 Jermaine Coward1
Jermaine Coward1 Angus Harding2*
Angus Harding2*- 1Mater Medical Research Institute, Princess Alexandra Hospital, Woolloongabba, QLD, Australia
- 2The University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, Brisbane, QLD, Australia
Tumor evolution presents a formidable obstacle that currently prevents the development of truly curative treatments for cancer. In this perspective, we advocate for the hypothesis that tumor cells with significantly elevated genomic content (polyploid tumor cells) facilitate rapid tumor evolution and the acquisition of therapy resistance in multiple incurable cancers. We appeal to studies conducted in yeast, cancer models, and cancer patients, which all converge on the hypothesis that polyploidy enables large phenotypic leaps, providing access to many different therapy-resistant phenotypes. We develop a flow-cytometry based method for quantifying the prevalence of polyploid tumor cells, and show the frequency of these cells in patient tumors may be higher than is generally appreciated. We then present recent studies identifying promising new therapeutic strategies that could be used to specifically target polyploid tumor cells in cancer patients. We argue that these therapeutic approaches should be incorporated into new treatment strategies aimed at blocking tumor evolution by killing the highly evolvable, therapy-resistant polyploid cell subpopulations, thus helping to maintain patient tumors in a drug sensitive state.
Coming to Terms with Cancer as a Rapidly Evolving System
It has long been appreciated that cancer is an evolutionary system (1). In this paradigm, individual cancer cells are the reproductive units within a tumor, with those cells that acquire a survival advantage through random genetic change being selected through multiple rounds of clonal expansion, during which they acquire further alterations that eventually combine to produce malignant phenotypes (1). The ability of a tumor to evolve solutions to selection pressures is a function of the selectable heritable variation that is present within the tumor, be it internal stressors such as low oxygen tumor micro-environments, or external stressors such as anti-cancer therapies (2–8). The paradigm of selectable heritable variation at the cellular level being a critical driver of cancer biology has been captured by the term tumor heterogeneity, and the emerging consensus is that tumor heterogeneity remains a fundamental obstacle preventing the development of truly curative anti-cancer therapies (2–8).
The introduction of efficacious targeted therapies highlighted the central role of evolution in cancer therapy failure. Patients with leukemia and lung cancer treated with specific inhibitors targeting oncogenic receptor tyrosine kinase (RTK) activity, eventually exhibit disease progression driven by point mutations within the oncogenic RTK that renders the tumor resistant to further therapy (9–11). Retrospective analysis revealed rare therapy-resistant mutants present in tumors prior to treatment initiation (12), confirming that in some cases targeted therapy selected for resistant clones that were already present within the tumor system. Melanoma offers a compelling case study of tumor evolution during targeted therapy. The identification of oncogenic mutations within the B-Raf kinase led to the development of specific inhibitors that initially display phenomenal clinical efficacy (13–16), which is swiftly followed by disease recurrence driven by rapidly evolving therapy resistance [reviewed in Ref. (17)].
Immunological based therapies are also vulnerable to therapy-resistance driven by tumor evolution, as revealed during a vaccine strategy trialed in adult patients with Glioblastoma. The vaccine therapy invokes a patient immune response that specifically targets the truncated, oncogenic EGFRvIII variant of the EGF receptor (18). The EGFRvIII variant is present in approximately one-third of Glioblastoma patients (19) and is an ideal target for anti-tumor immunotherapy because the constitutive activity of the EGFRvIII contributes to tumorigenicity, invasion and therapy resistance [reviewed in Ref. (18)]. Although the vaccine significantly increased overall survival time in treated patients whose tumors expressed the EGFRvIII receptor, disease recurrence occurred in all patients with most recurrent tumors losing EGFRvIII expression (18). EGFRvIII expression is typically heterogeneous in Glioblastoma tumors, and is only observed in a sub-population of tumor cells and rarely in the entire tumor (20, 21). The most plausible hypothesis is that the vaccine led to the immune-clearance of EGFRvIII expressing cells from patient tumors, but in the majority of cases it was the presence of viable EGFRvIII negative cells within the tumor that allowed immunological escape and rapid disease recurrence.
These and many other studies all converge on the hypothesis that long-term cancer patient survival requires the development of therapeutic strategies that actively suppress tumor evolution (2–8, 22). In this perspective, we propose that tumor cells containing an elevated genomic content (as described in Box 1) are key players in tumor evolution, and are therefore important therapeutic targets in preventing the acquisition of therapy resistance during treatment. We begin by summarizing seminal work conducted in yeast that characterizes how chromosomal gains facilitate rapid evolution under a wide variety of selection pressures. Next, we review recent work conducted in cancer, which show that chromosomal gains also underpin tumor initiation and the acquisition of therapy resistance in cancer patients. We then present an updated model of tumor evolution that highlights the central role of increasing ploidy in cancer initiation and disease progression.
Box 1. Definitions.
Polyploidy: An alteration of chromosomal number that is a multiple of the normal diploid (2n) complement.
Tetraploidy: A specific form of polyploidy that is a doubling of the normal diploid complement (i.e., 4n).
Aneuploidy: An alteration of chromosome number that is not a multiple of the diploid (2n) complement.
Hyperdiploidy: Having a chromosome number that is more than the diploid (2n) complement.
Around 90% of all solid tumors are aneuploid, and most aneuploidy tumors exhibit chromosomal gains and are therefore hyperdiploid (85). Many cancers have complex karyotypes [see for example Ref. (62)]. In this perspective, we have focused on subpopulations of cancer cells that have elevated genomic content relative to the tumor bulk, as a source of cells capable of rapid evolution. In the strict sense, these cells are grossly hyperdiploid relative to healthy, untransformed cells. However, we refer to them here simply as polyploid tumor cells (or pseudo-polyploid tumor cells) for the following reasons. (1) We are comparing these cells to the tumor bulk, and grossly hyperdiploid tumor sub-population of cells are typically polyploid (or close to polyploid) relative to the dominant aneuploid tumor karyotype, (2) many of the cited cell biology studies refer to this tumor cell sub-population as polyploid. We ask the reader to keep in mind that the “tumor polyploid cells” are in reality a genetically heterogeneous sub-population, which is composed of a variety of complex cancer karyotypes that are approximately polyploid relative to the dominant, aneuploid tumor cell population.
We finish the perspective showcasing recent studies that identify anti-polyploid compounds that we hope will provide a foundation for the development of efficacious chemopreventative and evolutionary suppressing cancer therapies of the future. Our goal is to focus research efforts on the development and translation of such novel anti-polyploid therapies to prevent and treat incurable cancers.
Hyperdiploidy and Polyploidy Facilitates Rapid Evolution: Lessons from Yeast That are Relevant to Cancer
How Increasing Genome Size Facilitates Rapid Evolution in Yeast
Serious systemic fungal infections continue to endanger patients with immunocompromised immune systems (23, 24). Anti-fungal azole drugs are the most commonly used therapy against superficial and systemic fungal infections due to their efficacy and safety (23). Fluconazole is a widely used azole that is orally and intravenously available and effective against Candida infections, and is used clinically to treat oropharyngeal and esophageal Candidas in HIV patients, invasive candidiasis, as well as fungal infections in the urinary tract and central nervous system (23). Prior to the HIV pandemic, fluconazole resistance was rare. However, the widespread use of fluconazole to treat HIV/AIDS patients has increased the incidence of fluconazole-resistant Candida isolates (25). Generally, resistance develops after administration of sub-optimal doses of fluconazole over long periods of time, but in 1992, Bossche et al. isolated a resistant Candida strain in a patient after only 9 days of fluconazole treatment (26), revealing circumstances under which the evolution of fluconazole therapy-resistance occurs astonishingly quickly. In a follow-up study examining the mechanisms underlying the rapidly acquired fluconazole resistance, it was found that the resistant strain expressed more cytochrome P-450 14α-lanosterol demethylase (the target for azole antifungals) due to duplication of the entire chromosome containing the CYPO51 gene (27). Subsequent studies have confirmed that chromosome duplication is an effective and widely utilized mechanism to evolve drug-resistance in fungal infections (28–31).
Increasing chromosome numbers also provides fitness advantages in other contexts. A powerful example of rapid adaptation through increasing genomic content was provided by Rancati et al. (32) when they experimentally perturbed cytokinesis by deleting the MYO1 gene in the yeast Saccharomyces cerevisiae, and then selected for mutant strains that had evolved a solution to MYO1 deletion to restore functional cytokinesis. Strikingly, they found that most of the evolved strains, including the 10 fittest isolates, displayed an increase in DNA content. Further, diploid strains evolved much faster than haploid strains. Together, these data suggest that polyploidization facilitated the rapid evolution of cytokinesis rescue, a finding reminiscent of the rapid evolution of therapy-resistance driven by polyploidy described in immunocompromised patients treated with anti-fungal therapies described above.
Hyperosmotic stress occurs when an organism is exposed to higher solute concentration outside the cell, leading to water loss and subsequent increases in intracellular ion and metabolite concentrations [reviewed in Ref. (33)]. Hyperosmotic stress is a common environmental stressor, and yeast have evolved a hyperosmotic stress response that is mediated by the high osmolarity glycerol (HOG) pathway, which activates genes involved in salt tolerance and adaptation (34). Wagner and colleagues investigated how yeast could evolve adaptations to hyperosmolarity stress in long-term evolution experiments, where three replicate Saccharomyces cerevisiae yeast populations were exposed to high-salt conditions for 300 generations (33). All three populations evolved a faster growth rate under high-salt conditions after selection compared to their ancestral cultures (33). DNA content analysis revealed that all three evolved lines had an increase in ploidy, suggesting that evolutionary adaptation to hyperosmotic stress is also facilitated through increasing genome size (33).
The Evolution Canyon originated in Israel 3–5 million years ago, and contains diverse micro-environments and has experienced minimal human disturbance, providing an excellent natural site to study evolutionary adaptations of many organisms (35). Chang et al. isolated and phenotypically characterized 14 diploid yeast strains collected from different micro-environments present within the Evolutionary Canyon (35). One of these strains was highly resistant to the metal copper. Strikingly, Chang et al. found that the copper-tolerant phenotype was the product of large-scale chromosomal rearrangements that increased the copy number of the CUP1 and CUP2, major genes involved in copper regulation (35). Additional copper-tolerance gene expression was up regulated by increased CUP2 copy number, showing that the increase in gene dosage both directly and indirectly contributes to the evolution of copper-tolerance. Surprisingly, when the tolerant strains were cultured in the absence of copper, a wild-type chromosome reappeared and was fixed within 300 generations. These findings reveal that “large-scale chromosomal rearrangements provide not only fast arising but also readily reversible sources of variation during early stages of adaptive evolution” (35).
Collectively, these studies reveal increasing chromosome content as a mechanism that facilitates the rapid evolution of yeast across many different selection pressures and environments. These include the rapid acquisition of therapy resistance in patients, rapid adaptation during experimental evolution, and the successful adaptation to selection pressures present in nature.
