 Yoshihiro Suzuki-Karasaki
Yoshihiro Suzuki-Karasaki Miki Suzuki-Karasaki3
Miki Suzuki-Karasaki3- 1Division of Physiology, Department of Biomedical Sciences, Nihon University School of Medicine, Tokyo, Japan
- 2Innovative Therapy Research Group, Nihon University Research Institute of Medical Science, Tokyo, Japan
- 3Department of Dermatology, Nihon University Surugadai Hospital, Tokyo, Japan
Conventional genotoxic anti-cancer drugs target the proliferative advantage of tumor cells over normal cells. This kind of approach lacks the selectivity of treatment to cancer cells, because most of the targeted pathways are essential for the survival of normal cells. As a result, traditional cancer treatments are often limited by undesirable damage to normal cells (side-effects). Ideal anti-cancer drugs are expected to be highly effective against malignant tumor cells with minimal cytotoxicity toward normal cells. Such selective killing can be achieved by targeting pathways essential for the survival of cancer cells, but not normal cells. As cancer cells are characterized by their resistance to apoptosis, selective apoptosis induction is a promising approach for selective killing of cancer cells. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising tumor-selective anti-cancer drug. However, the congenital and acquired resistance of some cancer cell types, including malignant melanoma cells, currently impedes effective TRAIL therapy, and an innovative approach that can override TRAIL resistance is urgently required. Apoptosis is characterized by cell shrinkage caused by disruption of the maintenance of the normal physiological concentrations of K+ and Na+ and intracellular ion homeostasis. The disrupted ion homeostasis leads to depolarization and apoptosis. Recent evidence suggests that depolarization is an early and prerequisite event during TRAIL-induced apoptosis. Moreover, diverse natural products and synthetic chemicals capable of depolarizing the cell membrane exhibit tumor-selective killing and TRAIL-sensitizing effects. Here, we discuss the role of depolarization in selective killing of cancer cells in connection with the emerging concept that oxidative stress is a critical mediator of mitochondrial and endoplasmic reticulum dysfunctions and serves as a tumor-selective target in cancer treatment.
Introduction
Despite remarkable progress in cancer biology and treatment over the past 50 years, malignant neoplasms are still highly threatening diseases for humans, as they are frequently resistant to traditional chemotherapy, radiotherapy, and immunotherapy with a poor prognosis. Conventional genotoxic anti-cancer drugs target the proliferative advantage of tumor cells over normal cells. This kind of approach lacks the selectivity of treatment to cancer cells, because most of the targeted pathways are essential for the survival of normal cells. As a result, traditional cancer treatments are often limited by undesirable damage to normal cells (side-effects). Ideal anti-cancer drugs are expected to be highly effective against malignant tumor cells with minimal cytotoxicity toward normal cells. Such selective killing can be achieved by targeting pathways essential for the survival of cancer cells, but not normal cells. As cancer cells are characterized by their resistance to apoptosis, selective apoptosis induction is a promising approach for selective killing of cancer cells.
A growing list of natural products and synthetic chemicals exhibit tumor-selective killing effects (Tables 1 and 2). Under optimal conditions, they exhibit considerable cytotoxicity toward malignant cells, while sparing non-transformed cells. In addition, when applied at non-toxic concentrations, some of them can sensitize cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) cytotoxicity (Table 3). In fact, overlapping mechanisms appear to be involved in the tumor-selective killing and TRAIL-sensitizing effects, and the generation of intracellular reactive oxygen species (ROS) is the most common cellular event documented in the literature. An emerging view is that owing to their active metabolism and oncogenic stimulation, most cancer cells exhibit elevated ROS generation that makes them more vulnerable than normal cells to additional oxidative stress, and that such vulnerability can be exploited to selectively kill these cells. We will discuss this concept in more detail later. Although diverse natural and synthetic chemical compounds have been shown to possess selective killing effects, this review focuses on TRAIL, because TRAIL and its related substances are promising tumor-selective anti-cancer drugs that are currently undergoing clinical trials.
