 Julie A. Wallace1,2†
Julie A. Wallace1,2† Jason R. Pitarresi1,2†
Jason R. Pitarresi1,2† Nandini Sharma1,2Marilly Palettas1,2
Nandini Sharma1,2Marilly Palettas1,2 Maria C. Cuitiño2Steven T. Sizemore2,3Lianbo Yu2,4
Maria C. Cuitiño2Steven T. Sizemore2,3Lianbo Yu2,4 Allen Sanderlin1,2Thomas J. Rosol2,5
Allen Sanderlin1,2Thomas J. Rosol2,5 Kamal D. Mehta1
Kamal D. Mehta1 Gina M. Sizemore1,2*Michael C. Ostrowski1,2*
Gina M. Sizemore1,2*Michael C. Ostrowski1,2*- 1Department of Molecular and Cellular Biochemistry, College of Medicine, The Ohio State University, Columbus, OH, USA
- 2Comprehensive Cancer Center, The Ohio State University, Columbus, OH, USA
- 3Department of Radiation Oncology, The Ohio State University, Columbus, OH, USA
- 4Center for Biostatistics, The Ohio State University, Columbus, OH, USA
- 5Department of Veterinary Clinical Sciences, College of Veterinary Medicine, The Ohio State University, Columbus, OH, USA
Protein kinase C beta (PKCβ) expression in breast cancer is associated with a more aggressive tumor phenotype, yet the mechanism for how PKCβ is pro-tumorigenic in this disease is still unclear. Interestingly, while it is known that PKCβ mediates angiogenesis, immunity, fibroblast function and adipogenesis, all components of the mammary tumor microenvironment (TME), no study to date has investigated whether stromal PKCβ is functionally relevant in breast cancer. Herein, we evaluate mouse mammary tumor virus–polyoma middle T-antigen (MMTV–PyMT) induced mammary tumorigenesis in the presence and absence of PKCβ. We utilize two model systems: one where PKCβ is deleted in both the epithelial and stromal compartments to test the global requirement for PKCβ on tumor formation, and second, where PKCβ is deleted only in the stromal compartment to test its role in the TME. MMTV–PyMT mice globally lacking PKCβ live longer and develop smaller tumors with decreased proliferation and decreased macrophage infiltration. Similarly, when PKCβ is null exclusively in the stroma, PyMT-driven B6 cells form smaller tumors with diminished collagen deposition. These experiments reveal for the first time a tumor promoting role for stromal PKCβ in MMTV–PyMT tumorigenesis. In corroboration with these results, PKCβ mRNA (Prkcb) is increased in fibroblasts isolated from MMTV–PyMT tumors. These data were confirmed in a breast cancer patient cohort. Combined these data suggest the continued investigation of PKCβ in the mammary TME is necessary to elucidate how to effectively target this signaling pathway in breast cancer.
Introduction
A role for protein kinase C (PKC) in cancer has been known for over 20 years when it was first recognized that phorbol esters promoted tumor formation through activation of the PKC family [reviewed in Ref. (1)]. There are 10 isozymes that make up this family of serine–threonine kinases, which is classified into three major groups by their mode of activation: the classical PKCs (PKCα, PKCβI, PKCβII, and PKCγ) are calcium and diacylglycerol (DAG) dependent; the novel PKCs (PKCδ, PKCε, PKCη, and PKCθ) are activated by DAG; and the atypical PKCs (PKCξ and PKCι) are independent of calcium and DAG. PKCβ is comprised of two splice variants, PKCβI and PKCβII, which are from the same gene (PRKCB) and differ only in their last 50 amino acids (2, 3). Differential PKC expression has been documented in essentially every histological cancer type including carcinomas (e.g., breast, colorectal), sarcomas (e.g., glioma), lymphomas (e.g., diffuse large B-cell lymphoma), and leukemias (e.g., B-cell chronic lymphocytic leukemia) [reviewed in Ref. (1, 4)]. Functionally, PKCs mediate various physiological processes such as proliferation, differentiation, apoptosis, cellular motility, and angiogenesis (1, 4).
Relevance for PKC in breast cancer was first reported in 1986 when Borner and colleagues observed that PKC is increased in more aggressive estrogen receptor-α (ERα) negative mammary tumor cells compared to their less aggressive ERα positive counterparts (5). This putative tumor promoting role was supported shortly thereafter by evidence of increased PKC enzymatic activity in breast tumor versus normal patient samples (6, 7). Evaluation of the specific PKC isozymes soon followed and PKCα (8–14), PKCβ (as discussed below), PKCδ (15, 16), PKCε (17), and PKCη (18, 19) were all found to have roles in breast cancer progression. Investigation of PKCβ, specifically, has revealed that both splice variants (PKCβI and PKCβII) are upregulated in breast tumor versus matched normal patient tissue (20), with cytoplasmic PKCβII associating with Ki-67 expression and HER2 positivity (21). Functionally, ectopic overexpression of either wild type (WT) or constitutively active PKCβI or PKCβII increases breast cancer cell line growth in vitro through upregulation of cyclin D1, while inhibition of kinase activity decreases growth as seen via introduction of dominant negative PKCβI or PKCβII, and through treatment with LY379196, a PKCβ selective inhibitor (22). Interestingly, even though PKCβII is associated with increased proliferation and highly aggressive HER2 positive disease, its expression does not correlate with patient survival in this cohort (21). An independent study also observed that PKCβ mRNA (PRKCB) correlates with increased grade and triple negative (ERα−/PR−/HER2−) disease, but not relapse-free survival (23).
This discrepancy between the in vitro breast cancer cell line and patient survival data may be due to PKCβ-mediating physiological processes within the breast tumor microenvironment (TME), which could cause the evaluation of PKCβ expression in whole tumor tissue, which contains both tumor and stroma, to be misleading. All epithelial-derived tumors (i.e., carcinomas) are surrounded by a complex TME that contains extracellular matrix proteins, endothelial cells, immune cells, and fibroblasts [reviewed in Ref. (24)]. Importantly, PKCβ function in non-epithelial cell types has been described in several model systems. Within the endothelial compartment of the TME, PKCβ is a well-known downstream effector of vascular endothelial growth factor (VEGF) signaling, which mediates angiogenesis (25–27). PKCβ is also involved in immunity through activation of NF-κB (28–30). Less is known about PKCβ specifically in tumor-associated fibroblasts, but it was recently shown that PKC is required for pancreatic cancer-associated fibroblast (CAF) invasiveness (31) as well as resistance to irradiation-induced apoptosis in primary human fibroblasts (32). PKCβ is, however, required for extracellular matrix and collagen deposition in rodent models of diabetes (33, 34). The breast TME also includes adipocytes and PKCβ null mice have decreased adipose tissue with altered lipid metabolism (35, 36). Combined, these studies suggest important roles for PKCβ in the breast TME and the need for further elucidation of PKCβ’s roles in this context.
Herein, we utilized PKCβ genomic knockout mice (37) to evaluate mammary tumorigenesis through the use of two model systems: one where PKCβ is deleted in both the mammary epithelium and the TME, and one where PKCβ is deleted only in the TME. We reveal for the first time a requirement for PKCβ in MMTV–PyMT (mouse mammary tumor virus–polyoma middle T-antigen) induced mammary tumor growth. MMTV–PyMT mice lacking PKCβ (Prkcb−/−) in both epithelial and stromal compartments have increased tumor latency with a decrease in tumor load and tumor volume. Decreased tumorigenesis in the MMTV–PyMT; Prkcb−/− mice is accompanied by diminished tumor proliferation and macrophage infiltration with angiogenesis seemingly unaffected. To test directly whether PKCβ in the TME is functionally relevant, we orthotopically injected B6 PyMT tumor cells, a MMTV–PyMT derived mammary tumor cell line (38), into WT or Prkcb−/− mice. Tumor volume was similarly decreased in the absence of stromal PKCβ confirming a requirement for PKCβ in the mammary cancer TME. Most interestingly, collagen deposition was decreased in this model. Moreover, fibroblasts isolated from the MMTV–PyMT tumors have dramatically higher levels of PKCβII mRNA (Prkcb) than WT mammary fibroblasts, whereas Prkcb in the MMTV–PyMT epithelium is decreased. This increase in stromal PRKCB is similarly observed in a human breast cancer patient cohort confirming the translational relevance of PKCβ function in the TME.
