 Elad Jacoby
Elad Jacoby Christopher D. Chien
Christopher D. Chien Terry J. Fry
Terry J. Fry- Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Leukemia remains the most common diagnosis in pediatric oncology and, despite dramatic progress in upfront therapy, is also the most common cause of cancer-related death in children. Much of the initial improvement in outcomes for acute lymphoblastic leukemia (ALL) was due to identification of cytotoxic agents that are active against leukemia followed by the recognition that combination of these cytotoxic agents and prolonged therapy are essential for cure. Recent data demonstrating lack of progress in patients for whom standard chemotherapy fails suggests that the ability to improve outcome for these children will not be dramatically impacted through more intensive or newer cytotoxic agents. Thus, much of the recent research focus has been in the area of improving our understanding of the genetics and the biology of leukemia. Although in vitro studies remain critical, given the complexity of a living system and the increasing recognition of the contribution of leukemia extrinsic factors such as the bone marrow microenvironment, in vivo models have provided important insights. The murine systems that are used can be broadly categorized into syngeneic models in which a murine leukemia can be studied in immunologically intact hosts and xenograft models where human leukemias are studied in highly immunocompromised murine hosts. Both of these systems have limitations such that neither can be used exclusively to study all aspects of leukemia biology and therapeutics for humans. This review will describe the various ALL model systems that have been developed as well as discuss the advantages and disadvantages inherent to these systems that make each particularly suitable for specific types of studies.
Syngeneic Mouse Models of ALL
The identification of genetic changes in acute lymphoblastic leukemia (ALL) such as chromosomal translocations, gene amplification, and gene inactivation/deletion has revolutionized our understanding of the potential drivers of leukemogenesis. These have served as a basis for development of murine models that recapitulate human ALL. Genetic modification of ALL associated genes in primary cells with subsequent transplantation into mice, generation of transgenic mice that modify ALL associated genes in the lymphoid compartment, and chemical carcinogen induced mouse models have all been employed to try to model human pediatric ALL. In this section, we will focus on syngeneic models that have been shown to develop ALL that employ the first two methodologies as they utilize reverse genetics, which is more relevant to what occurs in human ALL.
B Cell ALL Models
ETV6-RUNX1 (TEL-AML1)
The t(12;21)(p13;q22) chromosomal translocation encodes a novel fusion protein ETS variant 6 (ETV6)-runt-related transcription factor 1 (RUNX1) and is the most common rearrangement that occurs in ~25% of cases of pediatric B cell ALL (1). The ETV6-RUNX1 fusion protein consists of the basic helix loop helix (bHLH) domain of ETV6 combined with the transactivation domains of RUNX1. ETV6 belongs to the ETS family of transcription factors and has been found to have at least 20 different gene fusion partners and is also found to have other genetic alterations including deletions, point mutations, and promoter alterations in various leukemias (2). RUNX1 binds to DNA as part of the RUNX1/CBFβ transcription factor complex and the RUNX1 gene locus is commonly amplified in childhood ALL (3). Clinically, ETV6-RUNX1 expression is correlated with good prognosis (4).
Different groups have taken either bone marrow (BM) or fetal liver cells from mice and retrovirally transduced these cells to express ETV6-RUNX1 protein in an attempt to model human ALL. In three models, no incidence of leukemia was observed but increased numbers of precursor B cells were found with deficiencies of more mature B cells (5–7). In the BM models, increased ckit positive multi-potential progenitors were found with increased self-renewal potential of cells that expressed ETV6-RUNX1. In a fourth model of transduced BM, 2 of 9 mice that received translocation expressing BM developed ALL compared to 0 out of 20 control mice (8). Of the mice that had leukemia, one developed a T lineage ALL while the other developed a precursor B ALL (B ALL will be used to denote precursor B ALL throughout the rest of the review and a more mature B cell disease will be noted as such). In this same report, BM from p16INK4a/p19ARF mice were also transduced with ETV6-RUNX1 and six of eight mice developed leukemia while no control mice developed leukemia.
Transgenic mice have also been employed to try to model ETV6-RUNX1 leukemias. The first study utilized the immunoglobulin heavy chain enhancer (Eμ) to drive expression of ETV6-RUNX1 in mice, however no leukemia was observed (9). This result was corroborated in subsequent reports where no leukemia was observed despite the use of differing gene promoters to express the translocation. Pre-leukemic changes in the BM were shown in a second transgenic model as pre-pro-B cell numbers were increased in mice (10). Based on three other reports, it appears likely that a second genetic event is necessary for the development of leukemia as co-expression of other genes and mutations induced by irradiation or carcinogens increased the incidence of leukemia (11–13). These data are supported by the clinical observation that ETV6-RUNX1 has been detected at birth prior to the development of leukemia (14).
E2A-PBX1
The fusion protein consisting of the N-terminal transactivation domain of E2A protein and C terminal DNA binding homeobox domain of pre-B cell leukemia homeobox (PBX1) is expressed from the t(1;19)(q23;p13) and is detectable in 6% of childhood B ALL (15). E2A (transcription factor 3, TCF3) is a bHLH protein that codes for alternatively spliced transcripts for E12 and E47 proteins found to be required for B cell development (16). PBX1 was found to be necessary for lymphoid precursor development (17). Two groups have studied which domains are required for E2A-PBX1 transforming activity and found that the homeodomain of PBX1 which is responsible for binding to DNA is not necessary for oncogenesis (18, 19). However, the homeobox domain was important for blocking differentiation of myeloblasts (18). It was later found that a HOX cooperativity motif in PBX1 was needed to mediate transformation with E2A-PBX1 (20).
Initial attempts at modeling E2A-PBX1 driven leukemias were successful at causing myeloid leukemias and lymphomas, but not lymphoblastic leukemias. BM transduced with E2A-PBX1 from BALB/C mice led to the formation of acute myelogenous leukemia with an increase in immature blast cells in the BM (21). FVB transgenic mice in which E2A-PBX1 expression was driven with IgH V-gene promoter fused to Eμ died within 5 months of age of T cell lymphomas (22). These mice showed reduced numbers of T and B cell progenitors that were at 20% of normal levels. Though important to study the oncogenic potential of E2A-PBX1, these models did not reproduce the association of E2A-PBX1 expression seen in B ALL so the relevance of these model systems to human disease is not clear.
A later model succeeded in replicating E2A-PBX1 B ALL in mice (23). In this transgenic mouse, E2A-PBX1 is expressed under control of lymphoid specific Lck upstream sequence, Eμ enhancer, and TCR Vβ promoter. Interestingly, mice expressing the transgene that died at an earlier age were prone to developing T ALL in contrast to mice who developed leukemia at later timepoints, which succumbed to B ALL or mixed lineage ALL. E2A-PBX1 transgenic mice were crossed into a CD3ε−/− background, which led to 40% of mice acquiring B ALL. These mice had a long latency time before symptoms of disease were observed and this latency was reduced when combined with Hox gene overexpression by viral insertional mutagenesis. In addition to Hox genes, Pim1, Notch1, and downregulation of INK4A-ARF by Bmi-1 have also been shown to cooperate with E2A-PBX1 in oncogenesis suggesting the importance of secondary hits in the development of E2A-PBX1 driven leukemias (24–26).
E2A-HLF t(17;19)(q22;p13)
E2A-hepatic leukemia factor (HLF) protein is the result of t(17;19)(q22;p13) translocation most commonly found in adolescents as a high risk pro-B cell ALL, yet is a rare event (27). Similar to the E2A-PBX gene fusion, the N-terminal transactivation domains of E2A are joined to the basic leucine zipper dimerization domain of HLF. HLF is a member of the proline and acidic amino acid-rich basic leucine zipper (PAR bZip) transcription factor family and mice deficient in this protein develop without any cancer pathology (28). Both transactivation domains of E2A and the basic leucine zipper dimerization domain of HLF were required for transformation of NIH3T3 mouse fibroblasts (29). The transforming ability may in part be due to upregulation of LIM domain only 2 (LMO2) (30).
Two transgenic mouse models of E2A-HLF were developed with slightly different results. The first model used the Eμ enhancer with the SV40 promoter to drive expression of the translocation (31). In this report, they described changes in the precursor T cell populations in the thymus and spleen with mature splenic B cells expressing low levels of E2A-HLF. Sixty percent of mice became sick with lymphomas, 90% of which were derived from a T cell lineage, and the remaining 10% of B cell origin. A second model also used the Eμ enhancer with its corresponding promoter to express E2A-HLF (32). An increase in T cell apoptosis in the thymus and a block in the maturation of B cells in the spleen was observed. These mice showed incomplete penetrance with 5 of 26 mice developing T ALL.
A later study used BM transduction to study the leukemogenic inducing potential of E2A-HLF (33). BM was transduced with a murine stem cell retrovirus to express transgenes and grown in vitro on irradiated AC-6.21 stromal cells before transplantation. These transduced cells without additional mutations did not induce leukemia in non-irradiated hosts. However, BM cells that were transduced to express E2A-HLF with Bcl-2 and transplanted immediately into lethally irradiated hosts did develop B ALL, which is similar to the disease seen in human B ALL.
BCR-ABL
Breakpoint cluster region (BCR)-ABL is the protein product caused by the t(9;22)(q34;q11) translocation (also known as the Philadelphia chromosome) is most commonly found in CML, but also accounts for 20–40% of adult B ALL and 3–5% of childhood B ALL cases (34, 35). Expression of the translocation product is controlled by the BCR promoter, which is ubiquitously expressed throughout the body (36). BCR null mice show alterations in activity of neutrophils (37) while ABL1 knockout mice exhibit wasting of the spleen and thymus as well as significant loss of B and T cell numbers (38, 39). Two versions of the fusion protein associated with ALL are generated from different breakpoints leading to a 210-kDa protein (p210) and a 190-kDa protein (p190). The p210 isoform is detectable in 33% of adult Philadelphia chromosome positive (Ph+) ALL with the rest of cases expressing the shorter isoform. The p190 accounts for 90% of Ph+ ALL cases in childhood ALL. The BCR-ABL fusion protein is a constitutively active non-receptor tyrosine kinase due to removal of the domains of ABL1 that normally keep ABL1 in an inactive closed conformation (40) and this deregulation leads to transformation. There have been many reports to study the function of the BCR-ABL translocation in many leukemia models, however this section will focus on the work that pertains to studies of ALL in BCR-ABL model systems.