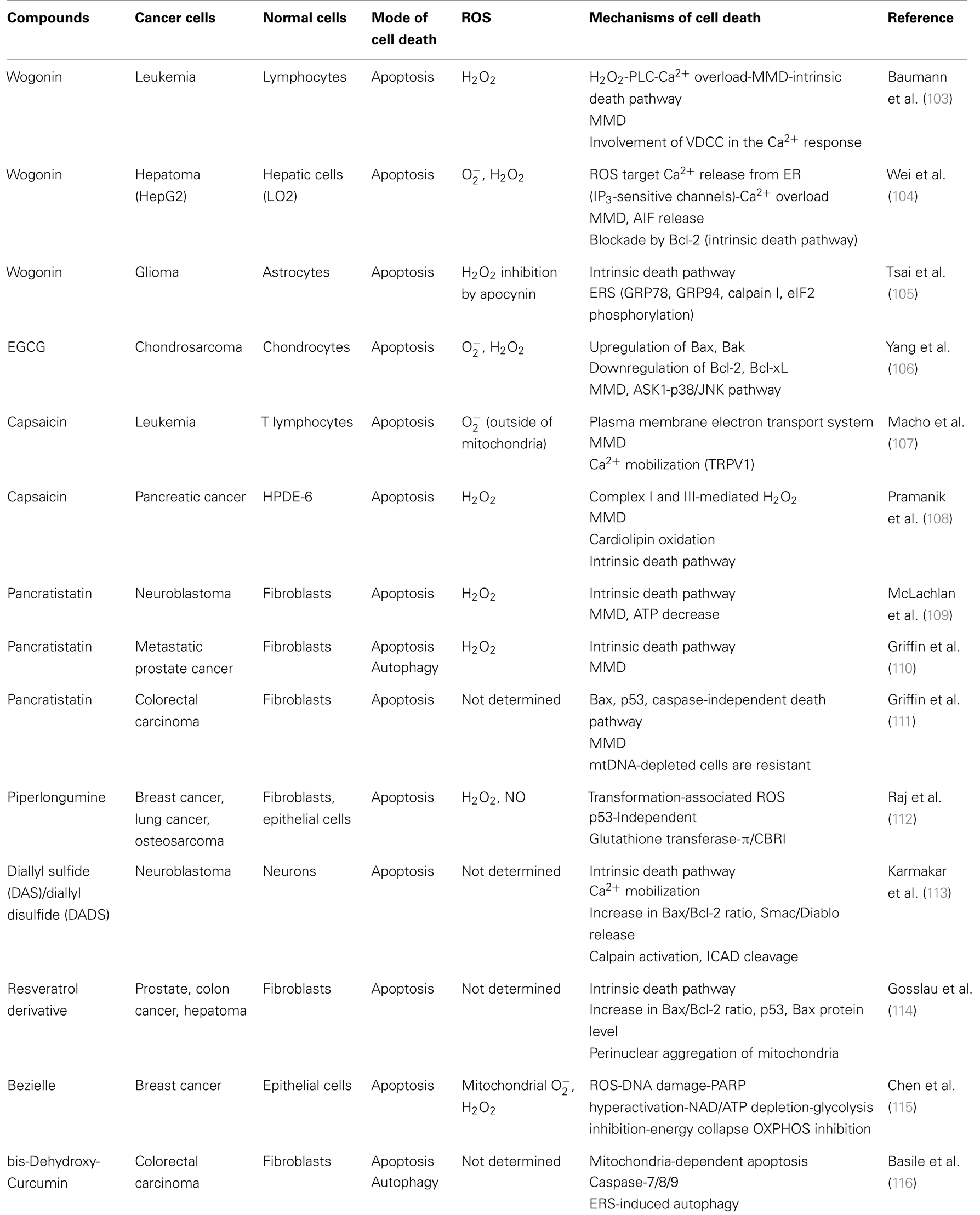
Table 1. Natural products with tumor-selective cytotoxicity.
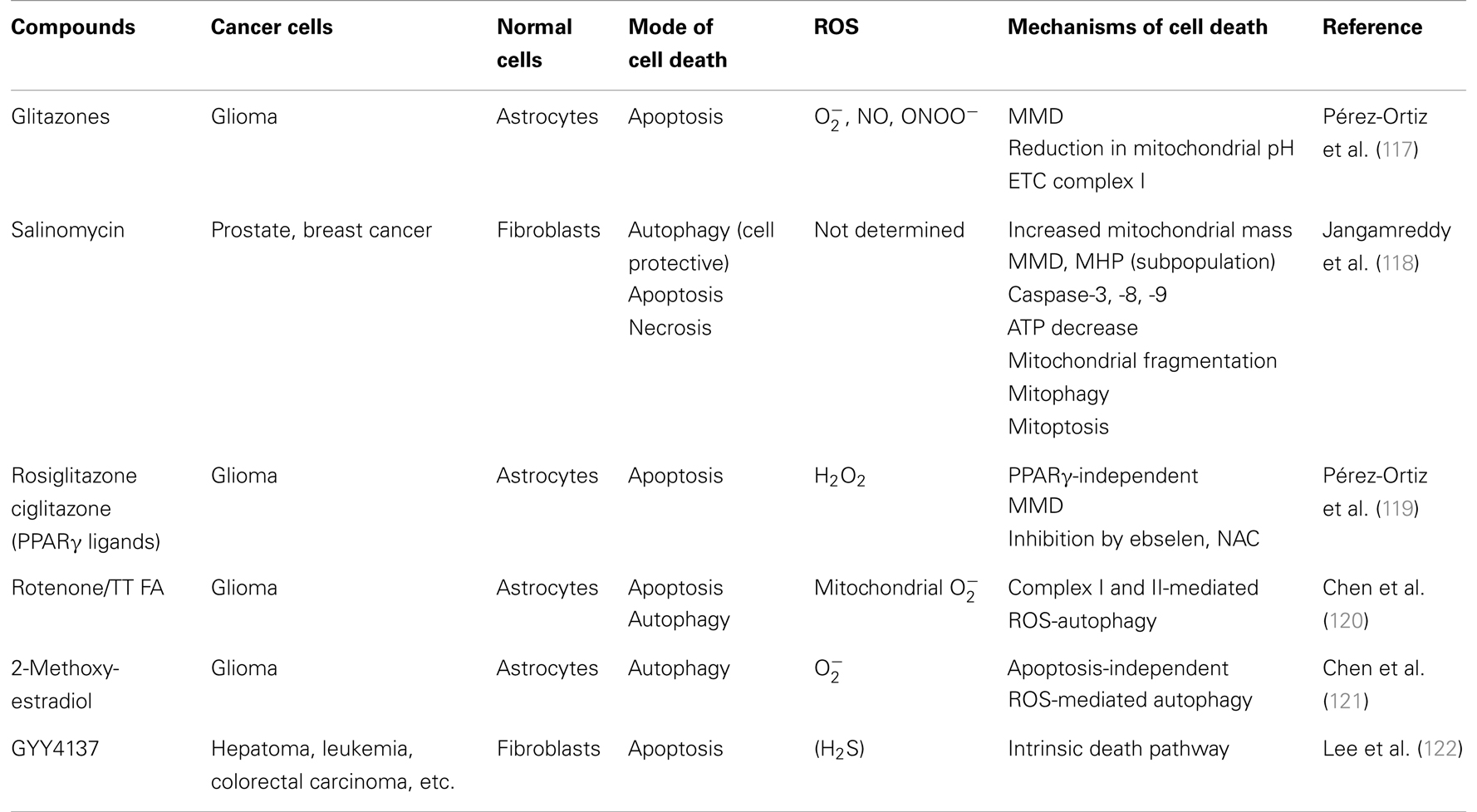
Table 2. Synthetic compounds with tumor-selective cytotoxicity.