Materials and Methods
Animal Breeding
All animal procedures were approved by The Ohio State University Institutional Animal Care and Use Committee (protocol #2007A0120-R2). Prkcb−/− (35, 37) and MMTV–PyMT mice (39) have been described. The MMTV–PyMT mice were bred into C57Bl/6 at least 10 generations. MMTV–PyMT males were bred with Prkcb+/− females to generate MMTV–PyMT; Prkcb+/− male progeny. The MMTV–PyMT; Prkcb+/− males were then bred with Prkcb+/− females to generate MMTV–PyMT; Prkcb+/+ and MMTV–PyMT; Prkcb−/−female progeny for tumor analysis. Genotyping primers and conditions for the Prkcb−/− mice have been previously reported (35). Genotyping primers and conditions for the MMTV–PyMT mice are described on The Jackson Laboratory website.
Tumor Studies and Fibroblast/Epithelial Isolation
MMTV–PyMT; Prkcb+/+ and MMTV–PyMT; Prkcb−/−mice were palpated starting at 3 months of age. Upon detection of palpable tumors, tumorigenesis was allowed to progress three additional weeks, upon which the tumors were harvested. At the time of harvest, tumor burden was measured as a percentage of the tumor weight relative to total body weight, and total tumor volume was calculated by caliper measurements (length × width × height). Each tumor was then either frozen or fixed in formalin for later histological analysis. For the orthotopic injection study, we injected 3 × 106 B6 PyMT tumor cells into the inguinal mammary fat pads of either Prkcb+/+ or Prkcb−/−mice. Tumors were monitored by weekly palpation. When the largest tumor reached 1 cm in diameter, a final measurement was determined by caliper and all tumors were harvested. B6 PyMT cells were a generous gift from Tsonwin Hai and have been described previously (38). B6 PyMT cells were maintained in vitro using DMEM/F-12 plus 10% fetal bovine serum plus penicillin/streptomycin. Isolation of mammary fibroblasts and epithelium was performed by gravity separation as has been described (40, 41).
Immunofluorescence and Histological Staining
Tumor tissue was fixed in formalin for 24 h, transferred to 70% ethanol, paraffin embedded, and sectioned (5 μm). Sections were rehydrated through xylenes and an ethanol series. Antigen retrieval was performed using DAKO antigen retrieval in a steamer for 30 min and sections were blocked with DAKO protein block. Primary detection of F4/80 (Molecular Probes), Ki-67 (DAKO), and MECA-32 (BD Pharmingen), was performed overnight at 4°C. Alexafluor-488 and Alexafluor-596 (Invitrogen) were used for secondary detection and DAPI as a nuclear counterstain. To quantify, five images (20× fields) were taken per mouse. Quantification of F4/80 and Ki-67 staining was done using the count tool in Adobe Photoshop CS5. Percent positivity of each stain was determined relative to the total number of cells as visualized by the DAPI counterstain. Quantification of MECA-32 staining was measured as percent positive area using Image J (42). Hematoxylin and eosin (H&E) and Masson’s Trichrome staining was performed by the Solid Tumor Pathology Core at OSU.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was isolated using Trizol reagent (Invitrogen), treated with Turbo DNase I (Ambion) and cDNA generated using Superscript III Reverse Transcriptase (Invitrogen), all per manufacturer’s recommendations. Prkcb (F = gaaactcgaacgcaaggaga; R = accg gtcgaagttttcagc; Probe #83) and Rpl4 (F = gatgagctgtatggcacttgg; R = cttgtgcatgggcaggtta; Probe #38) were detected using the Roche Universal Probe Library system.
Dataset and Statistical Analyses
The Karnoub breast cDNA microarray dataset of human breast tumors was retrieved from Oncomine (oncomine.org) for in silico analyses (43). PRKCB expression was compared between invasive ductal breast carcinoma samples versus normal breast tissue. Statistical significance was determined by Student’s t-test assuming a two-tailed distribution and equal variance. Kaplan–Meier survival curves were generated and statistical significance determined using log-rank. Tumor volume measurements and genetic marker staining was compared between two genetic groups. Log transformation of the data was performed if needed. Two-sample t-test or non-parametric Mann–Whitney test was applied after normality check. Type I error level was controlled at 0.05 level, and adjusted by Bonferroni’s method when multiple comparisons were made.
Results
Global PKCβ Deletion Reduces Mammary Tumorigenesis
Members of the PKC family are known to influence proliferation, differentiation, apoptosis, cellular motility, and angiogenesis (1, 4), leading us to hypothesize that loss of PKCβ would cause decreased mammary tumorigenesis in the MMTV–PyMT mammary tumor model. To evaluate the role for PKCβ in tumorigenesis, we bred the Prkcb−/− knockout mice with those harboring the MMTV–PyMT transgene, a well-characterized murine model of mammary tumor formation (44). We monitored tumor development and harvested tumors 3 weeks after initial palpation. Although the tumors between the two groups looked histologically similar by hematoxylin and eosin (H&E) staining (Figure 1A), MMTV–PyMT mice lacking PKCβ (MMTV–PyMT; Prkcb−/−) developed tumors at a longer latency when compared to control MMTV–PyMT; Prkcb+/+ mice (Figure 1B). The absence of Prkcb in the tumors was confirmed (Figure 1C). Not only did the absence of PKCβ delay tumor onset, but the tumors that developed in the MMTV–PyMT; Prkcb−/− mice were smaller as measured by both tumor load (Figure 1D) as well as total volume (Figure 1E). These data indicate that the global absence of PKCβ in this mouse model has a measurable impact on mammary tumorigenesis.