Early attempts to model ALL in transgenic mice used the metallothionein-1 promoter, which has broad tissue expression in the mouse, to express p190 BCR-ABL (41). This promoter was chosen as attempts to use the BCR promoter to drive expression of BCR-ABL, as occurs in human leukemia, led to embryonic lethality of mice. Of ten mice examined, two mice developed AML while six mice were diagnosed with B ALL. Though Heisterkamp et al. showed embryonic lethality using a BCR promoter, another report showed that a p190 BCR-ABL knock-in model, which has expression of p190 from the endogenous BCR gene locus, develops B ALL with high frequency in chimeric animals (42). In contrast, expression of p210 from the endogenous BCR locus does not result in neoplastic transformation of hematopoietic cells to form leukemia (43). Further work from the Heisterkamp group explored what happens when the p210 isoform of BCR-ABL is expressed using the metallothionein-1 promoter (44). p210 transgenic mice became sick from B, T, and myeloid leukemia with a long latency period while mice that expressed p190 only developed B ALL with a rapid disease course. A second (Honda et al.) publication also described a p210 BCR-ABL model driven by the same metallothionein-1 promoter (45). Interestingly, in this model two of six mice developed leukemia committed to the T cell lineage. The reasons underlying the difference in the spectrum of diseases seen in these p210 models are not clear, though differences in either strains of mice used or differences in promoter sequences used may contribute. Honda et al. created another BCR-ABL transgenic mouse that used the mouse Tec promoter to regulate p210 BCR-ABL expression (46). Tec is a cytoplasmic protein tyrosine kinase that is expressed in hematopoietic progenitor cells. Two of five founder mice developed ALL and the progeny of one of these mice developed CML at 1 year of age with some mice showing hallmarks of blast crisis in the lung. Another group took advantage of advances in tetracycline transactivator (tTA) proteins to regulate gene expression of p210 BCR-ABL (47). They used a “tet-off” system with the mouse mammary tumor virus long terminal repeat (MMTV-LTR) promoter, which controlled tTA protein. This allowed expression of p210 in the absence of tetracycline in the water of mice and repression of expression when mice were given tetracycline. In this model, mice developed B ALL and initiation and maintenance of ALL was dependent on p210 expression.
Bone marrow transduction studies have shown that BCR-ABL is capable of inducing multiple types of leukemia. Although the initial intention of this study was to model CML, expression of p210 via a myeloproliferative sarcoma virus (MPSV) in BM cells from BALB/c led not only to CML, but B and T ALL and macrophage derived tumor in 13 of 30 mice studied (48). A follow-up report using the same p210 BCR-ABL protein (b3a2) in BALB/c BM transduction/transplant studies, compared another p210 BCR-ABL protein (b2a2), resulting from an alternative breakpoint, when driven by a murine stem cell virus (MSCV) U3 promoter found that both gave rise to B ALL without any evidence of myeloid leukemia (49). Yet another group utilized a MPSV system to express p210 BCR-ABL in mouse BM and also observed a broad range of diseases in two different mouse strains (50). In both DBA/2 and C57BL/6 mice, macrophage and erythroid derived tumors were found, while in lymphoid derived tissues DBA/2 mice developed B cell lineage lymphomas while C57BL/6 mice developed T lineage lymphomas. In contrast to the previous report classical CML was not observed in either of these strains of mice. Another BALB/c BM transduction model was used with a Moloney murine leukemia virus (Mo-MuLV) to express p210 BCR-ABL with half of the animals developing CML like disease while the other half developed pre-B cell lymphomas (51). Another study using BALB/c mice used BM transduced with MSCV controlling expression of p190, p210, or p230 BCR-ABL, which is commonly associated with chronic neutrophilic leukemia (52). When BM from mice pre-treated with 5-fluoro uracil (5-FU), which enriches multi-potential myeloid cells in the BM, were transplanted to recipient mice all three isoforms of BCR-ABL gave rise to CML like disease in all mice with similar latency for development of disease. When the BM without 5-FU treatment was used for transduction and transplant, mice developed diseases of various lineages, which included not only CML like but also B ALL and macrophage lineage disease. The p190 isoform in the latter experiment showed more aggressiveness as these mice acquired disease at earlier timepoints with an increased incidence of B ALL. BM185 cells are another model system in which BALB/c BM is grown in early B cell progenitor Whitlock Witte cultures and transduced with p190 BCR-ABL. These cells when intravenously transplanted into mice developed B ALL with as little as 1000 cells in 3 weeks with 100% penetrance. (53). These findings underscore the complexity of modeling human disease as expression of the same cancer associated protein can have vastly different effects in mice and may be due to strain differences or use of alternative viral vectors. In addition, the cell of origin is another potential factor in disease variability highlighted by the fact that transplantation of a late onset CML like disease driven by p210 BCR-ABL in a BM transduction/transplant model led to not only myeloid disease, but also B and T ALL (54).
MLL Fusions
The mixed lineage leukemia (MLL) gene is most frequently found rearranged in infant ALL accounting for two-thirds of infant ALL cases and less frequently in AML and confers a worse prognosis (55, 56). There have been 121 rearrangements of MLL identified with 79 translocation partner genes characterized molecularly (57). The seven most common rearrangements involve AF4, AF9, ENL, AF10, ELL, AF6, or derive from partial tandem duplications of the MLL gene (MLL-PTD). The MLL-PTD are rare in acute lymphoblastic leukemia (58) and have not been modeled thus far and will not be covered in this review. Despite the rare instances of ALL in these models, we will highlight the work that has been done in mice with these common rearrangements that have been identified in ALL.
MLL-AF4
MLL-AF4/AFF1 is the product of t(4;11)(q21;q23) and account for about 50% of cases of MLL rearrangement. Mouse models have used various approaches to express the MLL-AF4 protein. A standard transgenic mouse using pronuclear injection of MSCV MLL-AF4 had B cell neoplasms found in the spleen, liver, lung, and blood after a long period of latency (59). Knock-in mouse models were also generated by using the MLL gene locus to express MLL-AF4. The first model used traditional gene targeting to fuse human AF4 to exon 7 of mouse MLL (60). These mice developed lymphoid and myeloid hyperplasia with the most common malignancies of B cell lineage at 22 months of age. The second knock-in mouse was an inverter mouse that utilized a Cre/Lox system to cause a recombination event to happen after Cre expression that would cause MLL-AF4 to be expressed instead of normal MLL (61). These mice developed more mature B cell neoplasms as opposed to B ALL from which the cDNA for AF4 was originally isolated to generate the mice. The last model made use of a Lox Stop Lox cassette to halt transcription of AF4 in the MLL locus prior to Cre recombination (62). MLL-AF4 was only expressed in cells in which Cre recombination occurred and led to ALL and AML in mice.
Bone marrow transduction has also been used to model MLL-AF4 leukemogenesis. In the Lox Stop Lox model, BM transduced with a transiently expressed Cre led to ALL, AML and MLL after transplant back into mice (62). In a separate report using MSCV MLL-AF4 transduced BM, leukemogenesis required expression of the normal MLL gene (63). Another study found that MLL-AF4 and its reciprocal translocation AF4-MLL were both able to cause ALL of all subtypes (B, T, mixed lineage) in mice that were transplanted with BM that expressed each alone or when co-expressed (64).
MLL-AF9
The t(9;11)(p22;q23) chromosomal rearrangement produces the MLL-AF9/MLLT3 leukemia associated protein. Two mouse models have been created to model MLL-AF9 leukemia. MLL-AF9 was targeted to the MLL locus in mice and expressed using native MLL gene regulatory elements (65). These mice only developed AML. In the second model, the investigators took a novel approach using Cre recombination where single LoxP sites were introduced into the endogenous MLL and AF9 genes to create Cre mediated interchromosomal recombination of MLL and AF9 (66). Chimeric mice generated using this strategy developed acute leukemias and in mice analyzed and was classified as AML. BM transduction/transplantation (tp) model AML has been recently reported (67). MLL-AF9 has also been transduced in granulocyte macrophage progenitors to produce AML (68).
MLL-ENL
MLL-ENL is the product of the t(11;19) (q23;p13.3) translocation. The only reported interchromosomal gene recombined mouse model of MLL-ENL developed myeloid leukemias (69). BM transduction models have been made but not all studies have involved transplant into mice. MSCV MLL-ENL was introduced into early hematopoietic progenitors and transplanted into mice, which led to myeloid leukemias (70). In a separate study, MSCV MLL-ENL transduced BM grown in ex vivo conditions favoring lymphopoiesis without a stromal feeder layer induced a continuously growing cell population (71). The proliferating cells were initially B220+CD19+, but after continual growth a B220+CD19− population dominated the culture. The B220+CD19− cells could induce splenomegaly and infiltrated the thymus of mice and despite being morphologically myeloid, they retained B220 positivity showing a mixed lineage phenotype. Later BM transduction models used conditional MLL-ENL. A mutated estrogen receptor fusion protein of MLL-ENL was used, but not introduced into animals so the resultant disease was not reported (72). A tetracycline regulatable MLL-ENL when used to transduce BM was able to induce AML and expression of MLL-ENL protein was required for leukemic growth even in leukemias that acquired secondary mutations (73).
MLL-AF10
MLL-AF10 is derived from the rearrangement t(10;11)(p13–15;q14–21). Only BM transduction studies have been done using this gene fusion. Two studies use the same MSCV based BM transduction with MLL-AF10 and both models develop myeloid lineage disease (74, 75). An additional model studied the cooperativity of MLL-AF10 with the activating mutation Kras G12C (76).
MLL-ELL
The only published study of MLL-ELL gene fusions utilized a MSCV based BM transduction strategy, where C terminal domain of ELL was shown to be required for transformation of myeloid progenitor cells. In this study, there was no data shown for transplantation of these transduced cells into mice but MLL-ELL expression caused immortalization of myeloid progenitors in vitro (77).
MLL-AF6
The only MLL-AF6 model of leukemia used BM transduction to demonstrate the requirement of histone methyltransferase Dot1l for MLL-AF6 induced leukemogenesis (78).
PAX 5 Deletions
Somatic mutations and deletions of PAX5 have been found in about 30% of pediatric B ALL samples (79). To date, only haploinsufficiency of PAX5 was shown to be important for induction of leukemia in an activated STAT5 B ALL mouse model (80).
IGH-Myc
Myc translocations occur in about 5% of both adult and childhood ALL (81). Transgenic mice that express Myc under control of IGH enhancers showed B cell lymphomas with leukemic involvement at later timepoints (82, 83). Knock-in of Myc into the native IGH locus induced B cell neoplasms (84). BM transduction studies using p53 null BM transduced with Myc led to B cell lymphomas (85).