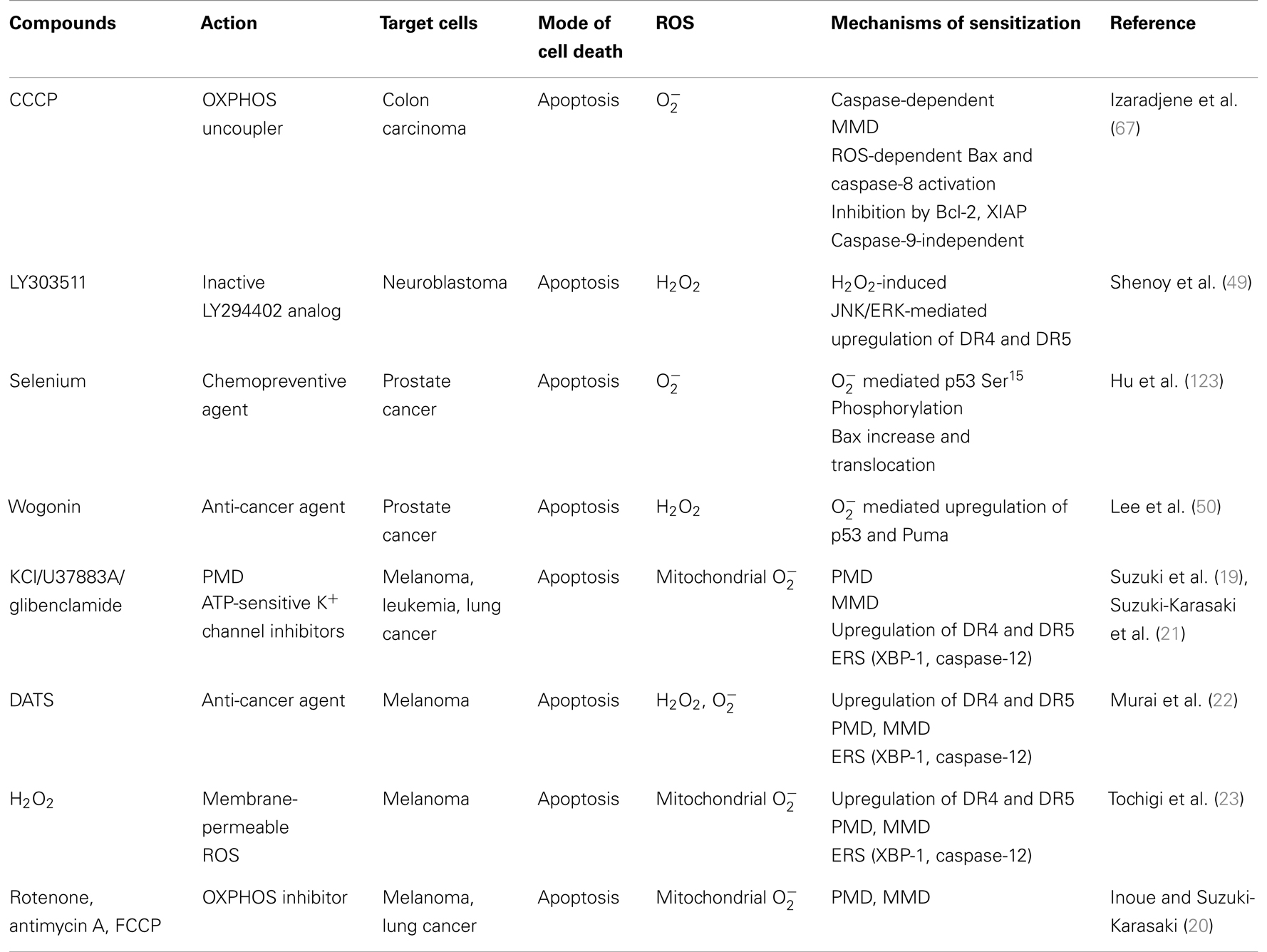
Table 3. Reactive oxygen species-generating TRAIL sensitizers.
