 C. Elizabeth Caldon
C. Elizabeth Caldon- 1Genome and Replication Stability Group, The Kinghorn Cancer Centre, Garvan Institute of Medical Research, Sydney, NSW, Australia
- 2St Vincent’s Clinical School, Faculty of Medicine, UNSW Australia, Sydney, NSW, Australia
Estrogen is necessary for the normal growth and development of breast tissue, but high levels of estrogen are a major risk factor for breast cancer. One mechanism by which estrogen could contribute to breast cancer is via the induction of DNA damage. This perspective discusses the mechanisms by which estrogen alters the DNA damage response (DDR) and DNA repair through the regulation of key effector proteins including ATM, ATR, CHK1, BRCA1, and p53 and the feedback on estrogen receptor signaling from these proteins. We put forward the hypothesis that estrogen receptor signaling converges to suppress effective DNA repair and apoptosis in favor of proliferation. This is important in hormone-dependent breast cancer as it will affect processing of estrogen-induced DNA damage, as well as other genotoxic insults. DDR and DNA repair proteins are frequently mutated or altered in estrogen responsive breast cancer, which will further change the processing of DNA damage. Finally, the action of estrogen signaling on DNA damage is also relevant to the therapeutic setting as the suppression of a DDR by estrogen has the potential to alter the response of cancers to anti-hormone treatment or chemotherapy that induces DNA damage.
DNA Damage Induced by Estrogen
Lifetime exposure to estrogen is a major risk factor for breast cancer. Elevated serum levels of estrogen are associated with a 2–2.5× greater risk of breast cancer development (1) and high levels of estrogen in the breast of postmenopausal women are associated with increased cancer risk (2). Estrogen signaling drives proliferation in the 60–70% of breast cancers that express the estrogen receptor, and adjuvant anti-estrogen therapy is prescribed to the majority of these patients to prevent breast cancer recurrence.
Estrogen signals through its two receptors, estrogen receptor α (ERα) and estrogen receptor β (ERβ). Only ERα is essential for breast development and activates pro-proliferative signaling in the normal breast and breast cancer, whereas ERβ generally antagonizes ERα in the breast (3). Upon estrogen binding ERα acts by parallel pathways to alter gene expression. ERα translocates to the nucleus to activate gene targets directly or in cooperation with co-activator proteins, or it can transactivate growth receptors to boost receptor tyrosine kinase signaling. These pathways converge to promote growth and proliferation and suppress apoptosis (3).
Despite the risks associated with estrogen exposure the exact mechanisms by which estrogen contributes to the initiation and progression of breast cancer remains elusive. However, a major mechanism is potentially the induction of DNA damage as estrogen treatment leads to double stranded DNA breaks and genomic instability (1, 4, 5). Early breast cancer lesions exhibit chromosomal instability and aneuploidy (6), and in rat models this is linked to estrogen exposure (7). Estrogen can induce DNA damage via the production of oxidative metabolites that cause DNA adducts, or other oxidative DNA damage, and this is supported by in vitro and animal model studies (1). The second explanation for estrogen-induced DNA damage is that hyperactivated estrogen signaling provokes excessive proliferation when pathways become dysregulated, and this theory has strong support from in vitro modeling and gene signatures in breast cancer (3). Excessive proliferation promotes DNA damage accumulation due to insufficient timely repair leading to replication fork stalling and possibly even double stranded DNA breaks (8). It is likely that both carcinogenic estrogen metabolites and deregulated estrogen signaling contribute to estrogen-induced DNA damage. In this perspective a third possibility is raised, that estrogen signaling suppresses the DNA damage response and DNA repair to allow the accumulation of genomic change conducive to tumorigenesis.
DNA Damage Response and DNA Repair Pathways Altered by Estrogen Signaling
DNA damage is recognized and processed by series of pathways called the “DNA damage response (DDR)”. The DDR assesses the scope and severity of DNA damage to initiate cell cycle arrest, senescence, repair, or in the case of irreparable damage, apoptosis. If repair is activated then a number of different repair mechanisms can be engaged [reviewed in Ref. (9)]. Small lesions of damaged or incorrectly inserted nucleotides are repaired by base excision repair (BER), nucleotide excision repair (NER), or mismatch repair (MMR). The more catastrophic double stranded breaks are repaired via non-homologous end-joining (NHEJ) or homologous recombination (HR). Small distorting lesions are extremely common so the pathways that repair these defects (BER, NER, and MMR) are also activated by constant genome surveillance, and repair is coupled to transcription and DNA replication.
The DDR signals through three main effector kinases, ATM, ATR, and DNA-PK. ATM and DNA-PK recognize double stranded breaks whereas ATR responds to single stranded regions that occur at stalled replication forks and double stranded break overhangs. The signaling pathways downstream of ATM, ATR, and DNA-PK involve a myriad of proteins, however there are a number of key effector proteins that include CHK1, CHK2, BRCA1, 53BP1, and MDC1 which signal to DNA repair coordinators such as BRCA2, PALB2 and to cell cycle checkpoints and the apoptotic machinery. The major tumor suppressor protein, p53, is activated downstream of ATM/ATR, and acts as a genome guardian to determine whether cells should arrest or apoptose. There is significant cross-talk between the various pathways depending on the nature and severity of the DNA damage.
The DDR is important to estrogen carcinogenesis as it dictates how estrogen-mediated damage is processed by breast cells. In prior genome wide studies of estrogen action, the major regulatory nodes of the ERα transcriptional program have included proliferation, growth, and apoptosis, but not the DDR or DNA repair (3). However, there is a growing body of literature, which identifies estrogen signaling as regulating key effector DDR proteins such as ATM, ATR, p53, BRCA1, and BRCA2, as well as direct interactions with the DNA repair machinery. This is significant not only for estrogen carcinogenesis, but also for the processing of any genotoxic insults by estrogen-responsive tissues. Described below are the most important interactions between ERα, the DDR, and DNA repair pathways (Figure 1). ERβ is not discussed in this perspective, but it should be noted that ERβ has opposing effects to ERα in many contexts (10), and this is also true of regulation of the DDR and DNA repair (11–13).