How Increasing Genome Size Changes Yeast Phenotypes
One important mechanism for rapid adaptation provided by chromosomal gain is increased gene expression due to elevated gene dosage. Multiple studies have confirmed that messenger RNA levels scale with chromosome copy number in aneuploid systems. Hughes et al. conducted expression profiling of yeast strains with characterized aneuploidy and showed that increased genomic content data “precisely mirrored the expression data in this region” (36), revealing that gene duplication leads to a commensurate increase in messenger expression (36). The second important finding from this study was that under experimental selection, large-scale gene duplications were shown to be the dominant adaptive response to loss-of-function deletions (36), providing early support for the hypothesis that increasing genomic content facilitates rapid adaptation.
In a later study, examining the effects of extra chromosomes on cell physiology and cell division in yeast, Torres et al. observed an approximate doubling of gene expression in duplicated chromosomes, with greater than 90% the amplified genes being expressed at a higher level (37). These data indicate that most genes are expressed in proportion to their gene copy number, and gene amplification results in a roughly proportional increase in gene expression (37).
The classic evolution study by Rancati et al. confirmed that on average there is a stoichiometric relationship between gene copy number and gene expression level, with gene expression levels from chromosomes roughly scaling with chromosome copy number (32). However, they noted that some gene expression levels deviated significantly from this trend, identifying outlier genes whose expression changed more than three standard deviations away from the stoichiometric trend (32). Further evidence suggests that the majority of outlier expression is caused by the increased expression of transcription factors (or their upstream regulators) caused by chromosomal copy number increase (32). Similarly, expression of the copper resistance gene CUP2 due to increase in gene dosage causes the expression of downstream genes, several of which also enhance resistance to copper (35). This reveals how a simple linear change in gene expression can generate a non-linear adaptive response through pathway amplification (35).
Changes in yeast chromosome numbers also increase protein expression levels in yeast cells. Pavelka et al. generated a panel of stable aneuploidy yeast strains to directly address this question (38). They found that chromosomal copy number changes in general caused proportional changes in gene expression and protein expression levels (38). Further, they found that yeast strains with similar karyotypes tend to display similar changes in global protein expression patterns (38). Interestingly, the Authors also identified outliers in gene and protein expression, however only a small fraction of the gene expression outliers overlapped with the protein expression outliers (38), revealing that gene dosage changes are likely to have complex effects on cellular phenotypes. The Authors applied a variety of selection pressures on euploid parent controls and aneuploid strains, revealing that aneuploid strains grew better under selection by generating rapid phenotypic variation, showing that aneuploidy that can provide fitness gains under diverse selection pressures (38).
Together, the data sets summarized above show that increasing DNA content modifies both gene and protein expression, in linear and non-linear ways, allowing cell populations to rapidly explore a wide range of heritable phenotypes. Thus, increasing ploidy enables yeast cells to experience large phenotypic leaps, which in turn facilitates rapid evolution to novel selection pressures (39).
How Increased Genomic Content Buffers Cells Against Deleterious Mutations
The mutator hypothesis proposes that mutations that increase genomic instability (the mutator phenotype) drives tumorigenesis by allowing cells to rapidly acquire the necessary number of mutations required for cellular transformation (40). The mutator phenotype was first proposed by Loeb to explain how tumors can accumulate the number of mutations necessary for tumorigenesis despite the extremely high accuracy with which mammalian cells replicate the genome (41, 42). One primary criticism of the mutator hypothesis is that most mutations are deleterious and therefore the mutator phenotype will accelerate the accumulation of mutations that reduce fitness, leading to negative clonal selection [(3) and references therein]. Although experimental and theoretical counters to this criticism have been provided [recently reviewed in Ref. (43)], one potentially important phenomenon that has been overlooked in this debate is the role of genome amplification in buffering eukaryotic cells against the effects of deleterious mutations.
Using adaptation to different laboratory environments as their selection pressure, Thompson et al. (44) compared the relative fitness of mismatch repair defective (mutator) strains of yeast within haploid and diploid yeast genetic backgrounds, with striking results. In the diploid genetic background, mutators displayed an advantage over non-mutators, and mutators that “win” adaptation experiments were on average fitter than non-mutator winners (44). In contrast mutators in the haploid background displayed no advantage when competed against haploid non-mutators and the haploid mutator winners were less fit than the haploid non-mutator controls (44). The most parsimonious explanation for this result is that most deleterious mutations are recessive, and are therefore buffered in the diploid yeast strain. Haploid yeast must bear the cost of deleterious mutations in full, which gives haploid yeast less time to accumulate beneficial mutations before the cumulative effects of deleterious mutations eliminates them from the population. An additional important observation from this study was the type of mutations that occurred with haploid versus diploid populations. The diploid mutators displayed a generalist class of beneficial mutation that provided a large selective advantage across a range of selection pressures (44). In contrast, haploid mutators displayed beneficial mutations whose advantage was limited to the specific stress they were selected under (44).
These results suggest two intriguing hypotheses. First, increases in ploidy may act co-operatively with a mutator phenotype by reducing the effect of deleterious mutations. Second, increased ploidy enables a mutator phenotype to generate “generalist” beneficial mutations that confer selective advantage across a wide range of stressors.
How Elevated Ploidy Drives the Evolution of Cancer
Aneuploidy and Tumorigenesis
The vast majority of cancers are aneuploidy, with around 90% of solid tumors and 75% of hematopoietic cancers having abnormal chromosome numbers (45). The high incidence of aneuploidy in cancer cells inspired Boveri over 100 years ago to propose the hypothesis that aneuploidy causes cancer (46). Consistent with this hypothesis, aneuploidy has been shown to precede transformation in a variety of cancers (47–54), and several studies provide both experimental and theoretical support for a fundamental role of aneuploidy during tumor initiation (55–59). Duesberg and colleagues have proposed that aneuploidy generates cancer causing karyotypes that are selected during the evolutionary process of tumor initiation and transformation (60–62). However, these ideas have been contested, in part because the aneuploidy model of tumor initiation is thought to downplay the established role of oncogenes in the process of transformation (63–65). Recent studies examining the role of polyploidy in tumor initiation may help incorporate the oncogenes and aneuploidy tumor initiation models into a single paradigm.
Evidence Supporting Tetraploid Cells Being the Cell of Origin in Tumor Initiation
Experimental evidence directly linking tetraploidy with tumor initiation was provided when Fujiwara et al. created a tetraploid cell population in p53-null mouse mammary epithelial cells (66). Tetraploid cells displayed a high level of tumorigenesis when injected into nude mice, in contrast to the diploid p53-null controls, which did not form tumors (66). Subsequent studies perturbing the mitotic spindle led to accumulation of tetraploid cells and a higher incidence of tumor formation, further supporting a central role of tetraploidy in tumor initiation (67, 68).
Tetraploidy potentially provides multiple beneficial functions during tumor initiation. First, a large body of evidence supports the hypothesis that tetraploidy acts as a gateway karyotype by inducing chromosomal instability (CIN), which leads to aneuploidy and the evolution of a transformed phenotype [reviewed in Ref. (69)]. Using several experimental models of telomere crisis, Davoli and de Lange recently demonstrated that endoreplication and mitotic failure created tetraploid cells during telomere crisis (70). Importantly, the resulting tetraploid cells displayed enhanced tumorigenic capacity relative to diploid controls in soft agar and mouse implantation assays (70). Finally, Davoli and de Lange then showed tumors that are initiated by tetraploid cells evolve more complex aneuploidy karyotypes in vivo, showing tetraploidy functions as a gateway mutation to aneuploidy (70).
Lv and colleagues used the spontaneous transformation of primary ovarian epithelial cells to provide compelling evidence for the role of tetraploidy as a gateway karyotype during tumorigenesis (71). Lv et al. generated primary cultures of mouse ovarian surface epithelial cells (MOSECs), which they continually subcultured for over 30 passages (71). Following ploidy status during culture revealed that the diploid cells underwent an intermediate tetraploid phase, and then evolved into aneuploid (near-tetraploid) cells (71). Tetraploidy was caused by cytokinesis failure in diploid cells, with the tetraploid cells subsequently experiencing chromosome mis-segregation during bipolar and multipolar mitosis to generate aneuploid progeny (71). When the lines were re-injected into mice, only late passage aneuploid cells formed tumors (71), showing that spontaneous transformation during long-term passaging likely involves a diploid–tetraploid–aneuploid transition caused by defects in mitosis.
Two recent studies have provided compelling support for the hypothesis that genome doubling facilitates the acquisition of a transformed phenotype in tumor initiation in human cancers. Examining neuroblastomas, Lundberg et al. combined karyotypic analyses of tumors with mathematical modeling and concluded that the loss of chromosomes from a tetraploid precursor cell was the most parsimonious hypothesis explaining the chromosomal numerical alterations present in neuroblastoma tumors (72). This conclusion was supported experimentally when it was shown that neuroblastoma lines displayed a high frequency of polyploidization events, and that clonal cultures with elevated genomic content generated aneuploid progeny with high frequency (72). Altogether these data suggest that polyploidy is a gateway cell state that facilitates the generation of aneuploidy and increases karyotypic complexity in neuroblastoma tumors (72).
More recently, Swanton and colleagues (73) systematically addressed the role of tetraploidy in colorectal cancer evolution (73). Colorectal cancers that had undergone genome doubling (i.e., tetraploid) displayed a significantly higher incidence of genomic instability than those cancers that began as diploids, with tetraploidization appearing to be an early event in the majority of colorectal cancers (73). Tetraploid clones were isolated from colorectal cancer lines, and these displayed a higher incidence of segregation errors during anaphase and increased chromosomal structural abnormalities relative to their cognate, diploid controls (73). Strikingly, daughter cells derived from diploid clones that had undergone a segregation error during mitosis frequently died or underwent cell-cycle arrest, whereas daughter generated from tetraploid clones after segregation error died much less frequently and continued to proliferate (73). These data provide direct experimental support for the hypothesis that tetraploidy endows tumor precursor cells with an elevated tolerance to CIN, facilitating the generation of aneuploidy and the evolution of a complex karyotype (74). Consistent with this model, genome doubling is associated with poor prognosis, being significantly associated with disease relapse (73).
In addition to increasing tolerance to aneuploidy and facilitating the evolution of a transformed karyotype, tetraploidy also helps overcome oncogene induced senescence. Aberrant activation of oncogenes such as Ras, Raf, or PI3-kinase triggers cellular senescence, which functions as a tumor suppressor by permanently restricting the proliferative capacity of cells (75–77). Activation of DNA-damage response pathways plays an important role during oncogene induced senescence (78–80), as does activation of p53 pathways (76, 81–83). Exploring how malignant cells overcome the senescent barrier, Zheng et al. used a mouse model of tumorigenesis discovered that cells that overcame tumorigenesis barriers to drive long-term proliferation in culture all displayed near-polyploid levels of aneuploidy (84). These near-polyploid cells overexpressed DNA repair genes to reduce the DNA-damage response, as well as methylating p53 promoter regions to silence p53 expression (84). These results indicate that polyploid cells may be able to overcome the oncogene induced senescence by increasing DNA repair activity and epigenetic reprograming of p53 expression (84).
