 Christian Stockmann
Christian Stockmann Dirk Schadendorf
Dirk Schadendorf Ralph Klose
Ralph Klose Iris Helfrich
Iris Helfrich- 1UMR 970, Paris Cardiovascular Research Center, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris, France
- 2Skin Cancer Unit, Dermatology Department, Medical Faculty, University Duisburg-Essen, Essen, Germany
Angiogenesis, the formation of new blood vessels, as well as inflammation with massive infiltration of leukocytes are hallmarks of various tumor entities. Various epidemiological, clinical, and experimental studies have not only demonstrated a link between chronic inflammation and cancer onset but also shown that immune cells from the bone marrow such as tumor-infiltrating macrophages significantly influence tumor progression. Tumor angiogenesis is critical for tumor development as tumors have to establish a blood supply in order to progress. Although tumor cells were first believed to fuel tumor angiogenesis, numerous studies have shown that the tumor microenvironment and infiltrating immune cell subsets are important for regulating the process of tumor angiogenesis. These infiltrates involve the adaptive immune system including several types of lymphocytes as well as cells of the innate immunity such as macrophages, neutrophils, eosinophils, mast cells, dendritic cells, and natural killer cells. Besides their known immune function, these cells are now recognized for their crucial role in regulating the formation and the remodeling of blood vessels in the tumor. In this review, we will discuss for each cell type the mechanisms that regulate the vascular phenotype and its impact on tumor growth and metastasis.
Introduction
Angiogenesis, which is the outgrowth of new vessels from pre-existing capillaries and post-capillary venules, is crucial embryonic development (1). In adults, angiogenesis occurs physiologically in the uterus during the menstrual cycle as well as in pathological conditions, such as the growth of malignant tumors.
In 1971, Folkman generated the hypothesis that tumor growth depends on the neoformation of blood vessels and, thus, inhibition of angiogenesis could prevent tumor progression (2). This work also defined the concept of “anti-angiogenesis” as the prevention of blood vessel recruitment to the tumor. The prediction was that tumors would not grow beyond a minimal size of 1–2 mm3 without perfusion and connection to the newly formed capillary network. Consistently, the majority of pre-clinical studies have shown effective inhibition of tumor growth by targeting angiogenic factors. However, the clinical outcome of anti-angiogenic treatment is rather modest as anti-angiogenic drugs improve survival by only a few months (3).
The net angiogenic activity depends on the balance of positive and negative modulators (4) that tightly coordinate the action of various molecules, including, extracellular matrix-degrading enzymes, cellular junction proteins, and cell adhesion receptors, which results in a migratory an invasive behavior of the angiogenic tumor endothelium. In healthy tissue though, the vasculature remains quiescent due to the dominance of negative regulators of angiogenesis (5). Hence, tumor angiogenesis depends on down-regulation of negative regulators as well as a shift toward positive regulators, which are mainly released by neoplastic cells and inflammatory cells that will ultimately lead to the growth of blood vessels (6).
In addition to their increase in density, tumor blood vessels are characterized by various structural and functional abnormalities including irregularities in size and shape, the absence of the typical vessel hierarchy or the distinct organization in arterioles, capillaries, and venules (7). Furthermore, they often exhibit a decreased mural cell coverage and/or abnormal basement membrane sleeves. The endothelial cells that constitute the vascular bed of tumors show a dramatically increased proliferation rate compared to normal endothelial cells resulting in a structurally aberrant and functionally defective vasculature. This distinct vascular phenotype is usually associated with increased permeability that allows the traffic of tumor cells into the circulation (8).
The process of angiogenesis involves a cascade of events including endothelial cell sprouting, the loss of mural cell-endothelial cell association as well as increased vessel permeability (8–10), and the value of vascular density to determine anti-angiogenic activity has been shown to be of limited use (11). Therefore, changes in the functionality of the vasculature are likely to be a more important readout of anti-angiogenic activity than just the presence of a vasculature (8). In fact, recent studies have shown that tumor blood flow and growth are decreased, whereas vessel count is increased (12–15), which further supports the notion that vascular function is more important than simple vessel counts. Indeed in most tumors, despite high vascular density, the blood supply is rather inefficient. Due to the fact that many features of the aberrant tumor vasculature are attributable to the abundance of angiogenic factors like Vascular Endothelial Growth Factor (VEGF), Jain and colleagues have hypothesized that anti-angiogenic therapy can temporarily “normalize” the vascular bed of tumors during the so called window of normalization. The definition of “vascular normalization” includes the reversion of vascular abnormalities (that are, increased permeability, tortuosity, and loss of pericytes) and redistribution of the blood flow with increased delivery of cytotoxic agents and oxygen during the normalization window (8). In fact, in a phase II study with glioblastoma patients, a VEGF receptor tyrosine kinase-inhibitor led to structural and functional to normalization of the tumor vasculature, as measured by MRI (16).
Recent studies gave insight into the causal role of host-derived soluble factors as well as tumor-associated host cells, for initiation and/or progression of cancer (17–20). Recent studies identified the paradox that some leukocytes have the potential to promote, rather than restrict, tumor growth (21, 22). Histological observations of multiple solid tumors revealed the presence of leukocytes within developing tumors as an attempt to eliminate transformed cells. Growing number of reports have implicated tumor-infiltrating immune cells as crucial mediators of cancer initiation and progression (17–19, 23). In addition, type and density of intra-tumoral immune cells have been validated as a reliable parameter for patient’s clinical outcome in certain types of cancer (24–26).
Leukocytes comprise diverse subsets of immune cells that can be separated into cells of the innate and adaptive immunity. The innate immune system consists of macrophages, granulocytes, mast cells, natural killer (NK) cells, and dendritic cells (DCs). Tissue-resident macrophages and mast cells recruit of additional leukocytes from the circulation into the inflamed tissue in response perturbed tissue homeostasis by secreting soluble cytokines and chemokines. Furthermore, DCs have the potential to cross-present antigens to adaptive immune cells, e.g., CD4+ T cells and B cells, which in turn undergo clonal expansion resulting in an adaptive immune response against the presented antigen (21).
However, an efficient immune response also depends on the appropriate distribution and positioning of immune cells within dynamic tissue microenvironments. This process is largely controlled by the vascular network and its interactions with circulating immune cells, particularly during pathological circumstances such as inflammation (27, 28). In consequence, vasculature modulated by inflammatory triggers, displays increased leakiness and enhanced leukocyte adhesiveness, resulting in endothelial cell activation, proliferation, and vascular sprouting (29, 30). Thus, there is a well-orchestrated interaction between inflammatory infiltrates and the endothelium. Recent reports further dissected the impact of different immune subsets for blood vessel neoformation and remodeling (20, 23, 31). They functionally contribute to tumor growth and progression by releasing pro-tumorigenic factors like cytokines and chemokines, extracellular matrix-degrading enzymes, reactive oxygen species, and other bioactive molecules, along with angiogenesis and tissue remodeling (20). The appreciation that immune cell-secreted factors might contribute to tumor angiogenesis and in consequence, potentially affect efficacy of anti-angiogenic therapy, identified these cells as a valuable target for anti-cancer strategies (18, 20, 32, 33). To illustrate the different forms of immune cell-EC communication, we will focus on each cell type of the innate and adaptive immunity and their implication on angiogenesis and vascular remodeling.
Innate Immunity
Macrophages
Macrophages are specialized phagocytes that are able to incorporate invading microbes and cell debris as well as to secrete release various immunomodulatory cytokines. They have a unique ability to adapt their phenotype to dynamically changing microenvironments that they encounter. The conventional phenotyping distinguishes M1 (classically activated) or M2 (alternatively activated) macrophages. The M1 phenotype is characterized as pro-inflammatory and is associated with T-helper-1 response and the secretion of bactericidal factors in response lipopolysaccharide and interferon γ (IFNγ) exposure. M2 macrophages exhibit a T-helper-2 cytokine expression pattern and are considered to be rather immunosuppressive (34).
The potential role of tumor-associated macrophages (TAMs) in modulating tumor angiogenesis was already proposed in the early 90s (35). After that, a variety of studies have shown that TAMs are often found in the surrounding of blood vessels of solid tumors (36–38). In addition, studies in human tumors demonstrate a positive correlation between blood vessel density and the number of TAMs in vessel areas (39, 40). The pro-angiogenic function of TAMs was also thoroughly investigated in animal cancer models. Accumulating evidences show that TAM depletion results in the decrease of tumor angiogenesis (31, 41), while TAM enhancement exhibits the opposing effect (42). For example, it has been shown that genetic depletion of macrophages in PyMT mammary tumor model delays the angiogenic switch, whereas restoring macrophage infiltration rescues the vessel phenotype (31).
In addition to the functional studies mentioned above, much interest has been given to the mechanistic insights on the pro-angiogenic function of TAMs. Hypoxia occurs quite frequently solid tumors, and macrophages are often attracted to the hypoxic areas of tumor site due to the secretion of hypoxia-induced chemoattractants by tumor cells. Such chemoattractants include VEGF, endothelin, endothelial monocyte activating polypeptide II (EMAP II) (43), and CCL2 (6). Once TAMs are attracted to the hypoxic areas, this microenvironment promotes the metabolic adaptation of TAMs to hypoxia by upregulating hypoxia-inducible factors (HIF)-1, HIF-2, and VEGF (44–46). VEGF-A functions as a potent mitogen for endothelial cells by binding to VEGFR1 and VEGFR2 (47). Genetic studies showed that TAM-derived VEGF-A is essential for angiogenesis in the PyMT mammary tumors (48). Restoring VEGF-A expression in macrophage-deficient PyMT tumor model induces the increase of tumor angiogenesis (48). These data indicate that VEGF is a key regulator of the pro-angiogenic activity of TAMs.