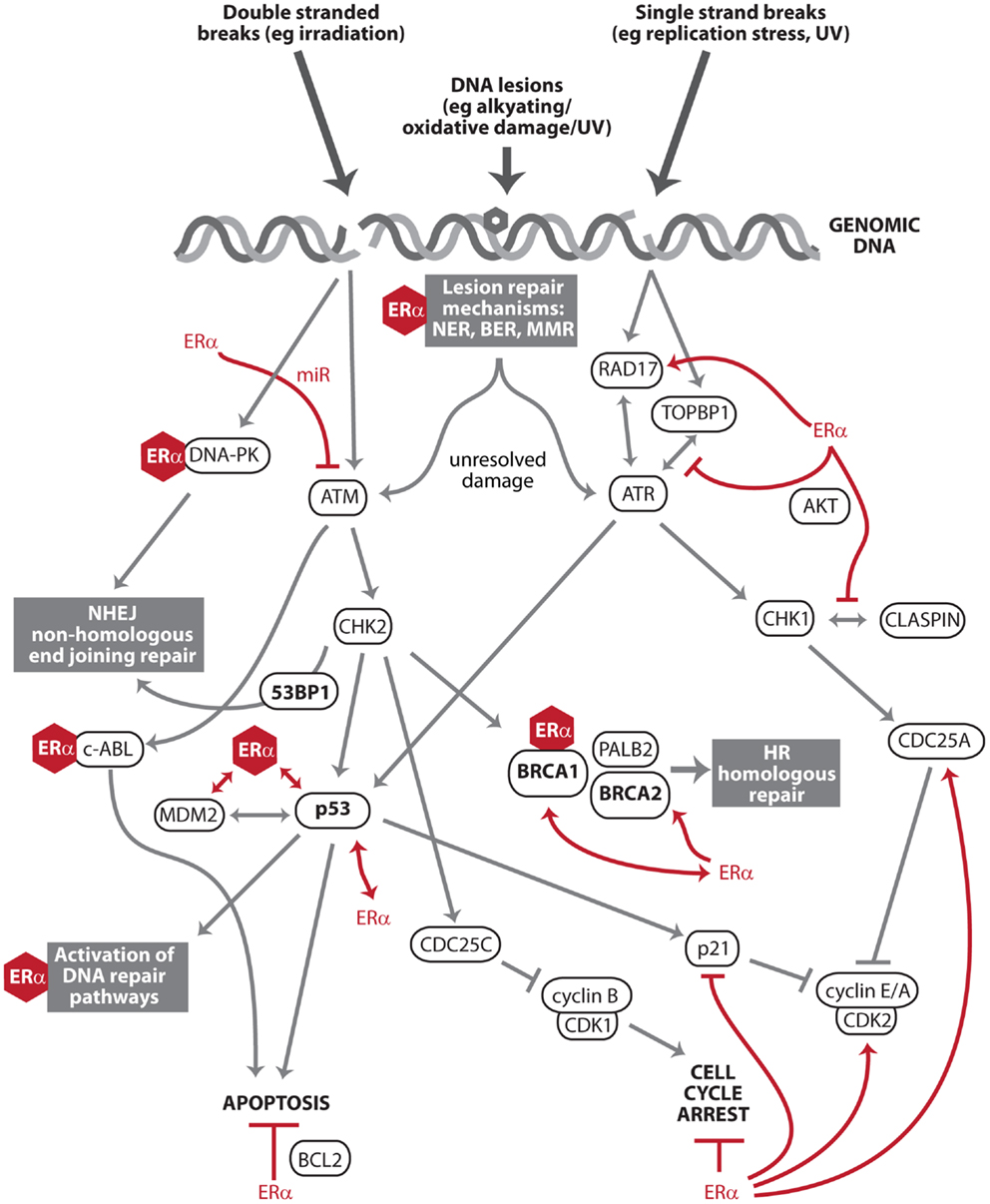
Figure 1. Key effectors of the DNA damage response and DNA repair that intersect with estrogen receptor α signaling. The DNA damage response (DDR) is a series of pathways that recognize and process DNA damage. After DNA damage recognition, signals are transduced and amplified through kinase activation (ATM, ATR, DNA-PK, CHK1, and CHK2) to downstream effectors (e.g., p53 and BRCA1) that facilitate DNA repair, apoptosis, and cell cycle arrest. Estrogen receptor α (ERα) exists in complex with multiple members of the DDR and DNA repair pathways (e.g., DNA-PK, BRCA1, p53, and MDM2). These protein:protein interactions are denoted by ERα represented as a hexagon. This includes c-Abl, a multi-functional regulator of the DDR and its downstream pathways (14). ERα also transcriptionally regulates or is regulated by other members of these pathways (e.g., ATM, ATR, CHK1, BRCA2, and DNA damage checkpoint protein Rad17), denoted by red lines. ERα signaling antagonizes two major endpoints of DDR action: apoptosis and cell cycle arrest (red lines).
Regulation of Effector Kinases ATM, ATR, and DNA-PK
ATM and ATR are key initiators of the DDR, and both are negatively regulated by ERα. ERα downregulates transcription of ATM via the activation of miR-18a and miR-106a (11). The ATR/CHK1 signal transduction cascade is suppressed by ERα-transactivated AKT phosphorylation of TOPBP1 to prevent an interaction with ATR at sites of DNA damage (15). AKT also phosphorylates CHK1 to prevent its interaction with co-activator CLASPIN (15). The downregulation of ATM and ATR by ERα interferes with the induction of cell cycle checkpoints so that cells continue to progress through the cell cycle after DNA damage, and DNA repair is delayed or not engaged (15, 16). Estrogen activity does not, however, preclude activation of the DDR. γ-H2Aχ foci form in response to estrogen-induced DNA damage, and the co-localization of Rad51 to these foci suggests the activation of HR (4).
While ERα negatively regulates both ATM and ATR, it is possible that ERα positively regulates DNA-PK mediated repair based on recent findings of DNA-PK regulation by the androgen receptor (AR). AR regulation of DNA-PK catalytic subunit (DNA-PKcs) promotes the repair of DNA double stranded breaks and resistance to DNA damage and DNA-PKcs likewise potentiates the function of AR (17). Like AR, ERα is in a complex with DNA-PK (18) and ERα is stabilized and its transcriptional function potentiated by DNA-PK (19), and by analogy to AR, ERα may also transactivate DNA-PK.