T Cell ALL
NOTCH1 Activation
NOTCH1 is a type I transmembrane receptor that is involved in signal transduction and is the only NOTCH gene involved in T ALL (86). Activation of the NOTCH1 pathway is found in over 50% of T ALL cases (87). In concordance with this clinical data, NOTCH1 mutation has been identified in two transgenic mouse T ALL models induced by KRas G12D (88, 89). No T ALL transgenic models have been shown to be solely driven by NOTCH1 mutations, but BM transduction/transplantation studies have shown that NOTCH1 activating mutants can drive T ALL formation. A L1601PΔP mutant, which has a L1601P and frameshift mutation in the PEST domain of NOTCH1 was shown to inefficiently induce T ALL in mice after transduction and transplantation into mice (90). A separate NOTCH1 mutant (ICN1) which consists of the transmembrane and intracellular domain of NOTCH1 was able to induce T ALL in BM cells transduced with ICN1 after transplant into mice (91). Transgenic mice expressing ICN1 in T cell progenitors have also been shown to develop T ALL (92).
Inactivation SCFFBW7
FBW7 is subunit of the SCF-type E3 ubiquitin ligases. FBW7 mutations are found in many human cancers such as T ALL, which corroborates its role as a tumor suppressor (93). Conditional inactivation of FBW7 in T cell lineage cells in mice caused thymic hyperplasia likely due to Myc accumulation (94).
TAL1 and LYL1 Mutations
TAL1 is a class II bHLH transcription factor that is a master regulator for commitment to the hematopoietic lineage. TAL1 mutations or translocations are found in about 25% of childhood T ALL (95, 96). Lck promoter driven TAL1 transgenic mice also develop lymphomas of a predominantly mixed T and B cell phenotype (97). Transgenic mice where a TAL1 DNA binding mutant is expressed still develop T cell leukemia/lymphoma (98). bHLH proteins are commonly found to interact with other bHLH proteins as heterodimers to regulate gene expression and a potential hypothesis for the role of TAL1 in T ALL is that overexpression of TAL1 and its mutants disrupt the normal gene program by interacting with other transcription factors that regulate T cell development such as E2A proteins (98).
LYL1 is also a class II bHLH transcription factor that heterodimerizes with class I bHLH transcription factors. Transgenic mice that express LYL1 developed T cell lymphomas positive for both CD4 and CD8 and mature B cell lymphomas (99). LYL1 is also proposed to alter transcription through its interaction with other transcription factors such as E2A (99).
LIM Domain Only 2
LIM domain only 2 is a cysteine rich protein that cooperates with other nuclear proteins to regulate gene transcription. Three transgenic models of LMO2 have been studied in relation to T ALL and differ in choice of promoter to drive expression of LMO2. Metallothionein-1 promoter led to 10% of LMO2 transgenic mice developing CD4/CD8 double positive, single positive, and double negative T cell neoplasms (100). Higher penetrance was seen in 75% of mice that expressed LMO2 under control of the CD2 promoter (101). A third mouse reported used Lck promoter to express LMO2 and showed cooperation with TAL1, a known binding partner to LMO2, to induce T ALL (102).
CALM-AF10
The translocation t(10;11)(p13;q14–21) creates the CALM-AF10 fusion protein. They are most commonly found in TCRγδ expressing T ALL. Vav driven CALM-AF10 in a transgenic mouse model led to mixed lineage ALL (103). BM transduced with MSCV driven CALM-AF10 and transplanted into mice led to leukemias that expressed predominantly myeloid markers Mac1 and Gr1 and more rarely B cell marker B220 alone or co-expressed with Mac1 and Gr1 (104). A recent study also showed that CALM-AF10 transformation is dependent on the histone methyltransferase Dot1l (105).
Summary of Syngeneic Mouse Models of ALL
There has been significant progress made in trying to model ALL in mice. The number of exogenous and endogenous promoters to control expression of leukemia associated genes and mutations has grown significantly and their tissue expression has been well characterized. Advances in conditional mouse technology have also been rapidly incorporated into various models with inducible models providing exquisite control of transgene expression in cell lineages. Through the use of BM transduction, a more rapid disease course is possible as secondary mutations can be introduced that cooperate with primary mutations. This is important because in the majority of ALL models long periods of latency are seen with expression of single leukemia associated aberrations in mice, which make study of these lesions difficult and time consuming. The development of these ALL models (summarized in Table 1) has contributed greatly to our understanding not only of pathways that underlie leukemogenesis, but also helps us identify potential therapeutic targets in these pathways.
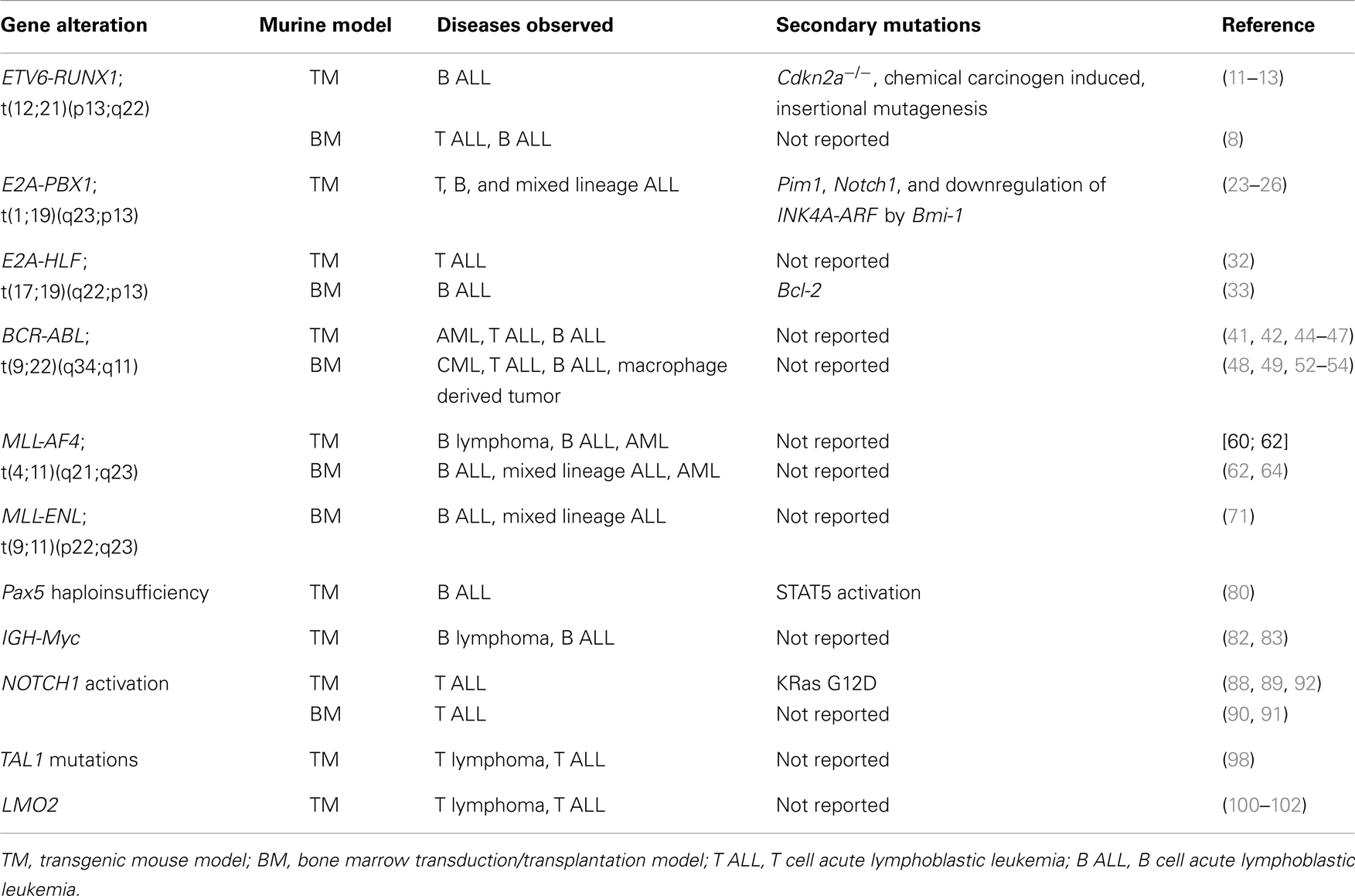
Table 1. Syngeneic mouse models of ALL.
Despite the promise in these models, there seems to be some discrepancies in the disease observed driven by these ALL associated genetic events. In trying to study ALL, expression of ALL associated genes/mutations/translocations has led to AML in a significant number of instances. This could be due to promoter choice, cell of origin or cross species protein expression. Various promoters have been used to try to express leukemia associated alterations in genes and promoter choice affects the cells that express those gene products. This would explain why the same leukemia factor expressed under control of alternative gene promoters leads to different diseases in mice than in humans. Additionally, there may be complications that arise in expressing human leukemia associated proteins in a mouse. Protein–protein interactions that exist in humans may be absent or different in mice and therefore proper reproduction of human disease in mice may become more difficult.
There are advantages for using syngeneic models of ALL. The contribution of the microenvironment to the development of many cancers is an important topic of study in carcinogenesis. Cytokines, growth factors, and cell–cell interactions provided by the local tumor microenvironment influence how cancers initiate and develop. Syngeneic models of ALL can be utilized to study the tumor microenvironment and its contribution to leukemogenesis. There are factors that contribute to leukemogenesis that do not signal across species and therefore would be important to study in a single species model. For example, the role of thymic stromal lymphopoietin receptor overexpression (TSLPR/CRLF2) in B ALL is being studied. TSLP, the ligand for TSLPR, does not signal cross species so study of TSLPR overexpressing leukemia must be pursued in a single species model. Syngeneic models of TSLPR overexpressing B ALL would allow study of disease course in the presence of ligand at physiologic doses that is not possible in a xenograft model. Expression of human TSLP in mice would be required to study human TSLPR overexpressing B ALL in xenograft model systems where mimicking physiologic levels of the ligand could be problematic.
Xenograft Models: Development of the Human ALL-Bearing Mouse
The pursuit of an animal model that would provide in vivo insights of direct relevance to human ALL has been undertaken by many groups using multiple types of immunodeficient mice to allow engraftment of human cells. The first candidate was the athymic nude mouse, developed in the late 1960s. Lack of T cells allowed engraftment of some human tissues and tumors, but intact NK cells and B cells prevented engraftment of human hematopoietic cells. Still, by conditioning mice with radiation, initial studies demonstrated some ability for leukemia cells to engraft (106–108). Nonetheless, engraftment of cells administered intravenously was extremely difficult. Thus, in these models, recipients were inoculated with leukemia cell lines in the intradermal, subcutaneous or intraperitoneal compartments, and at times required an addition of irradiated cells from a fibrosarcoma cell line in order to engraft (107). The results were ascites (106) or solid masses (108) of leukemia origin, which allowed studying of new drugs and therapeutic approaches. However, the leukemia lacked its “normal” BM niche and natural course in extramedullary organs. Thus, the study of homing, engraftment, persistence, course, and therapeutic approaches in the native leukemia niche was impossible in this model.