TRAIL and Cancer Cell Death
Tumor necrosis factor-related apoptosis-inducing ligand is a member of the TNF cytokine family. It binds to several distinct receptors, death receptor (DR) 4/TRAIL receptor 1 (TRAIL-R1), DR5/TRAIL-R2, TRAIL-R3, and TRAIL-R4 (1). Both DR4 and DR5 contain an intracellular death domain that is essential for the induction of apoptosis following receptor ligation. In contrast, despite their binding to TRAIL, neither TRAIL-R3 nor TRAIL-R4 mediates apoptosis owing to a complete or partial lack of the intracellular death domain. These receptors are regarded as decoy receptors (DcRs) (1, 2). TRAIL activates the extrinsic apoptotic pathway. Binding of TRAIL to DR4/DR5 induces their oligomerization and conformational changes in their death domains, resulting in the formation of a death-inducing signaling complex and subsequent activation of the initial caspase, caspase-8. In turn, activated caspase-8 activates the effector caspases, caspase-3/6/7 that execute the apoptotic process (3, 4). Activation of caspase-8 is also linked to the intrinsic (mitochondrial) apoptotic pathway. Activated caspase-8 can cleave and activate the pro-apoptotic Bcl-2-family molecule Bid. In turn, truncated Bid activates other Bcl-2-family molecules, Bax and Bak, resulting in their oligomerization and the formation of megachannels in the outer mitochondrial membrane (OMM). The release of cytochrome c through these Bax/Bak megachannels into the cytosol induces assembly of the apoptosome, representing the activation-platform for another initial caspase, caspase-9. Activated caspase-9 also promotes the activation of caspase-3/6/7, thereby providing a positive loop for caspase activation (3). Unlike TNF-α, TRAIL has been shown to induce apoptosis in cancer cells with minimal cytotoxicity toward non-transformed cells (4), although under certain circumstances, it enhances the cytotoxicity of several drugs to hepatocytes/liver and mast cells (5–9). Thus, TRAIL is a promising agent in cancer treatment with high selectivity. However, different cancer cell types such as malignant melanoma, glioma, and non-small cell lung cancer (NSCLC) cells are resistant to TRAIL treatment despite expressing DRs on their cell surface. Moreover, TRAIL-responsive tumors acquire a resistant phenotype that renders TRAIL therapy ineffective (10). Therefore, overcoming TRAIL resistance is necessary for effective TRAIL therapy, and small molecules that can potentiate TRAIL effectiveness are urgently required. Recently, much progress has been made in therapeutic intervention with TRAIL-related substances. Recombinant human TRAIL (rhTRAIL) or agonist monoclonal antibodies (mAbs) against DR4/DR5, collectively referred as to pro-apoptotic receptor agonists (PARAs), have been subjected to clinical trials for a variety of cancer cell types, including malignant melanoma and NSCLC cells. In addition, diverse chemical substances including ABT-737 and SM-164 are expected to potentiate the intrinsic death pathway by antagonizing natural inhibitors such as FLICE inhibitory protein, Bcl-2, Mcl-1, and survivin (11). Some recent clinical trials have demonstrated the safety and efficiency of combined treatments with PARAs and conventional genotoxic drugs, while the results of other clinical trials were disappointing and showed only modest effectiveness. PARAs such as dulanermin (rhTRAIL), mapatumumab (anti-DR4 mAb), conatumumab, CS-1008, or PRO95780 (anti-DR5 mAb), in combination with paclitaxel, carboplatin, or bevacizumab [anti-vascular endothelial growth factor (VEGF) mAb] were tested for their effects toward NSCLC patients in a randomized phase II trial, but showed only modest effects (11). Thus, induction of apoptosis by the intrinsic pathway does not appear to be a suitable target in the treatment of TRAIL-resistant cancer cells.
Dual Function of Depolarization in the Regulation of Apoptosis
Apoptosis is a fundamental physiological process characterized by the loss of cell volume (cell shrinkage), chromatin condensation, and internucleosomal DNA fragmentation. Cell shrinkage is a hallmark of apoptosis, which is caused by disruption of the maintenance of normal physiological concentrations of K+ and Na+ and intracellular ion homeostasis (12, 13). Loss of these monovalent ions has been reported to facilitate the loss of cell volume (apoptotic volume decrease) and caspase-3 activation (13). Cell shrinkage requires ion transport activity across the cell membrane, including Cl− and K+ channels. Impairment of ion channels or transporters responsible for Na+, K+, Cl−, and Ca2+ can disrupt intracellular ion homeostasis and lead to cell membrane depolarization and apoptosis. In fact, depolarization has been shown to be an early event in the apoptosis induced by diverse agents, including Fas, rotenone, and arsenic trioxide (14–16), and is considered to play an important pro-apoptotic role. In contrast, depolarization has also been shown to exhibit anti-apoptotic effects. Various membrane-depolarizing agents, including ouabain, tetraethylammonium (TEA), and veratridine, protect Purkinje cells against apoptosis (17). In addition, K+ loading and several K+ channel inhibitors protect various human tumor cells against staurosporine-induced apoptosis. Thus, depolarization can act in both pro-apoptotic and anti-apoptotic manners depending on the cell types and apoptotic stimuli involved. There is no general model that can depict the dual functions of depolarization.
