:10.1038/emboj.2010.271
24. Blodgett DM, De Zutter JK, Levine KB, Karim P, Carruthers A. Structural basis of GLUT1 inhibition by cytoplasmic ATP. J Gen Physiol (2007) 130:157–68. doi:10.1085/jgp.200709818
25. Chan TO, Zhang J, Rodeck U, Pascal JM, Armen RS, Spring M, et al. Resistance of Akt kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc Natl Acad Sci U S A (2011) 108:E1120–7. doi:10.1073/pnas.1109879108
26. Lin K, Lin J, Wu WI, Ballard J, Lee BB, Gloor SL, et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci Signal (2012) 5:ra37. doi:10.1126/scisignal.2002618
27. Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab (2007) 32:557–66. doi:10.1139/H07-026
28. Kumar A, Lawrence JC Jr, Jung DY, Ko HJ, Keller SR, Kim JK, et al. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes (2010) 59:1397–406. doi:10.2337/db09-1061
29. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science (2012) 335:1638–43. doi:10.1126/science.1215135
30. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature (2004) 431:200–5. doi:10.1038/nature02866
31. Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol (2001) 11:437–41. doi:10.1016/S0962-8924(01)81297-6
32. Treins C, Warne PH, Magnuson MA, Pende M, Downward J. Rictor is a novel target of p70 S6 kinase-1. Oncogene (2010) 29:1003–16. doi:10.1038/onc.2009.401
33. Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol (2010) 30:908–21. doi:10.1128/MCB.00601-09
34. Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol (2013) 15:1340–50. doi:10.1038/ncb2860
35. Lang SA, Hackl C, Moser C, Fichtner-Feigl S, Koehl GE, Schlitt HJ, et al. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim Biophys Acta (2010) 1803:435–42. doi:10.1016/j.bbamcr.2010.01.009
36. Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, et al. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and rictor. Dev Cell (2010) 18:592–604. doi:10.1016/j.devcel.2010.03.008
37. Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ (2012) 19:968–79. doi:10.1038/cdd.2011.179
38. Puig O, Tjian R. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev (2005) 19:2435–46. doi:10.1101/gad.1340505
39. Ide T, Shimano H, Yahagi N, Matsuzaka T, Nakakuki M, Yamamoto T, et al. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol (2004) 6:351–7. doi:10.1038/ncb1111
40. Tsunekawa S, Demozay D, Briaud I, McCuaig J, Accili D, Stein R, et al. FoxO feedback control of basal IRS-2 expression in pancreatic beta-cells is distinct from that in hepatocytes. Diabetes (2011) 60:2883–91. doi:10.2337/db11-0340
41. Iyer S, Ambrogini E, Bartell SM, Han L, Roberson PK, de Cabo R, et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest (2013) 123:3409–19. doi:10.1172/JCI68049
42. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab (2013) 18:726–39. doi:10.1016/j.cmet.2013.09.013
43. Graves LM, Duncan JS, Whittle MC, Johnson GL. The dynamic nature of the kinome. Biochem J (2013) 450:1–8. doi:10.1042/BJ20121456
44. Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell (2012) 47:349–58. doi:10.1016/j.molcel.2012.05.043
45. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol (2013) 9:712. doi:10.1038/msb.2013.65
46. Webb JD, Coleman ML, Pugh CW. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell Mol Life Sci (2009) 66:3539–54. doi:10.1007/s00018-009-0147-7
47. Fedotcheva NI, Sokolov AP, Kondrashova MN. Nonenzymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med (2006) 41:56–64. doi:10.1016/j.freeradbiomed.2006.02.012
48. Mohamed A, Deng X, Khuri FR, Owonikoko TK. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clin Lung Cancer (2014) 15:7–15. doi:10.1016/j.cllc.2013.09.001
49. Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood (2013) 122:3521–32. doi:10.1182/blood-2013-03-493163
50. Xiang L, Xie G, Liu C, Zhou J, Chen J, Yu S, et al. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim Biophys Acta (2013) 1833:2996–3005. doi:10.1016/j.bbamcr.2013.08.003
51. Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol (2014) 2:123–39. doi:10.1016/j.redox.2013.12.011