Altogether, these studies show that tetraploidy functions as a gateway phenotype that cooperates with oncogenes to induce cellular transformation in three ways. First, tetraploidy helps overcome oncogene induced senescence. Second, tetraploidy facilitates the acquisition of oncogenic karyotypes and phenotypes by inducing CIN leading to aneuploidy. Third tetraploidy buffers pre-malignant cells against the deleterious effects of chromosomal loss. Collectively, these findings go some way to explaining why the majority of human tumors contain a hyperdiploid karyotype (85).
Polyploidy Tumor Cells and the Evolution of Cancer Therapy Resistance
How Polyploidy Overcomes Therapy-Induced Senescence
Cancer cells can survive chemotherapy and radiotherapy by entering a reversible senescent state, called therapy-induced senescence (TIS), which is a senescent-like phenotype that displays many of the features of the normal physiological senescence phenotype (86). Even transformed cells lacking functional p53 and retinoblastoma protein (Rb) pathways retain the capacity to undergo TIS (87). TIS has been observed in vivo using both xenograft and transgenic cancer models (88, 89). Senescence markers have been observed from breast and lung cancer patient tumor specimens treated with chemotherapy, supporting the hypothesis that TIS is a clinically relevant cell fate in human cancer patients treated with cytotoxic therapies (90, 91).
Unfortunately TIS is not permanent, with rare cells being able to bypass TIS to re-enter the cell cycle and re-initiate tumor growth (90). One way cells overcome TIS is through the over-expression of the mitotic kinase CDK1, which phosphorylates the protein survivin to promote TIS escape and subsequent survival of cancer cells (92). In a follow-up study, Wang and colleagues went on to show that over-expression of CDK1 induced the formation of polyploid cells during TIS, and that these CDK expressing polyploid cells represent an important transition state through which escape from TIS preferentially occurs (92). Intriguingly, Wang et al. also reported that non-small cell lung cancer patients expressing markers of TIS following neo-adjuvant therapy had a significantly worse prognosis than patients who did not express TIS markers (92). Altogether, these data support a model whereby TIS provides an escape mechanism for tumor cells to avoid the toxic effects of chemotherapy to drive disease recurrence (92). Moreover, polyploid tumor cells are far more likely to overcome the TIS barrier, and polyploidy-mediated TIS escape represents an important new therapy-resistance mechanism in cancer patients undergoing a variety of chemotherapy regimes (92).
How Polyploidy Induces Infrequent Cell Cycle
Infrequent cell cycle is a well-established resistance mechanism against cytotoxic insult. Normal quiescent (G0) hematopoietic stem cells (HSCs) are resistant to the anti-proliferative chemotherapeutic agent 5-fluoro-uracil (5-FU) (93, 94), and become sensitive to 5-FU treatment when they are forced into a proliferative state by treatment with IFNα (95). Healthy HSCs can be protected from the effects of irradiation by increasing the proportion of HSCs in G0 through a variety of treatments in vivo (96–98). In cancer, the chemoprotective effect of cell-cycle-mediated drug-resistance is well-established (99). For example, Schmidt and colleagues demonstrated that colon adenocarcinoma cells arrested in G1 by over-expression of p27Kip1 are significantly more resistant to a variety of chemotherapeutic agents, including temozolomide (100). Using a mouse xenograft model, Naumov et al. showed that the DNA intercalating compound doxorubicin (DXR) effectively reduced the metastatic tumor burden but spared non-cycling tumor cells, which persisted during therapy and subsequently developed into metastases after DXR therapy was discontinued (101). More recently, label-retention has been used to phenotypically identify infrequently dividing cells that are resistant to chemotherapy from a variety of tumor types (102–105). Studies examining the cancer stem-cell phenotype have also shown that quiescence provides protection against cell death induced by DNA-damage agents (106, 107) and chemotherapy (108). Recently, a landmark study by Kreso et al. revealed how chemotherapy selects for minor, infrequently cycling subpopulations using lineage tracking in mouse models of cancer evolution (109). Collectively these studies provide strong support the hypothesis that infrequent cell cycle as a fundamental mechanism that contributes to the evolution of therapy resistance in cancer patients.
Recently, we identified a genetically diverse, polyploid tumor cell sub-population in Glioblastoma patients that is able to initiate and maintain tumor growth in vivo, and is resistant to cytotoxic therapy (110). Proliferation markers revealed that the polyploid tumor cell sub-population contain approximately three times more quiescent cells than the bulk near-diploid tumor population (110). Infrequently cycling cells retain the dye CFSE, and CFSE label-retention has been used to enrich for therapy-resistant, tumor-initiating cells in several tumor types [reviewed in Ref. (111)]. Polyploid tumor cells accumulate within the label-retaining sub-population of cells, providing a functional confirmation of their infrequent cell cycle (110). Altogether, these data show that increasing chromosome numbers provides a mechanism to generate infrequently cycling tumor cells, providing a general resistance mechanism against cytotoxic chemotherapy treatments designed to target actively cycling cells (110).
Why do polyploid tumor cells cycle less frequently? Seminal studies conducted in yeast show that increased transcription and translation caused by elevated genomic content causes cell-cycle delays during G1 (37). Murine embryonic fibroblasts (MEFs) containing extra chromosome copies also cycle less frequently (112, 113), likely due to changes in transcription and translation (85). In addition, polyploid tumor cells have a twofold larger cell volume compared to their diploid counterparts (110). Cell growth, cell size, and cell division are co-regulated to ensure cells are large enough to divide at mitosis (114). Studies in yeast reveal a size requirement for G1-S transition, with smaller cells delaying in G1 until a sufficient size was reached to maintain viable progeny after cell division (115, 116). Complementary studies in animal cells show that mammalian cells also delay in G1 to allow an appropriate cell size to be achieved (117, 118). A plausible hypothesis that combines both these observations is that the larger polyploid tumor cells arrest during G0/G1 to allow for a sufficient growth to occur before committing to division, which is hampered due to the increased transcription and translational demands placed on polyploid tumor cells by their elevated and unbalanced chromosomal copy number.
Thus increased ploidy provides a general resistance mechanism (that of infrequent cell cycle) to tumor cells, which are well-positioned to contribute to the rapid evolution of patient tumors during conventional chemotherapy and radiotherapy regimes.
The Role of Giant Polyploid Cells in Therapy Resistance and Tumor Repopulation after Therapy
Giant polyploid cells are formed if DNA replication is uncoupled from mitosis (119). This process has been termed the endocycle and is a characteristic of p53-null cells (120), which is further increased by exposure to radiation (121). It was thought that the process of endocycles was irreversible and the resulting giant polyploid cells represent a reproductive dead end (122). However two back-to-back manuscripts suggested that giant polyploid cells may provide an escape mechanism from severe genotoxic damage. The first study followed p53-null cells after genotoxic insult, noting that after delaying at G2/M for several days the cells enter endoreplication cycles that generate giant polyploid cells (123). Although the majority of giant polyploid cells die, a small subset survive that are able to produce viable progeny cells as determined using sensitive clonogenic assays (123). Viable giant polyploid cells appear to follow a defined path of chromosome re-organization that involves reconstructing nuclei into polyploidy “bouquets,” which subsequently return to an interphase state and separate into secondary nuclei (124). These secondary nuclei give rise to secondary cells in a manner reminiscent of the life-cycles of protozoa (124).
Looking at two forms of transformation, carcinogen-induced transformation of p53+/+ cell lines and spontaneous transformation of p53−/− cell lines Sundaram et al. reported a transformation process that involved giant polyploid cell intermediates (125). Here, the giant polyploid cells undergo a novel type of cell division that involves nuclear budding within the giant polyploid cells followed by intracellular cytokinesis to produce mononuclear daughter cells that bud off the parental giant polyploid mother cells (125). These mononuclear daughter cells are transformed, displaying anchorage-independent growth (a classical hallmark of cellular transformation) (125). A series of follow-up studies provided strong support for the hypothesis that a subset giant polyploid cells undergo some form of reductive division to produce small cells with near-diploid chromosomes that are proliferative and competent to re-initiate tumor growth (reviewed in Ref. (126)]. Interestingly, irradiated giant polyploid cells activate key meiotic genes that are involved in metaphase arrest, genetic recombination, and reductive divisions that occur during meiosis, indicating that giant polyploidy reductive divisions are likely “meiosis-like” in nature (127–130).
Puig et al. undertook a systematic study using xenograft in vivo models and in vitro approaches to characterize the role of giant polyploid cells in therapy response to cisplatin (131). Cisplatin treated tumors initially undergo shrinkage, and are increasingly populated with giant non-proliferating tumor cells that maintain DNA synthesis (131). After several weeks of latency tumor growth recurs, driven by a small fraction of proliferating cells (131). Cells treated in vitro using clinically relevant cisplatin doses also generate giant polyploid cells, a subset of which are able to generate colonies of rapidly cycling small diploid cells. This recapitulated the in vivo disease recurrence and suggested that giant polyploid cells are active contributors to disease progression after therapy (131). Intriguingly, the proliferative diploid cells generated from giant polyploidy cells have altered karyotypes and display increased resistance to cytotoxic drugs (131), suggesting for the first time that giant polyploid cells actively contribute to the evolution of therapy resistance.
Very recent work studying ovarian cancer has underscored the importance of giant polyploid cells in cancer disease progression and therapy resistance (132). Zhang et al. purified giant polyploid cells from established ovarian cancer lines and patient tumors, and confirmed that these cells can initiate tumors in vivo and are resistant to cisplatin cytotoxic therapy (132). Like previous studies, Zhang et al. confirmed that giant polyploidy cells cycle infrequently and generate smaller near-diploid progeny through budding and bursting mechanisms (132). In this way, giant polyploid cells are posited to function in a manner analogous to spores in lower organisms, surviving harsh conditions to facilitate rapid repopulation after stressful conditions have subsided (132, 133).
Polyploidy, EMT, and the Cancer Stem-Cell Phenotype
Cells with a primitive, undifferentiated phenotype tend to cycle infrequently and display enhanced DNA repair, making them difficult to kill using cytotoxic and genotoxic therapies that preferentially target actively cycling cells (134–136). The underlying drivers leading to the generation of a primitive phenotype in patient tumors remain incompletely understood. It has been reported that the frequency of CSC’s increases after treatment with genotoxic therapies (137–139). Salmina et al. tested the hypothesis whether polyploidy, which allows cells to survive cytotoxic therapy to continue proliferation, is also capable of endowing cells with a primitive cell phenotype (140). They found that irradiated giant polyploidy cells caused up regulation of the self-renewal stem-cell genes OCT4 and NANOG, and that the NANOG, OCT4, and SOX2 proteins were concentrated onto nuclear foci in giant polyploidy cells (140). The giant polyploid cells resisted apoptosis, overcame TIS, and transmitted the NANOG-OCT4-SOX2 self-renewal program to their progeny (140). Subsequently Lagadec et al. reported that ionizing radiation reprogramed differentiated breast cancer cells toward an undifferentiated CSC state (141). Strikingly, CSC reprograming only occurred within polyploidy subpopulations, and involved re-expression of the transcription factors OCT4, NANOG, sex determining region Y-box 2 and Klf4 (141). More recently, Zhang et al. demonstrated that ovarian cancer giant polyploid cells displayed the CSC properties of CD44+/CD133+ expression, generation of spheroids under serum-free culture conditions, increased tumorigenicity, and elevated therapy resistance (132).