Na+–K+-ATPase is a Key Player Connecting Death Ligands, Depolarization, and Apoptosis
The Na+ pump Na+–K+-ATPase mediates the export of three Na+ ions the import of two K+ ions, thereby maintaining high K+ and low Na+ intracellular concentrations. Diverse apoptotic stimuli including anti-Fas, A23187, and thapsigargin have been shown to induce depolarization without repolarization in Jurkat cells (14). This response is rapid (1–2 h after stimulation) and observed in a time-dependent manner. Unlike the depolarization observed in electrically excited cells, this apoptosis-associated depolarization is rather sustained (observed for throughout at least 8 h), as the depolarization lacks repolarization. These effects can be accounted for inactivation of Na+–K+-ATPase protein and its activity. Persistent depolarization caused by impairment of Na+–K+-ATPase is also associated with other apoptotic stimuli such as the mitochondrial toxins rotenone and squamocin (15). The persistent depolarization is observed not only in human leukemia cells such as Jurkat cells and U937 cells but also in human primary T cells, although primary cells are more resistant than leukemia cells to the effect. The persistent depolarization is pro-apoptotic, because some cardiac glycoside inhibitors of Na+–K+-ATPase such as ouabain and oleandrin, can sensitize human leukemia cells and NSCLC cells to apoptosis induced by anti-Fas, TRAIL, and mitochondrial toxins (14, 15, 18). These observations suggest that Na+–K+-ATPase is a key player connecting death ligands, depolarization, and apoptosis.
Role of Depolarization in TRAIL-Induced Apoptosis of Malignant Tumor Cells
Our recent work has shown that persistent depolarization also occurs in the early stage of TRAIL-induced apoptosis in malignant tumor cells. TRAIL dose- and time-dependently induces robust depolarization in human malignant tumor cells, including melanoma, leukemia, and lung cancer cells after a time lag of 2-4 h (19, 20). In all tumor cell lines tested, the magnitude of depolarization is correlated with their sensitivity to TRAIL. Importantly, the depolarization appears to be a prerequisite event for TRAIL-induced apoptosis, because TRAIL induces minimal depolarization in normal melanocytes, to which it shows minimal cytotoxicity despite their substantial cell surface expression of DR4 and DR5 (19). The pro-apoptotic role of depolarization is supported by the finding that high K+ loading sensitizes melanoma cells, but not melanocytes, to TRAIL. As ATP-sensitive K+ (KATP) channels mediate the efflux of intracellular K+ into the cytosol, thereby promoting plasma membrane hyperpolarization, inhibition of these channels is expected to induce depolarization and show similar effects. In fact, selective inhibitors of KATP channels such as glibenclamide and U37883A show similar TRAIL-sensitizing effects, while TEA, which mainly inhibits voltage-dependent K+ (Kv) and calcium-dependent K+ (KCa) channels, is ineffective, suggesting a specific role of KATP channels in the potentiation. In support of this view, the Kv channel-specific inhibitor α-dendrotoxin, KCa channel-specific inhibitor charybdotoxin, and mitochondrial KATP channel inhibitor 5-hydroxydecanoate have no such effects in melanoma and leukemia cells (19–21). As discussed below, the depolarization-mediated potentiation of apoptosis is associated with activation of the mitochondrial apoptotic pathway and intracellular ROS generation. Consistent with this view, glibenclamide has been shown to exert antitumor activity in human gastric cancer cells through the intrinsic pathway and ROS generation. Since Na+–K+-ATPase seems to play a key role in controlling death ligand-induced depolarization and apoptosis, the enzyme may also be involved in the depolarization induced by TRAIL. However, it is noted that among these death ligands, only TRAIL exerts tumor-selective cytotoxicity. Nonetheless, cardiac glycoside Na+–K+-ATPase inhibitors such as oleandrin (18) and KATP channel inhibitors share some biological effects including upregulation of DR4 and DR5. Thus, further studies are necessary to define the role of Na+–K+-ATPase in TRAIL-induced apoptosis in human cancer cells.