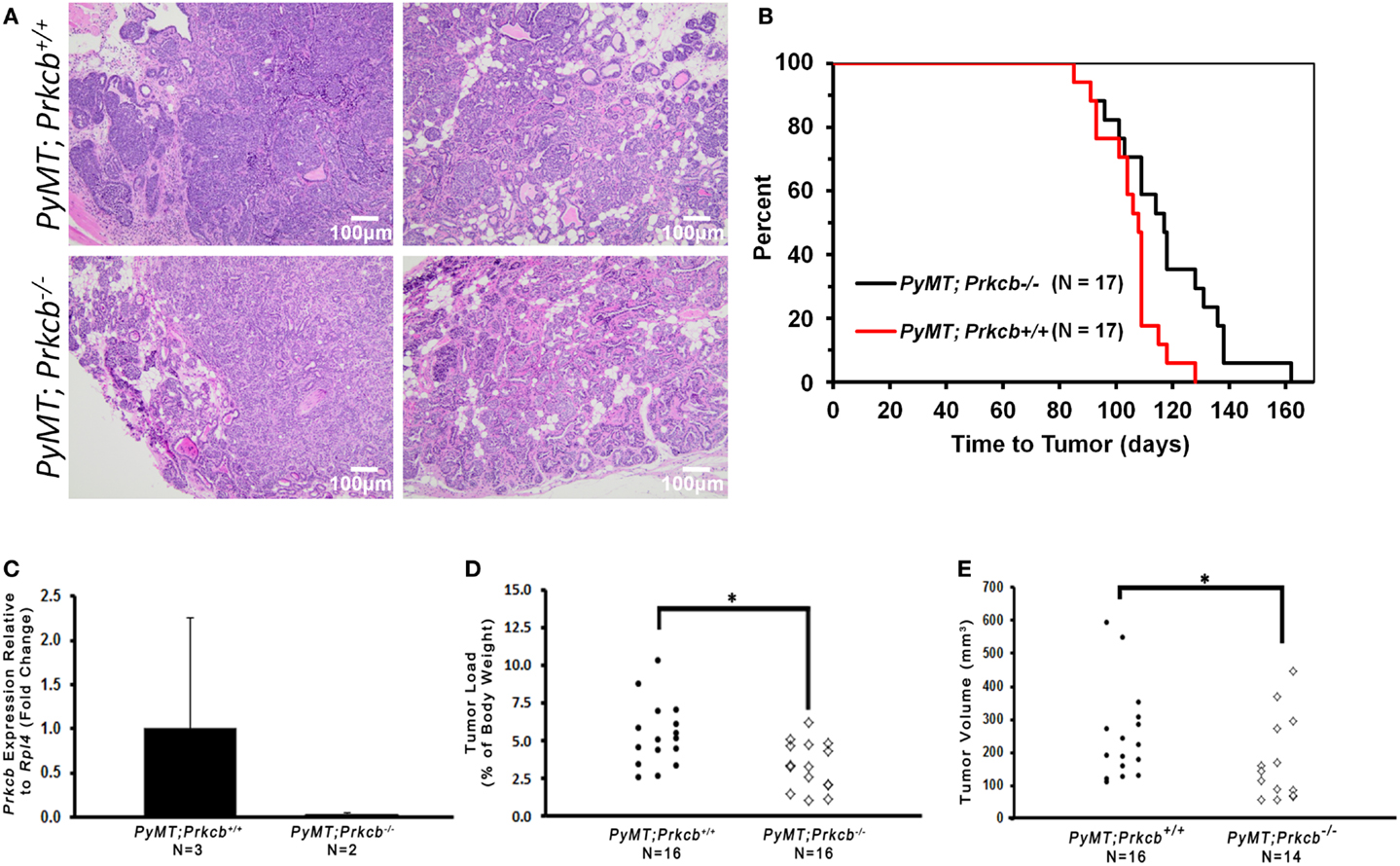
Figure 1. Global PKCβ deletion reduces mammary tumorigenesis.(A) Representative H&E stained mammary tumors from PyMT; Prkcb+/+ and PyMT; Prkcb−/−mice. Scale bar, 100 μm. (B) Kaplan–Meier curve comparing overall tumor-free survival of PyMT; Prkcb+/+ and PyMT; Prkcb−/−mice (n = 17 per group, *P < 0.02 obtained by log-rank). (C) Prkcb expression in mammary tumor cells of PyMT; Prkcb+/+ and PyMT; Prkcb−/−mice normalized to Rpl4 (n = 3 and 2, respectively, bars represent means + standard deviation). (D) Dissected tumor load (percentage of tumor mass relative to total body mass) from PyMT; Prkcb+/+ and PyMT; Prkcb−/−mice (n = 16 per group, *P < 0.05 obtained from Mann–Whitney test). (E) Dissected tumor volume (mm3) isolated from PyMT; Prkcb+/+ and PyMT; Prkcb−/−mice (n = 16 and 14, respectively, *P < 0.05 obtained from two-sample t-test of log based 10 transformation applied on original right skewed data).
Global PKCβ Deletion Decreases Tumor Cell Proliferation and Macrophage Recruitment, but Not Tumor Vasculature
To explain the delayed onset and decreased tumor size in mice lacking PKCβ, we stained for a proliferation marker (Ki-67) within the PyMT tumors of both genetic groups. We saw an observable decrease in the percentage of Ki-67 positive cells in MMTV–PyMT; Prkcb−/− mice (Figure 2A), indicating that PKCβ influences PyMT tumor progression by modulating tumor cell proliferation capacity. Given the recent knowledge that the TME can influence tumor cell proliferation (41), as well as the known function for PKCβ in endothelium and immune cells, we also investigated macrophage infiltration and tumor vasculature in the MMTV–PyMT tumors with and without PKCβ.We saw a significant decrease in percentage of F4/80 positive area, a macrophage marker, in MMTV–PyMT; Prkcb−/− tumors (Figure 2B), indicating that loss of PKCβ within macrophages influences their recruitment to the tumor site. We saw no measurable difference in tumor vascularization, as measured by MECA-32 staining, between the Prkcb−/− and WT groups (Figure 2C), demonstrating that the delayed tumor onset and growth observed in Prkcb−/− mice was independent of vasculature formation.
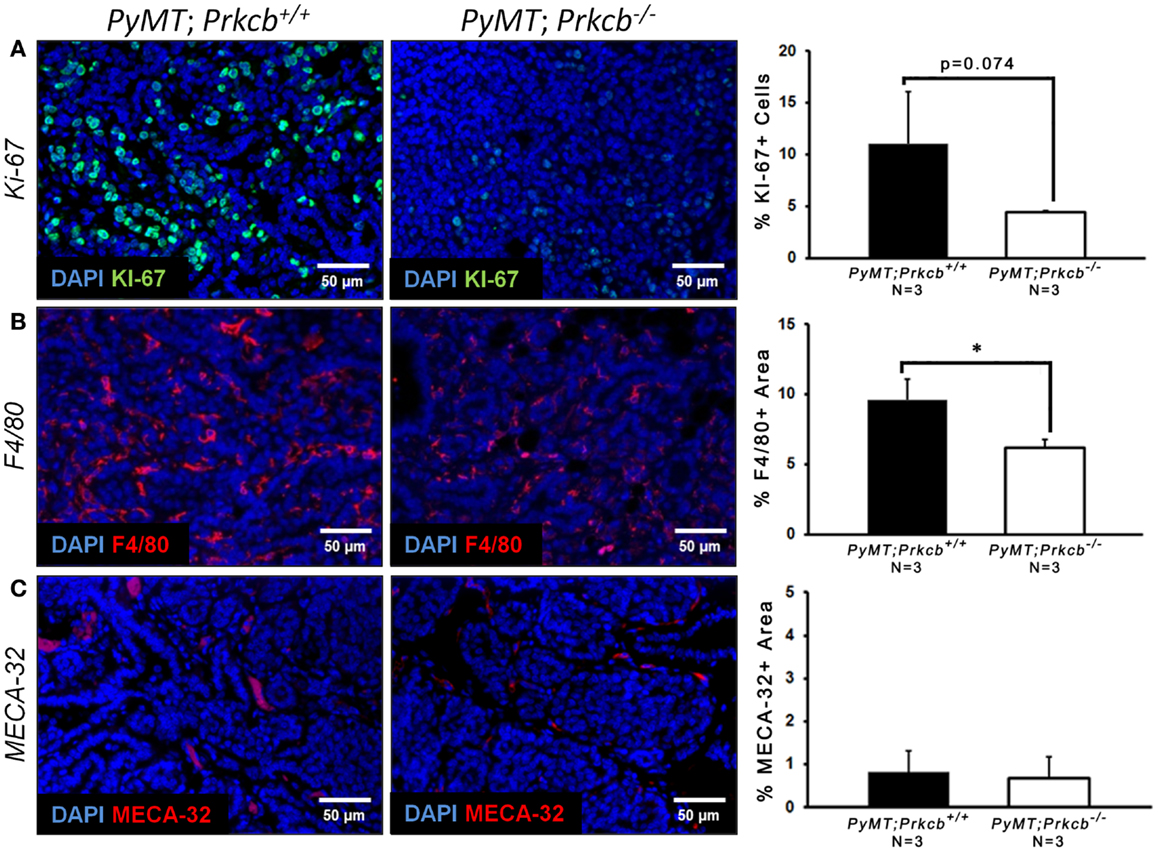
Figure 2. Global PKCβ deletion decreases tumor cell proliferation and macrophage recruitment, but not tumor vasculature. (A) Left: representative images of Ki-67 staining of proliferating cells within PyMT; Prkcb+/+ and PyMT; Prkcb−/−tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of Ki-67+ cells relative to total DAPI+ cells (n = 3 per group, bars represent means + standard deviation, *P = 0.074 obtained from two-sample t-test). (B) Left: representative images of F4/80 staining of macrophage cells within PyMT; Prkcb+/+ and PyMT; Prkcb−/−tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of F4/80+ area relative to total area of image (n = 3 per group, bars represent means + standard deviation, *P < 0.05 obtained from two-sample t-test). (C) Left: representative images of MECA-32 staining of tumor vasculature within PyMT; Prkcb+/+ and PyMT; Prkcb−/−tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of MECA-32+ area relative to total area of image (n = 3 per group, bars represent means + standard deviation, P > 0.05 obtained from two-sample t-test).