Cancer cells can also undergo epithelial to mesenchymal transition (EMT), where the cancer cells activate an evolutionarily conserved trans-differentiation program that is used during morphogenesis to convert differentiated epithelial cells into migratory mesenchymal cells [reviewed in Ref. (142)]. Cancer cells undergoing EMT not only adopt an invasive cell phenotype that can drive metastasis, but may also enter a drug refractory state due to epigenetic reprograming (142). Recent work has revealed that polyploidy facilitates EMT, with Zhang et al. showing that giant polyploid tumor cells gain a mesenchymal phenotype (132) that correlates with increased expression levels of EMT transcriptional factors (143). These data suggest that polyploidy can facilitate EMT, providing access to cell phenotype that is both invasive and resistant to a variety of therapies.
Together, these studies provide compelling support for the hypothesis that polyploidy drives the acquisition of undifferentiated, primitive cellular phenotypes in human cancer. These cell phenotypes can potentially increase therapy resistance, provide an elevated tumor initiation capacity, and increase both the invasive and metastatic potential of tumor cells.
Ploidy-Induced Escape from Targeted Anti-Cancer Therapies
Strong evidence supporting the role of polyploidy in evolving solutions to targeted therapy has come from mouse models of cancer. A defective spindle assembly checkpoint (SAC) results in “mitotic slippage,” where cells exit mitosis without undergoing anaphase or cytokinesis to produce a tetraploid cell [reviewed in Ref. (69)]. As essential component of the SAC is Mad2, and Mad2 over-expression commonly occurs in many human cancers and is associated with poor prognosis [reviewed in Ref. (68)]. Over-expression of Mad2 increases the frequency of mitotic slippage and tetraploidy (68, 69), and promotes tumorigenesis in mice (69). In a doxycycline-inducible K-Ras model of cancer, Sotillo and colleagues explored how Mad2 over-expression determined the tumors ability to escape inhibition of the primary oncogenic driver K-Ras (144). In these experiments, Sotillo et al. allowed K-Ras tumors to form in the presence or absence of Mad2, revealing that the presence of Mad2 expression increased the aggressiveness of the K-Ras tumor, as indicated by increased invasion, elevated proliferative index, and a significant decrease in overall survival (144). When doxycycline was removed, K-Ras and Mad2 expression was lost, leading to tumor regression in all animals. K-Ras only tumors recurred rarely, however the tumors expressing both K-Ras and Mad2 displayed a marked increase in recurrence rate, driven by activation of a variety of compensating transforming pathways (144). This finding supports the hypothesis that CIN increases the probability of disease relapse during targeted therapy by facilitating alternate pathway activation that allows tumor cells to avoid the effects of targeted therapy (144). Further, this study highlights how aneuploidy and oncogenes can act synergistically during tumor initiation and cancer evolution.
The proteasome inhibitor, bortezomib, has forged new horizons in the treatment of multiple myeloma (MM) (145). Although efficacious, bortezomib is non-curative for MM because patients eventually evolve therapy resistance. However, the underlying resistance mechanisms remain poorly understood (146). To begin to characterize resistance mechanisms, Balsas et al. generated bortezomib-resistant MM lines that displayed five to sixfold increased resistance to bortezomib (147). Unexpectedly, the target of bortezomib (PSMβ5, the β5 subunit of the proteasome) was not mutated, but was instead significantly overexpressed at both the mRNA and protein levels within resistant cells (147). In addition, the bortezomib-resistant cells had evolved a near-tetraploid genomic content, which also displayed cross-resistance to other chemically unrelated proteasome inhibitors (147). Together, these data provide direct support for the hypothesis that, as for yeast, increasing genomic content allows cancer cells to circumvent targeted therapy through over-expression of the therapy target.
An interesting and unwelcome twist to the use of targeted therapy came from study of Sharma et al. (148). When Sharma et al. treated several breast cancer lines with the tyrosine kinase inhibitor BMS-777607, they noted that the surviving cell population displayed elevated levels of polyploidy due to an increase in the incidence of failed cytokinesis caused by off-target inhibition of Aurora kinase B (148). They tested the surviving polyploidy cells for sensitivity toward a range of chemotherapeutics (doxorubicin, bleomycin, cisplatin, methotrexate, and paclitaxel), and found that the therapy-induced polyploidy cells were resistant to all classes of chemotherapies tested (148). This finding is reminiscent of an evolutionary study undertaken in yeast, where transiently targeting the function of Hsp90 protein led the chromosomal gains and the rapid evolution of therapy-resistance toward unrelated cytotoxic compounds (149).
Together these studies reveal that tumor cell polyploidy generates resistance toward targeted therapy. Of concern is the finding that treating tumor cells with targeted therapies can elevate levels of polyploidy in tumor cell populations, which then increases the risk of developing multi-drug-resistance within clinical settings.
Measuring the Prevalence of Polyploid Tumor Cells in Cancer
How many polyploid cells are there in patient tumors? Quantitation of polyploidy in patient cell lines and primary tumors is challenging due to the infrequent cell cycle of polyploid tumor cells. Cancer cell biologists have traditionally used a flow-cytometry approach, where they estimate the frequency of polyploidy by measuring the number of cells with greater than 4n DNA content. However, this approach can only detect polyploid cells that are actively cycling, because most of the polyploid tumor cells are tetraploid or near-tetraploid, and therefore remain indistinguishable from the G2/M cells of the “diploid” tumor population. More sophisticated metaphase analyses [for example those in Ref. (62)] are also likely to underestimate the frequency of polyploid tumor cells, because their infrequent cell cycle means they will be under-represented using a metaphase-dependent karyotypic analysis.
Flow-cytometry screens can utilize the expression of Cyclin-B1 to discriminate between the cycling tumor “diploid” cells that are transitioning through the G2/M phase of the cell cycle, from the polyploid tumor cells in that are in the G0/G1 phase of the cell cycle (Figure 1). The advantage of this assay is that it allows the assessment of large populations of cells (in this example, 100,000 single cells for each patient line were analyzed), and the inclusion of a control line in the same tube means that the ploidy levels of tumor cells relative to control cells can be estimated within the same assay under identical staining conditions, eliminating the confounding effects of cell number variation between tubes [Figure 1; Ref. (110)]. Here, we use the prodrug carboxyfluorescein diacetate succinimidyl ester (CFSE), which is converted by cellular esterase activity into a fluorescent compound covalently bound to proteins and retained within the cells (150). CFSE staining clearly delineates the CFSE-stained control from the unstained test cell populations, and the control and test cell populations are readily identified using standard flow-cytometry gating strategies (110). CFSE-stained control lines can be either healthy diploid cells to provide a more accurate estimate of DNA content [(110) and shown in Figure 1], or alternatively untreated tumor cells can be used to directly compare the effect of drug treatments on the prevalence of polyploidy during compound screening or pre-clinical testing. Because control cells are stained with CFSE immediately before fixation (110), the staining process has no effect on cell ploidy or viability and is therefore unlikely to generate Type I or Type II errors during compound screening.
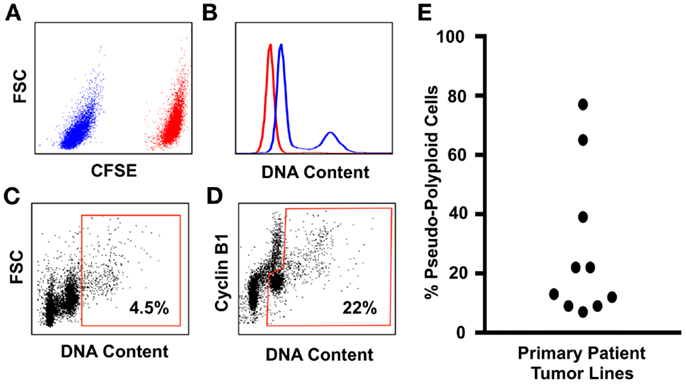
Figure 1. An improved flow-cytometry assay for measuring the prevalence of polyploidy in tumors cell populations. (A) Tumor cell samples are spiked with carboxyfluorescein diacetate succinimidylester (CFSE) stained primary neonatal foreskin fibroblasts (NFF) diploid control. The CFSE-negative tumor cells shown in blue are readily gated from the CFSE-high NFF diploid controls, shown in red. (B) DNA content of the Glioblastoma tumor cells (blue histogram) overlayed onto the NFF diploid control histogram, shown in red. Most Glioblastoma cell lines that we have studied are aneuploid with a slightly hyperdiploid DNA content, and contain a small sub-population of cells that are near-tetraploid with respect to the tumor bulk population (i.e., pseudo-polyploid). (C) A typical polyploidy flow-cytometry assessment utilized by many cancer cell biologists, who use the proportion of live single cells with greater than 4n DNA (shown within the red gate) as being representative of the total pseudo-polyploid population. In this example, 4.5% of the total cells are classed as pseudo-polyploid. (D) The same tumor sample assessed for pseudo-polyploidy using Cyclin-B1 staining to discriminate between the G2/M (the Cyclin-B1 high cells with a 4n DNA content) population of the pseudo-diploid bulk, from the pseudo-diploid G0/G1 population (the Cyclin-B1 low cells with a 4n DNA content). The pseudo-polyploid gate (shown in red) identifies both the cycling and the non-cycling pseudo-polyploid tumor cells, which make up approximately 22% of the total tumor cell population. (E) Ten low-passage primary patient glioblastoma cell lines, grown under serum-free tumorsphere conditions, assessed for pseudo-polyploidy using the Cyclin-B1 gating strategy from (D). In eight lines, the dominant cell population was aneuploidy with a near-diploid DNA content, with a sub-population of pseudo-polyploid cells that made up 5–38% of the total cell population. In contrast, two glioblastoma lines were pre-dominantly pseudo-polyploid (65 and 78%), with a small of near-diploid sub-population. Detailed staining protocols are provided in Ref. (110).
Using this method, we assessed the prevalence of polyploidy in 10 low-passage primary patient glioblastoma lines (Figure 1), cultured under tumorsphere conditions, a culture method that preserves the genotype, and phenotype of the original tumor (151). In 10 primary patient tumor lines, the lowest frequency of tumor cell polyploidy was 1 in 20 cells (i.e., 5% of the total tumor cell population were polyploid). To put this into context, it is estimated that a 1 g solid tumor contains 108–109 tumor cells (74, 152). If the lowest polyploid estimate of 5% is applied, then between 5 and 50 million rapidly evolving, therapy-resistant polyploid tumor cells will be present in brain cancer patients that have tumor volumes of 1 cm3.