If ERα does positively regulate DNA-PK, ERα may suppress DNA repair processes of higher fidelity (ATM- and ATR-mediated) in preference for DNA-PK-mediated NHEJ. This is consistent with observations of ERα activity leading to the accumulation of DNA damage (1) as it would sustain proliferation by not engaging the ATM/ATR pathways, while promoting DNA-PK-mediated NHEJ to maintain genome integrity. Toillon et al. found that estrogen treatment of irradiated breast cancer cells led to their sustained proliferation without any increase in p53 activation or apoptosis (20). This is consistent with a failure to activate ATM or ATR but the repair of DNA by DNA-PK mediated NHEJ.
BRCA1
BRCA1 is a downstream effector of the DDR that is recruited to sites of DNA damage, functions directly in HR, but also influences cell cycle arrest and other DNA repair pathways. There is strong evidence that BRCA1 limits estrogen-mediated tumorigenesis: Brca1 knockout mice show an enhanced proliferative response to estrogen treatment and accelerated development of preneoplastic mammary lesions (21), and the reduction of serum estrogen levels by oophorectomy protects carriers of the BRCA1 mutation against breast cancer (22). Indeed, BRCA1 has a negative effect on ERα, through direct binding to inhibit ERα-mediated gene transcription (23, 24), downregulation of ERα co-activator, p300 (25), reduced cross-talk from growth factor signaling (26), and potentially monoubiquitination (25, 27). These effects are antagonized by cyclin D1, a direct transcriptional target of ERα that is instrumental in estrogen-induced proliferation (28).
While BRCA1 suppresses ERα, ERα regulation of BRCA1 enhances BRCA1 function. Estrogen promotes transcription of BRCA1 via binding of an ERα/p300 complex (29), and stimulates the formation of a complex between ERα, CBP, and BRCA1 that facilitates double stranded break repair (30). Surprisingly, BRCA1 induces the transcription of ESR1 which encodes ERα, and the positive feedback between BRCA1 and ERα provides a rational explanation for why many BRCA1 negative cancers are ERα negative (31).
p53
Estrogen receptor α and p53 have a bi-directional relationship affecting both expression and function. The TP53 gene is transcriptionally activated by ERα (32, 33) and downstream of ERα-target, c-MYC (34), and ERα stabilizes the p53 protein (35). Despite ERα inducing higher levels of p53 it may not be active: in breast cancer cell lines estrogen induces cytoplasmic redistribution of p53 to reduce its transcriptional function (12, 36). ERα alters the p53 transcriptional program to reverse transcriptional activation and repression by p53, including downregulation of the p53-mediated apoptotic response induced by DNA damage (37). ERα represses p53-mediated transcription either through the recruitment of co-repressors (38) or via independent targeting and repression of p53 target gene sets (39). A separate subset of target genes for p53 activation is enhanced by ERα activity (37).
p53 and ERα exist in complex with MDM2, and this complex modulates the activity of p53 and ERα. MDM2 is a negative feedback regulator of p53 (40), whereas MDM2 positively regulates ERα transcriptional activity, most probably through direct MDM2:ERα interaction (41, 42). Conversely, the MDM2/p53/ERα ternary complex downregulates the activity of ERα by monoubiquitination, probably via the ubiquitin ligase activity of MDM2 (43). MDM2 may also downregulate ERα independently of p53 (43). In the presence of cellular stress, including UV-mediated DNA damage, p53 dissociates from MDM2 and this is associated with an increase in ERα levels and block of the estrogen-dependent downregulation of ERα (43). Paradoxically, while ERα represses p53-mediated transcription, ERα also protects p53 from repression by MDM2 (40), and estrogen treatment is necessary for a p53 response to be mounted in the mouse mammary gland against ionizing radiation (44).
p53 upregulates the expression of ESR1, but alters the transcriptional functions of ERα. p53 induction of ESR1 occurs following DNA damage such as irradiation (45). Like ERα modulation of p53 function, p53 alters the transcriptional program of ERα to repress certain estrogen responsive genes such as BRCA2, c-JUN, and BCL2 (37, 46). Indeed it appears that the combination of ERα and p53 induces a distinct transcriptional program compared to either ERα or p53 alone (47).
Overall this body of work suggests that estrogen and ERα upregulate but sequester p53, such that the DDR and DNA repair are suppressed in the presence of active estrogen signaling, but there is still some safeguard via p53. When estrogen treated breast cancer cells are irradiated there is partial activation of p53 and its downstream pathways, but the pro-proliferative effects of estrogen override any checkpoint-mediated cell cycle arrest (20). Conversely, in mouse models, p53 provides protection from lymph node hyperplasia and ductal carcinoma in situ (DCIS) induced by deregulated estrogen signaling (48).
DNA Repair Machinery
Estrogen receptor α interacts directly with DNA repair proteins with varying impact on DNA repair mechanisms and ERα function. This includes FEN1, MPG, APE1, and TDG of the BER pathway (49, 50), O(6)-methylguanine-DNA methyltransferase, which corrects mutagenic DNA lesion O(6)-methylguanine back to guanine (51), NHEJ repair proteins Ku70 and Ku86 in the context of gene transcription (18) and MSH2 of the MMR pathway (52). The binding of ERα to MPG enhances BER (53), while estrogen treatment upregulates or downregulates NER, depending on cell type (54, 55). The binding of repair proteins has different outcomes on ERα: MPG inhibits ERα-induced transcription and transactivation of signaling pathways (53), MSH2 and TDG transactivate ERα (50, 52), and the binding of FEN1 and APE1 to ERα has distinct effects on different ERα target genes (56, 57).
Estrogen receptor α interacts with other core DNA damage processing proteins, although the consequence for DNA repair or ERα action is unknown. Estrogen treatment upregulates BRCA2 (58) of the HR pathway, and through phosphorylation protects BRCA2 from degradation (59). ERα also directly interacts with DNA repair signaling and processing protein PARP-1 in the context of ERα-mediated gene transcription (18), which potentially affects ERα-regulated gene networks.