Introduction of the severe combined immunodeficiency (SCID) mouse on a CB17 background (109), was a major advance in the field, allowing marrow engraftment of human hematopoietic cells [reviewed by Shultz (110)]. These mice have a deletion in Prkdc, an essential protein involved in DNA repair following VDJ recombination in T and B cells. Hence, these mice lack adaptive immune cells, but retain NK cells as well as innate immunity; therefore, these models often required conditioning with sublethal irradiation to engraft human leukemia (111), though some groups overcame this need by repeated in vivo transfers of the leukemia cell line. An important breakthrough was the use of SCID mice to engraft patient derived primary leukemia cells following an IV injection (112). This resulted in engraftment and proliferation of the leukemia in vivo, with infiltration of the spleen, liver, kidney, lung, central nervous system, (112) lymph nodes and ovaries (113), thus imitating extramedullary involvement. Also, compared to nude mice, SCID mice engrafted leukemia following a lower inoculation dose (114).
Several attempts were made to genetically manipulate the SCID mouse to increase the engraftment ability of the SCID mouse. The SCID-beige mouse was developed by adding a mutation in the lysosome trafficking regulator (Lyst) to the Prkdcscid mutation, on a Balb/c or CB57/BL/6J background. This resulted in lack of T and B cells as well as neutropenia and reduced NK function; moreover, these strains avoided the late immune-reconstitution seen in SCID mice (115). This model can engraft leukemia and has been used by a number of groups in leukemia research (116).
The NOD-SCID mouse was generated by crossing the SCID mouse (Prkdcscid) to the non-obese diabetic (NOD/Lt) mouse background, resulting in reduced NK function as well as impaired innate immunity derived from the NOD mutation in a diabetes-free strain (110, 117). This mouse showed increased rate of engraftment of human hematopoietic cells in general, including ALL cell lines and patient samples (118, 119). Moreover, impaired NK function reduced the need for conditioning with radiation prior to leukemia administration, though conditioning with irradiation was still used by investigators (120, 121). This was of importance, since these mice have a tendency to develop spontaneous thymic lymphoma, and have generally a short life span [median survival of 257 days (117)]. Human leukemia seemed to have stable properties when passed to an NOD-SCID mouse, with stable antigenic markers, T- or B-cell receptor rearrangements, karyotype, as well as a stable rate of engraftment in subsequent passes of the leukemia (121, 122).
Crossing the common-gamma-chain (interleukin 2 receptor gamma) knockout to the NOD-SCID mouse generated the NOD/SCID/γC− (or NSG mouse), with multiple defects in both the innate and the adaptive immune system. Lack of NK cells omitted the need for irradiation prior to leukemia administration. NSG mice have superior engraftment of ALL cells in terms of time to engraftment (shorted by ~2 weeks compared with NOD-SCID mice), ability to detect blasts in the peripheral blood, and total number of cells required to engraft (123, 124). NSG mice can even be engrafted with as little as 1–100 cells derived from patients with poor-prognosis ALL (125). Again, as in the NOD-SCID model, patient derived B or T -lineage ALL engrafted in NSG mice have a stable phenotype and transcriptome, even after a several passes in vivo (126). The relative simplicity of engrafting human leukemia in these mice made the NSG model excellent for leukemia research.
In summary, the evolution of the immunocompromised mouse, with its ability to engraft human hematopoietic cells, provided in vivo models for studying human ALL. Nevertheless, there are some significant drawbacks in using these mice. Since they have profound immunodeficiency, they need to be housed in a protected pathogen-free environment. Moreover, they have an inherent tendency toward spontaneous lymphomas. Third, engraftment of human T cells (from BM transplant or immunotherapy experiments) results in high rate of lethal xenogeneic graft versus host disease (Xeno-GVHD).
Recently, inactivation of CD47 on a C57BL/6 mouse with Rag2 and Gamma-chain knockout (triple-knockout) allowed tolerance toward human hematopoietic stem cells. These triple-knockout mice had no GVHD at 29 weeks after transplantation, compared with one-third of NSG mice that develop GVHD at this point (127). This model has not been introduced to leukemia challenge, but it may be a promising avenue in the future, especially for immunotherapy models.
Utilizing these mouse strains (summarized in Table 2) to study leukemia require administration of leukemia cells, via intravenous route or direct into the marrow compartment (intratibial or intrafemoral) (128, 129). Commercially available cell lines or primary patient samples of ALL may be injected in xenogeneic mice, with different engraftment rates, depending on the mouse strain as well as the leukemia properties (discussed below).
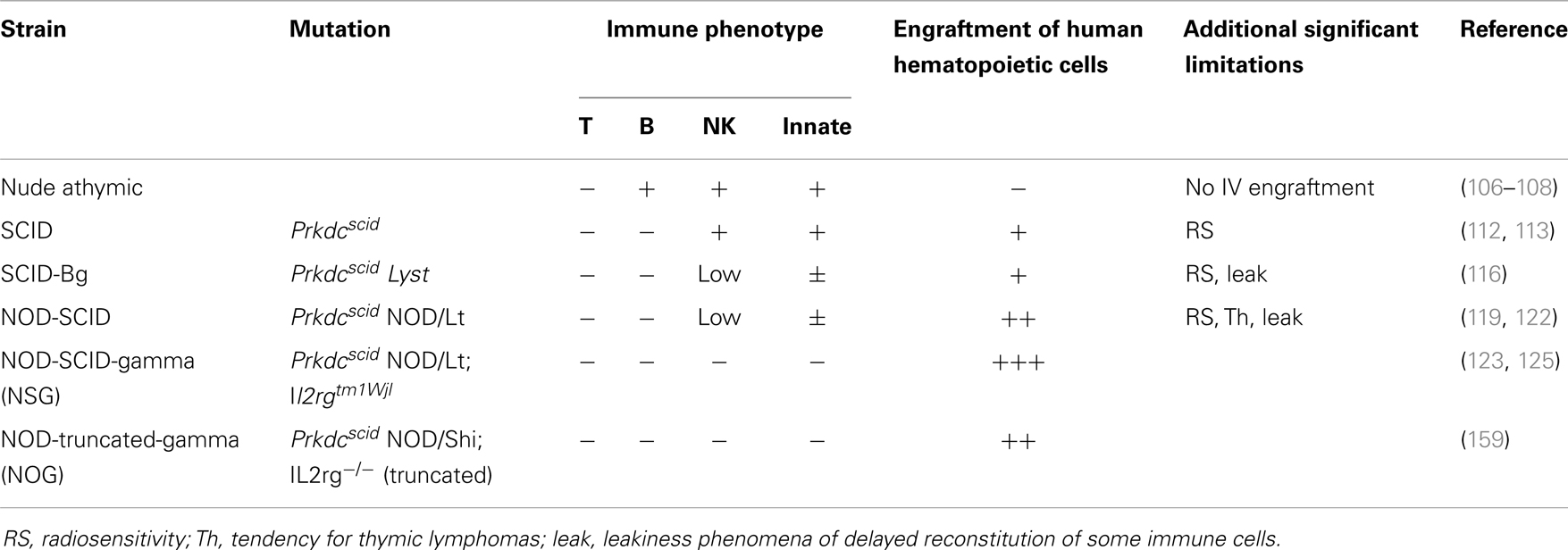
Table 2. Xenograft mouse strains used in human leukemia research.
Questions Addressed in the Xenogeneic Mouse Model
Microenvironment of ALL
The ability to engraft human leukemia in the murine BM provided an opportunity to study the leukemia niche within the marrow, as well as its progression and extramedullary involvement. Indeed, patient derived pre-B ALL cells that were intravenously administered to SCID mice caused multiple organ involvement, including hepatosplenomegaly, kidney and ovarian enlargement, as well as lymphadenopathy (112). Focusing on the BM niche is outside of this review’s scope, but some examples for utilizing the xenograft models in these studies can be given. For example, inhibition of CXCR4 in ALL samples administered to NOD-SCID mice showed increased sensitivity of the blasts compared with normal hematopoietic stem cells, pointing out the CXCR4–CXCL12 axis as significant in ALL homing (130). Messinger et al. searched for different adhesion molecules in ALL blasts in different organs of xenografts transplanted with primary patient samples of pre-B ALL. They identified VLA-4 and VLA-5 in the BM ALL blasts, but not in the liver or spleen, suggesting loss of these integrins may be a property of the leukemic cells prior to migration to extramedullary organs (131). CNS leukemia was studied initially in the SCID mouse. Eight days following IV administration of NALM-6 ALL cell line, PCR demonstrated evidence of CNS leukemia (132). This allowed experimental therapeutic tests of CNS leukemia.
Leukemia Stem Cell
The study of leukemia stem cells or leukemia initiating cells (LIC) utilized xenograft models as well, mainly by sorting cells based on surface markers and assessing capability of initiating leukemia in vivo. Initial studies focused on the hematopoietic stem cell markers CD34 and CD133, as well as lineage specific markers (CD10, CD19, CD22, CD38 for B cells, CD3 and CD7 for T cells). Cox et al. sorted patient derived T ALL cells. They found that CD34+/CD4− or CD34+/CD7− cells were capable of engraftment in NOD-SCID mice, with self-renewal and potential for secondary and tertiary engraftments, marking these as LIC (121). CD34+/CD7− cells had a higher engraftment rate in NSG mice compared with NOD/SCID mice (123), both resulting in engraftment, differentiation with similar immunophenotype to the original patient sample. In B-precursor ALL, CD34+CD19− and CD34+CD10− cells sorted from patients had long-term proliferation in NOD/SCID as well as NSG mice, compared with CD34+CD19+ cells, suggesting the prior sub-population to be LIC (123, 133). In contrary, other investigators have shown that CD34+CD19+CD38± cells sorted from patients’ BM were able to initiate leukemia in NSG mice, had self-renewal properties, and resulted in lethal CD19+ leukemia in the host. Transplantation of patient derived CD34+CD38−CD10−CD19− cells did not result in overt disease, suggesting the CD34+CD19+CD38± as the LIC (134). When sorting primary patient samples on CD133, CD133+/CD19− as well as CD133+/CD38− cells engrafted at low inoculating doses (5.4–6.5 × 103 cells), while CD34+CD19+ cells needed an almost 100-fold higher dose to engraft. Also, CD133+/CD19− cells were resistant to vincristine and dexamethasone, suggesting this phenotype as chemo-resistant leukemia stem cells (135). Even more controversy was introduced when, via intrafemoral injections of purely sorted leukemia cells, le Viseur et al. showed that different populations of the leukemia have potential to re-establish leukemia in subsequent transplantations to NOD/SCID mice (128).