An Integrated Model Explaining How Increased Genomic Content Facilitates Cancer Evolution
From the perspective of cancer as an evolutionary disease, we argue that the studies summarized above provide sufficient grounds for the development of an updated model of cancer that highlights a central role of polyploidy during tumorigenesis and disease progression (Figure 2). The hallmarks of cancer outlined by Hanahan and Weinberg (153) clearly highlight the selection pressures that must be overcome on the journey from pre-malignant lesion to full-blown cancer. The early selection pressures include apoptosis, senescence and terminal differentiation. We argue that pre-malignant polyploid cells are more likely to overcome these barriers than diploid pre-malignant cells. Polyploidy enables epigenetic silencing of p53 (84), reducing the probability of apoptosis and weakening the senescence barrier. Polyploidy also rewires the DNA-damage response (84), further subverting the senescence barrier and increasing the probability of pre-malignant polyploid cells re-entering the cell cycle (84). The vast majority of cells are terminally differentiated. Differentiated pre-malignant cells must somehow overcome the terminal differentiation program, revert to an undifferentiated phenotype, and reclaim the unlimited proliferative capacity of multi-potent stem cells (154). We now know that polyploidy facilitates acquisition of a primitive, stem cell like phenotype (132, 140, 141), although the underlying mechanisms remain incompletely uncharacterized.
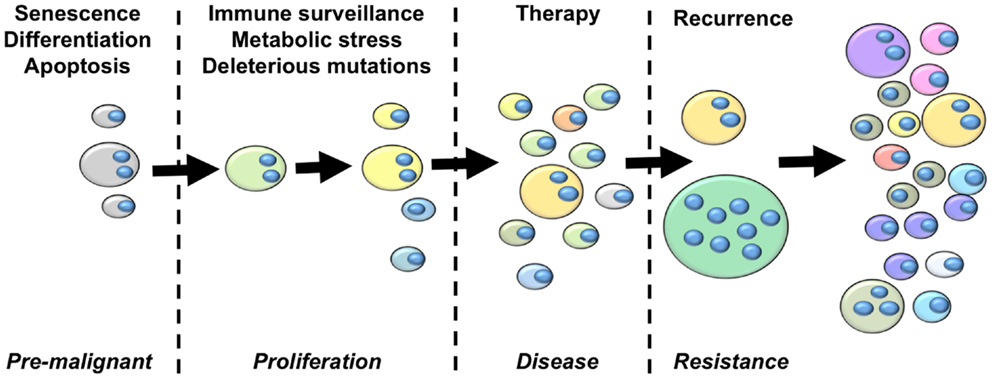
Figure 2. An integrated model of tumor evolution highlighting potential roles of polyploidy during cellular transformation. Here, we present a simplified view of disease progression, highlighting the role of polyploidy in overcoming selection pressures to drive the evolution of cellular transformation. The fist selections pressures pre-malignant lesions must overcome are those of apoptosis, senescence and terminal differentiation. Polyploidy enables adaptation to these barriers by silencing p53 remove p53-dependent pro-apoptotic and senescence signaling, rewiring the DNA-damage response to suppress p53-independent senescence programs, and enabling acquisition of primitive stem-cell phenotypes. Once a proliferative state is reached, polyploidy increases the acquisition of transforming mutations by increasing chromosomal instability and buffering the proliferative cells against the effects of deleterious mutations. Polyploidy also increases glycolysis, enabling survival in low oxygen environments, and enables EMT and the generation of invasive and metastatic phenotypes. Polyploid cells provide multiple mechanisms of therapy resistance, buffer the cancer genome against deleterious mutation resulting from genotoxic therapies, and generate primitive tumor-initiating phenotypes that are capable of driving disease recurrence. Throughout this process, tumor heterogeneity and karyotypic complexity increases, which in turn increase the heterogeneity and evolutionary capacity of the tumor.
Once pre-malignant cells circumvent these initial selection pressures to generate a proliferative phenotype, they must then acquire further transforming mutations to overcome subsequent selection pressures such as immune-surveillance, metabolic stressors, and the effects of deleterious mutation (154). Polyploidy is likely to facilitate the rapid acquisition of new transforming mutations in two ways. First, elevated ploidy reduces the lethality of deleterious mutations and chromosome loss (44, 73, 74). Second, polyploidy increases CIN (70, 71, 73, 141, 155, 156), which elevates karyotypic variation within the tumor cell population through large-scale genetic change (74). Hence, polyploidy enables both CIN and mutator phenotypes, thereby greatly increasing the speed at which proliferating tumor precursor cells can acquire the portfolio of mutations and the oncogenic karyotypes necessary for full-blown transformation (3, 61). In addition, polyploidy can help overcome metabolic stress by contributing to metabolic reprograming, invasion, and metastasis. Polyploid tumor cells display elevated levels of anaerobic glycolysis (110, 157) and are highly resistant to oxygen deprivation (132). Polyploid tumor cells also increase the expression of metastasis-related proteins (143), which may enable the acquisition of metastatic phenotypes by driving EMT (132).
Once disease presents and treatment commences, polyploid, and hyperdiploid cells remain key players driving the ongoing evolution of the patient disease. Elevated ploidy provides cells with multiple therapy-resistance mechanisms including infrequent cell cycle (110, 132), acquisition of primitive, therapy-resistant cell phenotypes (132, 158), over-expression of therapeutic targets leading to resistance (147), alternate pathway activation leading to therapy escape (144), as well as facilitating acquisition of the dreaded multi-drug resistant phenotype (148, 149). Polyploid tumor cells are created by cytotoxic and targeted therapies (149, 158–160), therefore the frequency of tumor cells with elevated ploidy is likely to significantly increase during therapy. Further, many front-line therapies are genotoxic mutagens. In this scenario, the therapy itself imparts a mutator phenotype onto the tumor, with polyploidy functioning as a genetic buffer to reduce the effects of deleterious mutations, increasing the probability of beneficial mutations surviving within the polyploidy sub-population to drive disease recurrence.
The adaptive capacity inherent to polyploidy cells means that even a small sub-population of surviving polyploid tumor cells are able to drive disease recurrence (39, 161). The capacity of polyploid tumor cells to repopulate post-therapy is likely to be significantly enhanced due to the ploidy-driven acquisition of a primitive cell phenotype with an elevated tumor-initiating capacity (132, 141), combined with a greatly reduced competition for resources due to the competing non-resistant tumor cells being killed off during therapy (162).
For these reasons, we predict that polyploid tumor cells play an integral role in disease recurrence and the acquisition of a therapy-resistant, increasingly malignant disease in patients during therapy.
New Therapeutic Strategies That Target Polyploidy and Hyperdiploid Tumor Cell Subpopulations
Experiments in yeast and cancer model systems have shown that the presence of polyploidy generates points of fragility within cellular systems that can be targeted using specific therapeutics (163–168). These studies provide the critical proof-of-principle that polyploidy is in fact a druggable phenotype. However, the strategies proposed in these pioneering studies target stress responses or mitotic machinery, which poses the risk of increasing polyploidy in surviving cells (148, 149). Fortunately, recent studies have identified new avenues for therapy development that could be used in conjunction with established therapies to inhibit the formation of polyploidy tumor cells and decrease the adaptive capacity of tumors in vivo.
Targeting Metabolism to Attack Polyploidy and Hyperdiploid Tumor Cells
Cell size scales linearly with DNA content in Eukaryotes (169–172). Cancer polyploid cells are proportionally larger than the euploid bulk population (110, 147), with giant polyploid cells being much larger than the euploid population (124). One potential consequence of large genome size and increased cell volume is a heightened metabolic demand, as bigger cells require more energy to grow to a sufficient cell volume to allow for cell doubling (117, 118). Further, the increased mRNA and protein expression caused by increased ploidy also demands more energy consumption (37). Consistent with increased genome size cell volume, and elevated transcription and translation, we noted polyploid tumor cells displayed a higher metabolic rate than the euploid control population (110).
The large cell size and increased metabolism of polyploid tumor cells may represent a point of fragility specific to the polyploid sub-population that could be exploited therapeutically. To test this hypothesis, we treated parental euploid and polyploid clonal cultures with the 2-deoxy-d-glucose, an established inhibitor of glycolysis (173–176). We found that the brain tumor polyploid cells were significantly more sensitive to the effects of glycolysis inhibition then the euploid parent control (110).
Critically, this increased dependence of polyploidy cells on glycolysis has been reported across several types of cancer. In acute myeloid leukemia (AML), Liu et al. demonstrated that targeting aurora kinases with specific inhibitors increased the prevalence of polyploidy in AML cells, and that AML polyploidy cells displayed increased glycolysis as measured by increased glucose uptake and lactic acid production (157). AML polyploidy cells were sensitive to the effects of 2-DG, suggesting that targeting metabolism may preferentially kill polyploidy tumor cells (157). mTOR is a conserved serine/threonine kinase that links cell signal transduction with cell metabolism and growth (177). Specific mTOR inhibitors promoted apoptosis and autophagy in polyploidy tumor cells and increased the efficacy of Aurora kinase inhibitors, confirming tumor metabolism as a viable point of therapeutic intervention against AML polyploidy tumor cells (157). Using breast cancer cells, Sharma et al. used the tyrosine kinase inhibitor BMS-777607 to induce polyploidization in breast cancer lines, and confirmed that therapy-induced polyploidy cells were resistant to the effects of a variety of chemotherapies (148). They then performed drug screens looking for additional inhibitors that specifically targeted polyploidy tumor cells, and identified that an inhibition of mTOR signaling prevented the formation of therapy-induced polyploidy and maintained the sensitivity of breast cancer cells toward the effects of chemotherapies (148). These results indicate that reducing polyploidy tumor cell formation by targeting metabolism may delay the evolution of therapy resistance (148). More recently, the same group revealed that BMS-777607 induced polyploidization in pancreatic cancer cells, which displayed pan-resistance to a range of chemotherapeutic compounds (160). Targeting metabolism using mTOR inhibitors reduced the formation of therapy-resistant polyploidy cells and synergized with BMS-770607, showing that for pancreatic tumor cells targeting tumor metabolism prevents the emergence of therapy-resistant polyploidy tumor cells (160).
Together, these results support the hypothesis that in brain, breast, leukemia, and pancreatic cancers, polyploid tumor cells have a commensurately higher metabolic requirement than euploid tumor cells, and that inhibiting metabolism is an effective therapeutic strategy to specifically target polyploid tumor cells to maintain tumors in a drug sensitive state.
Stimulating AMP Kinase Activity Using Resveratrol and Aspirin
Lissa et al. used an elegant high-throughput screening to screen a compound library for drugs that preferentially kill tetraploid cells, identifying resveratrol as an anti-tetraploid therapeutic agent (178). Resveratrol is an anti-fungal agent naturally occurring in grapes that has been reported to reduce tumor formation in a genetic mouse model of intestinal carcinoma when administered orally (179). Resveratrol stimulates AMPK activation by inhibiting phosphodiesterase 4 (PDE-4), allowing cAMP accumulation in cells and subsequent activation of protein kinase A (PKA) (180). Consistent with this being the primary mode of tetraploid killing, resveratrol treatment activated AMPK in tetraploid cells (180). Activation of AMPK using a separate PDE-4 inhibitor or over-expression of AMPK selectively killed tetraploid cells, whereas PKA inhibitors specifically blocked resveratrol killing (178). Aspirin (acetyl salicylate) and it is more active derivative salicylate also activate AMPK, and consistent with other AMPK activators were shown to selectively kill tetraploid cells (178). Using the mouse intestinal carcinoma cancer model, the Authors then confirmed that oral administration of either resveratrol or aspirin reduced the frequency of tetraploid intestinal epithelial cells (178), confirming that resveratrol and aspirin preferentially kill tetraploid cells in vivo using clinically relevant therapeutic doses (178).