Cell Cycle Checkpoints and Apoptosis
One of the most important functions of the DDR is to halt proliferation via the activation of cell cycle checkpoints or induce apoptosis. The effector proteins of these responses are not only targets of the DDR but as a set are antagonized by pro-proliferative ERα signaling. The DDR induces a G1/S phase arrest downstream of ATR via CDC25A inhibition of cyclin A/E/CDK2 complexes, and downstream of p53 via p21 inhibition of cyclin D/CDK4/6 and cyclin E/CDK2 complexes. A G2/M arrest is induced downstream of Chk1/Chk2 via activation of CDC25 phosphatases to inhibit cyclin B/Cdk1 complexes (60). ERα antagonizes cell cycle arrest by upregulating CCND1 (cyclin D1), CCNE2 (cyclin E2), and CDC25A, and downregulating CIP1 (p21) downstream of c-MYC (61–63). Likewise, p53 induces apoptosis by induction of FAS-R, BAX, PUMA, and NOXA (64), but ERα induces an anti-apoptotic signal including upregulation of BCL2 (65).
Consequently, active ERα signaling will antagonize the anti-proliferative and pro-apoptotic signals of the DRR. The outcome will be dictated by the strength of each signal, but ERα signaling is able to sustain proliferation in situations where otherwise DNA damage would have induced a cell cycle arrest and apoptosis (15, 20, 66).
Disruption of DDR and DNA Repair Pathways in Breast Cancer, and Their Association with ERα Status and Prognosis
DNA damage pathways are altered in breast cancer by mutation, changes in expression, amplification, and methylation, and as a class the DDR and DNA repair proteins are frequently altered in cancer and associated with poor prognosis. A survey of the literature shows that DDR pathways differ significantly between ERα positive and ERα negative breast cancer (Table 1). At least part of this change may be due to loss of ERα signaling, and certainly changes to p53, ATM, and TIMELESS (which functions in the ATR pathway) are consistent with the loss of ERα regulation of these genes/proteins. However, given that changes to DNA damage processing are a hallmark of cancer that contributes to tumor initiation, some of the changes no doubt precede loss of ERα, and may in fact contribute to its loss. This is exemplified in cancers with low BRCA1 and ERα, and BRCA1 loss is hypothesized to lead to ERα downregulation in breast cancer (31). Nevertheless, the presence or absence of DDR/DNA repair proteins will affect DNA repair in hormone-responsive cancers and the bi-directional regulation of the DDR/DNA repair and ERα. Likewise, the loss of ERα will affect the DDR/DNA repair in ERα negative cancers.
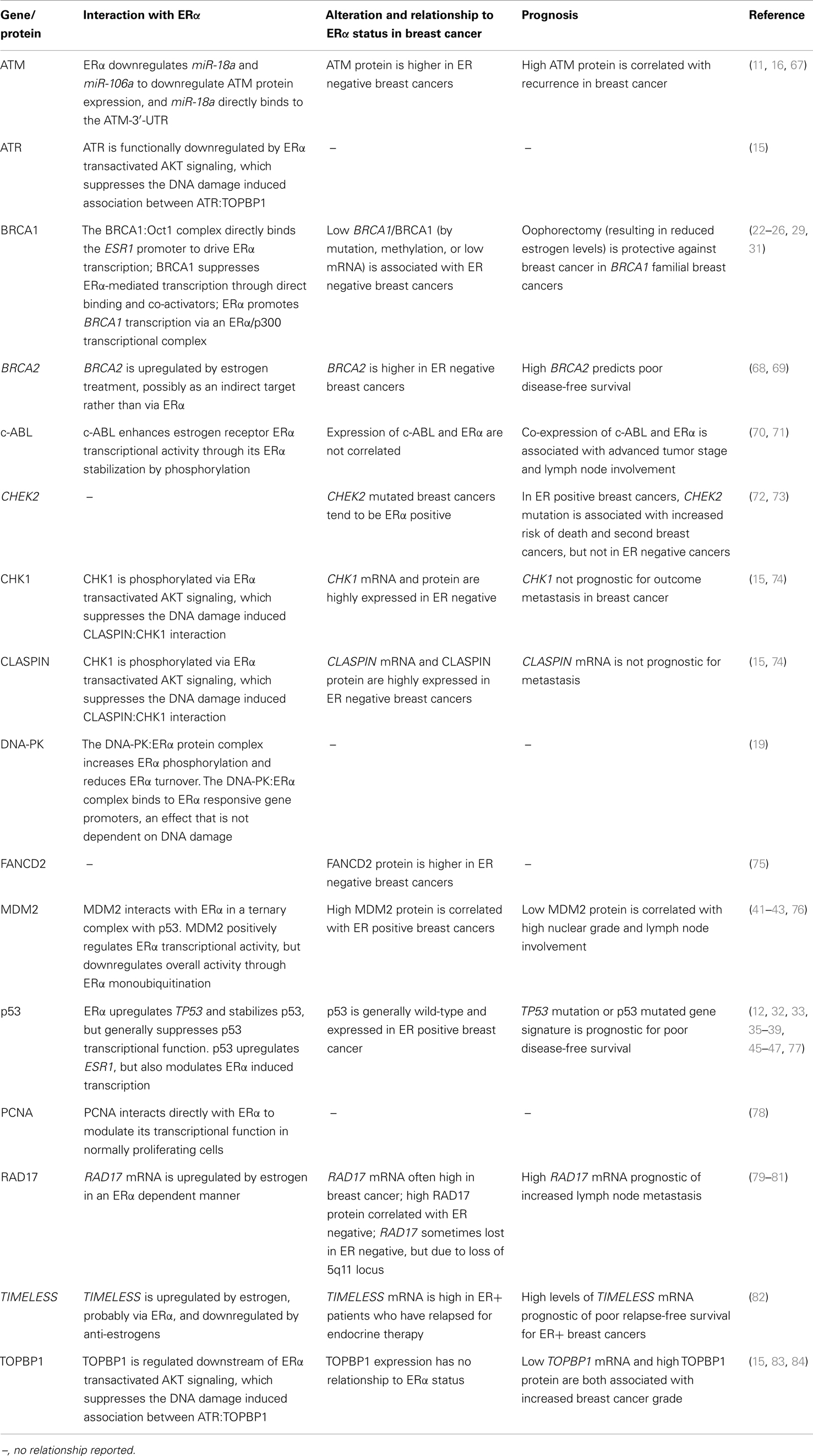
Table 1. DNA damage response and DNA repair genes altered in breast cancer and relationship to ERα status.