Loss of PKCβ in Stromal Compartments Decreases Tumor Volume and Collagen Deposition, but has no Effect on Tumor Cell Proliferation, Vascularization, or Macrophage Infiltration
To determine the importance of PKCβ signaling within the stroma, we orthotopically injected Prkcb−/− and WT mice with B6 PyMT-derived tumor cells. As seen with the genetic model, tumors developing in Prkcb−/− mice exhibited no obvious histological differences (Figure 3A), but did result in significantly smaller tumor volumes compared to WT controls (Figure 3B). This result, in conjunction with the results presented in Figure 1, confirms that stromal PKCβ is important in PyMT mammary tumor formation. In order to further assay the effect of stromal PKCβ deletion within the tumor and its microenvironment, tumors were also evaluated for histological alterations within the distinct cellular compartments. Interestingly, there was no difference in Ki-67, F4/80, or MECA-32 positivity between Prkcb−/− and WT mice (Figure 4). Thus, losing PKCβ signaling in the stroma decreased tumor size, but had no effect on tumor cell proliferation, vascularization, or macrophage recruitment. To evaluate whether stromal fibroblasts could be responsible for the observed decrease in tumor size in Prkcb−/− mice, we evaluated collagen deposition through Masson’s Trichrome staining. The absence of PKCβ specifically in the stroma resulted in significantly diminished collagen deposition while this decrease was not as pronounced in the global knockout model (Figure 5). These results imply that PKCβ signaling in fibroblasts is pro-tumorigenic in the mammary gland.
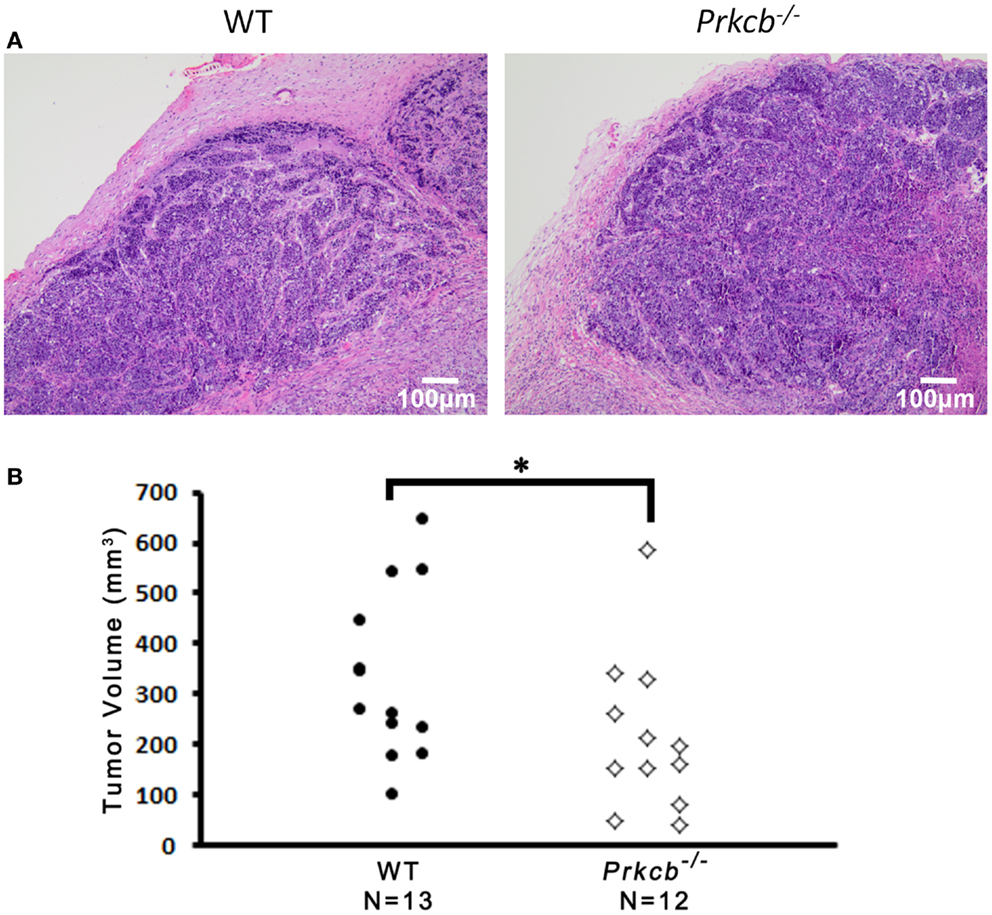
Figure 3. Loss of PKCβ in stromal compartments decreases tumor volume.(A) Representative H&E stained B6 PyMT tumors developed in wild type (WT) and Prkcb−/− mice. Scale bar, 100 μm. (B) Dissected B6 PyMT tumor volume isolated from wild type (WT) and Prkcb−/− mice (n = 13 and 12, respectively, *P < 0.05 obtained from Mann–Whitney test).
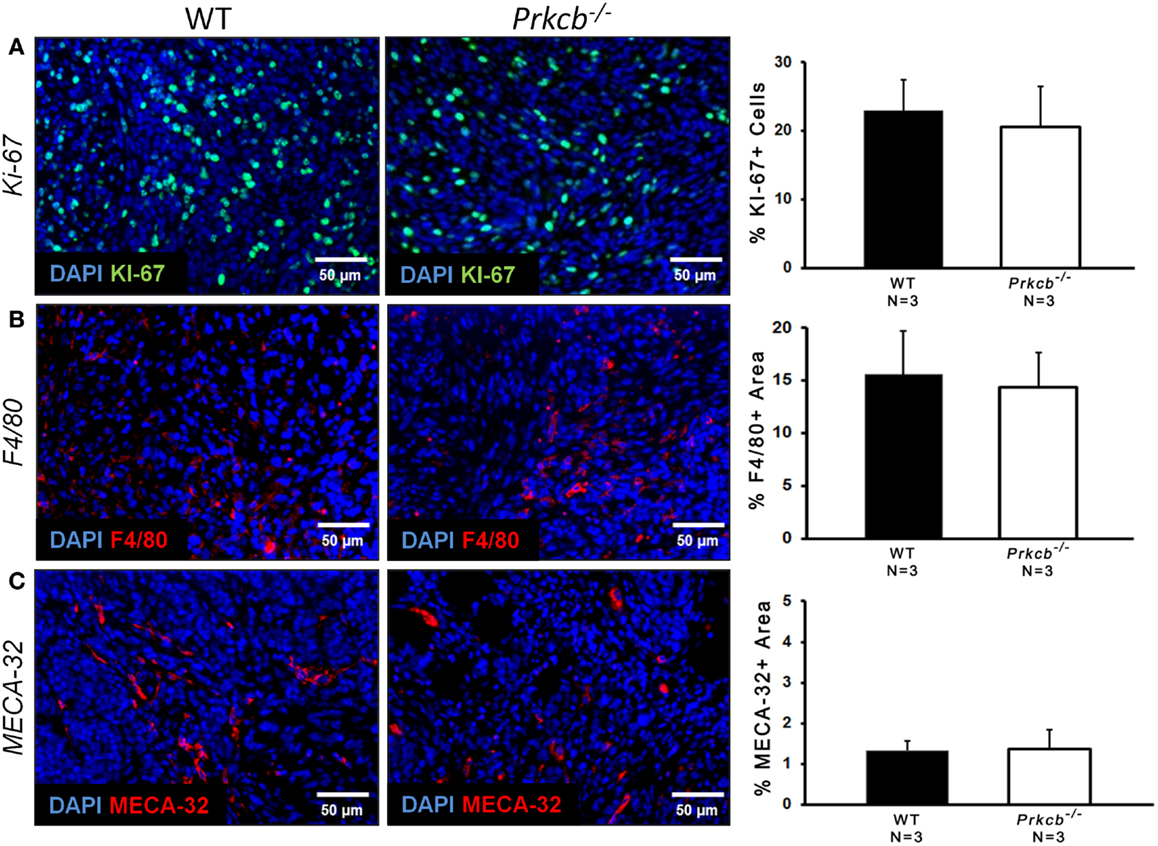
Figure 4. Loss of PKCβ in stromal compartments has no effect on tumor cell proliferation, vascularization, or macrophage infiltration. (A) Left: representative images of Ki-67 staining of proliferating cells within B6 PyMT tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of Ki-67+ cells relative to total DAPI+ cells (n = 3 per group, bars represent means + standard deviation, P > 0.05 obtained from two-sample t-test). (B) Left: representative images of F4/80 staining of macrophage cells within B6 PyMT tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of F4/80+ area relative to total area of image (n = 3 per group, bars represent means + standard deviation, P > 0.05 obtained from two-sample t-test). (C) Left: representative images of MECA-32 staining of B6 PyMT tumor vasculature in vivo. Scale bar, 50 μm. Right: graph of percentage of MECA-32+ area relative to total area of image (n = 3 per group, bars represent means + standard deviation, P > 0.05 obtained from two-sample t-test).