Together, these data show that activating AMPK using the natural products resveratrol and aspirin can be used to specifically target tetraploid tumor cells in vitro and in vivo, potentially explaining the cancer-preventing effects of these two compounds reported using mouse models of tumor initiation (181). These findings support the exciting hypothesis that targeting tetraploid malignant precursor cells may become an effective chemopreventative strategy for humans.
Conclusion and Future Directions
Cancer evolution has been intensely studied in recent years using cutting edge genomic approaches (182–185), and novel therapeutic strategies aimed at delaying tumor evolution are being developed using increasingly sophisticated and predictive computational models of tumor evolution (162, 186–189). Here, we have presented a complementary approach, which consists of identifying and therapeutically targeting the polyploid tumor cell subpopulations that are likely to facilitate rapid evolution. We have based our argument on evidence derived from yeast and cancer model systems, as well as from primary patient tumor samples, all of which support the hypothesis that polyploidy facilitates rapid evolution and the acquisition of therapy-resistant phenotypes in cancer patients.
We have also presented recent studies that identify promising new anti-polyploid therapeutic approaches, which could potentially be used to target polyploid tumor cells in cancer patients. We predict that the therapies stemming from these pioneering studies will be successfully translated and incorporated into novel anti-evolution therapies designed using systems biology approaches, which will significantly increase cancer patient lifespan by slowing the emergence of therapy resistance, as well as being used as chemopreventative agents to reduce the incidence of cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nowell PC. The clonal evolution of tumor cell populations. Science (1976) 194:23–8. doi: 10.1126/science.959840
2. Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer (2010) 103:1139–43. doi:10.1038/sj.bjc.6605912
3. Tian T, Olson S, Whitacre JM, Harding A. The origins of cancer robustness and evolvability. Integr Biol (Camb) (2010) 3:17–30. doi:10.1039/c0ib00046a
4. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer (2012) 12:487–93. doi:10.1038/nrc3298
5. Greaves M, Maley CC. Clonal evolution in cancer. Nature (2012) 481:306–13. doi:10.1038/nature10762
6. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer (2012) 12:323–34. doi:10.1038/nrc3261
7. Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med (2012) 4:127s110. doi:10.1126/scitranslmed.3003854
8. Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet (2012) 13:795–806. doi:10.1038/nrg3317
9. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science (2001) 293:876–80. doi:10.1126/science.1062538
10. Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell (2002) 2:117–25. doi:10.1016/S1535-6108(02)00096-X
11. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med (2005) 352:786–92. doi:10.1056/NEJMoa044238
12. Roche-Lestienne C, Preudhomme C. Mutations in the ABL kinase domain pre-exist the onset of imatinib treatment. Semin Hematol (2003) 40:80–2. doi:10.1053/shem.2003.50046
13. Flaherty KT, Puzanov I, Kim KB, Ribas A, Mcarthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med (2010) 363:809–19. doi:10.1056/NEJMoa1002011
14. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364:2507–16. doi:10.1056/NEJMoa1103782
15. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380:358–65. doi:10.1016/S0140-6736(12)60868-X
16. Young K, Minchom A, Larkin J. BRIM-1, -2 and -3 trials: improved survival with vemurafenib in metastatic melanoma patients with a BRAF(V600E) mutation. Future Oncol (2012) 8:499–507. doi:10.2217/fon.12.43
17. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med (2013) 19:1401–9. doi:10.1038/nm.3392
18. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28:4722–9. doi:10.1200/JCO.2010.28.6963
19. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A (1992) 89:2965–9. doi:10.1073/pnas.89.7.2965
20. Nishikawa R, Sugiyama T, Narita Y, Furnari F, Cavenee WK, Matsutani M. Immunohistochemical analysis of the mutant epidermal growth factor, deltaEGFR, in glioblastoma. Brain Tumor Pathol (2004) 21:53–6. doi:10.1007/BF02484510
21. Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev (2010) 24:1731–45. doi:10.1101/gad.1890510
22. Gatenby RA, Vincent TL. Application of quantitative models from population biology and evolutionary game theory to tumor therapeutic strategies. Mol Cancer Ther (2003) 2:919–27.
23. Rex JH, Rinaldi MG, Pfaller MA. Resistance of Candida species to fluconazole. Antimicrob Agents Chemother (1995) 39:1–8. doi:10.1128/AAC.39.1.1
24. Kronstad JW, Attarian R, Cadieux B, Choi J, D’Souza CA, Griffiths EJ, et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol (2011) 9:193–203. doi:10.1038/nrmicro2522
25. Johnson EM, Warnock DW, Luker J, Porter SR, Scully C. Emergence of azole drug resistance in Candida species from HIV-infected patients receiving prolonged fluconazole therapy for oral candidosis. J Antimicrob Chemother (1995) 35:103–14. doi:10.1093/jac/35.1.103
26. vanden Bossche H, Marichal P, Odds FC, Le Jeune L, Coene MC. Characterization of an azole-resistant Candida glabrata isolate. Antimicrob Agents Chemother (1992) 36:2602–10. doi:10.1128/AAC.36.12.2602
27. Marichal P, Vanden Bossche H, Odds FC, Nobels G, Warnock DW, Timmerman V, et al. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrob Agents Chemother (1997) 41:2229–37.
28. Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science (2006) 313:367–70. doi:10.1126/science.1128242
29. Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet (2009) 5:e1000705. doi:10.1371/journal.pgen.1000705
30. Sionov E, Lee H, Chang YC, Kwon-Chung KJ. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog (2010) 6:e1000848. doi:10.1371/journal.ppat.1000848
31. Kwon-Chung KJ, Chang YC. Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog (2012) 8:e1003022. doi:10.1371/journal.ppat.1003022
32. Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell (2008) 135:879–93. doi:10.1016/j.cell.2008.09.039
33. Dhar R, Sagesser R, Weikert C, Yuan J, Wagner A. Adaptation of Saccharomyces cerevisiae to saline stress through laboratory evolution. J Evol Biol (2011) 24:1135–53. doi:10.1111/j.1420-9101.2011.02249.x
34. Chellappan SP. HOG on the promoter: regulation of the osmotic stress response. Sci STKE (2001) 2001:e1. doi:10.1126/stke.2001.93.pe1
35. Chang SL, Lai HY, Tung SY, Leu JY. Dynamic large-scale chromosomal rearrangements fuel rapid adaptation in yeast populations. PLoS Genet (2013) 9:e1003232. doi:10.1371/journal.pgen.1003232
36. Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet (2000) 25:333–7. doi:10.1038/77116
37. Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science (2007) 317:916–24. doi:10.1126/science.1142210
38. Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature (2010) 468:321–5. doi:10.1038/nature09529
39. Chen G, Rubinstein B, Li R. Whole chromosome aneuploidy: big mutations drive adaptation by phenotypic leap. Bioessays (2012) 34:893–900. doi:10.1002/bies.201200069
40. Loeb LA. Cancer cells exhibit a mutator phenotype. Adv Cancer Res (1998) 72:25–56. doi:10.1016/S0065-230X(08)60699-5
41. Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res (1974) 34:2311–21.
42. Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res (1991) 51:3075–9.
43. Fox EJ, Prindle MJ, Loeb LA. Do mutator mutations fuel tumorigenesis? Cancer Metastasis Rev (2013) 32:353–61. doi:10.1007/s10555-013-9426-8
44. Thompson DA, Desai MM, Murray AW. Ploidy controls the success of mutators and nature of mutations during budding yeast evolution. Curr Biol (2006) 16:1581–90. doi:10.1016/j.cub.2006.06.070
45. Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol (2006) 18:658–67. doi:10.1016/j.ceb.2006.10.002
46. Boveri T. Über mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Neu Folge (1902) 35:67–90.
47. Dooley TP, Mattern VL, Moore CM, Porter PA, Robinson ES, Vandeberg JL. Cell lines derived from ultraviolet radiation-induced benign melanocytic nevi in Monodelphis domestica exhibit cytogenetic aneuploidy. Cancer Genet Cytogenet (1993) 71:55–66. doi:10.1016/0165-4608(93)90202-W
48. Ried T, Heselmeyer-Haddad K, Blegen H, Schröck E, Auer G. Genomic changes defining the genesis, progression, and malignancy potential in solid human tumors: a phenotype/genotype correlation. Genes Chromosomes Cancer (1999) 25:195–204. doi:10.1002/(SICI)1098-2264(199907)25:3<195::AID-GCC1>3.0.CO;2-8
49. Ai H, Barrera JE, Meyers AD, Shroyer KR, Varella-Garcia M. Chromosomal aneuploidy precedes morphological changes and supports multifocality in head and neck lesions. Laryngoscope (2001) 111:1853–8. doi:10.1097/00005537-200110000-00034
50. Medina D. Biological and molecular characteristics of the premalignant mouse mammary gland. Biochim Biophys Acta (2002) 1603:1–9. doi:10.1016/S0304-419X(02)00053-7
51. Doak SH, Jenkins GJ, Parry EM, Griffiths AP, Baxter JN, Parry JM. Differential expression of the MAD2, BUB1 and HSP27 genes in Barrett’s oesophagus – their association with aneuploidy and neoplastic progression. Mutat Res (2004) 547:133–44. doi:10.1016/j.mrfmmm.2003.12.009
52. Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer (2004) 109:157–62. doi:10.1002/ijc.11691
53. Amiel A, Gronich N, Yukla M, Suliman S, Josef G, Gaber E, et al. Random aneuploidy in neoplastic and pre-neoplastic diseases, multiple myeloma, and monoclonal gammopathy. Cancer Genet Cytogenet (2005) 162:78–81. doi:10.1016/j.cancergencyto.2005.03.006
54. Cardoso J, Molenaar L, De Menezes RX, Van Leerdam M, Rosenberg C, Möslein G, et al. Chromosomal instability in MYH-and APC-mutant adenomatous polyps. Cancer Res (2006) 66:2514–9. doi:10.1158/0008-5472.can-05-2407
55. Li R, Yerganian G, Duesberg P, Kraemer A, Willer A, Rausch C, et al. Aneuploidy correlated 100% with chemical transformation of Chinese hamster cells. Proc Natl Acad Sci U S A (1997) 94:14506–11. doi:10.1073/pnas.94.26.14506
56. Duesberg P, Rausch C, Rasnick D, Hehlmann R. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci U S A (1998) 95:13692–7. doi:10.1073/pnas.95.23.13692
57. Rasnick D, Duesberg P. How aneuploidy affects metabolic control and causes cancer. Biochem J (1999) 340:621–30. doi:10.1042/0264-6021:3400621
58. Duesberg P, Li R, Rasnick D, Rausch C, Willer A, Kraemer A, et al. Aneuploidy precedes and segregates with chemical carcinogenesis. Cancer Genet Cytogenet (2000) 119:83–93. doi:10.1016/S0165-4608(99)00236-8
59. Li R, Sonik A, Stindl R, Rasnick D, Duesberg P. Aneuploidy vs. gene mutation hypothesis of cancer: recent study claims mutation but is found to support aneuploidy. Proc Natl Acad Sci U S A (2000) 97:3236–41. doi:10.1073/pnas.97.7.3236
60. Li L, Mccormack AA, Nicholson JM, Fabarius A, Hehlmann R, Sachs RK, et al. Cancer-causing karyotypes: chromosomal equilibria between destabilizing aneuploidy and stabilizing selection for oncogenic function. Cancer Genet Cytogenet (2009) 188:1–25. doi:10.1016/j.cancergencyto.2008.08.016
61. Nicholson JM, Duesberg P. On the karyotypic origin and evolution of cancer cells. Cancer Genet Cytogenet (2009) 194:96–110. doi:10.1016/j.cancergencyto.2009.06.008
62. Duesberg P, McCormack A. Immortality of cancers: a consequence of inherent karyotypic variations and selections for autonomy. Cell Cycle (2013) 12:783. doi:10.4161/cc.23720
63. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature (1999) 400:464–8. doi:10.1038/22780
64. Zimonjic D, Brooks MW, Popescu N, Weinberg RA, Hahn WC. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res (2001) 61:8838–44.