Perspectives
Estrogen receptor signaling is not typically thought to influence DNA repair as the literature has focused on its classic nodes of action of proliferation, growth, and apoptosis. The evidence, however, is overwhelming that ERα signaling has an impact on DNA damage processing through its regulation of ATM, ATR, DNA-PK, p53, BRCA1, BRCA2, and the DNA repair machinery. Given that estrogen can cause DNA damage, this raises a vital question of how estrogen receptor signaling processes the DNA damage caused by estrogen action. For example, does it dampen damage responses in favor of continuing proliferation, or does it act as a sentinel against DNA damage? Overall, estrogen receptor activity appears to downplay the response to DNA damage while simultaneously promoting proliferation. Consequently sustained ERα signaling may be permissive of the accumulation of genomic change from low level DNA damage that contributes to tumor initiation. Some of the major effectors of the DDR (e.g., p53 and BRCA1) do have negative feedback on the estrogen receptor, as does active DNA repair. Thus in the face of serious DNA damage ERα signaling is downregulated to protect the cell from continuing proliferation, and potentially allow full engagement in the DDR.
Several critical experiments will clarify whether active ERα signaling overrides the DDR. These include co-treatment with estrogen and different DNA damaging agents to determine the extent to which the DDR is activated and how ERα promoter binding is affected by DNA damage. This should incorporate the titration of doses of DNA damage to determine if there is a tipping point between sustained proliferation due to ERα action, and engagement of the DDR and DNA repair. Since ERα has cross-talk with both BRCA1 and p53, the combinatorial effects should be considered by simultaneously activating ERα signaling and treating with DNA damage in the context of BRCA1 and p53 ablation. Finally, it is a priority to investigate the effect of ERα on its binding partners DNA-PK, PCNA, and PARP-1 in the context of DNA damage.
The role of ERα in modulating DNA damage has important clinical implications. Anti-estrogen treatment is the mainstay of adjuvant therapy for breast cancer, but the most common therapy, Tamoxifen, is itself a source of DNA damage (85), and this damage has been detected in patients and is implicated in endometrial cancer (86). Tamoxifen has agonist effects through ERα in the endometrium (87) so it is interesting to speculate that Tamoxifen therapy induces DNA damage and disturbs a balance between estrogen signaling and the DDR in the endometrium to detrimental effect. Chemotherapies and radiation therapy induce DNA damage, so ERα may suppress the DDR to reduce the efficacy of these treatments. Indeed, patients with ER positive breast cancers have significantly lower response rates to chemotherapy than those with ER negative cancers (88), and in vitro studies suggest this is dependent on ERα action (89–91). Co-administration of anti-estrogens and radiation therapy or chemotherapy appears to enhance therapy cytotoxicity and a likely explanation is that anti-estrogen treatment prevents pro-proliferative bypass of cytotoxicity by estrogen (66, 90). Conversely, estrogen receptor action is needed for sustained p53 expression to allow the induction of apoptosis by chemotherapeutic doxorubicin (92), and good prognosis ERα positive breast cancers generally express p53. Consequently, the pro-apoptotic arm of the DDR appears compromised in some circumstances by the complete inhibition of ERα signaling. Further understanding of the cross-talk between ERα and DNA damage processing will provide crucial information to guide drug, radiation therapy, and hormone combination treatment of breast cancer patients.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Dr. Andrew Burgess and Dr. Tim Molloy for their comments. C. Elizabeth Caldon is supported by a National Breast Cancer Foundation Australia Postdoctoral Fellowship (PF-11-04) and Cancer Institute NSW Career Development Fellowship (13/CDF/1-05).
References
1. Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med (2006) 354(3):270–82. doi:10.1056/NEJMra050776
2. Thomas HV, Reeves GK, Key TJ. Endogenous estrogen and postmenopausal breast cancer: a quantitative review. Cancer Causes Control (1997) 8(6):922–8. doi:10.1023/A:1018476631561
3. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer (2009) 9(9):631–43. doi:10.1038/nrc2713
4. Williamson LM, Lees-Miller SP. Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis (2011) 32(3):279–85. doi:10.1093/carcin/bgq255
5. Liehr JG. Is estradiol a genotoxic mutagenic carcinogen? Endocr Rev (2000) 21(1):40–54. doi:10.1210/er.21.1.40
6. O’Connell P, Pekkel V, Allred DC, Fuqua SAW, Osborne CK, Clark GM. Analysis of loss of heterozygosity in 399 premalignant breast lesions at 15 genetic loci. J Natl Cancer Inst (1998) 90(9):697–703. doi:10.1093/jnci/90.9.697
7. Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci U S A (2004) 101(52):18123–8. doi:10.1073/pnas.0408273101
8. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science (2008) 319(5868):1352–5. doi:10.1126/science.1140735
9. Pallis A, Karamouzis M. DNA repair pathways and their implication in cancer treatment. Cancer Metastasis Rev (2010) 29(4):677–85. doi:10.1007/s10555-010-9258-8
10. Risbridger GP, Davis ID, Birrell SN, Tilley WD. Breast and prostate cancer: more similar than different. Nat Rev Cancer (2010) 10(3):205–12. doi:10.1038/nrc2795
11. Guo X, Yang C, Qian X, Lei T, Li Y, Shen H, et al. Estrogen receptor α regulates ATM expression through miRNAs in breast cancer. Clin Cancer Res (2013) 19(18):4994–5002. doi:10.1158/1078-0432.ccr-12-3700