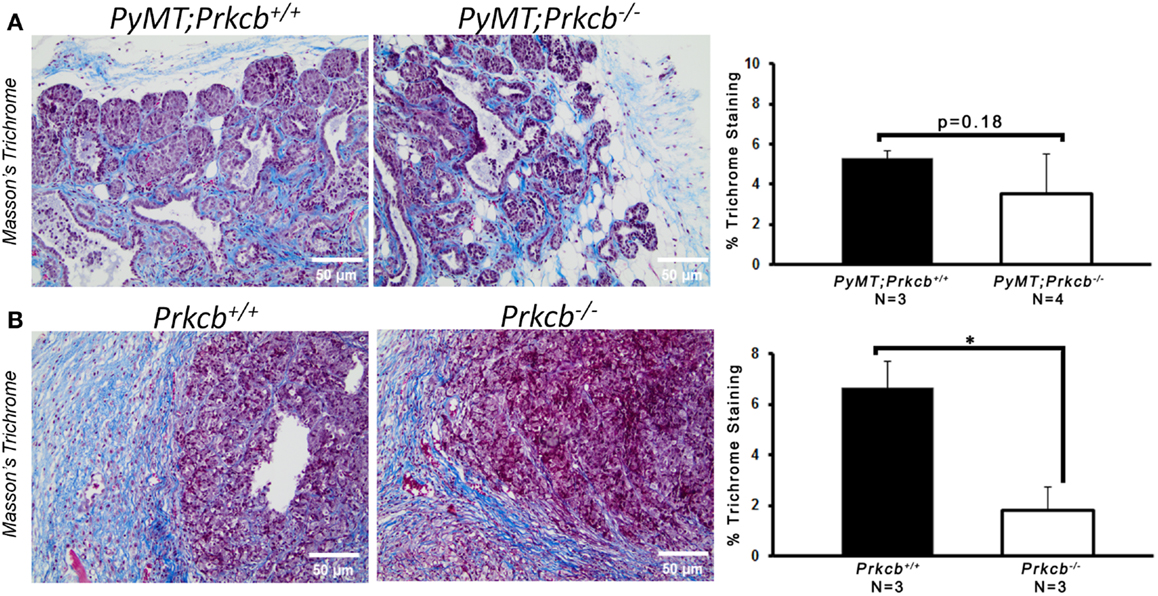
Figure 5. Loss of PKCβ in stromal compartments decreases collagen deposition. (A) Left: representative images of Masson’s Trichrome staining within PyMT; Prkcb+/+ and PyMT; Prkcb−/−tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of Trichrome collagen (blue) staining relative to total area (n = 3 and 4, respectively, bars represent means + standard deviation, P = 0.18 obtained from two-sample t-test). (B) Left: representative images of Masson’s Trichrome staining within B6 PyMT tumors in vivo. Scale bar, 50 μm. Right: graph of percentage of Trichrome collagen (blue) staining relative to total area. n = 3 per group, bars represent means + standard deviation, P < 0.05 obtained from two-sample t-test).
PKCβ is Increased in PyMT Tumor-Associated Fibroblasts and Patient Breast Cancer Stroma
Given the decreased collagen deposition in the absence of stromal PKCβ as just discussed, in addition to previously described roles for PKCβ in fibroblast function (31–34), we further hypothesized that MMTV–PyMT tumorigenesis may be, at least in part, due to aberrant PKCβ expression in the mammary fibroblasts. To this end, we isolated primary mammary tumor epithelial cells and tumor-associated fibroblasts from PyMT mice 2 weeks after palpable tumor formation. We also isolated mammary epithelial cells and fibroblasts from age-matched WT littermate controls. We assayed the expression level of Prkcb mRNA in both cellular compartments by quantitative real-time PCR (qRT-PCR), and observed a significant decrease in Prkcb expression in mammary epithelial cells derived from PyMT tumors (Figure 6A), possibly indicating that epithelial PKCβ is tumor suppressive during PyMT-driven tumor formation. In contrast, a dramatic increase in Prkcb mRNA in PyMT tumor-associated fibroblasts was observed compared to WT mammary fibroblasts (Figure 6B). These data, in combination with the decreased tumor formation we observed in Prkcb−/− mice, suggest a pro-tumorigenic role for PKCβ in mammary tumor fibroblasts. To confirm these findings in human disease, we evaluated PRKCB in a publicly available breast cancer dataset [Karnoub et al. (43)] comparing stroma isolated from invasive ductal breast carcinoma compared to normal breast tissue. In this dataset, PRKCB is significantly increased in breast cancer stroma versus normal confirming the translational relevance of PKCβ in the breast TME (Figure 6C).