Role of the Mitochondrial Death Pathway in the Effects of Depolarization
In addition to high K+ and KATP channel inhibitors, different natural and synthetic chemicals have recently been identified as powerful potentiators of TRAIL-induced apoptosis in malignant melanoma cells regardless of their diverse chemical structures and biological targets. These include diallyltrisulfide (DATS), a major garlic organosulfur compound (22), the cell-permeable oxidant H2O2 (23), and mitochondrial metabolic inhibitors such as rotenone, antimycin A, and FCCP (20). Moreover, when employed at higher concentrations and longer exposures, all of these chemicals kill malignant cells while sparing normal cells. Strikingly, all of these agents also can commonly induce robust depolarization prior to apoptosis, supporting a universal role for depolarization in the potentiation of TRAIL-induced apoptosis and tumor-selective killing. In contrast, agents that do not affect cell survival such as TEA, have no effect on DR5 expression, suggesting that upregulation of DR5 expression is a common target of these membrane-depolarizing agents for potentiating apoptosis. This view is also in agreement with previous studies demonstrating that amplification of TRAIL-induced apoptosis by diverse agents, including thapsigargin, tunicamycin, and 2-deoxy-d-glucose, is associated with upregulation of DR5 expression (24–26). The intrinsic mitochondrial pathway plays a crucial role in amplifying TRAIL-induced apoptosis, and collapse of the mitochondrial membrane potential (ΔΨm) is considered to be a hallmark of this pathway, although it remains a matter of debate whether this event is a cause or a result of permeabilization of the OMM (27, 28). Permeabilization of the OMM by pro-apoptotic Bcl-2 family proteins promotes the release of a number of apoptogenic factors, such as cytochrome c, endonuclease G, second mitochondrial activator of caspases (SMAC), Omi/HtrA2, and apoptosis-inducing factor (AIF), from the inner mitochondrial membrane (IMM) space into the cytosol, and these apoptogenic proteins promote activation of the caspase cascade, thereby leading to apoptosis. Cytochrome c interacts with the apoptotic peptidase activating factor 1 (Apaf-1), leading to the formation of the multimeric apoptosome in the presence of ATP/dATP (29). The apoptosome acts as a platform for activation of the initiator caspase, caspase-9, which subsequently cleaves and activates the effector caspases, caspases-3 and -7. A cytochrome c-independent apoptosis pathway has also been defined, and this pathway requires proteins such as endonuclease G and AIF to carry out apoptosis. Although the molecular mechanisms underlying the OMM permeabilization are poorly understood, it is widely accepted that the mitochondrial permeability transition (MPT), which was originally defined as a sudden increase in IMM permeability to solutes with molecular masses of ~1,500 Da, is involved. It is now believed that opening of putative megachannels referred to as the mitochondrial permeability transition pores (mPTPs) plays pivotal roles (30, 31). mPTPs are high-conductance non-specific pores in the IMM composed of proteins that link the IMM and OMM. Several mitochondrial proteins localized in the IMM and OMM, such as voltage-dependent anion channels (VDACs), adenine nucleotide translocase (ANT), hexokinase, peripheral benzodiazepine receptors, and cyclophilin-D are thought to constitute the mPTP. Under physiological conditions, the proteins in the OMM and IMM that constitute mPTPs are thought to be in close proximity to one another and in a closed or low-conductance conformation, although mPTPs have not been isolated and the components of these complexes remain controversial (32–34). When an mPTP changes to an open conformation, water and solutes with molecular masses of up to 1,500 Da enter the mitochondrial matrix, resulting in osmotic swelling of the mitochondrion. It is thought that when multiple mPTPs open concurrently and extensive mitochondrial swelling takes place, physical disorganization of the OMM occurs and mitochondrial apoptogenic proteins are released, thereby triggering apoptosis (27). Hence, much attention has been paid to the potential role of mPTPs as a target for anti-cancer and chemopreventive agents (27, 28). TRAIL induces both ΔΨm collapse and caspase-3/7 activation in melanoma and leukemia cells and the sensitization of TRAIL-induced apoptosis by membrane-depolarizing agents is associated with their enhancement, indicating the involvement of the intrinsic pathway. Although glibenclamide and U37883A are potent KATP channel inhibitors, glibenclamide targets sulfonylurea receptors (SURs) while U37883A is non-SUR drug. It is noted that glibenclamide and U37883A have different effects on TRAIL-induced cell death depending on TRAIL sensitivity. In TRAIL-resistant A375 melanoma cells, U37883A can potentiate TRAIL-induced cell death as rapidly as within 24 h, while glibenclamide is ineffective, despite considerable potentiation of TRAIL-induced ΔΨm collapse and caspase-3/7 activation. This cell death is mainly caused by apoptosis. However, after 72 h of treatment, glibenclamide alone can induce considerable apoptotic and necrotic cell death and potentiates TRAIL effectiveness. However, both U37883A and glibenclamide can enhance apoptosis in TRAIL-sensitive Jurkat leukemia cells during the initial 24 h (20). Thus, these observations suggest that: (i) TRAIL and the two KATP channel inhibitors can induce different modes of cell death depending on the basal cellular sensitivity to TRAIL and experimental conditions such as exposure time, which is supported by the fact that substantial necrotic cell death is associated with the late cell death, but not the early cell death; (ii) SURs may play differential roles in the two modes of cell death in TRAIL-resistant cells; and (iii) activation of mitochondrial death pathway is insufficient for complete overriding of TRAIL resistance in malignant cells. This view coincides with the findings in clinical trials that the combined use of PARAs and other conventional chemotherapeutic drugs exhibit only modest effects toward TRAIL-resistant cancers, although they can induce substantial activation of the intrinsic death pathway (11). Consequently, the potentiation of TRAIL-induced apoptosis in TRAIL-resistant cancer cells by membrane-depolarizing agents may involve another cell death pathway.