Interestingly, a study using an in vivo myeloid cell-specific deletion of VEGF and tested its impact on vessel density and tumor progression in various murine tumor models in order to determine the role of myeloid cell-derived VEGF in this context (49). In the MMTV-PyMT model of mammary tumorigenesis increased vascular density was found as tumors progressed to malignancy, consistent with an “angiogenic switch.” However, in mutant mice with a deletion of VEGF restricted to myeloid cells, the malignancy-associated increase in vascularization, thus the “angiogenic switch,” did not occur. Along with impaired angiogenesis a decrease in vessel length and reduced vessel tortuosity was observed in the absence of myeloid cell-derived VEGF. Although, VEGF protein levels did not vary in tumor lysates from wild type and mutant animals, loss of myeloid-derived VEGF caused an approximately 50% reduction in VEGFR2 phosphorylation, suggesting that myeloid cell-derived VEGF plays an unique role in tumor vascularization, that cannot be compensated for by VEGF from other sources within the tumor. Noteworthy, the onset of tumor growth was not affected by the lack VEGF in myeloid cells. However, surprisingly mutant mice had a significantly higher tumor burden at endpoint than their wild type littermates and along with this a higher number of proliferating cells, indicating that tumors develop at a more rapid pace in the absence of myeloid cell-derived VEGF. Furthermore, the loss of VEGF expression in myeloid cells resulted in a marked increase in the level of pericyte coverage, indicating vascular normalization and suggesting that VEGF expression from infiltrating myeloid cells is essential for intra-tumoral loss of vessel pericyte association. Interestingly, vessel permeability was also reduced in tumors from mutant animals, representing another indicator of vascular normalization. Consistent with the vascular changes and the concept of vascular normalization loss of myeloid-derived VEGF increased the efficacy of chemotherapeutic treatment (49).
Further studies suggested that hypoxia also upregulates the expression and secretion of ADM by macrophages (50), which are often regulated by HIF and VEGF (51, 52). A recent study showed that TAM-induced endothelial cell migration and tubule formation are inhibited by treatment with an ADM neutralizing antibody (53). These findings demonstrate that ADM can function as a novel pivotal factor of TAMs in facilitating tumor angiogenesis. TAMs also have the ability to release a number of other pro-angiogenic factors, including growth factors [such as PlGF, basic-fibroblast growth factor (b-FGF), M-CSF, PDGF, heparin-binding epidermal growth factor (HB-EGF), macrophage-inhibitory factor (MIF), platelet activating factor (PAF), and TGF-β], and cytokines (such as IL-1, IL-8, TNF-α, and MCP-1) (54, 55). Recent studies have increased our understanding about TAM-derived factors involved in angiogenesis. In solid tumors, the hypoxic condition often induces apoptosis of tumor cells (56). The apoptotic tumor cells can up-regulate prostaglandin E2 (PGE2) production from macrophages to promote angiogenesis (57). Semaphorin 4D (Sema4D) is a pro-angiogenic molecule that acts through its receptor, plexin B1 (58). In the tumor microenvironment, TAMs are the major source of Sema4D, which is critical for tumor angiogenesis and vessel maturation, as demonstrated by the impaired angiogenesis and vessel maturation in Sema4D knockout mice (59). In addition to producing pro-angiogenic factors in the hypoxic condition, TAMs also promote angiogenesis by downregulating the expression of angiogenesis inhibitors, such as vasohibin-2 (60).
Apart from the secretion of pro-angiogenic factors, TAMs also express a number of angiogenesis-modulating enzymes, such as COX-2, iNOS, and various matrix metalloproteinases (45, 61–64). For instance, TAM-derived MMP-9 is required for angiogenesis in a model of human cervical cancer (62). Cathepsin proteases are also implicated in human tumor progression (65). In the tumor microenvironment, TAMs represent an important source of cathepsins in pancreatic cancer and mammary tumor. Ablation of TAM-derived cathepsin B or S in these tumors impairs tumor angiogenesis, suggesting their critical roles in mediating TAMs effects on angiogenesis (66).
Recently, it has also been proposed that circulating monocytes transdifferentiate into endothelial cells and thereby contributing to tumor angiogenesis (67). However, whether recruited monocytes/macrophages significantly contribute to the formation of the tumor vasculature by this mechanisms remains to be further determined.
In summary, when TAMs are attracted to the hypoxic areas of tumor site, they produce a large body of pro-angiogenic factors in addition to angiogenesis-modulating enzymes, under the regulation of specific signaling pathways (i.e., NF-κB and mTOR) and transcription factors (i.e., HIFs and Stat3), which contribute to tumor angiogenesis. On the other hand, targeting angiogenic factors in TAMs may also promote tumor vessel normalization. A number of findings support the concept that TAMs are educated by tumor cells and tumor microenvironment, and “re-education” of TAMs is now emerging as a novel strategy for cancer therapies via tumor angiogenesis inhibition and vessel normalization.
Neutrophil Granulocytes
These phagocytes represent the largest population of blood leukocytes and are critical for the initial inflammatory reaction to invading microbes. Neutrophil infiltration has been reported in various cancer entities (68) and neutrophils are particularly abundant in the invasive front of the tumor (69, 70).
The CXC chemokine system plays a crucial role in neutrophil recruitment and transmigration via activation of the receptors CXCR1 and/or CXCR2 (68, 71–73). Particular attention has been paid to CXCL8 (IL-8), which is highly expressed in a large number of cancer types (74–76). Furthermore, CXCL8 expression in patients with bronchioloalveolar carcinoma correlates positively the number of tumor-associated neutrophils as well as with a poor prognosis for the patients (70). Likewise, in a mouse model of CXCL8-overexpressing (human) ovarian carcinoma, the tumors show increase infiltration of neutrophils (77). However, it is important to mention that there is redundancy within the CXC chemokine system. Hence, it is likely that a complex crosstalk between different CXC chemokines and the activation of their cognate CXC regulates the recruitment of neutrophil granulocytes to the tumor.
Various mouse models have shown that neutrophils are crucially involved in the process of tumor angiogenesis. Antibody-mediated depletion of neutrophils impaired angiogenesis in mice inoculated with CXCL8-containing matrigel plugs (78) as well as in the transgenic RIPK1-TAG2 mouse model of pancreatic carcinoma (79). Furthermore, tumor-infiltrating neutrophils are express high levels MMP-9 (80), and therefore could foster angiogenesis by releasing angiogenic factors from the extracellular matrix (81).
In addition, neutrophils are able to release angiogenic molecules like VEGF upon activation to induce vascular remodeling. However, whereas hypoxia induces the upregulation of VEGF expression in TAMs, neutrophil VEGF-release remains unaffected by the oxygen levels (82). In contrast, exposure to TNFα triggers the release of VEGF directly from neutrophils (83) and furthermore, TNFα induces the production of the angiogenic chemokines CXCL8 and CXCL1 (84, 85). Finally, CXCL8 can trigger neutrophil MMP-9 release, which in turn generates a highly active form of CXCL8 by means of protein cleavage (86), thereby creating feed forward loop involving release of angiogenic cytokines and additional neutrophil recruitment.
Mast Cells
Infiltrates of mast cells have been observed in solid tumors as well as hematological malignancies (87). Mast cells are able to release an array of angiogenic factors, including fibroblast growth factor (FGF)-2 and VEGF (88, 89). In experimentally induced tumors, mast cell infiltration precedes the angiogenic switch and the development of carcinomas from dysplastic cells (90–92).
Mast cell recruitment depends on the secretion of tumor-cell-derived soluble factors of which the stem cell factor (SCF) is considered to be the most important (93, 94). In addition FGF-2, VEGF, platelet-derived endothelial cell growth factor (PD-ECGF), RANTES, monocyte chemotactic protein (MCP)-1, adenosine, and adrenomedullin have been reported to play a role in mast cell trafficking (95–97).
In the transgenic mouse model human papilloma virus (HPV) 16-induced carcinogenesis showed mast cell accumulation around hyperplastic and dysplastic cells that preceded the onset of angiogenesis and malignant transformation (98). Infiltrating mast cells were indentified as important sources of MMP-9, tryptase, and chymase and noteworthy, in this setting angiogenesis was abrogated by mast cell-deficiency, highlighting the importance of mast cell-driven angiogenesis in squamous cell carcinogenesis. (98).
Likewise, in colon carcinomas that develop from premalignant polyps, the adenomateous polyps are characterized by high numbers of mast cells. Remarkably, existing polyps show significant remission upon mast cell depletion (99). Furthermore, the presence of mast cell and particularly their ability to degranulate has been shown to be indispensable for tumor progression in a Myc-driven model of pancreatic cancer. In contrast, preventing the degranulation of mast cells within the tumor stroma leads to rapid apoptosis of tumor cells as well as vascular endothelial cells (100).
Several experiments have shown that the expression of another important angiogenic factor, angiopoietin-1 (Ang-1) by mast cells drives neoangiogenesis (101). A close correlation between the presence of mast cells, neovascularization, and tumor progression has been shown for various tumor entities, including plasmacytoma, in mammary carcinoma (102, 103), colon cancer (104), and cervical cancer (105). In the latter tumor-associated mast cells are tryptase-positive and their number increases number along with vascular density during the transition from cervical dysplasia to invasive carcinoma of the cervix (106). Furthermore, accumulation of VEGF-expressing mast cells has been documented in laryngeal, pulmonary neoplasms, and malignant melanoma (107–114) and in the latter one this correlated with a poor prognosis (115). In esophageal and endometrial cancer as well as in hematological malignancies including B-cell non-Hodgkin’s lymphomas, multiple myeloma, myelodysplastic syndrome, and B-cell chronic lymphocytic leukemia, the degree of mast cell infiltration and vascular density have been shown to be of prognostic value (116–122).
Eosinophil Granulocytes
Eosinophils are specialized in defending the body against parasites by releasing granules loaded with highly cationic proteins and play a crucial role during allergies (123).
Accumulation of eosinophils has been documented in various tumor types including nasopharyngeal (124) and oral squamous cell carcinomas (125), tumors of the gastrointestinal tract (104), and lymphomas (126). However, whether the presence of eosinophils represents a positive or negative prognostic factor is tumor entity-dependent. Recruitment of eosinophils to the tumor depends on the chemokine CCL11, which is highly selective for this cell type (127, 128). Depending on the tumor context, tumor cells, fibroblasts, and endothelial cells in the tumor stroma as well eosinophils themselves have been identified as important sources for CCL11 within the tumor.