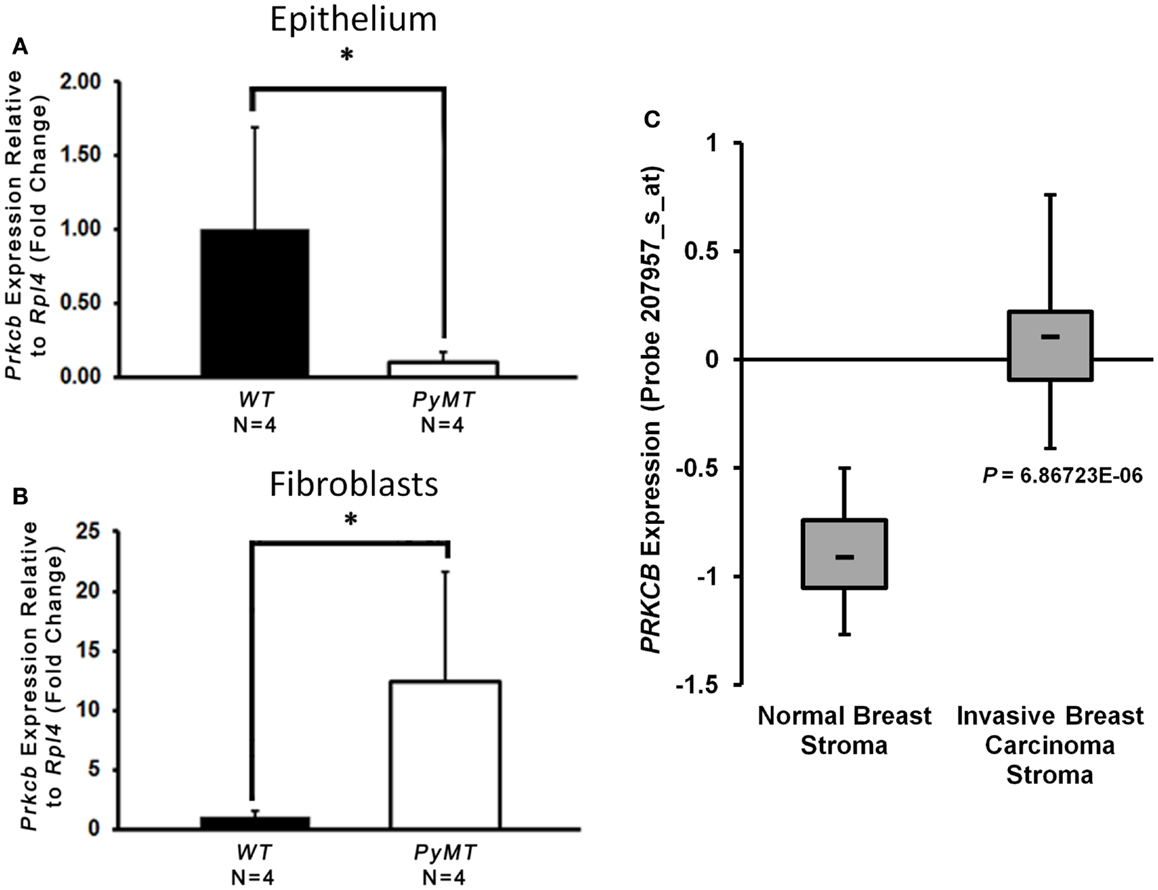
Figure 6. PKCβ is increased in PyMT tumor-associated fibroblasts and patient breast cancer stroma. (A) Prkcb expression in primary mammary fibroblasts normalized to Rpl4 and relative to WT. Cells isolated from PyMT mice 2-weeks after first palpable tumor and WT littermate controls (n = 4, bars represent means + standard deviation, *P < 0.05 obtained from two-sample t-test). (B) Prkcb expression in primary mammary epithelial cells normalized to Rpl4 and relative to WT. Cells isolated from PyMT mice 2-weeks after first palpable tumor and WT littermate controls (n = 4, bars represent means + standard deviation, *P < 0.05 obtained from two-sample t-test). (C) Box and whisker plot of PRKCB expression in invasive ductal breast carcinoma stroma compared to normal breast stroma in the Karnoub breast dataset (normal, n = 15; IDC, n = 7, *P < 10−5).
Discussion
The PKC family is well-documented to have a variety of roles in cancer [reviewed in Ref. (1, 4)] with PKCβ functioning specifically in a number of cancer types including in breast (20–23, 45). Prior evaluation of PKCβ in breast cancer has utilized overexpression studies in vitro (20, 45) and expression analyses in patient tumor tissue: both by immunohistochemistry of tumor sections (21) and through whole tumor extraction (20, 23). While the in vitro breast cancer cell line data are highly suggestive of a tumor promoting role for PKCβ in breast cancer, tumor expression at both RNA (23) and protein (21) levels does not correlate with breast cancer patient survival. Given that PKCβ is required for VEGF-induced angiogenesis (25–27), immunity via NF-κB signaling (28–30), pro-tumorigenic fibroblast properties (31, 32), and adipocyte lipid metabolism (35, 36), it is likely that our understanding of how PKCβ functions in breast tumorigenesis requires further evaluation of its role in the breast TME.
In this study, we took advantage of the genomic Prkcb−/− mouse (37). These mice are viable allowing for investigation of mammary tumorigenesis in the absence of PKCβ either throughout the whole animal as we investigated in MMTV–PyMT mice with and without PKCβ, or through transplantation approaches where we injected a MMTV–PyMT derived tumor cell line (B6 PyMT cells) into either WT or PKCβ null mammary fat pads. The Prkcb−/− mice were crossed with the MMTV–PyMT mouse model of breast cancer, which is a valuable translational tool given that tumor formation in these mice recapitulates disease progression as seen in humans (44). Furthermore, the importance of the mammary TME in MMTV–PyMT-induced tumorigenesis has been well-described. Alterations in signaling from the endothelium (46), fibroblasts (41, 47, 48), immune cells (49–52), and adipocytes (53–55) have all been shown to alter tumor growth and/or metastatic progression in these mice. Moreover, increased stromal collagen deposition hastens MMTV–PyMT tumor development and metastatic spread (56).
As described herein, MMTV–PyMT; Prkcb−/− mice develop smaller tumors at a longer latency than their MMTV–PyMT; Prkcb+/+ counterparts. This decrease in tumorigenesis is associated with a reduction in proliferation and macrophage recruitment. Tumor-associated macrophages in breast cancer act in a pro-tumor manner by upregulation of M2 functions such as promoting angiogenesis, remodeling of the tumor matrix, and suppressing the adaptive immune response (57, 58). Furthermore, M2 macrophages are thought to have an immunomodulatory function that activates tumor cell proliferation (59) suggesting a possible mechanistic connection between the observed differences in proliferation and macrophage number upon loss of PKCβ. Surprisingly, even with the well-described role for PKCβ in VEGF signaling (25–27), there was no alteration in the tumor vasculature in PKCβ null PyMT mice. Similar findings were observed in the injection model. However, in this case we saw no difference in proliferation, macrophage recruitment, or angiogenesis, but did see a decrease in collagen deposition. These results imply a mechanistic role for PKCβ signaling in fibroblast activity, specifically in the synthesis of collagen. It is important to note that this model only evaluates tumor progression, not initiation, since the B6 PyMT cells are already transformed. Future studies are required using Prkcb floxed mice to conditionally delete PKCβ in the endothelial (Tie2-cre), macrophage (Lys-cre), and fibroblast (Fsp-cre) specific cellular compartments. These mice can then be crossed with the MMTV–PyMT as well as other murine models of breast cancer to investigate how PKCβ alters mammary tumor initiation. While this investigation is well beyond the scope of the current study, it will be necessary to truly define the specific breast TME component where PKCβ has the most tumor altering effect.
Support for a pro-tumorigenic role for PKCβ in the breast TME is evidenced by the significantly upregulated expression of Prkcb in fibroblasts isolated from MMTV–PyMT mammary tumors when compared to WT mammary glands. Increased PRKCB in breast tumor stroma versus normal stroma was confirmed in a breast cancer patient cohort. Interestingly, we observed a significant decrease in Prkcb expression in the epithelial fraction of the MMTV–PyMT tumors. The concomitant decrease of Prkcb in epithelium and increase in the stroma suggests that whole tumor analysis of PKCβ either at the mRNA or protein level could be misinterpreted as showing no observable difference in expression. Likewise, observable differences may be reflective of expression changes in specific cellular populations confounding any predictive power of PKCβ. This is supported by the fact that similar to published studies (21, 23), we observed no significant correlation of PRKCB with patient outcome in several publicly available breast cancer datasets (data not shown). Given its role in the TME as described herein, PKCβ expression can only be truly defined through immunohistological evaluation of whole tumor sections in order to delineate tumor versus stromal expression. Pursuing whether PKCβ expression is predictive in breast cancer is critical because a number of PKCβ selective inhibitors have shown efficacy in pre-clinical and clinical trials for a multitude of cancer types (4, 60). To date, no studies have proven efficacious in breast cancer patients, but this could be due to the absence of a proper biomarker as tumor expression of PKCβ has not been evaluated in trial participants [(61, 62); clinicaltrials.gov]. Continued investigation into the mechanism of PKCβ function in breast tumor stroma through the use of the conditional knockout models as described above will undoubtedly translate to a greater understanding of how to clinically evaluate PKCβ tumor expression and ultimately use this aberrant signaling pathway as a therapeutic target.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We graciously thank Tsonwin Hai for the B6 PyMT cells as well as the Solid Tumor Pathology Core at OSU for their technical expertise. This work was supported by the National Institutes of Health (PO1CA09719 to Michael C. Ostrowski) and an OSU CCC Pelotonia Postdoctoral Fellowship (Gina M. Sizemore).