12. Lewandowski SA, Thiery J, Jalil A, Leclercq G, Szczylik C, Chouaib S. Opposite effects of estrogen receptors alpha and beta on MCF-7 sensitivity to the cytotoxic action of TNF and p53 activity. Oncogene (2005) 24(30):4789–98. doi:10.1038/sj.onc.1208595
13. Thomas C, Strom A, Lindberg K, Gustafsson J-A. Estrogen receptor beta decreases survival of p53-defective cancer cells after DNA damage by impairing G2/M checkpoint signaling. Breast Cancer Res Treat (2011) 127(2):417–27. doi:10.1007/s10549-010-1011-z
14. Meltser V, Ben-Yehoyada M, Shaul Y. c-Abl tyrosine kinase in the DNA damage response: cell death and more. Cell Death Differ (2011) 18(1):2–4. doi:10.1038/cdd.2010.132
15. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell (2009) 20(14):3374–89. doi:10.1091/mbc.E09-01-0085
16. Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J, et al. miR-18a Impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One (2011) 6(9):e25454. doi:10.1371/journal.pone.0025454
17. Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov (2013) 3(11):1254–71. doi:10.1158/2159-8290.cd-13-0108
18. Ju B-G, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, et al. A topoisomerase IIß-mediated dsDNA break required for regulated transcription. Science (2006) 312(5781):1798–802. doi:10.1126/science.1127196
19. Medunjanin S, Weinert S, Schmeisser A, Mayer D, Braun-Dullaeus RC. Interaction of the double-strand break repair kinase DNA-PK and estrogen receptor-α. Mol Biol Cell (2010) 21(9):1620–8. doi:10.1091/mbc.E09-08-0724
20. Toillon R-A, Magné N, Laïos I, Castadot P, Kinnaert E, Van Houtte P, et al. Estrogens decrease γ-ray-induced senescence and maintain cell cycle progression in breast cancer cells independently of p53. Int J Radiat Oncol Biol Phys (2007) 67(4):1187–200. doi:10.1016/j.ijrobp.2006.11.040
21. Jones LP, Tilli MT, Assefnia S, Torre K, Halama ED, Parrish A, et al. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERα-negative and ERα-positive mammary preneoplasia and cancer. Oncogene (2007) 27(6):794–802. doi:10.1038/sj.onc.1210674
22. Rebbeck TR. Prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers. Eur J Cancer (2002) 38(Suppl 6):15–7. doi:10.1016/S0959-8049(02)00269-1
23. Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene (2001) 20(1):77–87. doi:10.1038/sj.onc.1204073
24. Xu J, Fan S, Rosen EM. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology (2005) 146(4):2031–47. doi:10.1210/en.2004-0409
25. Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-α. Mol Endocrinol (2010) 24(1):76–90. doi:10.1210/me.2009-0218
26. Razandi M, Pedram A, Rosen EM, Levin ER. BRCA1 inhibits membrane estrogen and growth factor receptor signaling to cell proliferation in breast cancer. Mol Cell Biol (2004) 24(13):5900–13. doi:10.1128/mcb.24.13.5900-5913.2004
27. La Rosa P, Pesiri V, Marino M, Acconcia F. 17β-Estradiol-induced cell proliferation requires estrogen receptor (ER) α monoubiquitination. Cell Signal (2011) 23(7):1128–35. doi:10.1016/j.cellsig.2011.02.006
28. Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y, et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor α activity. Cancer Res (2005) 65(15):6557–67. doi:10.1158/0008-5472.can-05-0486
29. Jeffy BD, Hockings JK, Kemp MQ, Morgan SS, Hager JA, Beliakoff J, et al. An estrogen receptor-α/p300 complex activates the BRCA-1 promoter at an AP-1 site that binds Jun/Fos transcription factors: repressive effects of p53 on BRCA-1 transcription. Neoplasia (2005) 7(9):873–82. doi:10.1593/neo.05256
30. Crowe D, Lee M. New role for nuclear hormone receptors and coactivators in regulation of BRCA1-mediated DNA repair in breast cancer cell lines. Breast Cancer Res (2006) 8(1):R1. doi:10.1186/bcr1416
31. Hosey AM, Gorski JJ, Murray MM, Quinn JE, Chung WY, Stewart GE, et al. Molecular basis for estrogen receptor α deficiency in BRCA1-linked breast cancer. J Natl Cancer Inst (2007) 99(22):1683–94. doi:10.1093/jnci/djm207
32. Hurd C, Khattree N, Alban P, Nag K, Jhanwar SC, Dinda S, et al. Hormonal regulation of the p53 tumor suppressor protein in T47D human breast carcinoma cell line. J Biolo Chem (1995) 270(48):28507–10. doi:10.1074/jbc.270.48.28507
33. Hurd C, Khattree N, Dinda S, Alban P, Moudgil VK. Regulation of tumor suppressor proteins, p53 and retinoblastoma, by estrogen and antiestrogens in breast cancer cells. Oncogene (1997) 15(8):991–5. doi:10.1038/sj.onc.1201233
34. Hurd C, Dinda S, Khattree N, Moudgil VK. Estrogen-dependent and independent activation of the P1 promoter of the p53 gene in transiently transfected breast cancer cells. Oncogene (1999) 18(4):1067–72. doi:10.1038/sj.onc.1202398
35. Okumura N, Saji S, Eguchi H, Hayashi S-I, Saji S, Nakashima S. Estradiol stabilizes p53 protein in breast cancer cell line, MCF–7. Cancer Sci (2002) 93(8):867–73. doi:10.1111/j.1349-7006.2002.tb01331.x
36. Molinari AM, Bontempo P, Schiavone EM, Tortora V, Verdicchio MA, Napolitano M, et al. Estradiol induces functional inactivation of p53 by intracellular redistribution. Cancer Res (2000) 60(10):2594–7.
37. Berger C, Qian Y, Chen X. The p53-estrogen receptor loop in cancer. Curr Mol Med (2013) 13(8):1229–40. doi:10.2174/15665240113139990065
38. Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A (2010) 107(34):15081–6. doi:10.1073/pnas.1009575107
39. Bailey ST, Shin H, Westerling T, Liu XS, Brown M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc Natl Acad Sci U S A (2012) 109(44):18060–5. doi:10.1073/pnas.1018858109
40. Liu G, Schwartz JA, Brooks SC. Estrogen receptor protects p53 from deactivation by human double minute-2. Cancer Res (2000) 60(7):1810–4.