With regard to their angiogenic function, it has been suggested that eosinophil-stimulated proliferation and migration of endothelial cells is at least partially mediated by VEGF (129). Indeed, in vitro-cultured eosinophils release VEGF with their granules and the secretion of these granules is triggered by IL-5 (130, 131). However, whether this takes place to the same extent in the tumor microenvironment awaits further experimental evidence.
In addition to VEGF, eosinophil granules contain a diverse array of molecules that promote angiogenesis, including b-FGF, IL6, CXCL8, GM-CSF, PDGF, TGFβ (132), and MMP-9 (133). These angiogenic responses and the release of these molecules occur upon stimulation with TNF-α and CCL11 (133, 134). Interestingly, eosinophils preferentially infiltrate into hypoxic areas of the tumor (135). Therefore, degranulation of eosinophils and secretion of angiogenic factors in the tumor microenvironment might deliver the angiogenic signal specifically the hypoxic regions of the tumor.
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSC) represent a subset of immature progenitor cells for myeloid cells (136). MDSC can be roughly divided into CD11b+Gr1hi (alternatively LY6G+LY6Chi), which are reminiscent of immature neutrophils, and those that are CD11b+Gr1low (LY6G+LY6Clow) and present a monocyte-like phenotype (137, 138). MDSCs have a strong immunosuppressive function (137, 139, 140) and potently inhibit T and NK cell activity as well as DC maturation (141, 142).
Myeloid-derived suppressor cells can be detected in tumors as well as in the circulation of cancer patients and their numbers correlate with cancer stage (142–144). Noteworthy, therapy with cytotoxic agents can further increase the burden of circulating MDSCs, indicating that this cell type might play a role in treatment failure (143). Likewise, MDSCs can be found in various murine tumor models (140, 145–147) where they represent up to 5% of the cells (136).
Upon stimulation with G-CSF, CD11b+Gr1+ cells can directly contribute to vessel neoformation by releasing the protein Bv8 (146) with and its interaction via its receptors EG-VEGRF/PKR-1 and EG-VEGFR/PKR-2 (148). In addition, Bv8 can stimulate the mobilization of granulocytes and monocytes (149). Neutralization of Bv8 results in reduced vessel density and impaired tumor growth in xenograft tumor models as well as in transgenic model of pancreatic cancer (146, 150). Besides the suppression of the anti-tumor activity of T and NK cells by means of arginase 1 and inducible nitric oxide synthase (iNoS) (151–153), MDSCs can foster tumor growth by releasing MMPs that increase the bioavailability of VEGF within the tumor microenvironment (136, 147). Particularly, MMP-9 seems to play an important role in tumor vascularization since vessel formation and tumor growth was impaired in the presence of MMP-9-deficient MDSCs (136).
Interestingly, some MDSC populations are found in juxtaposition to tumor blood vessels, indicating that MDSC are actively retained in the perivascular area whereas other MDSCs seem to transdifferentiate into endothelial cell-like cells including increased expression of CD31 and VEGFR2 (136) and integrate into tumor vasculature. Hence, it will be key to identify the mechanisms that guide MDSC positioning within the tumor as well as the signaling pathways that regulate MDSC transdifferentiation.
TIE2-Expressing Monocytes
A unique feature of the recently discovered TIE2-expressing monocytes (TEM) in contrast to other monocyte populations is that they express the angiopoietin receptor TIE2 (154–157). However, TEM are different from TIE2-expressing circulating endothelial cells or endothelial progenitors cells (157). The presence of TEM has been described in various human tumor entities (157) as well as in different mouse models of cancer (154). TEM recruitment is largely regulated by the TIE2 ligand, angiopoietin-2 (ANGPT2) (155, 157). Tumor-infiltrating TEM have been shown to localize in close proximity to blood vessels and to hypoxic areas of the tumor (154, 157). However, it is not known whether this distribution pattern is due to differential ANGPT2 expression (158, 159) within the tumor.
The localization of TEM adjacent to tumor blood vessels indicated that these cells might have profound impact on the process of tumor angiogenesis. Indeed, selective ablation of TEM from the tumor microenvironment reduced angiogenesis and impaired growth in gliomas (154) without affecting the recruitment of TAM or neutrophils into these tumors. Remarkably, despite the fact that TEM numbers within the tumor are lower than those of TAM and granulocytes, TEM showed significant contribution to vessel neoformation, further indicating that this cell type is a potent driver of tumor angiogenesis (154). Recently, it has been shown that TEM transmit the angiogenic signal at least partially by the expression of b-FGF (154). However, the mechanisms by which TEM exert they pro-angiogenic function are still matter of debate and subject of current studies.
Natural Killer Cells
Natural killer cells are cells of the innate immunity that arise from a common lymphoid progenitor cell. These cells are characterized by a high cytolytic capacity against transformed cancer cells. In addition to their important role in immunosurveillance, NK cells can contribute to neovascularization, particularly in the uterus. In humans, uterine NK cells express high levels of CD56 and low levels of CD16 (CD56bright CD16dim) and can infiltrate the uterus in large numbers. These uterine NK cells show a highly angiogenic phenotype and contribute to the physiological vascular remodeling in the uterus during the secretory phase of menstrual cycle as well as during pregnancy (160).
However, the contribution of NK cells to the process of tumor angiogenesis has not been thoroughly dissected yet. A recent study showed that the CD56(+)CD16(−) NK subset in non-small cell lung cancer patients, which represents the predominant NK subset in tumors, was associated with VEGF, placental growth factor (PIGF), and interleukin-8 (IL-8)/CXCL8 production. Peripheral blood CD56(+)CD16(−) NK cells from patients with the squamous cell carcinoma subtype showed higher VEGF and PlGF production compared to those from patients with adenocarcinoma and controls. This suggests that NK cells in non-small cell lung cancer act as pro-angiogenic cells (161).
Furthermore, a recent study identified (NCR) NKp46-expressing lymphoid tissue inducer cells to play an important role in IL-12 mediated tumor rejection. Interestingly, tumor rejection by these cells did neither depend a cytokine response involving IFN-γ, IL-22, lymphotoxin, or IL-17 nor perforin-dependent cytotoxic activity. Instead, NKp46+ lymphoid tissue inducer cells induced the expression of various adhesion receptors by tumor endothelium thereby facilitating the infiltration of other pro-angiogenic leukocytes into the tumor (162).
Yet, the precise role for NK cells in tumor angiogenesis remains to be defined. Given the crucial impact of NK cells on the phenotype of the uterine vasculature, it will be important to define the role of NK cells for vascular remodeling of the tumor vasclature by combining NK cell-specific deletions of angiogenic factors with murine models of cancer.
Dendritic Cells
Dendritic cells play a pivotal role in tuning the adaptive immune response owing to their highly specialized function of antigen presentation and the ability to trigger both primary T- and B-cell responses. DC can be roughly divided into two subpopulations: myeloid DC (MDC) and plasmacytoid DC (PDC) (163, 164).
Myeloid DC in the bone marrow are immature dendritic cells with a high phagocytic potential. The maturation of these cells is usually initiated upon antigen processing, which also leads to homing of DC to secondary lymphoid tissues. In a subsequent step DC can trigger the activation of antigen-specific T cells. Interestingly, recent studies could show that soluble factors derived from the tumor can interfere with this maturation process and impair the development of mature DC (165, 166). Consistent with this, tumors frequently exhibit an accumulation of immature DC and only very few mature MDC (167, 168). Among the tumor-derived factors that potentially recruit immature DC to the tumor, VEGF (165, 169), β-defensin (170), CXCL12 (171), HGF (172), and CXCL8 (173) have been suggested.
Tumor-associated DC can directly drive tumor angiogenesis through the release of pro-angiogenic cytokines such as TNFα, CXCL8, and osteopontin (171, 173). Moreover, these factors stimulate other cells including monocytes to release pro-angiogenic molecules such as IL-1 (174–176). Furthermore, recent work indicates that immature DC which coexpress DC and endothelial markers represent a reservoir of endothelial progenitor cells. After transdifferentiation into endothelial-like cells, these cells are able to integrate into the vasculature and thereby foster tumor angiogenesis (177). Interestingly, the expression of endothelial cell markers in DC and the process of transdifferentiation seem to be controlled by the angiogenic factors VEGF and oncostatin M (170, 178).
Among the tumor-derived factors that might be responsible for the angiogenic phenotype of immature DC, VEGF has been most extensively studied (169, 179). However, other tumor-derived factors like HGF (172), TGFβ (180), prostaglandin E2 (181), lactate (182), and osteopontin (183) are also involved in the suppression of DC maturation and the induction of pro-angiogenic properties. Conversely, immature DC can increase the expression of VEGF and CXCL8 upon hypoxic challenge (184), which might exert pro-angiogenic function in the tumor microenvironment (169, 173, 185).