65. Marx J. Debate surges over the origins of genomic defects in cancer. Science (2002) 297:544–6. doi:10.1126/science.297.5581.544
66. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature (2005) 437:1043–7. doi:10.1038/nature04217
67. Castillo A, Morse HC III, Godfrey VL, Naeem R, Justice MJ. Overexpression of Eg5 causes genomic instability and tumor formation in mice. Cancer Res (2007) 67:10138–47. doi:10.1158/0008-5472.CAN-07-0326
68. Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell (2007) 11:9–23. doi:10.1016/j.ccr.2006.10.019
69. Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci (2008) 121:3859–66. doi:10.1242/jcs.039537
70. Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell (2012) 21:765–76. doi:10.1016/j.ccr.2012.03.044
71. Lv L, Zhang T, Yi Q, Huang Y, Wang Z, Hou H, et al. Tetraploid cells from cytokinesis failure induce aneuploidy and spontaneous transformation of mouse ovarian surface epithelial cells. Cell Cycle (2012) 11:2864–75. doi:10.4161/cc.21196
72. Lundberg G, Jin Y, Sehic D, Ora I, Versteeg R, Gisselsson D. Intratumour diversity of chromosome copy numbers in neuroblastoma mediated by on-going chromosome loss from a polyploid state. PLoS One (2013) 8:e59268. doi:10.1371/journal.pone.0059268
73. Dewhurst SM, Mcgranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov (2014) 4:175–85. doi:10.1158/2159-8290.CD-13-0285
74. Rasnick D. Aneuploidy theory explains tumor formation, the absence of immune surveillance, and the failure of chemotherapy. Cancer Genet Cytogenet (2002) 136:66–72. doi:10.1016/S0165-4608(01)00665-3
75. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature (2005) 436:660–5. doi:10.1038/nature03841
76. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature (2005) 436:725–30. doi:10.1038/nature03918
77. Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, Van Der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature (2005) 436:720–4. doi:10.1038/nature03890
78. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature (2005) 434:864–70. doi:10.1038/nature03482
79. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature (2006) 444:633–7. doi:10.1038/nature05268
80. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature (2006) 444:638–42. doi:10.1038/nature05327
81. Serrano M, Lin AW, Mccurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88:593–602. doi:10.1016/S0092-8674(00)81902-9
82. Ventura A, Kirsch DG, Mclaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature (2007) 445:661–5. doi:10.1038/nature05541
83. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature (2007) 445:656–60. doi:10.1038/nature05529
84. Zheng L, Dai H, Zhou M, Li X, Liu C, Guo Z, et al. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat Commun (2012) 3:815. doi:10.1038/ncomms1825
85. Weaver BA, Cleveland DW. The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell (2008) 14:431–3. doi:10.1016/j.ccr.2008.11.011
86. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst (2010) 102:1536–46. doi:10.1093/jnci/djq364
87. Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res (2005) 65:2795–803. doi:10.1158/0008-5472.CAN-04-1270
88. Elmore LW, Rehder CW, Di X, Mcchesney PA, Jackson-Cook CK, Gewirtz DA, et al. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem (2002) 277:35509–15. doi:10.1074/jbc.M205477200
89. Christov KT, Shilkaitis AL, Kim ES, Steele VE, Lubet RA. Chemopreventive agents induce a senescence-like phenotype in rat mammary tumours. Eur J Cancer (2003) 39:230–9. doi:10.1016/S0959-8049(02)00497-5
91. Elmore LW, Di X, Dumur C, Holt SE, Gewirtz DA. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: implications for treatment response. Clin Cancer Res (2005) 11:2637–43. doi:10.1158/1078-0432.CCR-04-1462
92. Wang Q, Wu PC, Dong DZ, Ivanova I, Chu E, Zeliadt S, et al. Polyploidy road to therapy-induced cellular senescence and escape. Int J Cancer (2012) 132:1505–15. doi:10.1002/ijc.27810
93. Randall TD, Weissman IL. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood (1997) 89:3596–606.
94. Wilson A, Laurenti E, Oser G, Van Der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell (2008) 135:1118–29. doi:10.1016/j.cell.2008.10.048
95. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458:904–8. doi:10.1038/nature07815
96. Davis TA, Mungunsukh O, Zins S, Day RM, Landauer MR. Genistein induces radioprotection by hematopoietic stem cell quiescence. Int J Radiat Biol (2008) 84:713–26. doi:10.1080/09553000802317778
97. Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest (2010) 120:2528–36. doi:10.1172/JCI41402
98. Forristal CE, Winkler IG, Nowlan B, Barbier V, Walkinshaw G, Levesque JP. Pharmacologic stabilization of HIF-1alpha increases hematopoietic stem cell quiescence in vivo and accelerates blood recovery after severe irradiation. Blood (2013) 121:759–69. doi:10.1182/blood-2012-02-408419
99. Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res (2001) 7:2168–81.
100. Schmidt M, Lu Y, Parant JM, Lozano G, Bacher G, Beckers T, et al. Differential roles of p21(Waf1) and p27(Kip1) in modulating chemosensitivity and their possible application in drug discovery studies. Mol Pharmacol (2001) 60:900–6.
101. Naumov GN, Townson JL, Macdonald IC, Wilson SM, Bramwell VH, Groom AC, et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat (2003) 82:199–206. doi:10.1023/B:BREA.0000004377.12288.3c
102. Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res (2008) 10:R25. doi:10.1186/bcr1982
103. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis (2009) 26:611–23. doi:10.1007/s10585-009-9260-0
104. Kusumbe AP, Bapat SA. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res (2009) 69:9245–53. doi:10.1158/0008-5472.CAN-09-2802
105. Moore N, Houghton J, Lyle S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev (2012) 21:1822–30. doi:10.1089/scd.2011.0477
106. Chen J, Li Y, Yu TS, Mckay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488:522–6. doi:10.1038/nature11287
107. Chen Y, Li D, Wang D, Liu X, Yin N, Song Y, et al. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J Cell Biochem (2012) 113:3643–52. doi:10.1002/jcb.24228
108. Kobayashi S, Yamada-Okabe H, Suzuki M, Natori O, Kato A, Matsubara K, et al. LGR5-positive colon cancer stem cells interconvert with drug-resistant LGR5-negative cells and are capable of tumor reconstitution. Stem Cells (2012) 30:2631–44. doi:10.1002/stem.1257
109. Kreso A, O’Brien CA, Van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science (2013) 339:543–8. doi:10.1126/science.1227670
110. Donovan P, Cato K, Legaie R, Jayalath R, Olsson G, Hall B, et al. Hyperdiploid tumor cells increase phenotypic heterogeneity within Glioblastoma tumors. Mol Biosyst (2014) 10:741–58. doi:10.1039/c3mb70484j
111. Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain (2011) 134:1331–43. doi:10.1093/brain/awr081
112. Rao CV, Yang Y-M, Swamy MV, Liu T, Fang Y, Mahmood R, et al. Colonic tumorigenesis in BubR1±ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A (2005) 102:4365–70. doi:10.1073/pnas.0407822102
113. Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science (2008) 322:703–9. doi:10.1126/science.1160058
114. Turner JJ, Ewald JC, Skotheim JM. Cell size control in yeast. Curr Biol (2012) 22:R350–9. doi:10.1016/j.cub.2012.02.041
115. Johnston GC, Pringle JR, Hartwell LH. Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp Cell Res (1977) 105:79–98. doi:10.1016/0014-4827(77)90154-9
116. Martin-Castellanos C, Blanco MA, De Prada JM, Moreno S. The puc1 cyclin regulates the G1 phase of the fission yeast cell cycle in response to cell size. Mol Biol Cell (2000) 11:543–54. doi:10.1091/mbc.11.2.543
117. Dolznig H, Grebien F, Sauer T, Beug H, Mullner EW. Evidence for a size-sensing mechanism in animal cells. Nat Cell Biol (2004) 6:899–905. doi:10.1038/ncb1166
118. Kafri R, Levy J, Ginzberg MB, Oh S, Lahav G, Kirschner MW. Dynamics extracted from fixed cells reveal feedback linking cell growth to cell cycle. Nature (2013) 494:480–3. doi:10.1038/nature11897
119. Nagl W. Cdc2-kinases, cyclins, and the switch from proliferation to polyploidization. Protoplasma (1995) 188:143–50. doi:10.1007/BF01280365
120. Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, et al. A p53-dependent mouse spindle checkpoint. Science (1995) 267:1353–6. doi:10.1126/science.7871434
121. Peled A, Schwartz D, Elkind NB, Wolkowicz R, Li R, Rotter V. The role of p53 in the induction of polyploidity of myelomonocytic leukemic M1/2 cells. Oncogene (1996) 13:1677–85.