41. Saji S, Okumura N, Eguchi H, Nakashima S, Suzuki A, Toi M, et al. MDM2 enhances the function of estrogen receptor α in human breast cancer cells. Biochem Biophys Res Commun (2001) 281(1):259–65. doi:10.1006/bbrc.2001.4339
42. Brekman A, Singh K, Polotskaia A, Kundu N, Bargonetti J. A p53-independent role of Mdm2 in estrogen-mediated activation of breast cancer cell proliferation. Breast Cancer Res (2011) 13(1):R3. doi:10.1186/bcr2804
43. Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, et al. Differential regulation of estrogen receptor α turnover and transactivation by MDM2 and stress-inducing agents. Cancer Res (2007) 67(11):5513–21. doi:10.1158/0008-5472.can-07-0967
44. Becker KA, Lu S, Dickinson ES, Dunphy KA, Mathews L, Schneider SS, et al. Estrogen and progesterone regulate radiation-induced p53 activity in mammary epithelium through TGF-β-dependent pathways. Oncogene (2005) 24(42):6345–53. doi:10.1038/sj.onc.1208787
45. Shirley SH, Rundhaug JE, Tian J, Cullinan-Ammann N, Lambertz I, Conti CJ, et al. Transcriptional regulation of estrogen receptor-α by p53 in human breast cancer cells. Cancer Res (2009) 69(8):3405–14. doi:10.1158/0008-5472.can-08-3628
46. Liu G, Schwartz JA, Brooks SC. p53 down-regulates ER-responsive genes by interfering with the binding of ER to ERE. Biochem Biophys Res Commun (1999) 264(2):359–64. doi:10.1006/bbrc.1999.1525
47. Lion M, Bisio A, Tebaldi T, De Sanctis V, Menendez D, Resnick MA, et al. Interaction between p53 and estradiol pathways in transcriptional responses to chemotherapeutics. Cell Cycle (2013) 12(8):1211–24. doi:10.4161/cc.24309
48. Díaz-Cruz ES, Furth PA. Deregulated estrogen receptor α and p53 heterozygosity collaborate in the development of mammary hyperplasia. Cancer Res (2010) 70(10):3965–74. doi:10.1158/0008-5472.can-09-3450
49. Schultz-Norton JR, Ziegler YS, Nardulli AM. ERα-associated protein networks. Trends Endocrinol Metab (2011) 22(4):124–9. doi:10.1016/j.tem.2010.11.005
50. Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, et al. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor α. J Biol Chem (2003) 278(40):38586–92. doi:10.1074/jbc.M304286200
51. Teo AKC, Oh HK, Ali RB, Li BFL. The modified human DNA repair enzyme O 6-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol Cell Biol (2001) 21(20):7105–14. doi:10.1128/mcb.21.20.7105-7114.2001
52. Wada-Hiraike O, Yano T, Nei T, Matsumoto Y, Nagasaka K, Takizawa S, et al. The DNA mismatch repair gene hMSH2 is a potent coactivator of oestrogen receptor α. Br J Cancer (2005) 92(12):2286–91. doi:10.1038/sj.bjc.6602614
53. Likhite VS, Cass EI, Anderson SD, Yates JR, Nardulli AM. Interaction of estrogen receptor α with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J Biol Chem (2004) 279(16):16875–82. doi:10.1074/jbc.M313155200
54. Boulay F, Perdiz D. 17β-estradiol modulates UVB-induced cellular responses in estrogen receptors positive human breast cancer cells. J Photochem Photobiol B (2005) 81(3):143–53. doi:10.1016/j.jphotobiol.2005.05.008
55. Evans MD, Butler JM, Nicoll K, Cooke MS, Lunec J. 17β-oestradiol attenuates nucleotide excision repair. FEBS Lett (2003) 535(1–3):153–8. doi:10.1016/S0014-5793(02)03898-X
56. Curtis CD, Thorngren DL, Ziegler YS, Sarkeshik A, Yates JR, Nardulli AM. Apurinic/apyrimidinic endonuclease 1 alters estrogen receptor activity and estrogen-responsive gene expression. Mol Endocrinol (2009) 23(9):1346–59. doi:10.1210/me.2009-0093
57. Schultz-Norton JR, Walt KA, Ziegler YS, McLeod IX, Yates JR, Raetzman LT, et al. The deoxyribonucleic acid repair protein Flap endonuclease-1 modulates estrogen-responsive gene expression. Mol Endocrinol (2007) 21(7):1569–80. doi:10.1210/me.2006-0519
58. Spillman MA, Bowcock AM. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene (1996) 13(8):1639–45.
59. Malone JL, Nelson AC, Lieberman R, Anderson S, Holt JT. Oestrogen-mediated phosphorylation and stabilization of BRCA2 protein in breast. J Pathol (2009) 217(3):380–8. doi:10.1002/path.2458
60. Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol (2006) 18(2):185–91. doi:10.1016/j.ceb.2006.02.003
61. Ru Lee W, Chen C-C, Liu S, Safe S. 17β-estradiol (E2) induces cdc25A gene expression in breast cancer cells by genomic and non-genomic pathways. J Cell Biochem (2006) 99(1):209–20. doi:10.1002/jcb.20902
62. Caldon CE, Sergio CM, Schutte J, Boersma MN, Sutherland RL, Carroll JS, et al. Estrogen regulation of cyclin E2 requires cyclin D1 but not c-Myc. Mol Cell Biol (2009) 29(17):4623–39. doi:10.1128/mcb.00269-09
63. Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem (2006) 97(2):261–74. doi:10.1002/jcb.20690
64. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett (2013) 332(2):237–48. doi:10.1016/j.canlet.2012.01.007
65. Gompel A, Somaï S, Chaouat M, Kazem A, Kloosterboer HJ, Beusman I, et al. Hormonal regulation of apoptosis in breast cells and tissues. Steroids (2000) 65(10–11):593–8. doi:10.1016/S0039-128X(00)00172-0
66. Toillon RA, Magné N, Laïos I, Lacroix M, Duvillier H, Lagneaux L, et al. Interaction between estrogen receptor alpha, ionizing radiation and (anti-) estrogens in breast cancer cells. Breast Cancer Res Treat (2005) 93(3):207–15. doi:10.1007/s10549-005-5148-0
67. Tommiska J, Bartkova J, Heinonen M, Hautala L, Kilpivaara O, Eerola H, et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene (2007) 27(17):2501–6. doi:10.1038/sj.onc.1210885
68. Egawa C, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S. Quantitative analysis of BRCA1 and BRCA2 mRNA expression in sporadic breast carcinomas and its relationship with clinicopathological characteristics. Jpn J Cancer Res (2001) 92(6):624–30. doi:10.1111/j.1349-7006.2001.tb01140.x