Adaptive Immunity
B Cells
The impact of B cells on inflammation-associated cancer development remains to be further explored. High numbers of B lymphocytes have been found in aggregates with other immune cells at the inflammatory site in tumor tissues of various human cancers (186). The intra-tumoral presence of B cells together with CD8+ T cells has been correlated with enhanced survival in patients of ovarian (187) and non-small lung cancer (188) in contrast to tumor tissue with exclusively one cell population, and beneficial effects of B-cell-mediated antibody production have been shown to result in better prognosis for patients of medullary breast cancer (189). Beside the beneficial effect of B cells on anti-cancer immunity, mechanistic in vivo studies also identified a cancer-promoting role of this cell type. Increased immunoglobulin deposition, triggered by B cells, promote enhanced recruitment of immune cells into premalignant skin and, in consequence, resulted in malignant progression during chronic inflammation in experimental skin cancer models (190, 191). Exemplarily, adoptive transfer of B cells into B- and T-cell deficient mice has been shown to restore the phenotype of activated tumor vasculature (190). Quite recently, Yang and colleagues identified the interplay between B cells with ECs via the signal transducer and activator of transcription 3 (STAT3) (192) an established and critical mediator of tumor angiogenesis caused by its potential to regulate VEGF expression (193–195). By using different experimental tumor models they could show that B cells differ in their function in dependence of STAT3 expression. STAT3 was persistently activated in tumor-infiltrating B cells during tumor growth. Adoptive transfer of intrinsic activated STAT3-expressing B lymphocytes into implanted Rag1−/− mice, lacking mature T or B cells resulted, contributed to tumor growth and progression whereas in turn, adding STAT3-deficient B cells to the tumor microenvironment resulted in reduced tumor development. Furthermore, the impact of Stat3 activity in B cells for tumor progression was accompanied by enhanced tumor angiogenesis representing increased numbers of tumor-associated blood vessels (192). Further analyses identified the upregulation of several STAT3-downstream pro-angiogenic molecules such as VEGF in ECs after reciprocal interaction with STAT3-activated B cells (192). Nevertheless, B cells also have the potential to modulate tumor angiogenesis via interaction with myeloid cells. It is well known that in progressing tumors, TAMs generally represent the M2-like polarization, which is characterized by low inflammatory but high tissue remodeling and pro-angiogenic potential (34, 196). The polarization of TAMs is orchestrated by tumor- and host-derived cytokines and chemokines (6). In the majority of human cancers high amounts of TAMs in tumor infiltrates correlates with bad prognosis and reduced overall survival (197, 198). A recent study using a HPV-driven mouse model of squamous cell carcinoma indicates that B-cell-produced antibodies have a key role in macrophage-driven tumor progression by interaction and activation of Fcγ receptors on both tumor-resident and recruited myeloid cells. As a result, immune complex-triggered TAMs recruit myofibroblasts via macrophage-derived IL-1 into the tumor site, which in consequence, promote tumor angiogenesis (199). These studies pointed out the significance of B-cell-mediated pathways for therapeutic intervention in patients with chronic inflammatory disease. Initial clinical trials targeting B cells and IL-1 are currently running (200, 201) and may provide more insights into defining the diversity of cancer-related inflammatory response in humans, as well as may offer new innovative anti-tumor strategies.
T Cells
Circulating T lymphocytes interact with human vascular ECs that express class I and II MHC-peptide complexes but also a variety of different co-stimulatory molecules on their surface by attachment and transmigration through capillaries (202). So, whenever foreign peptides, such as microbial pathogens, are presented by endothelial MHC molecules, this contact-dependent interaction offers the opportunity to trigger circulating T-cell response. Undeniable, the MHC expression patterns vary among species and tissue. Exemplarily, the expression level of MHC molecules in non-lymphoid tissues has been found to be much higher than on other cells (203) and also class II MHC molecules have been detected on ECs throughout the human microvasculature and veins but its expression varies on arteries dependent on anatomic location (204). However, T cells can directly regulate the level of MHC expression via IFN-γ secretion (205, 206) but also influence the regulatory function of ECs namely the regulation of blood vessel formation and remodeling, blood flow, permselectivity, blood fluidity, and hemostasis (207). Although T cells are not a source of classical angiogenic modulators such as VEGF or angiopoietin-2, they can directly synthesize b-FGF and heparin-binding epidermal-like growth factor (HB-EGF), acting in a pro-angiogenic manner (208). On the other hand, also inhibitory properties of T-cell-derived cytokines such as TNF, TGF-β, and INF-γ on angiogenic processes have been reported in vitro and in vivo (209–212). Paradoxically, TNF can also act in a pro-angiogenic manner through induction of sphingosine-1-phosphate, which in turn interacts with Edg family receptors on ECs (213). Via surface-bound molecules like TNF, FasL, or Trail, T cells can also induce cell-contact-dependent apoptosis of ECs (214). In accordance with TNF-induced apoptosis, killing via FasL appears to require sensitization of the endothelium, e.g., by IFN-γ-induced upregulation of Fas and pro-caspase-8 (214).
T-cell-secreted TNF can also influence the blood fluidity by converting EC from an anti-thrombotic to a pro-thrombotic state via production of pro-coagulant proteins such as tissue factor (TF) and plasminogen activator inhibitor-1 (215). In parallel, TNF has the potential to diminish thrombomodulin expression by transcription inhibition (216). The physiological synergy between activating responses, such as the induction of TF expression, and dysfunctional responses like the loss of thrombomodulin which, in consequence, up-regulates fibrin deposition on the surface of vascular ECs may underlie pathophysiological processes such as intravascular thrombosis in a variety of vascular diseases.
T-cell-derived cytokines also allow T cells to influence the cytoskeletal rearrangement in EC by stimulation of gap junction formation, resulting in reduced vascular permselectivity of cultured ECs (217), but also cytokine-independent T-cell contact-dependent vascular leakage has been reported (218) of which the mechanism behind is already unknown. Heterotypic gap junctions between T cells and ECs have also been reported during the course of autoimmune inflammation (219).
Importantly, T-cell-derived TNF, IL-1, and INF-γ have been implicated in the regulation of the inhibitory molecule programed cell death-1 ligand (PD-L1) (220). This is of high importance since a successful anti-tumor immunotherapy requires not only activated tumor antigen-specific T cells, but also the access of T-cell to the malignant compartment by a vascular network. Recent studies suggest that lower dosage of anti-angiogenic treatment may be pave the way forward a “normalized” tumor vessel situation, which, in turns, result in a more effective strategy to recondition the tumor immune microenvironment for anti-cancer immunotherapies in a clinical setting such as blockage of immune checkpoints. For example, the ongoing clinical trial using an anti-PD-L1 antibody in combination with a high dose of bevacizumab (anti-VEGF antibody) in patients with advanced solid tumors might shed some light on this interaction (see ClinicalTrails.gov, NCT01633970).
Conclusion
It is now recognized that the immune cell compartment within the tumor is a major driver of angiogenesis and vascular remodeling in addition to the tumor cell itself. As summarized in Figure 1, every immune cell type identified so far has been shown to impact the process of tumor angiogenesis either directly or indirectly. Furthermore, many angiogenic signaling pathways like VEGF are shared by the different cell types (Figure 1) so that targeting the angiogenic signal in one cell type could be compensated by another cell type. Similarly, the angiogenic signal can be transmitted by different factors. Therefore, inhibiting one factor might lead to the compensatory upregulation of another angiogenic molecule resulting in a rather modest effect on net angiogenic activity. Hence, overall angiogenic activity within a tumor is influenced by many different immune cell types and an even larger repertoire of angiogenic factors. It will be the future challenge to dissect out and understand how the interplay between all these different sources of pro-angiogenic stimuli is orchestrated. On the other hand, delivery of some angiogenic factors by certain immune cell subsets seems to play a strictly non-redundant role at least in a context-dependent manner. It is therefore of utmost importance to identify the players that provide exclusive angiogenic signals to the tumor microenvironment.
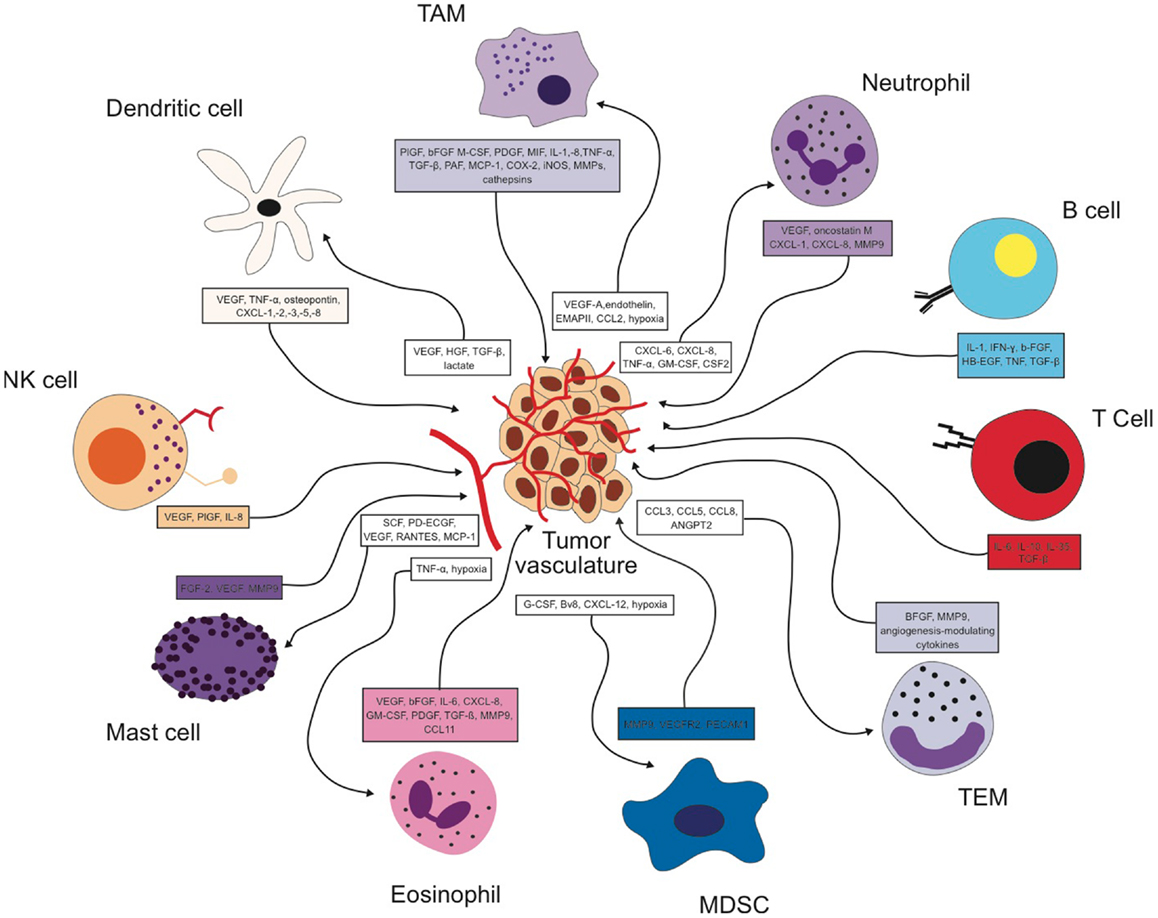
Figure 1. Reciprocal interactions between different immune cell types and the tumor vasculature in the tumor microenvironment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ribatti D. Genetic and epigenetic mechanisms in the early development of the vascular system. J Anat (2006) 208(2):139–52. doi:10.1111/j.1469-7580.2006.00522.x
2. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med (1971) 285(21):1182–6. doi:10.1056/NEJM197111182852108
3. Ellis LM, Fidler IJ. Finding the tumor copycat. Therapy fails, patients don’t. Nat Med (2010) 16(9):974–5. doi:10.1038/nm0910-974
4. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell (1996) 86(3):353–64. doi:10.1016/S0092-8674(00)80108-7
5. Ribatti D, Nico B, Crivellato E, Roccaro AM, Vacca A. The history of the angiogenic switch concept. Leukemia (2007) 21(1):44–52. doi:10.1038/sj.leu.2404402
6. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8(8):618–31. doi:10.1038/nrc2444
7. Ribatti D, Nico B, Crivellato E, Vacca A. The structure of the vascular network of tumors. Cancer Lett (2007) 248(1):18–23. doi:10.1016/j.canlet.2006.06.007
8. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science (2005) 307(5706):58–62. doi:10.1126/science.1104819
9. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell (2005) 8(4):299–309. doi:10.1016/j.ccr.2005.09.005
10. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol (2002) 20(21):4368–80. doi:10.1200/JCO.2002.10.088
11. Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us. J Natl Cancer Inst (2002) 94(12):883–93. doi:10.1093/jnci/94.12.883
12. Hicklin DJ. Promoting angiogenesis to a fault. Nat Biotechnol (2007) 25(3):300–2. doi:10.1038/nbt0307-300
13. Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, et al. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res (2007) 67(23):11244–53. doi:10.1158/0008-5472.CAN-07-0969
14. Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature (2006) 444(7122):1032–7. doi:10.1038/nature05355
15. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature (2006) 444(7122):1083–7. doi:10.1038/nature05313
16. Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell (2007) 11(1):83–95. doi:10.1016/j.ccr.2006.11.021
17. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell (2005) 7(3):211–7. doi:10.1016/j.ccr.2005.02.013
18. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357(9255):539–45. doi:10.1016/S0140-6736(00)04046-0
19. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer (2001) 1(1):46–54. doi:10.1038/35094059
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
21. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi:10.1038/nrc1782
22. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer (2005) 5(4):263–74. doi:10.1038/nrc1586
23. de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol (2006) 13:118–37. doi:10.1159/000092969
24. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
25. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
26. Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol (2007) 19(2):209–16. doi:10.1016/j.coi.2007.01.001
27. Cook-Mills JM, Deem TL. Active participation of endothelial cells in inflammation. J Leukoc Biol (2005) 77(4):487–95. doi:10.1189/jlb.0904554
28. Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol (2007) 178(10):6017–22.
29. Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood (1998) 91(10):3527–61.
30. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med (1995) 1(1):27–31. doi:10.1038/nm0195-27
31. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66(23):11238–46. doi:10.1158/0008-5472.CAN-06-1278
32. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol (2002) 196(3):254–65. doi:10.1002/path.1027
33. Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell (2005) 7(6):513–20. doi:10.1016/j.ccr.2005.05.024
34. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol (2010) 22(2):231–7. doi:10.1016/j.coi.2010.01.009
35. Sunderkotter C, Goebeler M, Schulze-Osthoff K, Bhardwaj R, Sorg C. Macrophage-derived angiogenesis factors. Pharmacol Ther (1991) 51(2):195–216. doi:10.1016/0163-7258(91)90077-Y
36. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res (1996) 56(20):4625–9.
37. Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol (1997) 150(5):1723–34.
38. Ohno S, Ohno Y, Suzuki N, Kamei T, Koike K, Inagawa H, et al. Correlation of histological localization of tumor-associated macrophages with clinicopathological features in endometrial cancer. Anticancer Res (2004) 24(5C):3335–42.
39. Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia (2002) 7(2):177–89. doi:10.1023/A:1020304003704
40. Onita T, Ji PG, Xuan JW, Sakai H, Kanetake H, Maxwell PH, et al. Hypoxia-induced, perinecrotic expression of endothelial Per-ARNT-Sim domain protein-1/hypoxia-inducible factor-2alpha correlates with tumor progression, vascularization, and focal macrophage infiltration in bladder cancer. Clin Cancer Res (2002) 8(2):471–80.
41. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med (2001) 193(6):727–40. doi:10.1084/jem.193.6.727
42. Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res (2010) 16(13):3420–30. doi:10.1158/1078-0432.CCR-09-2904
43. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood (2004) 104(8):2224–34. doi:10.1182/blood-2004-03-1109
44. Burke B, Tang N, Corke KP, Tazzyman D, Ameri K, Wells M, et al. Expression of HIF-1alpha by human macrophages: implications for the use of macrophages in hypoxia-regulated cancer gene therapy. J Pathol (2002) 196(2):204–12. doi:10.1002/path.1029
45. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 66(2):605–12. doi:10.1158/0008-5472.CAN-05-4005
46. Murdoch C, Lewis CE. Macrophage migration and gene expression in response to tumor hypoxia. Int J Cancer (2005) 117(5):701–8. doi:10.1002/ijc.21422
47. Squadrito ML, De Palma M. Macrophage regulation of tumor angiogenesis: implications for cancer therapy. Mol Aspects Med (2011) 32(2):123–45. doi:10.1016/j.mam.2011.04.005
48. Lin EY, Li JF, Bricard G, Wang W, Deng Y, Sellers R, et al. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol (2007) 1(3):288–302. doi:10.1016/j.molonc.2007.10.003
49. Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature (2008) 456(7223):814–8. doi:10.1038/nature07445
50. Cejudo-Martín P, Morales-Ruiz M, Ros J, Navasa M, Fernández-Varo G, Fuster J, et al. Hypoxia is an inducer of vasodilator agents in peritoneal macrophages of cirrhotic patients. Hepatology (2002) 36(5):1172–9. doi:10.1053/jhep.2002.36371
51. Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood (2009) 114(4):844–59. doi:10.1182/blood-2008-12-195941
52. Oladipupo S, Hu S, Kovalski J, Yao J, Santeford A, Sohn RE, et al. VEGF is essential for hypoxia-inducible factor-mediated neovascularization but dispensable for endothelial sprouting. Proc Natl Acad Sci U S A (2011) 108(32):13264–9. doi:10.1073/pnas.1101321108
53. Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res (2011) 17(23):7230–9. doi:10.1158/1078-0432.CCR-11-1354
54. Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol (2006) 80(6):1183–96. doi:10.1189/jlb.0905495
55. Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell (2007) 131(3):463–75. doi:10.1016/j.cell.2007.08.038
56. Weinmann M, Jendrossek V, Handrick R, Güner D, Goecke B, Belka C. Molecular ordering of hypoxia-induced apoptosis: critical involvement of the mitochondrial death pathway in a FADD/caspase-8 independent manner. Oncogene (2004) 23(21):3757–69. doi:10.1038/sj.onc.1207481
57. Brecht K, Weigert A, Hu J, Popp R, Fisslthaler B, Korff T, et al. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J (2011) 25(7):2408–17. doi:10.1096/fj.10-179473
58. Conrotto P, Valdembri D, Corso S, Serini G, Tamagnone L, Comoglio PM, et al. Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood (2005) 105(11):4321–9. doi:10.1182/blood-2004-07-2885
59. Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A, et al. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J Exp Med (2008) 205(7):1673–85. doi:10.1084/jem.20072602
60. Shen Z, Kauttu T, Seppänen H, Vainionpää S, Ye Y, Wang S, et al. Vasohibin-1 and vasohibin-2 expression in gastric cancer cells and TAMs. Med Oncol (2012) 29(4):2718–26. doi:10.1007/s12032-012-0212-1
61. Klimp AH, Hollema H, Kempinga C, van derZee AG, de Vries EG, Daemen T. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res (2001) 61(19):7305–9.
62. Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest (2004) 114(5):623–33. doi:10.1172/JCI200422087
63. Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, et al. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol (2003) 163(4):1233–43. doi:10.1016/S0002-9440(10)63483-9
64. Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature (2007) 446(7136):690–4. doi:10.1038/nature05656
65. Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer (2006) 6(10):764–75. doi:10.1038/nrc1949
66. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev (2010) 24(3):241–55. doi:10.1101/gad.1874010
67. Kuwana M, Okazaki Y, Kodama H, Satoh T, Kawakami Y, Ikeda Y. Endothelial differentiation potential of human monocyte-derived multipotential cells. Stem Cells (2006) 24(12):2733–43. doi:10.1634/stemcells.2006-0026
68. Eck M, Schmausser B, Scheller K, Brändlein S, Müller-Hermelink HK. Pleiotropic effects of CXC chemokines in gastric carcinoma: differences in CXCL8 and CXCL1 expression between diffuse and intestinal types of gastric carcinoma. Clin Exp Immunol (2003) 134(3):508–15. doi:10.1111/j.1365-2249.2003.02305.x
69. Nielsen BS, Timshel S, Kjeldsen L, Sehested M, Pyke C, Borregaard N, et al. 92 kDa type IV collagenase (MMP-9) is expressed in neutrophils and macrophages but not in malignant epithelial cells in human colon cancer. Int J Cancer (1996) 65(1):57–62. doi:10.1002/(SICI)1097-0215(19960103)65:1<57::AID-IJC10>3.0.CO;2-F
70. Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol (1998) 152(1):83–92.
71. Arenberg DA, Keane MP, DiGiovine B, Kunkel SL, Morris SB, Xue YY, et al. Epithelial-neutrophil activating peptide (ENA-78) is an important angiogenic factor in non-small cell lung cancer. J Clin Invest (1998) 102(3):465–72. doi:10.1172/JCI3145
72. Gijsbers K, Gouwy M, Struyf S, Wuyts A, Proost P, Opdenakker G, et al. GCP-2/CXCL6 synergizes with other endothelial cell-derived chemokines in neutrophil mobilization and is associated with angiogenesis in gastrointestinal tumors. Exp Cell Res (2005) 303(2):331–42. doi:10.1016/j.yexcr.2004.09.027
73. Luan J, Shattuck-Brandt R, Haghnegahdar H, Owen JD, Strieter R, Burdick M, et al. Mechanism and biological significance of constitutive expression of MGSA/GRO chemokines in malignant melanoma tumor progression. J Leukoc Biol (1997) 62(5):588–97.
74. Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev (2001) 12(4):375–91. doi:10.1016/S1359-6101(01)00016-8
75. Shi Q, Xiong Q, Le X, Xie K. Regulation of interleukin-8 expression by tumor-associated stress factors. J Interferon Cytokine Res (2001) 21(8):553–66. doi:10.1089/10799900152547812
76. Xu L, Xie K, Mukaida N, Matsushima K, Fidler IJ. Hypoxia-induced elevation in interleukin-8 expression by human ovarian carcinoma cells. Cancer Res (1999) 59(22):5822–9.
77. Lee LF, Hellendall RP, Wang Y, Haskill JS, Mukaida N, Matsushima K, et al. IL-8 reduced tumorigenicity of human ovarian cancer in vivo due to neutrophil infiltration. J Immunol (2000) 164(5):2769–75.
78. Benelli R, Morini M, Carrozzino F, Ferrari N, Minghelli S, Santi L, et al. Neutrophils as a key cellular target for angiostatin: implications for regulation of angiogenesis and inflammation. FASEB J (2002) 16(2):267–9.
79. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A (2006) 103(33):12493–8. doi:10.1073/pnas.0601807103
80. Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell (2000) 103(3):481–90. doi:10.1016/S0092-8674(00)00139-2
81. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol (2000) 2(10):737–44. doi:10.1038/35036374
82. Koehne P, Willam C, Strauss E, Schindler R, Eckardt KU, Bührer C. Lack of hypoxic stimulation of VEGF secretion from neutrophils and platelets. Am J Physiol Heart Circ Physiol (2000) 279(2):H817–24.
83. McCourt M, Wang JH, Sookhai S, Redmond HP. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch Surg (1999) 134(12):1325–31; discussion 1331–2. doi:10.1001/archsurg.134.12.1325
84. Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res (2005) 65(19):8896–904. doi:10.1158/0008-5472.CAN-05-1734
85. Cassatella MA. Neutrophil-derived proteins: selling cytokines by the pound. Adv Immunol (1999) 73:369–509. doi:10.1016/S0065-2776(08)60791-9
86. Van denSteen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood (2000) 96(8):2673–81.
87. Ribatti D, Vacca A, Nico B, Crivellato E, Roncali L, Dammacco F. The role of mast cells in tumour angiogenesis. Br J Haematol (2001) 115(3):514–21. doi:10.1046/j.1365-2141.2001.03202.x
88. Qu Z, Huang X, Ahmadi P, Stenberg P, Liebler JM, Le AC, et al. Synthesis of basic fibroblast growth factor by murine mast cells. Regulation by transforming growth factor beta, tumor necrosis factor alpha, and stem cell factor. Int Arch Allergy Immunol (1998) 115(1):47–54. doi:10.1159/000023829
89. Qu Z, Liebler JM, Powers MR, Galey T, Ahmadi P, Huang XN, et al. Mast cells are a major source of basic fibroblast growth factor in chronic inflammation and cutaneous hemangioma. Am J Pathol (1995) 147(3):564–73.
90. Kessler DA, Langer RS, Pless NA, Folkman J. Mast cells and tumor angiogenesis. Int J Cancer (1976) 18(5):703–9. doi:10.1002/ijc.2910180520
91. Dabbous MK, Walker R, Haney L, Carter LM, Nicolson GL, Woolley DE. Mast cells and matrix degradation at sites of tumour invasion in rat mammary adenocarcinoma. Br J Cancer (1986) 54(3):459–65. doi:10.1038/bjc.1986.198
92. Flynn EA, Schwartz JL, Shklar G. Sequential mast cell infiltration and degranulation during experimental carcinogenesis. J Cancer Res Clin Oncol (1991) 117(2):115–22. doi:10.1007/BF01613134
93. Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1± mast cells. J Clin Invest (2003) 112(12):1851–61. doi:10.1172/JCI19195
94. Zhang W, Stoica G, Tasca SI, Kelly KA, Meininger CJ. Modulation of tumor angiogenesis by stem cell factor. Cancer Res (2000) 60(23):6757–62.
95. Conti P, Pang X, Boucher W, Letourneau R, Reale M, Barbacane RC, et al. Impact of Rantes and MCP-1 chemokines on in vivo basophilic cell recruitment in rat skin injection model and their role in modifying the protein and mRNA levels for histidine decarboxylase. Blood (1997) 89(11):4120–7.
96. Feoktistov I, Ryzhov S, Goldstein AE, Biaggioni I. Mast cell-mediated stimulation of angiogenesis: cooperative interaction between A2B and A3 adenosine receptors. Circ Res (2003) 92(5):485–92. doi:10.1161/01.RES.0000061572.10929.2D
97. Zudaire E, Martínez A, Garayoa M, Pío R, Kaur G, Woolhiser MR, et al. Adrenomedullin is a cross-talk molecule that regulates tumor and mast cell function during human carcinogenesis. Am J Pathol (2006) 168(1):280–91. doi:10.2353/ajpath.2006.050291
98. Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev (1999) 13(11):1382–97. doi:10.1101/gad.13.11.1382
99. Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A (2007) 104(50):19977–82. doi:10.1073/pnas.0704620104
100. Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med (2007) 13(10):1211–8. doi:10.1038/nm1649
101. Nakayama T, Yao L, Tosato G. Mast cell-derived angiopoietin-1 plays a critical role in the growth of plasma cell tumors. J Clin Invest (2004) 114(9):1317–25. doi:10.1172/JCI200422089
102. Bowrey PF, King J, Magarey C, Schwartz P, Marr P, Bolton E, et al. Histamine, mast cells and tumour cell proliferation in breast cancer: does preoperative cimetidine administration have an effect? Br J Cancer (2000) 82(1):167–70. doi:10.1054/bjoc.1999.0895
103. Hartveit F. Mast cells and metachromasia in human breast cancer: their occurrence, significance and consequence: a preliminary report. J Pathol (1981) 134(1):7–11. doi:10.1002/path.1711340103
104. Nielsen HJ, Hansen U, Christensen IJ, Reimert CM, Brünner N, Moesgaard F. Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. J Pathol (1999) 189(4):487–95. doi:10.1002/(SICI)1096-9896(199912)189:4<487::AID-PATH484>3.0.CO;2-I
105. Graham RM, Graham JB. Mast cells and cancer of the cervix. Surg Gynecol Obstet (1966) 123(1):3–9.
106. Benítez-Bribiesca L, Wong A, Utrera D, Castellanos E. The role of mast cell tryptase in neoangiogenesis of premalignant and malignant lesions of the uterine cervix. J Histochem Cytochem (2001) 49(8):1061–2. doi:10.1177/002215540104900816
107. Sawatsubashi M, Yamada T, Fukushima N, Mizokami H, Tokunaga O, Shin T. Association of vascular endothelial growth factor and mast cells with angiogenesis in laryngeal squamous cell carcinoma. Virchows Arch (2000) 436(3):243–8. doi:10.1007/s004280050037
108. Imada A, Shijubo N, Kojima H, Abe S. Mast cells correlate with angiogenesis and poor outcome in stage I lung adenocarcinoma. Eur Respir J (2000) 15(6):1087–93. doi:10.1034/j.1399-3003.2000.01517.x
109. Takanami I, Takeuchi K, Naruke M. Mast cell density is associated with angiogenesis and poor prognosis in pulmonary adenocarcinoma. Cancer (2000) 88(12):2686–92. doi:10.1002/1097-0142(20000615)88:12<2686::AID-CNCR6>3.0.CO;2-6
110. Tomita M, Matsuzaki Y, Onitsuka T. Effect of mast cells on tumor angiogenesis in lung cancer. Ann Thorac Surg (2000) 69(6):1686–90. doi:10.1016/S0003-4975(00)01160-7
111. Dvorak AM, Mihm MC Jr, Osage JE, Dvorak HF. Melanoma. An ultrastructural study of the host inflammatory and vascular responses. J Invest Dermatol (1980) 75(5):388–93. doi:10.1111/1523-1747.ep12523627
112. Reed JA, McNutt NS, Bogdany JK, Albino AP. Expression of the mast cell growth factor interleukin-3 in melanocytic lesions correlates with an increased number of mast cells in the perilesional stroma: implications for melanoma progression. J Cutan Pathol (1996) 23(6):495–505. doi:10.1111/j.1600-0560.1996.tb01441.x
113. Tóth-Jakatics R, Jimi S, Takebayashi S, Kawamoto N. Cutaneous malignant melanoma: correlation between neovascularization and peritumor accumulation of mast cells overexpressing vascular endothelial growth factor. Hum Pathol (2000) 31(8):955–60. doi:10.1053/hupa.2000.16658
114. Ribatti D, Vacca A, Ria R, Marzullo A, Nico B, Filotico R, et al. Neovascularisation, expression of fibroblast growth factor-2, and mast cells with tryptase activity increase simultaneously with pathological progression in human malignant melanoma. Eur J Cancer (2003) 39(5):666–74. doi:10.1016/S0959-8049(02)00150-8
115. Ribatti D, Ennas MG, Vacca A, Ferreli F, Nico B, Orru S, et al. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur J Clin Invest (2003) 33(5):420–5. doi:10.1046/j.1365-2362.2003.01152.x
116. Elpek GO, Gelen T, Aksoy NH, Erdogan A, Dertsiz L, Demircan A, et al. The prognostic relevance of angiogenesis and mast cells in squamous cell carcinoma of the oesophagus. J Clin Pathol (2001) 54(12):940–4. doi:10.1136/jcp.54.12.940
117. Ribatti D, Finato N, Crivellato E, Marzullo A, Mangieri D, Nico B, et al. Neovascularization and mast cells with tryptase activity increase simultaneously with pathologic progression in human endometrial cancer. Am J Obstet Gynecol (2005) 193(6):1961–5. doi:10.1016/j.ajog.2005.04.055
118. Ribatti D, Nico B, Vacca A, Marzullo A, Calvi N, Roncali L, et al. Do mast cells help to induce angiogenesis in B-cell non-Hodgkin’s lymphomas? Br J Cancer (1998) 77(11):1900–6. doi:10.1038/bjc.1998.316
119. Fukushima N, Satoh T, Sano M, Tokunaga O. Angiogenesis and mast cells in non-Hodgkin’s lymphoma: a strong correlation in angioimmunoblastic T-cell lymphoma. Leuk Lymphoma (2001) 42(4):709–20. doi:10.3109/10428190109099333
120. Ribatti D, Vacca A, Nico B, Quondamatteo F, Ria R, Minischetti M, et al. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br J Cancer (1999) 79(3–4):451–5. doi:10.1038/sj.bjc.6690070
121. Ribatti D, Molica S, Vacca A, Nico B, Crivellato E, Roccaro AM, et al. Tryptase-positive mast cells correlate positively with bone marrow angiogenesis in B-cell chronic lymphocytic leukemia. Leukemia (2003) 17(7):1428–30. doi:10.1038/sj.leu.2402970
122. Molica S, Vacca A, Crivellato E, Cuneo A, Ribatti D. Tryptase-positive mast cells predict clinical outcome of patients with early B-cell chronic lymphocytic leukemia. Eur J Haematol (2003) 71(2):137–9. doi:10.1034/j.1600-0609.2003.00110.x
123. Gleich GJ, Adolphson CR, Leiferman KM. The biology of the eosinophilic leukocyte. Annu Rev Med (1993) 44:85–101. doi:10.1146/annurev.me.44.020193.000505
124. Looi LM. Tumor-associated tissue eosinophilia in nasopharyngeal carcinoma. A pathologic study of 422 primary and 138 metastatic tumors. Cancer (1987) 59(3):466–70. doi:10.1002/1097-0142(19870201)59:3<466::AID-CNCR2820590319>3.0.CO;2-P
125. Dorta RG, Landman G, Kowalski LP, Lauris JR, Latorre MR, Oliveira DT. Tumour-associated tissue eosinophilia as a prognostic factor in oral squamous cell carcinomas. Histopathology (2002) 41(2):152–7. doi:10.1046/j.1365-2559.2002.01437.x
126. Teruya-Feldstein J, Jaffe ES, Burd PR, Kingma DW, Setsuda JE, Tosato G. Differential chemokine expression in tissues involved by Hodgkin’s disease: direct correlation of eotaxin expression and tissue eosinophilia. Blood (1999) 93(8):2463–70.