Role of the Endoplasmic Reticulum Death Pathway in the Effects of Depolarization
The emerging concept is that, besides mitochondria, the endoplasmic reticulum (ER) is another key player in the regulation of apoptosis induced by a variety of death stimuli, including TRAIL. Disparate perturbations in their normal ER functions, such as accumulation of unfolded or misfolded proteins, ER lipid imbalances or changes in the redox balance, or Ca2+ conditions in the ER lumen, trigger ER stress (ERS) (35–38). The cells then activate ERS responses, called the unfolded protein response (UPR), to alleviate the stress, but an excessive and prolonged UPR leads to apoptosis (36–38). The UPR involves transcription-dependent upregulation of ER-resident chaperones, and the ER chaperone glucose-related protein 78 (GRP78; also known as Bip) is thought to be a primary sensor in this response. Upon ERS, GRP78 dissociates from ER transmembrane proteins, such as inositol requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6), to bind to unfolded or misfolded proteins, resulting in aggregation of the transmembrane proteins and their activation. Activated IRE 1 splices the mRNA for X-box-binding protein-1 (XBP-1) to allow translation of the mature spliced form of XBP-1 protein, which acts as a transcription factor and mediates the transcriptional upregulation of numerous genes involved in ER function as well as TRAIL-R2 (25). In support of the role of ERS in TRAIL-induced apoptosis, TRAIL induces the splicing of XBP-1, and membrane-depolarizing agents including high K+, U37883A (19, 21), DATS (22), H2O2 (23), and mitochondrial inhibitors such as antimycin A and FCCP (20) all commonly potentiate this effect, although none of these agents except H2O2 alone can induce the splicing. Only glibenclamide cannot potentiate TRAIL-induced XBP-1 activation, although it upregulates GRP78 expression in accordance with its minimal effect on TRAIL-induced cell death during the initial 24 h (19). These observations are interesting because GRP78 acts as an anti-apoptotic factor in the cells, since downregulation of GRP78 expression by small interfering RNA (siRNA) administration potentiates the apoptosis induced by diverse drugs including cisplatin, adriamycin (25), fenretinide [N-(4-hydroxyphenyl) retinamide], a synthetic derivative of retinoic acid, and bortezomib, a 26S proteasome inhibitor (39). Taken together, it is strongly suggested that pro-apoptotic ERS responses including GRP78 downregulation, XBP-1 upregulation and processing are activated by depolarization and play an important role in TRAIL-sensitization and tumor-selective killing.
Dual Role of ROS in Apoptosis
Reactive oxygen species, such as superoxide anions (), H2O2, and hydroxyl radicals (•OH), are the products of normal metabolism in virtually all aerobic organisms. Low physiological levels of ROS function as second messengers in intracellular signaling and are required for normal cell function, while excessive ROS cause damage to multiple macromolecules, impair cell function, and promote apoptotic or necrotic cell death including those induced by DR ligands (40, 41). ROS levels are controlled by the antioxidant defense system, including the antioxidant enzymes manganese- or copper–zinc-containing superoxide dismutase (SOD), which catalyzes the dismutation of into H2O2, and catalase and glutathione peroxidase, which degrade H2O2. Thus, the balance between the machinery for ROS generation (prooxidant system) and the machinery for ROS scavenging (antioxidant system) is a critical determinant of whether the cell fate is proliferation or death. Indeed, generation of ROS is also associated with the apoptosis induced by DR ligands such as Fas (42–45) and TNF-α (46, 47). TRAIL has been shown to induce the generation of intracellular ROS, including H2O2, which may be critical in regulating the responses of cancer cells to TRAIL (48). A variety of natural and synthetic compounds capable of increasing the intracellular H2O2 level, including LY35001 (49), wogonin (50), and diallyl polysulfides (51, 52) potentiate TRAIL cytotoxicity toward different human malignant cells (Table 3). Moreover, direct application of H2O2 alone induces apoptosis or potentiates death ligand-induced apoptosis in different cell types (23, 53). In addition, various antioxidants such as N-acetyl-l-cysteine (NAC) and manganese SOD, and catalase block TNF-α- and Fas-induced apoptosis (54, 55). These observations suggest that ROS are important mediators of death ligand-induced apoptosis. Both NADPH oxidase (42, 45, 47) and mitochondria (43, 46) have been implicated as the cellular sources of ROS generated by TNF-α- or Fas-mediated signaling in various cell types. Conversely, ROS or prooxidative conditions are protective under certain circumstances (56). Thus, ROS seem to exhibit both pro-apoptotic and anti-apoptotic functions. Hence, the role of H2O2 in DR-mediated cell death remains controversial. To date there is no general model that can explain the dual role of ROS in apoptosis. However, accumulating evidence suggests that ROS control another form of cell death, autophagy (57, 58), which protects cancer cells against apoptosis. This emerging concept is interesting because it may explain that ROS exert their anti-apoptotic function through the control of cancer cell autophagy. However, this issue is not the focus of the present review, as it has been comprehensively reviewed by others (59–61).