Studies of identical twins with leukemia suggested similar LIC with either TEL-AML1 or BCL-ABL translocations (136, 137). To study whether the fusion proteins resulting from these translocations may generate a self-renewing pre-leukemic clone, human cord blood cells transduced with TEL/AML1 were transplanted into NOD/SCID mice, resulting in CD34+CD38−/lowCD19+ population, similar to pre-leukemic phase seen in identical twins of the patients (129).
Marker for Aggressiveness/Prognosis
Given the diverse nature of pediatric leukemia, a search for prognostic factors, which, at diagnosis, would allow a tailored therapy with escalation or de-escalation of treatment, is an ongoing mission. Several investigators attempted to use xenograft models to answer this question, by injecting primary leukemic cells or BM from leukemia patients into these mice.
Initial studies in SCID mice showed some correlation with prolonged remissions in patients whose cells did not engraft (111, 138, 139), but this was not consistent, nor was it consistently observed in the NOD-SCID mouse (120, 140). In a large cohort of patient derived pre-B ALL, only samples from low-risk patients showed correlation between engraftment rate (time to leukemia, TTL) in NOD-SCID mice and duration of remission (140, 141). Similar observations were made in the NSG mouse, with lower engraftment rate from patients who did not relapse (126). When assessing samples from relapsed patients, these tended to grow at a faster rate compared with samples from diagnosis when engrafted in SCID mice (111). The TTL or rate of engraftment of the relapsed sample in the NOD-SCID mouse correlated with remission duration (120). In vivo sensitivity to drugs, as assessed by survival of leukemia bearing mice following drug administration vs. saline, was also tested as a prognostic marker. Sensitivity to vincristine and dexamethasone, but not methotrexate, correlated with remission rate of patient derived ALL engrafted in NOD-SCID mice (120, 122). In general, these studies suggest that rate of engraftment in xenogeneic models may be used as a prognostic factor, but this method is complicated, expensive, and too difficult to be utilized in many centers.
Therapeutic Approaches
Most of the chemotherapy drugs used in treating pediatric ALL were developed prior to these in vivo models. However, in the past two decades, several novel therapies have utilized the xenograft model as an essential part of the pre-clinical evaluation. In an in vitro drug test, the target cells are continuously exposed to the tested compound. An in vivo model required scheduled doses, which have different pharmacodynamic and pharmacokinetic properties, similar to clinical administration of drugs (142). Treatment models use survival, or bioluminescence readings, as measures to evaluate in vivo treatment efficacy. This review is focused on the modeling system and will not comprehensively address all treatment approaches. Thus, we will discuss only selected therapies tested in the xenogeneic models, including chemotherapy, biologic agents, and immunotherapy.
New chemotherapy agents, as topotecan, have been evaluated in the SCID mouse. SCID mice had a long-term remission following topotecan administration. Also, the pharmacokinetics of topotecan in mice was similar to the pharmacokinetics in patients (143). mTOR inhibitors are also increasingly used in different pediatric tumors. Mouse models of ALL demonstrated that the mTOR inhibitor sirolimus was able to prolong survival of mice bearing ALL (144). Temsirolimus, another mTOR inhibitor, significantly prolonged survival of NOD-SCID mice injected with patient derived ALL and was found to be synergistic with methotrexate (145).
As previously mentioned, leukemic blasts require CXCR4 to stay in their BM niche. CXCR4 antagonists (Plerixafor or AMD11070), used for hematopoietic stem cell mobilization, were found to increase the proportion of actively cycling ALL cells in the peripheral blood of NOD-SCID mice, prolong survival of NSG mice, and increase either strain’s sensitivity to vincristine (130, 146).
Histone DeAcetylase (HDAC) inhibitors are also novel cancer medications. Balb/c-Rag−/−gammaC−/− mice bearing patient derived T or B ALL were treated with LBH589, a HDAC inhibitor. This treatment resulted in prolonged survival of the mice compared with standard vincristine and dexamethasone treated mice, while combination of all three therapies prolonged survival even further (147). Other novel therapies based on specific leukemia biology, such as CBP/catenin inhibitor (ICG-001) (148), interleukin 27 administration (149), spleen-tyrosine kinase (SYK) inhibitors (150), anti-connective tissue growth factor (CTGF) monoclonal antibody (151) for pre-B ALL, and anti-Notch1 monoclonal antibodies or for T ALL (152), were all shown to prolong survival and/or reduce engraftment in NOD-SCID or NSG mice, with or without additional chemotherapy.
Ricin-conjugated immunotoxin were tested in SCID mice and shown to induce a prolonged remission compared with controls (114). Messinger et al. combined an anti CD19 immunotoxin conjugated to poke-weed antiviral protein with several chemotherapeutic agents, and showed that only combination with cytarabine led to prolonged survival in SCID mice (153). More recently, a combination of two ricin-conjugated monoclonal antibodies (against CD19 and CD22) was shown to be effective in combination with cytarabine against ALL-bearing NSG mice (154).
Use of adoptive-T cell based immunotherapy is an increasing focus of basic as well as clinical research. As these are cell-based therapies that proliferate in vivo, xenograft models were an essential part of describing these therapies, and of the pre-clinical evaluation. Cytotoxic T lymphocytes (CTLs) designed to target specific leukemia cells, resulted in remission of NOD/SCID mice bearing ALL (155). There is a growing interest in genetically engineered chimeric-antigen-receptor (CAR) T cells. Ability to track leukemic progression with luciferase-transduced leukemia cell lines was used to assess in vivo activity of CAR-T cells. A CD19-CAR administered to NOD/SCID mice inoculated with Daudi cells (156), and to SCID-Beige mice inoculated with NALM-6 cells (116), resulted in disease remission, as did a CD22 CAR in NSG mice inoculated with NALM-6 cells (157). These models can be used to assess treatment efficacy and treatment failures (escape mechanisms) on ALL cell lines as well as primary samples, and were able to identify the periodontal region as a new sanctuary site of leukemia, in which homing of the CAR-T cells was inadequate (116). Some major disadvantages of these models can also be elucidated when testing cell-based therapies: lack of a native immune system limits true studies of engraftment and persistence of adoptively transferred cells, in an environment without competition and/or native cytokine support. Also, there is an inherent limited survival of human T cells on some models (SCID-Beige mice) (116) and graft versus host disease frequently arises from the adoptively transferred human T cells against the mouse (158).
Summary
As described in this review, there are multiple murine models in current ALL research. The xenogeneic model, especially the NOD-SCID and NSG strains, has superb ability to engraft human hematopoietic cells and human leukemia, and may serve as an appropriate platform for in vivo research of human-derived leukemia, utilizing either cell lines or patient derived cells. Given the diverse landscape of acute leukemia as far as genetic abnormalities, the ability to engraft patient derived cells is a major advantage of xenogeneic models. Also, they may serve as in vivo platforms for drug testing against ALL. Moreover, the relative simplicity of using bioluminescent imaging of leukemia (cell lines or patient samples) in xenogeneic mice for evaluation of disease has provided a method to detect proliferation and progression of disease without sacrificing multiple mice (144). Though bioluminescent imaging has been used in syngeneic models of other cancers, it has not been successfully utilized in leukemia.
Major limitations of these models include limited life span and even more – lack of a native immune system, which is crucial to this model’s engraftment properties. The immune system’s role in the biology of leukemia development and ALL niche cannot be assessed in these models. Also, novel therapies, as immunomodulatory drugs or bispecific T cell engagers (BiTE) antibodies, are based on the immune system and cannot be completely assessed in this model.
The syngeneic model allows the study of leukemia in an immunocompetent host. This model can be used extensively to study the leukemia microenvironment in different organs. Though these models are driven by common human ALL genetic rearrangements or oncogene overexpression, in mice (as in humans) these translocations are not sufficient for leukemogenesis. Thus, these mice have additional germline alterations, which may not represent the additional hits in human disease. Also, these are limited models that cannot match the full spectrum of human acute leukemias. Moreover, given that these leukemia models are of murine origin, many drugs developed for human leukemia cannot be tested given the species barrier.
In summary, murine models play an essential part of leukemia research as well as therapeutic development. The benefits and limitations of each model should be weighed when addressing a specific study question. Altogether, the development of both xenogeneic as well as syngeneic models create many opportunities to answer fundamental questions of leukemia biology, and serves as a platform for the development of novel therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
1. Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia (1995) 9:1985–9.
2. Bohlander SK. ETV6: a versatile player in leukemogenesis. Semin Cancer Biol (2005) 15:162–74. doi: 10.1016/j.semcancer.2005.01.008
3. Dal Cin P, Atkins L, Ford C, Ariyanayagam S, Armstrong SA, George R, et al. Amplification of AML1 in childhood acute lymphoblastic leukemias. Genes Chromosomes Cancer (2001) 30:407–9. doi:10.1002/1098-2264(2001)9999:9999<::AID-GCC1107>3.0.CO;2-C
4. McLean TW, Ringold S, Neuberg D, Stegmaier K, Tantravahi R, Ritz J, et al. TEL/AML-1 dimerizes and is associated with a favorable outcome in childhood acute lymphoblastic leukemia. Blood (1996) 88:4252–8.
5. Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, et al. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene (2005) 24:7579–91. doi:10.1038/sj.onc.1208931
6. Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood (2004) 103:3890–6. doi:10.1182/blood-2003-10-3695
7. Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc Natl Acad Sci U S A (2004) 101:8443–8. doi:10.1073/pnas.0402063101
8. Bernardin F, Yang Y, Cleaves R, Zahurak M, Cheng L, Civin CI, et al. TEL-AML1, expressed from t(12;21) in human acute lymphocytic leukemia, induces acute leukemia in mice. Cancer Res (2002) 62:3904–8.
9. Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet (2001) 130:93–104. doi:10.1016/S0165-4608(01)00518-0
10. Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest (2009) 119:826–36. doi:10.1172/JCI36428
11. Li M, Jones L, Gaillard C, Binnewies M, Ochoa R, Garcia E, et al. Initially disadvantaged, TEL-AML1 cells expand and initiate leukemia in response to irradiation and cooperating mutations. Leukemia (2013) 27:1570–3. doi:10.1038/leu.2013.15
12. Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, et al. TEL-AML1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell Stem Cell (2009) 5:43–53. doi:10.1016/j.stem.2009.04.019
13. van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, et al. Modeling the evolution of ETV6-RUNX1-induced B-cell precursor acute lymphoblastic leukemia in mice. Blood (2011) 118:1041–51. doi:10.1182/blood-2011-02-338848
14. Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet (1999) 354:1499–503. doi:10.1016/S0140-6736(99)09403-9
15. Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood (1996) 87:1211–24.
16. Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell (1994) 79:875–84. doi:10.1016/0092-8674(94)90076-0
17. Sanyal M, Tung JW, Karsunky H, Zeng H, Selleri L, Weissman IL, et al. B-cell development fails in the absence of the Pbx1 proto-oncogene. Blood (2007) 109:4191–9. doi:10.1182/blood-2006-10-054213
18. Kamps MP, Wright DD, Lu Q. DNA-binding by oncoprotein E2a-Pbx1 is important for blocking differentiation but dispensable for fibroblast transformation. Oncogene (1996) 12:19–30.
19. Monica K, LeBrun DP, Dedera DA, Brown R, Cleary ML. Transformation properties of the E2a-Pbx1 chimeric oncoprotein: fusion with E2a is essential, but the Pbx1 homeodomain is dispensable. Mol Cell Biol (1994) 14:8304–14.
20. Chang CP, de Vivo I, Cleary ML. The Hox cooperativity motif of the chimeric oncoprotein E2a-Pbx1 is necessary and sufficient for oncogenesis. Mol Cell Biol (1997) 17:81–8.
21. Kamps MP, Baltimore D. E2A-Pbx1, the t(1;19) translocation protein of human pre-B-cell acute lymphocytic leukemia, causes acute myeloid leukemia in mice. Mol Cell Biol (1993) 13:351–7.
22. Dedera DA, Waller EK, LeBrun DP, Sen-Majumdar A, Stevens ME, Barsh GS, et al. Chimeric homeobox gene E2A-PBX1 induces proliferation, apoptosis, and malignant lymphomas in transgenic mice. Cell (1993) 74:833–43. doi:10.1016/0092-8674(93)90463-Z
23. Bijl J, Sauvageau M, Thompson A, Sauvageau G. High incidence of proviral integrations in the Hoxa locus in a new model of E2a-PBX1-induced B-cell leukemia. Genes Dev (2005) 19:224–33. doi:10.1101/gad.1268505
24. Feldman BJ, Reid TR, Cleary ML. Pim1 cooperates with E2a-Pbx1 to facilitate the progression of thymic lymphomas in transgenic mice. Oncogene (1997) 15:2735–42. doi:10.1038/sj.onc.1201670
25. Feldman BJ, Hampton T, Cleary ML. A carboxy-terminal deletion mutant of Notch1 accelerates lymphoid oncogenesis in E2A-PBX1 transgenic mice. Blood (2000) 96:1906–13.
26. Smith KS, Chanda SK, Lingbeek M, Ross DT, Botstein D, van Lohuizen M, et al. Bmi-1 regulation of INK4A-ARF is a downstream requirement for transformation of hematopoietic progenitors by E2a-Pbx1. Mol Cell (2003) 12:393–400. doi:10.1016/S1097-2765(03)00277-6
27. Seidel MG, Look AT. E2A-HLF usurps control of evolutionarily conserved survival pathways. Oncogene (2001) 20:5718–25. doi:10.1038/sj.onc.1204591
28. Gachon F, Fonjallaz P, Damiola F, Gos P, Kodama T, Zakany J, et al. The loss of circadian PAR bZip transcription factors results in epilepsy. Genes Dev (2004) 18:1397–412. doi:10.1101/gad.301404
29. Yoshihara T, Inaba T, Shapiro LH, Kato JY, Look AT. E2A-HLF-mediated cell transformation requires both the trans-activation domains of E2A and the leucine zipper dimerization domain of HLF. Mol Cell Biol (1995) 15:3247–55.
30. Hirose K, Inukai T, Kikuchi J, Furukawa Y, Ikawa T, Kawamoto H, et al. Aberrant induction of LMO2 by the E2A-HLF chimeric transcription factor and its implication in leukemogenesis of B-precursor ALL with t(17;19). Blood (2010) 116:962–70. doi:10.1182/blood-2009-09-244673
31. Smith KS, Rhee JW, Naumovski L, Cleary ML. Disrupted differentiation and oncogenic transformation of lymphoid progenitors in E2A-HLF transgenic mice. Mol Cell Biol (1999) 19:4443–51.
32. Honda H, Inaba T, Suzuki T, Oda H, Ebihara Y, Tsuiji K, et al. Expression of E2A-HLF chimeric protein induced T-cell apoptosis, B-cell maturation arrest, and development of acute lymphoblastic leukemia. Blood (1999) 93:2780–90.
33. Smith KS, Rhee JW, Cleary ML. Transformation of bone marrow B-cell progenitors by E2a-Hlf requires coexpression of Bcl-2. Mol Cell Biol (2002) 22:7678–87. doi:10.1128/MCB.22.21.7678-7688.2002
34. Gleissner B, Gokbuget N, Bartram CR, Janssen B, Rieder H, Janssen JW, et al. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood (2002) 99:1536–43. doi:10.1182/blood.V99.5.1536
35. Ribeiro RC, Abromowitch M, Raimondi SC, Murphy SB, Behm F, Williams DL. Clinical and biologic hallmarks of the Philadelphia chromosome in childhood acute lymphoblastic leukemia. Blood (1987) 70:948–53.
36. Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature (1985) 315:758–61. doi:10.1038/315758a0
37. Voncken JW, van Schaick H, Kaartinen V, Deemer K, Coates T, Landing B, et al. Increased neutrophil respiratory burst in bcr-null mutants. Cell (1995) 80:719–28. doi:10.1016/0092-8674(95)90350-X
38. Schwartzberg PL, Stall AM, Hardin JD, Bowdish KS, Humaran T, Boast S, et al. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell (1991) 65:1165–75. doi:10.1016/0092-8674(91)90012-N
39. Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell (1991) 65:1153–63. doi:10.1016/0092-8674(91)90011-M
40. Harrison SC. Variation on an Src-like theme. Cell (2003) 112:737–40. doi:10.1016/S0092-8674(03)00196-X
41. Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature (1990) 344:251–3. doi:10.1038/344251a0
42. Castellanos A, Pintado B, Weruaga E, Arevalo R, Lopez A, Orfao A, et al. A BCR-ABL(p190) fusion gene made by homologous recombination causes B-cell acute lymphoblastic leukemias in chimeric mice with independence of the endogenous bcr product. Blood (1997) 90:2168–74.
43. Foley SB, Hildenbrand ZL, Soyombo AA, Magee JA, Wu Y, Oravecz-Wilson KI, et al. Expression of BCR/ABL p210 from a knockin allele enhances bone marrow engraftment without inducing neoplasia. Cell Rep (2013) 5:51–60. doi:10.1016/j.celrep.2013.08.037
44. Voncken JW, Kaartinen V, Pattengale PK, Germeraad WT, Groffen J, Heisterkamp N. BCR/ABL P210 and P190 cause distinct leukemia in transgenic mice. Blood (1995) 86:4603–11.
45. Honda H, Fujii T, Takatoku M, Mano H, Witte ON, Yazaki Y, et al. Expression of p210bcr/abl by metallothionein promoter induced T-cell leukemia in transgenic mice. Blood (1995) 85:2853–61.
46. Honda H, Oda H, Suzuki T, Takahashi T, Witte ON, Ozawa K, et al. Development of acute lymphoblastic leukemia and myeloproliferative disorder in transgenic mice expressing p210bcr/abl: a novel transgenic model for human Ph1-positive leukemias. Blood (1998) 91:2067–75.
47. Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet (2000) 24:57–60. doi:10.1038/71691
48. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science (1990) 247:824–30. doi:10.1126/science.2406902
49. Uchida N, Hanawa H, Dan K, Inokuchi K, Shimada T. Leukemogenesis of b2a2-type p210 BCR/ABL in a bone marrow transplantation mouse model using a lentiviral vector. J Nippon Med Sch (2009) 76:134–47. doi:10.1272/jnms.76.134
50. Elefanty AG, Hariharan IK, Cory S. bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J (1990) 9:1069–78.
51. Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci U S A (1990) 87:6649–53. doi:10.1073/pnas.87.17.6649
52. Li S, Ilaria RL Jr, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med (1999) 189:1399–412. doi:10.1084/jem.189.9.1399
53. Stripecke R, Skelton DC, Gruber T, Afar D, Pattengale PK, Witte ON, et al. Immune response to Philadelphia chromosome-positive acute lymphoblastic leukemia induced by expression of CD80, interleukin 2, and granulocyte-macrophage colony-stimulating factor. Hum Gene Ther (1998) 9:2049–62. doi:10.1089/hum.1998.9.14-2049
54. Gishizky ML, Johnson-White J, Witte ON. Efficient transplantation of BCR-ABL-induced chronic myelogenous leukemia-like syndrome in mice. Proc Natl Acad Sci U S A (1993) 90:3755–9. doi:10.1073/pnas.90.8.3755
55. Pui CH, Behm FG, Downing JR, Hancock ML, Shurtleff SA, Ribeiro RC, et al. 11q23/MLL rearrangement confers a poor prognosis in infants with acute lymphoblastic leukemia. J Clin Oncol (1994) 12:909–15.
57. Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, et al. The MLL recombinome of acute leukemias in 2013. Leukemia (2013) 27:2165–76. doi:10.1038/leu.2013.135
58. Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. MLL partial tandem duplications in acute leukemia cell lines. Leukemia (2003) 17:980–1. doi:10.1038/sj.leu.2402911
59. Tamai H, Miyake K, Takatori M, Miyake N, Yamaguchi H, Dan K, et al. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia (2011) 25:888–91. doi:10.1038/leu.2011.15
60. Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood (2006) 108:669–77. doi:10.1182/blood-2005-08-3498
61. Metzler M, Forster A, Pannell R, Arends MJ, Daser A, Lobato MN, et al. A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene (2006) 25:3093–103. doi:10.1038/sj.onc.1209636
62. Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell (2008) 14:355–68. doi:10.1016/j.ccr.2008.10.001
63. Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell (2010) 17:148–59. doi:10.1016/j.ccr.2009.12.034
64. Bursen A, Schwabe K, Ruster B, Henschler R, Ruthardt M, Dingermann T, et al. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood (2010) 115:3570–9. doi:10.1182/blood-2009-06-229542
65. Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell (1996) 85:853–61. doi:10.1016/S0092-8674(00)81269-6
66. Collins EC, Pannell R, Simpson EM, Forster A, Rabbitts TH. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep (2000) 1:127–32. doi:10.1093/embo-reports/kvd021
67. Xu SM, Yang Y, Zhou M, Zhao XJ, Qin Y, Zhang PL, et al. Establishment of the retrovirus-mediated murine model with MLL-AF9 leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi (2013) 21:1126–32. doi:10.7534/j.issn.1009-2137.2013.05.008
68. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature (2006) 442:818–22. doi:10.1038/nature04980
69. Forster A, Pannell R, Drynan LF, McCormack M, Collins EC, Daser A, et al. Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell (2003) 3:449–58. doi:10.1016/S1535-6108(03)00106-5
70. Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J (1997) 16:4226–37. doi:10.1093/emboj/16.14.4226
71. Zeisig BB, Garcia-Cuellar MP, Winkler TH, Slany RK. The oncoprotein MLL-ENL disturbs hematopoietic lineage determination and transforms a biphenotypic lymphoid/myeloid cell. Oncogene (2003) 22:1629–37. doi:10.1038/sj.onc.1206104
72. Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol (2003) 24:617–28. doi:10.1128/MCB.24.2.617-628.2004
73. Horton SJ, Walf-Vorderwulbecke V, Chatters SJ, Sebire NJ, de Boer J, Williams O. Acute myeloid leukemia induced by MLL-ENL is cured by oncogene ablation despite acquisition of complex genetic abnormalities. Blood (2009) 113:4922–9. doi:10.1182/blood-2008-07-170480
74. DiMartino JF. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood (2002) 99:3780–5. doi:10.1182/blood.V99.10.3780
75. Fu JF, Hsu CL, Shih LY. MLL/AF10(OM-LZ)-immortalized cells expressed cytokines and induced host cell proliferation in a mouse bone marrow transplantation model. Int J Cancer (2010) 126:1621–9. doi:10.1002/ijc.24867
76. Fu JF, Yen TH, Chen Y, Huang YJ, Hsu CL, Liang DC, et al. Involvement of Gpr125 in the myeloid sarcoma formation induced by cooperating MLL/AF10(OM-LZ) and oncogenic KRAS in a mouse bone marrow transplantation model. Int J Cancer (2013) 133:1792–802. doi:10.1002/ijc.28195
77. DiMartino JF, Miller T, Ayton PM, Landewe T, Hess JL, Cleary ML, et al. A carboxy-terminal domain of ELL is required and sufficient for immortalization of myeloid progenitors by MLL-ELL. Blood (2000) 96:3887–93.
78. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood (2013) 121:2533–41. doi:10.1182/blood-2012-11-465120
79. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature (2007) 446:758–64. doi:10.1038/nature05690
80. Heltemes-Harris LM, Willette MJ, Ramsey LB, Qiu YH, Neeley ES, Zhang N, et al. Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med (2011) 208:1135–49. doi:10.1084/jem.20101947
81. Faderl S, O’Brien S, Pui CH, Stock W, Wetzler M, Hoelzer D, et al. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer (2010) 116:1165–76. doi:10.1002/cncr.24862
82. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature (1985) 318:533–8. doi:10.1038/318533a0
83. Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med (1988) 167:353–71. doi:10.1084/jem.167.2.353
84. Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, et al. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res (2005) 65:1306–15. doi:10.1158/0008-5472.CAN-04-0268
85. Yu D, Thomas-Tikhonenko A. A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene (2002) 21:1922–7. doi:10.1038/sj.onc.1205244
86. Aster JC, Pear WS, Blacklow SC. Notch signaling in leukemia. Annu Rev Pathol (2008) 3:587–613. doi:10.1146/annurev.pathmechdis.3.121806.154300
87. Weng AP, Ferrando AA, Lee W, Morris JPT, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306:269–71. doi:10.1126/science.1102160
88. Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood (2008) 112:3373–82. doi:10.1182/blood-2008-03-147587
89. Kong G, Du J, Liu Y, Meline B, Chang YI, Ranheim EA, et al. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J Biol Chem (2013) 288:18219–27. doi:10.1074/jbc.M113.475376
90. Chiang MY, Xu L, Shestova O, Histen G, L’Heureux S, Romany C, et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest (2008) 118:3181–94. doi:10.1172/JCI35090
91. D’Altri T, Gonzalez J, Aifantis I, Espinosa L, Bigas A. Hes1 expression and CYLD repression are essential events downstream of Notch1 in T-cell leukemia. Cell Cycle (2011) 10:1031–6. doi:10.4161/cc.10.7.15067
92. Beverly LJ, Capobianco AJ. Perturbation of Ikaros isoform selection by MLV integration is a cooperative event in NotchIC-induced T cell leukemogenesis. Cancer Cell (2003) 3:551–64. doi:10.1016/S1535-6108(03)00137-5
93. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer (2008) 8:83–93. doi:10.1038/nrc2290
94. Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, et al. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med (2007) 204:2875–88. doi:10.1084/jem.20062299
95. Aplan PD, Jones CA, Chervinsky DS, Zhao X, Ellsworth M, Wu C, et al. An scl gene product lacking the transactivation domain induces bony abnormalities and cooperates with LMO1 to generate T-cell malignancies in transgenic mice. EMBO J (1997) 16:2408–19. doi:10.1093/emboj/16.9.2408
96. Aplan PD, Lombardi DP, Reaman GH, Sather HN, Hammond GD, Kirsch IR. Involvement of the putative hematopoietic transcription factor SCL in T-cell acute lymphoblastic leukemia. Blood (1992) 79:1327–33.
97. Condorelli GL, Facchiano F, Valtieri M, Proietti E, Vitelli L, Lulli V, et al. T-cell-directed TAL-1 expression induces T-cell malignancies in transgenic mice. Cancer Res (1996) 56:5113–9.
98. O’Neil J, Billa M, Oikemus S, Kelliher M. The DNA binding activity of TAL-1 is not required to induce leukemia/lymphoma in mice. Oncogene (2001) 20:3897–905. doi:10.1038/sj.onc.1204519
99. Zhong Y, Jiang L, Hiai H, Toyokuni S, Yamada Y. Overexpression of a transcription factor LYL1 induces T- and B-cell lymphoma in mice. Oncogene (2007) 26:6937–47. doi:10.1038/sj.onc.1210494
100. Neale GA, Rehg JE, Goorha RM. Ectopic expression of rhombotin-2 causes selective expansion of CD4-CD8- lymphocytes in the thymus and T-cell tumors in transgenic mice. Blood (1995) 86:3060–71.
101. Larson RC, Fisch P, Larson TA, Lavenir I, Langford T, King G, et al. T cell tumours of disparate phenotype in mice transgenic for Rbtn-2. Oncogene (1994) 9:3675–81.
102. Draheim KM, Hermance N, Yang Y, Arous E, Calvo J, Kelliher MA. A DNA-binding mutant of TAL1 cooperates with LMO2 to cause T cell leukemia in mice. Oncogene (2011) 30:1252–60. doi:10.1038/onc.2010.495
103. Caudell D, Zhang Z, Chung YJ, Aplan PD. Expression of a CALM-AF10 fusion gene leads to Hoxa cluster overexpression and acute leukemia in transgenic mice. Cancer Res (2007) 67:8022–31. doi:10.1158/0008-5472.CAN-06-3749
104. Deshpande AJ, Cusan M, Rawat VP, Reuter H, Krause A, Pott C, et al. Acute myeloid leukemia is propagated by a leukemic stem cell with lymphoid characteristics in a mouse model of CALM/AF10-positive leukemia. Cancer Cell (2006) 10:363–74. doi:10.1016/j.ccr.2006.08.023
105. Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia (2013) 27:813–22. doi:10.1038/leu.2012.327
106. Watanabe S, Shimosato Y, Kameya T, Kuroki M, Kitahara T, Minato K, et al. Leukemic distribution of a human acute lymphocytic leukemia cell line (Ichikawa strain) in nude mice conditioned with whole-body irradiation. Cancer Res (1978) 38:3494–8.
107. Ziegler HW, Frizzera G, Bach FH. Successful transplantation of a human leukemia cell line into nude mice: conditions optimizing graft acceptance. J Natl Cancer Inst (1982) 68:15–8.
108. Hara H, Luo Y, Haruta Y, Seon BK. Efficient transplantation of human non-T-leukemia cells into nude mice and induction of complete regression of the transplanted distinct tumors by ricin A-chain conjugates of monoclonal antibodies SN5 and SN6. Cancer Res (1988) 48:4673–80.
109. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (1983) 301:527–30. doi:10.1038/301527a0
110. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol (2007) 7:118–30. doi:10.1038/nri2017
111. Kamel-Reid S, Letarte M, Doedens M, Greaves A, Murdoch B, Grunberger T, et al. Bone marrow from children in relapse with pre-B acute lymphoblastic leukemia proliferates and disseminates rapidly in scid mice. Blood (1991) 78:2973–81.
112. Kamel-Reid S, Letarte M, Sirard C, Doedens M, Grunberger T, Fulop G, et al. A model of human acute lymphoblastic leukemia in immune-deficient SCID mice. Science (1989) 246:1597–600. doi:10.1126/science.2595371
113. Uckun FM, Downing JR, Chelstrom LM, Gunther R, Ryan M, Simon J, et al. Human t(4;11)(q21;q23) acute lymphoblastic leukemia in mice with severe combined immunodeficiency. Blood (1994) 84:859–65.
114. Kawata A, Yoshida M, Okazaki M, Yokota S, Barcos M, Seon BK. Establishment of new SCID and nude mouse models of human B leukemia/lymphoma and effective therapy of the tumors with immunotoxin and monoclonal antibody: marked difference between the SCID and nude mouse models in the antitumor efficacy of monoclonal antibody. Cancer Res (1994) 54:2688–94.
115. Mosier DE, Stell KL, Gulizia RJ, Torbett BE, Gilmore GL. Homozygous scid/scid;beige/beige mice have low levels of spontaneous or neonatal T cell-induced B cell generation. J Exp Med (1993) 177:191–4. doi:10.1084/jem.177.1.191
116. Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res (2007) 13:5426–35. doi:10.1158/1078-0432.CCR-07-0674
117. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol (1995) 154:180–91.