52. Bassil MS, Gougeon R. Muscle protein anabolism in type 2 diabetes. Curr Opin Clin Nutr Metab Care (2013) 16:83–8. doi:10.1097/MCO.0b013e32835a88ee
53. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal (2012) 18:522–55. doi:10.1089/ars.2011.4391
54. Satoh T, McKercher SR, Lipton SA. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med (2013) 66:45–57. doi:10.1016/j.freeradbiomed.2013.11.002
55. Talukder JR, Boyd B, Griffin A, Jaima A, Rajendran VM. Inflammatory cytokine TNF-alpha inhibits Na(+)-glutamine cotransport in intestinal epithelial cells. Can J Physiol Pharmacol (2013) 91:275–84. doi:10.1139/cjpp-2011-0488
56. Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Chen SY, et al. Glutamate released by Japanese encephalitis virus-infected microglia involves TNF-alpha signaling and contributes to neuronal death. Glia (2012) 60:487–501. doi:10.1002/glia.22282
57. Schriever SC, Deutsch MJ, Adamski J, Roscher AA, Ensenauer R. Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. J Nutr Biochem (2013) 24:824–31. doi:10.1016/j.jnutbio.2012.04.018
58. Hitosugi T, Chen J. Post-translational modifications and the Warburg effect. Oncogene (2013). doi:10.1038/onc.2013.406
59. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science (2012) 334:1278–83. doi:10.1126/science.1211485
60. Gruning NM, Rinnerthaler M, Bluemlein K, Mulleder M, Wamelink MM, Lehrach H, et al. Pyruvate kinase triggers a metabolic feedback loop that controls redox metabolism in respiring cells. Cell Metab (2011) 14:415–27. doi:10.1016/j.cmet.2011.06.017
61. Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol (2012) 76:325–34. doi:10.1101/sqb.2012.76.010900
62. Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A (2012) 109:6904–9. doi:10.1073/pnas.1204176109
63. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis (2013) 4:e877. doi:10.1038/cddis.2013.393
64. Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle (2013) 12:1371–84. doi:10.4161/cc.24092
65. Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle (2013) 12:2723–32. doi:10.4161/cc.25695
66. Han S, Auger C, Thomas SC, Beites CL, Appanna VD. Mitochondrial biogenesis and energy production in differentiating murine stem cells: a functional metabolic study. Cell Reprogram (2013) 16:84–90. doi:10.1089/cell.2013.0049
67. Hsieh JY, Chen SH, Hung HC. Functional roles of the tetramer organization of malic enzyme. J Biol Chem (2009) 284:18096–105. doi:10.1074/jbc.M109.005082
68. Hunnewell MG, Forbes NS. Active and inactive metabolic pathways in tumor spheroids: determination by GC-MS. Biotechnol Prog (2009) 26:789–96. doi:10.1002/btpr.360
69. Meyer FM, Stulke J. Malate metabolism in Bacillus subtilis: distinct roles for three classes of malate-oxidizing enzymes. FEMS Microbiol Lett (2012) 339:17–22. doi:10.1111/1574-6968.12041
70. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature (2013) 493:689–93. doi:10.1038/nature11776
71. Merritt ME, Harrison C, Sherry AD, Malloy CR, Burgess SC. Flux through hepatic pyruvate carboxylase and phosphoenolpyruvate carboxykinase detected by hyperpolarized 13C magnetic resonance. Proc Natl Acad Sci U S A (2011) 108:19084–9. doi:10.1073/pnas.1111247108
72. Testa G, Biasi F, Poli G, Chiarpotto E. Calorie restriction and dietary restriction mimetics: a strategy for improving healthy aging and longevity. Curr Pharm Des (2013).
Keywords: Pasteur, nitrogen, glucose, mTOR, IIS, growth
Citation: Vadlakonda L, Reddy VDK, Pasupuleti M and Reddanna P (2014) The Pasteur’s dictum: nitrogen promotes growth and oxygen reduces the need for sugar. Front. Oncol. 4:51. doi: 10.3389/fonc.2014.00051
Received: 09 November 2013; Accepted: 03 March 2014;
Published online: 17 March 2014.
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Francesco Massimo Lasorsa, Consiglio Nazionale delle Ricerche, ItalyCopyright: © 2014 Vadlakonda, Reddy, Pasupuleti and Reddanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:bHZhZGxha29uZGFAZ21haWwuY29t
†Present address: Mukesh Pasupuleti, Central Drug Research Institute, Lucknow, India