122. Weibezahn KF, Lohrer H, Herrlich P. Double-strand break repair and G2 block in Chinese hamster ovary cells and their radiosensitive mutants. Mutat Res (1985) 145:177–83. doi:10.1016/0167-8817(85)90025-2
123. Illidge TM, Cragg MS, Fringes B, Olive P, Erenpreisa JA. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int (2000) 24:621–33. doi:10.1006/cbir.2000.0557
124. Erenpreisa JA, Cragg MS, Fringes B, Sharakhov I, Illidge TM. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol Int (2000) 24:635–48. doi:10.1006/cbir.2000.0558
125. Sundaram M, Guernsey DL, Rajaraman MM, Rajaraman R. Neosis: a novel type of cell division in cancer. Cancer Biol Ther (2004) 3:207–18. doi:10.4161/cbt.3.2.663
126. Wheatley D. Growing evidence of the repopulation of regressed tumours by the division of giant cells. Cell Biol Int (2008) 32:1029–30. doi:10.1016/j.cellbi.2008.06.001
127. Kalejs M, Ivanov A, Plakhins G, Cragg MS, Emzinsh D, Illidge TM, et al. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer (2006) 6:6. doi:10.1186/1471-2407-6-6
128. Erenpreisa J, Cragg MS, Salmina K, Hausmann M, Scherthan H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp Cell Res (2009) 315:2593–603. doi:10.1016/j.yexcr.2009.05.011
129. Ianzini F, Kosmacek EA, Nelson ES, Napoli E, Erenpreisa J, Kalejs M, et al. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res (2009) 69:2296–304. doi:10.1158/0008-5472.CAN-08-3364
130. Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J (2010) 29:1272–84. doi:10.1038/emboj.2010.11
131. Puig PE, Guilly MN, Bouchot A, Droin N, Cathelin D, Bouyer F, et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int (2008) 32:1031–43. doi:10.1016/j.cellbi.2008.04.021
132. Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene (2014) 33:116–28. doi:10.1038/onc.2013.96
133. Erenpreisa J, Cragg MS. Cancer: a matter of life cycle? Cell Biol Int (2007) 31:1507–10. doi:10.1016/j.cellbi.2007.08.013
134. Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle (2006) 5:2862–6. doi:10.4161/cc.5.24.3573
135. Moncharmont C, Levy A, Gilormini M, Bertrand G, Chargari C, Alphonse G, et al. Targeting a cornerstone of radiation resistance: cancer stem cell. Cancer Lett (2012) 322:139–47. doi:10.1016/j.canlet.2012.03.024
136. Ogawa K, Yoshioka Y, Isohashi F, Seo Y, Yoshida K, Yamazaki H. Radiotherapy targeting cancer stem cells: current views and future perspectives. Anticancer Res (2013) 33:747–54.
137. Bao S, Wu Q, Mclendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444:756–60. doi:10.1038/nature05236
138. Phillips TM, Mcbride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst (2006) 98:1777–85. doi:10.1093/jnci/djj495
139. Lagadec C, Vlashi E, Della Donna L, Meng Y, Dekmezian C, Kim K, et al. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res (2010) 12:R13. doi:10.1186/bcr2479
140. Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, Klymenko T, et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp Cell Res (2010) 316:2099–112. doi:10.1016/j.yexcr.2010.04.030
141. Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells (2012) 30:833–44. doi:10.1002/stem.1058
142. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene (2010) 29:4741–51. doi:10.1038/onc.2010.215
143. Zhang S, Mercado-Uribe I, Hanash S, Liu J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS One (2013) 8:e80120. doi:10.1371/journal.pone.0080120
144. Sotillo R, Schvartzman J-M, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature (2010) 464:436–40. doi:10.1038/nature08803
145. Merchionne F, Perosa F, Dammacco F. New therapies in multiple myeloma. Clin Exp Med (2007) 7:83–97. doi:10.1007/s10238-007-0134-y
146. Orlowski RZ. Novel agents for multiple myeloma to overcome resistance in phase III clinical trials. Semin Oncol (2013) 40:634–51. doi:10.1053/j.seminoncol.2013.07.007
147. Balsas P, Galan-Malo P, Marzo I, Naval J. Bortezomib resistance in a myeloma cell line is associated to PSMbeta5 overexpression and polyploidy. Leuk Res (2012) 36:212–8. doi:10.1016/j.leukres.2011.09.011
148. Sharma S, Zeng JY, Zhuang CM, Zhou YQ, Yao HP, Hu X, et al. Small-molecule inhibitor BMS-777607 induces breast cancer cell polyploidy with increased resistance to cytotoxic chemotherapy agents. Mol Cancer Ther (2013) 12:725–36. doi:10.1158/1535-7163.MCT-12-1079
149. Chen G, Bradford WD, Seidel CW, Li R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature (2012) 482:246–50. doi:10.1038/nature10795
150. Lyons AB, Blake SJ, Doherty KV. Flow cytometric analysis of cell division by dilution of CFSE and related dyes. Curr Protoc Cytom (2013) Chapter 9:Unit911. doi:10.1002/0471142956.cy0911s64
151. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell (2006) 9:391–403. doi:10.1016/j.ccr.2006.03.030
152. Del Monte U. Does the cell number 10 (9) still really fit one gram of tumor tissue. Cell Cycle (2009) 8:505–6. doi:10.4161/cc.8.3.7608
153. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi:10.1016/S0092-8674(00)81683-9
154. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
155. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol (2009) 10:478–87. doi:10.1038/nrm2718
156. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet (2012) 13:189–203. doi:10.1038/nrg3123
157. Liu LL, Long ZJ, Wang LX, Zheng FM, Fang ZG, Yan M, et al. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Mol Cancer Res (2013) 11:1326–36. doi:10.1158/1541-7786.MCR-13-0172
158. Lagadec C, Vlashi E, Alhiyari Y, Phillips TM, Bochkur Dratver M, Pajonk F. Radiation-induced Notch signaling in breast cancer stem cells. Int J Radiat Oncol Biol Phys (2013) 87:609–18. doi:10.1016/j.ijrobp.2013.06.2064
159. Zhang S, Mercado-Uribe I, Liu J. Tumor stroma and differentiated cancer cells can be originated directly from polyploid giant cancer cells induced by paclitaxel. Int J Cancer (2013) 134:508–18. doi:10.1002/ijc.28319
160. Zeng JY, Sharma S, Zhou YQ, Yao HP, Hu X, Zhang R, et al. Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol Cancer Ther (2014) 13:37–48. doi:10.1158/1535-7163.MCT-13-0242
161. Telleria CM. Repopulation of ovarian cancer cells after chemotherapy. Cancer Growth Metastasis (2013) 6:15–21. doi:10.4137/CGM.S11333
162. Gatenby RA, Brown J, Vincent T. Lessons from applied ecology: cancer control using an evolutionary double bind. Cancer Res (2009) 69:7499–502. doi:10.1158/0008-5472.CAN-09-1354
163. Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L, et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One (2007) 2:e1337. doi:10.1371/journal.pone.0001337
164. Rello-Varona S, Vitale I, Kepp O, Senovilla L, Jemaa M, Metivier D, et al. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle (2009) 8:1030–5. doi:10.4161/cc.8.7.7950
165. Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, et al. Identification of aneuploidy-tolerating mutations. Cell (2010) 143:71–83. doi:10.1016/j.cell.2010.08.038
166. Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell (2011) 144:499–512. doi:10.1016/j.cell.2011.01.017
167. Marxer M, Foucar CE, Man WY, Chen Y, Ma HT, Poon RY. Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle (2012) 11:2567–77. doi:10.4161/cc.20947
168. Roh M, Van Der Meer R, Abdulkadir SA. Tumorigenic polyploid cells contain elevated ROS and ARE selectively targeted by antioxidant treatment. J Cell Physiol (2012) 227:801–12. doi:10.1002/jcp.22793
169. Gregory TR. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev Camb Philos Soc (2001) 76:65–101. doi:10.1111/j.1469-185X.2000.tb00059.x
170. Petrov DA. Mutational equilibrium model of genome size evolution. Theor Popul Biol (2002) 61:531–44. doi:10.1006/tpbi.2002.1605
171. Cavalier-Smith T. Economy, speed and size matter: evolutionary forces driving nuclear genome miniaturization and expansion. Ann Bot (2005) 95:147–75. doi:10.1093/aob/mci010
172. Chan YH, Marshall WF. Scaling properties of cell and organelle size. Organogenesis (2010) 6:88–96. doi:10.4161/org.6.2.11464
173. Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-d-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer (2002) 87:805–12. doi:10.1038/sj.bjc.6600547
174. Gupta S, Farooque A, Adhikari JS, Singh S, Dwarakanath BS. Enhancement of radiation and chemotherapeutic drug responses by 2-deoxy-d-glucose in animal tumors. J Cancer Res Ther (2009) 5(Suppl 1):S16–20. doi:10.4103/0973-1482.55135
175. Kalia VK, Prabhakara S, Narayanan V. Modulation of cellular radiation responses by 2-deoxy-d-glucose and other glycolytic inhibitors: implications for cancer therapy. J Cancer Res Ther (2009) 5(Suppl 1):S57–60. doi:10.4103/0973-1482.55145
176. Zhang F, Aft RL. Chemosensitizing and cytotoxic effects of 2-deoxy-d-glucose on breast cancer cells. J Cancer Res Ther (2009) 5(Suppl 1):S41–3. doi:10.4103/0973-1482.55140
177. Fan QW, Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr Top Microbiol Immunol (2010) 347:279–96. doi:10.1007/82_2010_67
178. Lissa D, Senovilla L, Rello-Varona S, Vitale I, Michaud M, Pietrocola F, et al. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc Natl Acad Sci U S A (2014) 111:3020–5. doi:10.1073/pnas.1318440111
179. Schneider Y, Duranton B, Gosse F, Schleiffer R, Seiler N, Raul F. Resveratrol inhibits intestinal tumorigenesis and modulates host-defense-related gene expression in an animal model of human familial adenomatous polyposis. Nutr Cancer (2001) 39:102–7. doi:10.1207/S15327914nc391_14
180. Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell (2012) 148:421–33. doi:10.1016/j.cell.2012.01.017
181. Mahmoud NN, Dannenberg AJ, Mestre J, Bilinski RT, Churchill MR, Martucci C, et al. Aspirin prevents tumors in a murine model of familial adenomatous polyposis. Surgery (1998) 124:225–31. doi:10.1016/S0039-6060(98)70124-2
182. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, Mcindoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature (2011) 472:90–4. doi:10.1038/nature09807
183. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature (2012) 481:506–10. doi:10.1038/nature10738
184. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366:883–92. doi:10.1056/NEJMoa1113205
185. Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature (2012) 482:529–33. doi:10.1038/nature10825
186. Coldman AJ, Goldie JH. A stochastic model for the origin and treatment of tumors containing drug-resistant cells. Bull Math Biol (1986) 48:279–92. doi:10.1007/BF02459682
187. Foo J, Michor F. Evolution of resistance to targeted anti-cancer therapies during continuous and pulsed administration strategies. PLoS Comput Biol (2009) 5:e1000557. doi:10.1371/journal.pcbi.1000557
188. Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med (2011) 3:90ra59. doi:10.1126/scitranslmed.3002356
Keywords: polyploidy, hyperdiploidy, tumor evolution, therapy resistance, tumor initiation, cancer stem cell, aneuploidy, chromosomal instability
Citation: Coward J and Harding A (2014) Size does matter: why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 4:123. doi: 10.3389/fonc.2014.00123
Received: 01 April 2014; Paper pending published: 27 April 2014;
Accepted: 11 May 2014; Published online: 26 May 2014.
Edited by:
Megan Chircop, Children’s Medical Research Institute, AustraliaReviewed by:
Markus A. N. Hartl, University of Innsbruck, AustriaLuisa Lanfrancone, European Institute of Oncology, Italy
Copyright: © 2014 Coward and Harding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angus Harding, The University of Queensland Diamantina Institute, Translational Research Institute, Princess Alexandra Hospital, 37 Kent Street, Woolloongabba, Brisbane, QLD 4102, Australia e-mail:YS5oYXJkaW5nMUB1cS5lZHUuYXU=