References
1. Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer (2007) 7(4):281–94. doi: 10.1038/nrc2110
2. Ono Y, Kikkawa U, Ogita K, Fujii T, Kurokawa T, Asaoka Y, et al. Expression and properties of two types of protein kinase C: alternative splicing from a single gene. Science (1987) 236(4805):1116–20. doi:10.1126/science.3576226
3. Kawakami T, Kawakami Y, Kitaura J. Protein kinase C beta (PKC beta): normal functions and diseases. J Biochem (2002) 132(5):677–82. doi:10.1093/oxfordjournals.jbchem.a003273
4. Bosco R, Melloni E, Celeghini C, Rimondi E, Vaccarezza M, Zauli G. Fine tuning of protein kinase C (PKC) isoforms in cancer: shortening the distance from the laboratory to the bedside. Mini Rev Med Chem (2011) 11(3):185–99. doi:10.2174/138955711795049899
5. Borner C, Wyss R, Regazzi R, Eppenberger U, Fabbro D. Immunological quantitation of phospholipid/Ca2+-dependent protein kinase of human mammary carcinoma cells: inverse relationship to estrogen receptors. Int J Cancer (1987) 40(3):344–8. doi:10.1002/ijc.2910400310
6. O’Brian C, Vogel VG, Singletary SE, Ward NE. Elevated protein kinase C expression in human breast tumor biopsies relative to normal breast tissue. Cancer Res (1989) 49(12):3215–7.
7. Gordge PC, Hulme MJ, Clegg RA, Miller WR. Elevation of protein kinase A and protein kinase C activities in malignant as compared with normal human breast tissue. Eur J Cancer (1996) 32A(12):2120–6. doi:10.1016/S0959-8049(96)00255-9
8. Ways DK, Kukoly CA, deVente J, Hooker JL, Bryant WO, Posekany KJ, et al. MCF-7 breast cancer cells transfected with protein kinase C-alpha exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. J Clin Invest (1995) 95(4):1906–15. doi:10.1172/JCI117872
9. Chisamore MJ, Ahmed Y, Bentrem DJ, Jordan VC, Tonetti DA. Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase C alpha. Clin Cancer Res (2001) 7(10):3156–65.
10. Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer (2000) 83(6):782–91. doi:10.1054/bjoc.2000.1326
11. Tonetti DA, Morrow M, Kidwai N, Gupta A, Badve S. Elevated protein kinase C alpha expression may be predictive of tamoxifen treatment failure. Br J Cancer (2003) 88(9):1400–2. doi:10.1038/sj.bjc.6600923
12. Tan M, Li P, Sun M, Yin G, Yu D. Upregulation and activation of PKC alpha by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC alpha and Src inhibitors. Oncogene (2006) 25(23):3286–95. doi:10.1038/sj.onc.1209361
13. Lonne GK, Cornmark L, Zahirovic IO, Landberg G, Jirstrom K, Larsson C. PKCalpha expression is a marker for breast cancer aggressiveness. Mol Cancer (2010) 9:76. doi:10.1186/1476-4598-9-76
14. Kim J, Thorne SH, Sun L, Huang B, Mochly-Rosen D. Sustained inhibition of PKCalpha reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene (2011) 30(3):323–33. doi:10.1038/onc.2010.415
15. Assender JW, Gee JM, Lewis I, Ellis IO, Robertson JF, Nicholson RI. Protein kinase C isoform expression as a predictor of disease outcome on endocrine therapy in breast cancer. J Clin Pathol (2007) 60(11):1216–21. doi:10.1136/jcp.2006.041616
16. McKiernan E, O’Brien K, Grebenchtchikov N, Geurts-Moespot A, Sieuwerts AM, Martens JW, et al. Protein kinase C delta expression in breast cancer as measured by real-time PCR, western blotting and ELISA. Br J Cancer (2008) 99(10):1644–50. doi:10.1038/sj.bjc.6604728
17. Pan Q, Bao LW, Kleer CG, Sabel MS, Griffith KA, Teknos TN, et al. Protein kinase C epsilon is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anticancer therapy. Cancer Res (2005) 65(18):8366–71. doi:10.1158/0008-5472.CAN-05-0553
18. Masso-Welch PA, Winston JS, Edge S, Darcy KM, Asch H, Vaughan MM, et al. Altered expression and localization of PKC eta in human breast tumors. Breast Cancer Res Treat (2001) 68(3):211–23. doi:10.1023/A:1012265703669
19. Karp G, Abu-Ghanem S, Novack V, Mermershtain W, Ariad S, Sion-Vardy N, et al. Localization of PKCeta in cell membranes as a predictor for breast cancer response to treatment. Onkologie (2012) 35(5):260–6. doi:10.1159/000338443
20. Ali S, Al-Sukhun S, El-Rayes BF, Sarkar FH, Heilbrun LK, Philip PA. Protein kinases C isozymes are differentially expressed in human breast carcinomas. Life Sci (2009) 84(21–22):766–71. doi:10.1016/j.lfs.2009.03.007
21. Gokmen-Polar Y, Mehta R, Tuzmen S, Mousses S, Thorat MA, Sanders KL, et al. Differential subcellular expression of protein kinase C betaII in breast cancer: correlation with breast cancer subtypes. Breast Cancer Res Treat (2010) 124(2):327–35. doi:10.1007/s10549-010-0733-2
22. Li H, Weinstein IB. Protein kinase C beta enhances growth and expression of cyclin D1 in human breast cancer cells. Cancer Res (2006) 66(23):11399–408. doi:10.1158/0008-5472.CAN-06-2386
23. Awadelkarim KD, Callens C, Rosse C, Susini A, Vacher S, Rouleau E, et al. Quantification of PKC family genes in sporadic breast cancer by qRT-PCR: evidence that PKCiota/lambda overexpression is an independent prognostic factor. Int J Cancer (2012) 131(12):2852–62. doi:10.1002/ijc.27600
24. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432(7015):332–7. doi:10.1038/nature03096
25. Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, et al. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest (1996) 98(9):2018–26. doi:10.1172/JCI119006
26. Yoshiji H, Kuriyama S, Ways DK, Yoshii J, Miyamoto Y, Kawata M, et al. Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res (1999) 59(17):4413–8.
27. Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, et al. Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci USA (2002) 99(2):721–6. doi:10.1073/pnas.022644499
28. Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, et al. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol (2002) 3(8):780–6. doi:10.1038/ni823
29. Saijo K, Mecklenbrauker I, Santana A, Leitger M, Schmedt C, Tarakhovsky A. Protein kinase C beta controls nuclear factor kappaB activation in B cells through selective regulation of the IkappaB kinase alpha. J Exp Med (2002) 195(12):1647–52. doi:10.1084/jem.20020408
30. Shinohara H, Kurosaki T. Comprehending the complex connection between PKCbeta, TAK1, and IKK in BCR signaling. Immunol Rev (2009) 232(1):300–18. doi:10.1111/j.1600-065X.2009.00836.x
31. Goicoechea SM, Garcia-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene (2013) 33(10):1265–73. doi:10.1038/onc.2013.68
32. Bluwstein A, Kumar N, Leger K, Traenkle J, Oostrum J, Rehrauer H, et al. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis (2013) 4:e498. doi:10.1038/cddis.2013.15
33. Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, et al. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes (2003) 52(2):512–8. doi:10.2337/diabetes.52.2.512
34. Meier M, Park JK, Overheu D, Kirsch T, Lindschau C, Gueler F, et al. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes (2007) 56(2):346–54. doi:10.2337/db06-0891
35. Bansode RR, Huang W, Roy SK, Mehta M, Mehta KD. Protein kinase C deficiency increases fatty acid oxidation and reduces fat storage. J Biol Chem (2008) 283(1):231–6. doi:10.1074/jbc.M707268200
36. Huang W, Bansode R, Mehta M, Mehta KD. Loss of protein kinase C beta function protects mice against diet-induced obesity and development of hepatic steatosis and insulin resistance. Hepatology (2009) 49(5):1525–36. doi:10.1002/hep.22815
37. Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, et al. Immunodeficiency in protein kinase cbeta-deficient mice. Science (1996) 273(5276):788–91. doi:10.1126/science.273.5276.788
38. Wolford CC, McConoughey SJ, Jalgaonkar SP, Leon M, Merchant AS, Dominick JL, et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest (2013) 123(7):2893–906. doi:10.1172/JCI64410
39. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol (1992) 12(3):954–61.
40. Soule HD, McGrath CM. A simplified method for passage and long-term growth of human mammary epithelial cells. In vitro Cell Dev Biol (1986) 22(1):6–12. doi:10.1007/BF02623435
41. Wallace JA, Li F, Balakrishnan S, Cantemir-Stone CZ, Pecot T, Martin C, et al. Ets2 in tumor fibroblasts promotes angiogenesis in breast cancer. PLoS One (2013) 8(8):e71533. doi:10.1371/journal.pone.0071533
42. Abramoff MD, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int (2004) 11:36–42.
43. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature (2007) 449(7162):557–63. doi:10.1038/nature06188
44. Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol (2003) 163(5):2113–26. doi:10.1016/S0002-9440(10)63568-7
45. Manni A, Buckwalter E, Etindi R, Kunselman S, Rossini A, Mauger D, et al. Induction of a less aggressive breast cancer phenotype by protein kinase C-alpha and -beta overexpression. Cell Growth Differ (1996) 7(9):1187–98.
46. Gibby K, You WK, Kadoya K, Helgadottir H, Young LJ, Ellies LG, et al. Early vascular deficits are correlated with delayed mammary tumorigenesis in the MMTV-PyMT transgenic mouse following genetic ablation of the NG2 proteoglycan. Breast Cancer Res (2012) 14(2):R67. doi:10.1186/bcr3174
47. Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, et al. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res (2013) 73(17):5336–46. doi:10.1158/0008-5472.CAN-13-0012
48. Szabova L, Chrysovergis K, Yamada SS, Holmbeck K. MT1-MMP is required for efficient tumor dissemination in experimental metastatic disease. Oncogene (2008) 27(23):3274–81. doi:10.1038/sj.onc.1210982
49. Ricciardelli C, Frewin KM, Tan Ide A, Williams ED, Opeskin K, Pritchard MA, et al. The ADAMTS1 protease gene is required for mammary tumor growth and metastasis. Am J Pathol (2011) 179(6):3075–85. doi:10.1016/j.ajpath.2011.08.021
50. Grum-Schwensen B, Klingelhofer J, Grigorian M, Almholt K, Nielsen BS, Lukanidin E, et al. Lung metastasis fails in MMTV-PyMT oncomice lacking S100A4 due to a T-cell deficiency in primary tumors. Cancer Res (2010) 70(3):936–47. doi:10.1158/0008-5472.CAN-09-3220
51. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16(2):91–102. doi:10.1016/j.ccr.2009.06.018
52. Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res (2006) 66(10):5242–50. doi:10.1158/0008-5472.CAN-05-4463
53. Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest (2005) 115(5):1163–76. doi:10.1172/JCI23424
54. Lam JB, Chow KH, Xu A, Lam KS, Liu J, Wong NS, et al. Adiponectin haploinsufficiency promotes mammary tumor development in MMTV-PyMT mice by modulation of phosphatase and tensin homolog activities. PLoS One (2009) 4(3):e4968. doi:10.1371/journal.pone.0004968
55. Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, et al. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res (2009) 15(10):3265–76. doi:10.1158/1078-0432.CCR-08-2649
56. Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med (2008) 6:11. doi:10.1186/1741-7015-6-11
57. Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia (2002) 7(2):177–89. doi:10.1023/A:1020304003704
58. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev (2006) 25(3):315–22. doi:10.1007/s10555-006-9001-7
59. Baay M, Brouwer A, Pauwels P, Peeters M, Lardon F. Tumor cells and tumor-associated macrophages: secreted proteins as potential targets for therapy. Clin Dev Immunol (2011) 2011:565187. doi:10.1155/2011/565187
60. Sledge GW Jr., Gokmen-Polar Y. Protein kinase C-beta as a therapeutic target in breast cancer. Semin Oncol (2006) 33(3 Suppl 9):S15–8. doi:10.1053/j.seminoncol.2006.03.019
61. Mina L, Krop I, Zon RT, Isakoff SJ, Schneider CJ, Yu M, et al. A phase II study of oral enzastaurin in patients with metastatic breast cancer previously treated with an anthracycline and a taxane containing regimen. Invest New Drugs (2009) 27(6):565–70. doi:10.1007/s10637-009-9220-1
62. Clemons M, Joy AA, Abdulnabi R, Kotliar M, Lynch J, Jordaan JP, et al. Phase II, double-blind, randomized trial of capecitabine plus enzastaurin versus capecitabine plus placebo in patients with metastatic or recurrent breast cancer after prior anthracycline and taxane therapy. Breast Cancer Res Treat (2010) 124(1):177–86. doi:10.1007/s10549-010-1152-0
Keywords: protein kinase C beta, breast cancer, mammary neoplasms (experimental), tumor microenvironment, stroma, fibroblasts
Citation: Wallace JA, Pitarresi JR, Sharma N, Palettas M, Cuitiño MC, Sizemore ST, Yu L, Sanderlin A, Rosol TJ, Mehta KD, Sizemore GM and Ostrowski MC (2014) Protein Kinase C beta in the tumor microenvironment promotes mammary tumorigenesis. Front. Oncol. 4:87. doi: 10.3389/fonc.2014.00087
Received: 17 January 2014; Paper pending published: 15 February 2014;
Accepted: 08 April 2014; Published online: 23 April 2014.
Edited by:
Jozsef Dudas, Medical University Innsbruck, AustriaReviewed by:
Alexandre Arcaro, University of Bern, SwitzerlandSilvia Pastorekova, Slovak Academy of Sciences, Slovakia
Copyright: © 2014 Wallace, Pitarresi, Sharma, Palettas, Cuitiño, Sizemore, Yu, Sanderlin, Rosol, Mehta, Sizemore and Ostrowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gina M. Sizemore, Department of Molecular and Cellular Biochemistry, The Comprehensive Cancer Center, The Ohio State University, 570A Biomedical Research Tower, 460 West, 12th Avenue, Columbus, OH 43210, USA e-mail:Z2luYS5zaXplbW9yZUBvc3VtYy5lZHU=;
Michael C. Ostrowski, Department of Molecular and Cellular Biochemistry, The Comprehensive Cancer Center, The Ohio State University, 598 Biomedical Research Tower, 460 West, 12th Avenue, Columbus, OH 43210, USA e-mail:bWljaGFlbC5vc3Ryb3dza2lAb3N1bWMuZWR1
†Julie A. Wallace and Jason R. Pitarresi have contributed equally to this work.