118. Borgmann A, Baldy C, von Stackelberg A, Beyermann B, Fichtner I, Nurnberg P, et al. Childhood all blasts retain phenotypic and genotypic characteristics upon long-term serial passage in NOD/SCID mice. Pediatr Hematol Oncol (2000) 17:635–50. doi:10.1080/08880010050211349
119. Nijmeijer BA, Mollevanger P, van Zelderen-Bhola SL, Kluin-Nelemans HC, Willemze R, Falkenburg JH. Monitoring of engraftment and progression of acute lymphoblastic leukemia in individual NOD/SCID mice. Exp Hematol (2001) 29:322–9. doi:10.1016/S0301-472X(00)00669-X
120. Lock RB, Liem N, Farnsworth ML, Milross CG, Xue C, Tajbakhsh M, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood (2002) 99:4100–8. doi:10.1182/blood.V99.11.4100
121. Cox CV, Martin HM, Kearns PR, Virgo P, Evely RS, Blair A. Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood (2007) 109:674–82. doi:10.1182/blood-2006-06-030445
122. Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood (2004) 103:3905–14. doi:10.1182/blood-2003-08-2911
123. Diamanti P, Cox CV, Blair A. Comparison of childhood leukemia initiating cell populations in NOD/SCID and NSG mice. Leukemia (2012) 26:376–80. doi:10.1038/leu.2011.212
124. Agliano A, Martin-Padura I, Mancuso P, Marighetti P, Rabascio C, Pruneri G, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int J Cancer (2008) 123:2222–7. doi:10.1002/ijc.23772
125. Morisot S, Wayne AS, Bohana-Kashtan O, Kaplan IM, Gocke CD, Hildreth R, et al. High frequencies of leukemia stem cells in poor-outcome childhood precursor-B acute lymphoblastic leukemias. Leukemia (2010) 24:1859–66. doi:10.1038/leu.2010.184
126. Woiterski J, Ebinger M, Witte KE, Goecke B, Heininger V, Philippek M, et al. Engraftment of low numbers of pediatric acute lymphoid and myeloid leukemias into NOD/SCID/IL2Rcgammanull mice reflects individual leukemogenecity and highly correlates with clinical outcome. Int J Cancer (2013) 133:1547–56. doi:10.1002/ijc.28170
127. Lavender KJ, Pang WW, Messer RJ, Duley AK, Race B, Phillips K, et al. BLT-humanized C57BL/6 Rag2-/-gammac-/-CD47-/- mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood (2013) 122:4013–20. doi:10.1182/blood-2013-06-506949
128. le Viseur C, Hotfilder M, Bomken S, Wilson K, Rottgers S, Schrauder A, et al. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell (2008) 14:47–58. doi:10.1016/j.ccr.2008.05.015
129. Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science (2008) 319:336–9. doi:10.1126/science.1150648
130. Welschinger R, Liedtke F, Basnett J, Dela Pena A, Juarez JG, Bradstock KF, et al. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp Hematol (2013) 41: 293–302.e1. doi:10.1016/j.exphem.2012.11.004
131. Messinger Y, Chelstrom L, Gunther R, Uckun FM. Selective homing of human leukemic B-cell precursors to specific lymphohematopoietic microenvironments in SCID mice: a role for the beta 1 integrin family surface adhesion molecules VLA-4 and VLA-5. Leuk Lymphoma (1996) 23:61–9. doi:10.3109/10428199609054803
132. Gunther R, Chelstrom LM, Tuel-Ahlgren L, Simon J, Myers DE, Uckun FM. Biotherapy for xenografted human central nervous system leukemia in mice with severe combined immunodeficiency using B43 (anti-CD19)-pokeweed antiviral protein immunotoxin. Blood (1995) 85:2537–45.
133. Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood (2004) 104:2919–25. doi:10.1182/blood-2004-03-0901
134. Kong Y, Yoshida S, Saito Y, Doi T, Nagatoshi Y, Fukata M, et al. CD34+CD38+CD19+ as well as CD34+CD38-CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia (2008) 22:1207–13. doi:10.1038/leu.2008.83
135. Cox CV, Diamanti P, Evely RS, Kearns PR, Blair A. Expression of CD133 on leukemia-initiating cells in childhood ALL. Blood (2009) 113:3287–96. doi:10.1182/blood-2008-04-154187
136. Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc Natl Acad Sci U S A (1998) 95:4584–8. doi:10.1073/pnas.95.8.4584
137. Cazzaniga G, van Delft FW, Lo Nigro L, Ford AM, Score J, Iacobucci I, et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood (2011) 118:5559–64. doi:10.1182/blood-2011-07-366542
138. Uckun FM, Sather H, Reaman G, Shuster J, Land V, Trigg M, et al. Leukemic cell growth in SCID mice as a predictor of relapse in high-risk B-lineage acute lymphoblastic leukemia. Blood (1995) 85:873–8.
139. Uckun FM. Severe combined immunodeficient mouse models of human leukemia. Blood (1996) 88:1135–46.
140. Meyer LH, Eckhoff SM, Queudeville M, Kraus JM, Giordan M, Stursberg J, et al. Early relapse in ALL is identified by time to leukemia in NOD/SCID mice and is characterized by a gene signature involving survival pathways. Cancer Cell (2011) 19:206–17. doi:10.1016/j.ccr.2010.11.014
141. Meyer LH, Debatin KM. Diversity of human leukemia xenograft mouse models: implications for disease biology. Cancer Res (2011) 71:7141–4. doi:10.1158/0008-5472.CAN-11-1732
142. Guihard S, Peyrouze P, Cheok MH. Pharmacogenomic considerations of xenograft mouse models of acute leukemia. Pharmacogenomics (2012) 13:1759–72. doi:10.2217/pgs.12.158
143. Uckun FM, Stewart CF, Reaman G, Chelstrom LM, Jin J, Chandan-Langlie M, et al. In vitro and in vivo activity of topotecan against human B-lineage acute lymphoblastic leukemia cells. Blood (1995) 85:2817–28.
144. Barrett DM, Seif AE, Carpenito C, Teachey DT, Fish JD, June CH, et al. Noninvasive bioluminescent imaging of primary patient acute lymphoblastic leukemia: a strategy for preclinical modeling. Blood (2011) 118:e112–7. doi:10.1182/blood-2011-04-346528
145. Teachey DT, Sheen C, Hall J, Ryan T, Brown VI, Fish J, et al. mTOR inhibitors are synergistic with methotrexate: an effective combination to treat acute lymphoblastic leukemia. Blood (2008) 112:2020–3. doi:10.1182/blood-2008-02-137141
146. Parameswaran R, Yu M, Lim M, Groffen J, Heisterkamp N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia (2011) 25:1314–23. doi:10.1038/leu.2011.76
147. Vilas-Zornoza A, Agirre X, Abizanda G, Moreno C, Segura V, De Martino Rodriguez A, et al. Preclinical activity of LBH589 alone or in combination with chemotherapy in a xenogeneic mouse model of human acute lymphoblastic leukemia. Leukemia (2012) 26:1517–26. doi:10.1038/leu.2012.31
148. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene (2014) 33, 2169–78. doi:10.1038/onc.2013.169
149. Canale S, Cocco C, Frasson C, Seganfreddo E, Di Carlo E, Ognio E, et al. Interleukin-27 inhibits pediatric B-acute lymphoblastic leukemia cell spreading in a preclinical model. Leukemia (2011) 25:1815–24. doi:10.1038/leu.2011.158
150. Uckun FM, Qazi S, Cely I, Sahin K, Shahidzadeh A, Ozercan I, et al. Nanoscale liposomal formulation of a SYK P-site inhibitor against B-precursor leukemia. Blood (2013) 121:4348–54. doi:10.1182/blood-2012-11-470633
151. Lu H, Kojima K, Battula VL, Korchin B, Shi Y, Chen Y, et al. Targeting connective tissue growth factor (CTGF) in acute lymphoblastic leukemia preclinical models: anti-CTGF monoclonal antibody attenuates leukemia growth. Ann Hematol (2014) 93:485–92. doi:10.1007/s00277-013-1939-2
152. Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia (2014) 28:278–88. doi:10.1038/leu.2013.183
153. Messinger Y, Yanishevski Y, Avramis VI, Ek O, Chelstrom LM, Gunther R, et al. Treatment of human B-cell precursor leukemia in SCID mice using a combination of the investigational biotherapeutic agent B43-PAP with cytosine arabinoside. Clin Cancer Res (1996) 2:1533–42.
154. Barta SK, Zou Y, Schindler J, Shenoy N, Bhagat TD, Steidl U, et al. Synergy of sequential administration of a deglycosylated ricin A chain-containing combined anti-CD19 and anti-CD22 immunotoxin (Combotox) and cytarabine in a murine model of advanced acute lymphoblastic leukemia. Leuk Lymphoma (2012) 53:1999–2003. doi:10.3109/10428194.2012.679267
155. Nijmeijer BA. An animal model for human cellular immunotherapy: specific eradication of human acute lymphoblastic leukemia by cytotoxic T lymphocytes in NOD/scid mice. Blood (2002) 100:654–60. doi:10.1182/blood.V100.2.654
156. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res (2006) 66:10995–1004. doi:10.1158/0008-5472.CAN-06-0160
157. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood (2013) 121:1165–74. doi:10.1182/blood-2012-06-438002
158. Ali N, Flutter B, Sanchez Rodriguez R, Sharif-Paghaleh E, Barber LD, Lombardi G, et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS One (2012) 7:e44219. doi:10.1371/journal.pone.0044219
Keywords: acute lymphoblastic leukemia, xenografts, murine model, immunotherapy, adoptive, syngeneic
Citation: Jacoby E, Chien CD and Fry TJ (2014) Murine models of acute leukemia: important tools in current pediatric leukemia research. Front. Oncol. 4:95. doi: 10.3389/fonc.2014.00095
Received: 01 March 2014; Accepted: 18 April 2014;
Published online: 07 May 2014.
Edited by:
Alan Wayne, Children’s Hospital Los Angeles, USAReviewed by:
Yong-Mi Kim, Children’s Hospital Los Angeles, USAAlan Wayne, Children’s Hospital Los Angeles, USA
Copyright: © 2014 Jacoby, Chien and Fry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terry J. Fry, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 10 Center Drive, Room 1W-3952, Bethesda, MD 20816, USA e-mail:ZnJ5dEBtYWlsLm5paC5nb3Y=
†Elad Jacoby and Christopher D. Chien have contributed equally to this work.