127. Jose PJ, Adcock IM, Griffiths-Johnson DA, Berkman N, Wells TN, Williams TJ, et al. Eotaxin: cloning of an eosinophil chemoattractant cytokine and increased mRNA expression in allergen-challenged guinea-pig lungs. Biochem Biophys Res Commun (1994) 205(1):788–94. doi:10.1006/bbrc.1994.2734
128. Daugherty BL, Siciliano SJ, DeMartino JA, Malkowitz L, Sirotina A, Springer MS. Cloning, expression, and characterization of the human eosinophil eotaxin receptor. J Exp Med (1996) 183(5):2349–54. doi:10.1084/jem.183.5.2349
129. Puxeddu I, Alian A, Piliponsky AM, Ribatti D, Panet A, Levi-Schaffer F. Human peripheral blood eosinophils induce angiogenesis. Int J Biochem Cell Biol (2005) 37(3):628–36. doi:10.1016/j.biocel.2004.09.001
130. Horiuchi T, Weller PF. Expression of vascular endothelial growth factor by human eosinophils: upregulation by granulocyte macrophage colony-stimulating factor and interleukin-5. Am J Respir Cell Mol Biol (1997) 17(1):70–7. doi:10.1165/ajrcmb.17.1.2796
131. Simson L, Ellyard JI, Dent LA, Matthaei KI, Rothenberg ME, Foster PS, et al. Regulation of carcinogenesis by IL-5 and CCL11: a potential role for eosinophils in tumor immune surveillance. J Immunol (2007) 178(7):4222–9.
132. Munitz A, Levi-Schaffer F. Eosinophils: ‘new’ roles for ‘old’ cells. Allergy (2004) 59(3):268–75. doi:10.1111/j.1398-9995.2003.00442.x
133. Ohno I, Ohtani H, Nitta Y, Suzuki J, Hoshi H, Honma M, et al. Eosinophils as a source of matrix metalloproteinase-9 in asthmatic airway inflammation. Am J Respir Cell Mol Biol (1997) 16(3):212–9. doi:10.1165/ajrcmb.16.3.9070604
134. Salcedo R, Young HA, Ponce ML, Ward JM, Kleinman HK, Murphy WJ, et al. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J Immunol (2001) 166(12):7571–8.
135. Cormier SA, Taranova AG, Bedient C, Nguyen T, Protheroe C, Pero R, et al. Pivotal advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J Leukoc Biol (2006) 79(6):1131–9. doi:10.1189/jlb.0106027
136. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6(4):409–21. doi:10.1016/j.ccr.2004.08.031
137. Movahedi K, Guilliams M, Van denBossche J, Van denBergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi:10.1182/blood-2007-07-099226
138. Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood (2008) 111(12):5457–66. doi:10.1182/blood-2008-01-136895
139. Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood (2000) 96(12):3838–46.
140. Kusmartsev S, Gabrilovich DI. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol Immunother (2002) 51(6):293–8. doi:10.1007/s00262-002-0280-8
141. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425; author reply 426. doi:10.1158/0008-5472.CAN-06-3037
142. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166(1):678–89.
143. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58(1):49–59. doi:10.1007/s00262-008-0523-4
144. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology (2008) 135(1):234–43. doi:10.1053/j.gastro.2008.03.020
145. Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood (2003) 102(6):2138–45. doi:10.1182/blood-2003-01-0190
146. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature (2007) 450(7171):825–31. doi:10.1038/nature06348
147. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13(1):23–35. doi:10.1016/j.ccr.2007.12.004
148. Masuda Y, Takatsu Y, Terao Y, Kumano S, Ishibashi Y, Suenaga M, et al. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem Biophys Res Commun (2002) 293(1):396–402. doi:10.1016/S0006-291X(02)00239-5
149. LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci U S A (2004) 101(48):16813–8. doi:10.1073/pnas.0407697101
150. Shojaei F, Singh M, Thompson JD, Ferrara N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci U S A (2008) 105(7):2640–5. doi:10.1073/pnas.0712185105
151. Dolcetti L, Marigo I, Mantelli B, Peranzoni E, Zanovello P, Bronte V. Myeloid-derived suppressor cell role in tumor-related inflammation. Cancer Lett (2008) 267(2):216–25. doi:10.1016/j.canlet.2008.03.012
152. Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol (2007) 601:213–23. doi:10.1007/978-0-387-72005-0_22
153. Sinha P, Clements VK, Miller S, Ostrand-Rosenberg S. Tumor immunity: a balancing act between T cell activation, macrophage activation and tumor-induced immune suppression. Cancer Immunol Immunother (2005) 54(11):1137–42. doi:10.1007/s00262-005-0703-4
154. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell (2005) 8(3):211–26. doi:10.1016/j.ccr.2005.08.002
155. Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol (2007) 178(11):7405–11.
156. Nowak G, Karrar A, Holmén C, Nava S, Uzunel M, Hultenby K, et al. Expression of vascular endothelial growth factor receptor-2 or Tie-2 on peripheral blood cells defines functionally competent cell populations capable of reendothelialization. Circulation (2004) 110(24):3699–707. doi:10.1161/01.CIR.0000143626.16576.51
157. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood (2007) 109(12):5276–85. doi:10.1182/blood-2006-10-053504
158. Gu J, Yamamoto H, Ogawa M, Ngan CY, Danno K, Hemmi H, et al. Hypoxia-induced up-regulation of angiopoietin-2 in colorectal cancer. Oncol Rep (2006) 15(4):779–83.
159. Stratmann A, Risau W, Plate KH. Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol (1998) 153(5):1459–66. doi:10.1016/S0002-9440(10)65733-1
160. Zhang J, Chen Z, Smith GN, Croy BA. Natural killer cell-triggered vascular transformation: maternal care before birth? Cell Mol Immunol (2011) 8(1):1–11. doi:10.1038/cmi.2010.38
161. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia (2013) 15(2):133–42.
162. Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol (2010) 11(11):1030–8. doi:10.1038/ni.1947
163. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol (2004) 5(12):1219–26. doi:10.1038/ni1141
164. O’Neill HC, Wilson HL, Quah B, Abbey JL, Despars G, Ni K. Dendritic cell development in long-term spleen stromal cultures. Stem Cells (2004) 22(4):475–86. doi:10.1634/stemcells.22-4-475
165. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2(10):1096–103. doi:10.1038/nm1096-1096
166. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202(7):919–29. doi:10.1084/jem.20050463
167. Troy AJ, Summers KL, Davidson PJ, Atkinson CH, Hart DN. Minimal recruitment and activation of dendritic cells within renal cell carcinoma. Clin Cancer Res (1998) 4(3):585–93.
168. Fricke I, Gabrilovich DI. Dendritic cells and tumor microenvironment: a dangerous liaison. Immunol Invest (2006) 35(3–4):459–83. doi:10.1080/08820130600803429
169. Dikov MM, Oyama T, Cheng P, Takahashi T, Takahashi K, Sepetavec T, et al. Vascular endothelial growth factor effects on nuclear factor-kappaB activation in hematopoietic progenitor cells. Cancer Res (2001) 61(5):2015–21.
170. Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med (2004) 10(9):950–8. doi:10.1038/nm1097
171. Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res (2004) 64(16):5535–8. doi:10.1158/0008-5472.CAN-04-1272
172. Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, et al. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol (2005) 175(7):4745–53.
173. Feijoó E, Alfaro C, Mazzolini G, Serra P, Peñuelas I, Arina A, et al. Dendritic cells delivered inside human carcinomas are sequestered by interleukin-8. Int J Cancer (2005) 116(2):275–81. doi:10.1002/ijc.21046
174. Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, et al. Activation of human dendritic cells through CD40 cross-linking. J Exp Med (1994) 180(4):1263–72. doi:10.1084/jem.180.4.1263
175. Naldini A, Leali D, Pucci A, Morena E, Carraro F, Nico B, et al. Cutting edge: IL-1beta mediates the proangiogenic activity of osteopontin-activated human monocytes. J Immunol (2006) 177(7):4267–70.
176. Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A (2003) 100(5):2645–50. doi:10.1073/pnas.0437939100
177. Conejo-Garcia JR, Buckanovich RJ, Benencia F, Courreges MC, Rubin SC, Carroll RG, et al. Vascular leukocytes contribute to tumor vascularization. Blood (2005) 105(2):679–81. doi:10.1182/blood-2004-05-1906
178. Gottfried E, Kreutz M, Haffner S, Holler E, Iacobelli M, Andreesen R, et al. Differentiation of human tumour-associated dendritic cells into endothelial-like cells: an alternative pathway of tumour angiogenesis. Scand J Immunol (2007) 65(4):329–35. doi:10.1111/j.1365-3083.2007.01903.x
179. Laxmanan S, Robertson SW, Wang E, Lau JS, Briscoe DM, Mukhopadhyay D. Vascular endothelial growth factor impairs the functional ability of dendritic cells through Id pathways. Biochem Biophys Res Commun (2005) 334(1):193–8. doi:10.1016/j.bbrc.2005.06.065
180. Alard P, Clark SL, Kosiewicz MM. Mechanisms of tolerance induced by TGF beta-treated APC: CD4 regulatory T cells prevent the induction of the immune response possibly through a mechanism involving TGF beta. Eur J Immunol (2004) 34(4):1021–30. doi:10.1002/eji.200324547
181. Pockaj BA, Basu GD, Pathangey LB, Gray RJ, Hernandez JL, Gendler SJ, et al. Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Ann Surg Oncol (2004) 11(3):328–39. doi:10.1245/ASO.2004.05.027
182. Puig-Kröger A, Pello OM, Selgas R, Criado G, Bajo MA, Sánchez-Tomero JA, et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. J Leukoc Biol (2003) 73(4):482–92. doi:10.1189/jlb.0902451
183. Konno S, Eckman JA, Plunkett B, Li X, Berman JS, Schroeder J, et al. Interleukin-10 and Th2 cytokines differentially regulate osteopontin expression in human monocytes and dendritic cells. J Interferon Cytokine Res (2006) 26(8):562–7. doi:10.1089/jir.2006.26.562
184. Ricciardi A, Elia AR, Cappello P, Puppo M, Vanni C, Fardin P, et al. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res (2008) 6(2):175–85. doi:10.1158/1541-7786.MCR-07-0391
185. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92(11):4150–66.
186. Nelson BH. CD20+ B cells: the other tumor-infiltrating lymphocytes. J Immunol (2010) 185(9):4977–82. doi:10.4049/jimmunol.1001323
187. Milne K, Köbel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, et al. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS One (2009) 4(7):e6412. doi:10.1371/journal.pone.0006412
188. Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res (2008) 14(16):5220–7. doi:10.1158/1078-0432.CCR-08-0133
189. Lim KH, Telisinghe PU, Abdullah MS, Ramasamy R. Possible significance of differences in proportions of cytotoxic T cells and B-lineage cells in the tumour-infiltrating lymphocytes of typical and atypical medullary carcinomas of the breast. Cancer Immun (2010) 10:3.
190. de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell (2005) 7(5):411–23. doi:10.1016/j.ccr.2005.04.014
191. Mantovani A. B cells and macrophages in cancer: yin and yang. Nat Med (2011) 17(3):285–6. doi:10.1038/nm0311-285
192. Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, et al. B cells promote tumor progression via STAT3 regulated-angiogenesis. PLoS One (2013) 8(5):e64159. doi:10.1371/journal.pone.0064159
193. Gong W, Wang L, Yao JC, Ajani JA, Wei D, Aldape KD, et al. Expression of activated signal transducer and activator of transcription 3 predicts expression of vascular endothelial growth factor in and angiogenic phenotype of human gastric cancer. Clin Cancer Res (2005) 11(4):1386–93. doi:10.1158/1078-0432.CCR-04-0487
194. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest (2008) 118(10):3367–77. doi:10.1172/JCI35213
195. Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene (2003) 22(3):319–29. doi:10.1038/sj.onc.1206122
196. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi:10.1016/j.cell.2010.03.014
197. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362(10):875–85. doi:10.1056/NEJMoa0905680
198. Torisu H, Ono M, Kiryu H, Furue M, Ohmoto Y, Nakayama J, et al. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int J Cancer (2000) 85(2):182–8. doi:10.1002/(SICI)1097-0215(20000115)85:2%3C182::AID-IJC6%3E3.0.CO;2-M
199. Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell (2010) 17(2):121–34. doi:10.1016/j.ccr.2009.12.019
200. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi:10.1146/annurev.immunol.021908.132612
201. Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev (2010) 29(2):317–29. doi:10.1007/s10555-010-9229-0
202. Pober JS, Tellides G. Participation of blood vessel cells in human adaptive immune responses. Trends Immunol (2012) 33(1):49–57. doi:10.1016/j.it.2011.09.006
203. Natali PG, De Martino C, Quaranta V, Nicotra MR, Frezza F, Pellegrino MA, et al. Expression of Ia-like antigens in normal human nonlymphoid tissues. Transplantation (1981) 31(1):75–8. doi:10.1097/00007890-198101000-00017
204. Turner RR, Beckstead JH, Warnke RA, Wood GS. Endothelial cell phenotypic diversity. In situ demonstration of immunologic and enzymatic heterogeneity that correlates with specific morphologic subtypes. Am J Clin Pathol (1987) 87(5):569–75.
205. Pober JS, Gimbrone MA Jr, Cotran RS, Reiss CS, Burakoff SJ, Fiers W, et al. Ia expression by vascular endothelium is inducible by activated T cells and by human gamma interferon. J Exp Med (1983) 157(4):1339–53. doi:10.1084/jem.157.4.1339
206. Pober JS, Gimbrone MAJr, Lapierre LA, Mendrick DL, Fiers W, Rothlein R, et al. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. J Immunol (1986) 137(6):1893–6.
207. Choi J, Enis DR, Koh KP, Shiao SL, Pober JS. T lymphocyte-endothelial cell interactions. Annu Rev Immunol (2004) 22:683–709. doi:10.1146/annurev.immunol.22.012703.104639
208. Blotnick S, Peoples GE, Freeman MR, Eberlein TJ, Klagsbrun M. T lymphocytes synthesize and export heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor, mitogens for vascular cells and fibroblasts: differential production and release by CD4+ and CD8+ T cells. Proc Natl Acad Sci U S A (1994) 91(8):2890–4. doi:10.1073/pnas.91.8.2890
209. Fathallah-Shaykh HM, Zhao LJ, Kafrouni AI, Smith GM, Forman J. Gene transfer of IFN-gamma into established brain tumors represses growth by antiangiogenesis. J Immunol (2000) 164(1):217–22.
210. Friesel R, Komoriya A, Maciag T. Inhibition of endothelial cell proliferation by gamma-interferon. J Cell Biol (1987) 104(3):689–96. doi:10.1083/jcb.104.3.689
211. Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol (1988) 106(4):1375–84. doi:10.1083/jcb.106.4.1375
212. Sato N, Goto T, Haranaka K, Satomi N, Nariuchi H, Mano-Hirano Y, et al. Actions of tumor necrosis factor on cultured vascular endothelial cells: morphologic modulation, growth inhibition, and cytotoxicity. J Natl Cancer Inst (1986) 76(6):1113–21.
213. Xia P, Gamble JR, Rye KA, Wang L, Hii CS, Cockerill P, et al. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci U S A (1998) 95(24):14196–201. doi:10.1073/pnas.95.24.14196
214. Li JH, Kirkiles-Smith NC, McNiff JM, Pober JS. TRAIL induces apoptosis and inflammatory gene expression in human endothelial cells. J Immunol (2003) 171(3):1526–33.
215. Martin NB, Jamieson A, Tuffin DP. The effect of interleukin-4 on tumour necrosis factor-alpha induced expression of tissue factor and plasminogen activator inhibitor-1 in human umbilical vein endothelial cells. Thromb Haemost (1993) 70(6):1037–42.
216. Conway EM, Rosenberg RD. Tumor necrosis factor suppresses transcription of the thrombomodulin gene in endothelial cells. Mol Cell Biol (1988) 8(12):5588–92.
217. Stolpen AH, Guinan EC, Fiers W, Pober JS. Recombinant tumor necrosis factor and immune interferon act singly and in combination to reorganize human vascular endothelial cell monolayers. Am J Pathol (1986) 123(1):16–24.
218. Damle NK, Doyle LV. Ability of human T lymphocytes to adhere to vascular endothelial cells and to augment endothelial permeability to macromolecules is linked to their state of post-thymic maturation. J Immunol (1990) 144(4):1233–40.
219. Raine CS, Cannella B, Duijvestijn AM, Cross AH. Homing to central nervous system vasculature by antigen-specific lymphocytes. II. Lymphocyte/endothelial cell adhesion during the initial stages of autoimmune demyelination. Lab Invest (1990) 63(4):476–89.
Keywords: microenvironment, immune cells, leukocytes, endothelial cells, angiogenesis
Citation: Stockmann C, Schadendorf D, Klose R and Helfrich I (2014) The impact of the immune system on tumor: angiogenesis and vascular remodeling. Front. Oncol. 4:69. doi: 10.3389/fonc.2014.00069
Received: 15 January 2014; Paper pending published: 13 February 2014;
Accepted: 20 March 2014; Published online: 08 April 2014.
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Angel Porgador, Ben-Gurion University of the Negev, IsraelJamie Lynn Sturgill, Virginia Commonwealth University, USA
Copyright: © 2014 Stockmann, Schadendorf, Klose and Helfrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Stockmann, UMR 970, Paris Cardiovascular Research Center, Institut National de la Santé et de la Recherche Médicale (INSERM), 56 rue Leblanc, Paris 75015, France e-mail:Y2hyaXN0aWFuLnN0b2NrbWFubkBpbnNlcm0uZnI=