69. Egawa C, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S. High BRCA2 mRNA expression predicts poor prognosis in breast cancer patients. Int J Cancer (2002) 98(6):879–82. doi:10.1002/ijc.10231
70. He X, Zheng Z, Song T, Wei C, Ma H, Ma Q, et al. c-Abl regulates estrogen receptor α transcription activity through its stabilization by phosphorylation. Oncogene (2010) 29(15):2238–51. doi:10.1038/onc.2009.513
71. Zhao H, Ou-Yang F, Chen IF, Hou MF, Yuan SS, Chang HL, et al. Enhanced resistance to tamoxifen by the c-ABL proto-oncogene in breast cancer. Neoplasia (2010) 12(3):214–23. doi:10.1593/neo.91576
72. Cybulski C, Huzarski T, Byrski T, Gronwald J, Debniak T, Jakubowska A, et al. Estrogen receptor status in CHEK2-positive breast cancers: implications for chemoprevention. Clin Genet (2009) 75(1):72–8. doi:10.1111/j.1399-0004.2008.01111.x
73. Weischer M, Nordestgaard BG, Pharoah P, Bolla MK, Nevanlinna H, Van’t Veer LJ, et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J Clin Oncol (2012) 30(35):4308–16. doi:10.1200/jco.2012.42.7336
74. Verlinden L, Vanden Bempt I, Eelen G, Drijkoningen M, Verlinden I, Marchal K, et al. The E2F-regulated gene Chk1 Is highly expressed in triple-negative estrogen receptor-/progesterone receptor-/HER-2- breast carcinomas. Cancer Res (2007) 67(14):6574–81. doi:10.1158/0008-5472.can-06-3545
75. Groep P, Hoelzel M, Buerger H, Joenje H, Winter J, Diest P. Loss of expression of FANCD2 protein in sporadic and hereditary breast cancer. Breast Cancer Res Treat (2008) 107(1):41–7. doi:10.1007/s10549-007-9534-7
76. Hori M, Shimazaki J, Inagawa S, Itabashi M, Hori M. Overexpression of MDM2 oncoprotein correlates with possession of estrogen receptor alpha and lack of MDM2 mRNA splice variants in human breast cancer. Breast Cancer Res Treat (2002) 71(1):77–84. doi:10.1023/a:1013350419426
77. Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA (2005) 102(38):13550–5. doi:10.1073/pnas.0506230102
78. Schultz-Norton JR, Gabisi VA, Ziegler YS, McLeod IX, Yates JR, Nardulli AM. Interaction of estrogen receptor α with proliferating cell nuclear antigen. Nucleic Acids Res (2007) 35(15):5028–38. doi:10.1093/nar/gkm533
79. Kataoka A, Sadanaga N, Mimori K, Ueo H, Barnard GF, Sugimachi K, et al. Overexpression of hRad17 mRNA in human breast cancer: correlation with lymph node metastasis. Clin Cancer Res (2001) 7(9):2815–20.
80. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol (2008) 26(15):2568–81. doi:10.1200/jco.2007.13.1748
81. Secreto FJ, Monroe DG, Dutta S, Ingle JN, Spelsberg TC. Estrogen receptor α/β isoforms, but not βcx, modulate unique patterns of gene expression and cell proliferation in Hs578T cells. J Cell Biochem (2007) 101(5):1125–47. doi:10.1002/jcb.21205
82. Tozlu-Kara S, Roux V, Andrieu C, Vendrell J, Vacher S, Lazar V, et al. Oligonucleotide microarray analysis of estrogen receptor α-positive postmenopausal breast carcinomas: identification of HRPAP20 and TIMELESS as outstanding candidate markers to predict the response to tamoxifen. J Mol Endocrinol (2007) 39(4):305–18. doi:10.1677/jme-07-0001
83. Going JJ, Nixon C, Dornan ES, Boner W, Donaldson MM, Morgan IM. Aberrant expression of TopBP1 in breast cancer. Histopathology (2007) 50(4):418–24. doi:10.1111/j.1365-2559.2007.02622.x
84. Forma E, Krzeslak A, Bernaciak M, Romanowicz-Makowska H, Brys M. Expression of TopBP1 in hereditary breast cancer. Mol Biol Rep (2012) 39(7):7795–804. doi:10.1007/s11033-012-1622-z
85. Wozniak K, Kolacinska A, Blasinska-Morawiec M, Morawiec-Bajda A, Morawiec Z, Zadrozny M, et al. The DNA-damaging potential of tamoxifen in breast cancer and normal cells. Arch Toxicol (2007) 81(7):519–27. doi:10.1007/s00204-007-0188-3
86. Hemminki K, Rajaniemi H, Lindahl B, Moberger B. Tamoxifen-induced DNA adducts in endometrial samples from breast cancer patients. Cancer Res (1996) 56(19):4374–7.
88. Berry DA, Cirrincione C, Henderson IC, Citron ML, Budman DR, Goldstein LJ, et al. Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA (2006) 295(14):1658–67. doi:10.1001/jama.295.14.1658
89. Pritchard JE, Dillon PM, Conaway MR, Silva CM, Parsons SJ. A mechanistic study of the effect of doxorubicin/adriamycin on the estrogen response in a breast cancer model. Oncology (2012) 83(6):305–20. doi:10.1159/000341394
90. Sui M, Jiang D, Hinsch C, Fan W. Fulvestrant (ICI 182,780) sensitizes breast cancer cells expressing estrogen receptor α to vinblastine and vinorelbine. Breast Cancer Res Treat (2010) 121(2):335–45. doi:10.1007/s10549-009-0472-4
91. Tokuda E, Seino Y, Arakawa A, Saito M, Kasumi F, Hayashi S-I, et al. Estrogen receptor-α directly regulates sensitivity to paclitaxel in neoadjuvant chemotherapy for breast cancer. Breast Cancer Res Treat (2012) 133(2):427–36. doi:10.1007/s10549-011-1758-x
Keywords: estrogen receptor, DNA damage response, breast cancer, p53, BRCA1, DNA repair, tamoxifen, DDR
Citation: Caldon CE (2014) Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front. Oncol. 4:106. doi: 10.3389/fonc.2014.00106
Received: 19 March 2014; Accepted: 28 April 2014;
Published online: 14 May 2014.
Edited by:
Daniel Speidel, Children’s Medical Research Institute, AustraliaReviewed by:
Lisa Wiesmüller, Ulm University, GermanyJennifer Anne Byrne, The Children’s Hospital at Westmead, Australia
Copyright: © 2014 Caldon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Elizabeth Caldon, Genome and Replication Stability Group, The Kinghorn Cancer Centre, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, Sydney, NSW 2010, Australia e-mail:bC5jYWxkb25AZ2FydmFuLm9yZy5hdQ==