Role of Mitochondrial in Cancer Cell Apoptosis
Another possible explanation for the dual role of ROS in TRAIL-induced apoptosis is that different oxidant species mediate reciprocal effects. Indeed, the generation of various ROS and reactive nitrogen species including H2O2, , nitric oxide (NO), and peroxynitrite (ONOO−) is associated with selective killing (Tables 1 and 2) and TRAIL-sensitization (Table 3) in cancer cells. To define the oxidant species mediating TRAIL-induced apoptosis in cancer cells, we analyzed ROS generation using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and dihydroethidine (DHE) after TRAIL treatment. DCFH-DA is rapidly taken up by cells and hydrolyzed into DCFH by cellular esterase activity. DCFH is then converted into the fluorescent compound DCF by oxidation mediated by H2O2, ONOO−, and •OH, but not . Conversely, DHE undergoes two-electron oxidation to form DNA-binding ethidium bromide in a reaction that is relatively specific for . Consequently, DCFH-DA and DHE have been widely used to detect intracellular H2O2 and , respectively in various cell types (62–64). TRAIL treatment results in the generation of intracellular H2O2 and in Jurkat leukemia and A375 melanoma cells (20). The time courses of the generation of these two oxidants are different in that the H2O2 generation is rapid (detected within 30 min) and transient (declining to the basal level within 1 h), while the generation is initially detected at 2 h and persistent (increasing for another 2 h), and the latter, but not the former, is correlated with apoptosis. In support of the role of , Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBaP), a SOD mimetic that can scavenge and ONOO−, but not NO, abolishes TRAIL-induced apoptosis and DHE responses (20). Moreover, analyses using MitoSOX, which is localized to mitochondria and thereby serve as a selective probe for in these organelles (65), revealed that TRAIL dose-dependently induces generation within the mitochondria, which is blocked by MnTBaP. ONOO− can also oxidize DCFH, and MnTBaP was reported to selectively scavenge ONOO− over (66). However, ONOO− seems to play a minor role in TRAIL-induced apoptosis, because: (i) TRAIL induces no substantial NO production for up to at least 4 h; and (ii) synthesized ONOO− causes minimal cell death, although it can dose-dependently cause DCFH oxidation. The observations that MnTBaP blocks TRAIL-induced ΔΨm collapse and caspase-3/7 activation indicate that is the main participator in the mitochondrial dysfunction. In addition, MnTBaP reduces TRAIL-induced XBP-1 activation (20). These observations suggest that mitochondrial ROS (mROS) mediate both mitochondrial and ER dysfunctions during TRAIL-induced apoptosis.
Studies on the effects of exogenously applied H2O2 have provided another line of evidence for the role of mROS in mediating apoptosis. Direct application of H2O2 causes apoptosis in TRAIL-resistant melanoma cells and sensitizes these cells to TRAIL (23). The potentiation of apoptosis mainly occurs through the mitochondrial and ERS pathways, as shown by ΔΨm collapse, caspase-3/7 activation, GRP-78 downregulation, and XBP-1 activation. Strikingly, after TRAIL treatment, the intracellular H2O2 level increases rapidly (within 10–30 min) but transiently (declining to the basal level at 1 h), while the intracellular level increases over time for at least 4 h. MnTBaP, but not catalase, can block the increase and apoptosis in parallel, indicating that mainly mediates the apoptosis. In addition, a robust increase in MitoSOX signals is observed in parallel with the increase and MnTBaP blocks this oxidative response, suggesting an increase within the mitochondria. Moreover, H2O2 induces ΔΨm collapse, caspase-3/7 activation, and XBP-1 activation, all of which are blocked by MnTBaP treatment. These observations suggest that exogenously applied H2O2 can stimulate generation within mitochondria and that the mROS mediate the mitochondrial and ER dysfunctions. In addition, H2O2 causes minimal intracellular and mROS generation in melanocytes in parallel with its minimal cytotoxic effect (23). Taken together, these findings strongly suggest a pro-apoptotic role of mROS in cancer cell apoptosis.
In normal resting cells, 0.1–2% of electrons carried by the mitochondrial electron transport chain (ETC) leak from this pathway and form . Impaired mitochondrial metabolism causes robust leakage of free electrons, thereby resulting in ROS generation within the organelles. Consequently, mitochondrial metabolic inhibitors serve as powerful tools for studying the role of mROS in a given biological response. Studies using Jurkat cells and A375 melanoma cells as a model have revealed that in both cell types, rotenone, an inhibitor of complex I, is the most potent at inducing apoptosis, while FCCP, a classic uncoupler of oxidative phosphorylation (OXPHOS) is less effective and complex III inhibitor antimycin A is ineffective. However, all of these compounds markedly potentiate TRAIL-induced apoptosis and mROS generation (20, 21). The three metabolic inhibitors can also potentiate TRAIL-induced activation of caspase-3/7 and XBP-1, and both effects are blocked by MnTBaP, indicating that mROS generated by the ETC mediate both mitochondrial and ER dysfunctions during TRAIL-induced apoptosis (20). These findings are similar to a previous report that CCCP, another OXPHOS uncoupler, enhances TRAIL-induced apoptosis in TRAIL-resistant human colon carcinoma cell lines (RKO, HT29, and HCT8), while CCCP alone has little effect on apoptosis or release of pro-apoptotic factors from mitochondria (67). CCCP also enhances TRAIL-induced caspase-8, Bid, caspase-9 activation, and Bax conformational change and translocation to mitochondria. CCCP treatment results in the generation of ROS as determined by DHE, and the synergistic effect of CCCP on the TRAIL-induced apoptotic pathway is abrogated by the non-specific ROS scavenger NAC. It is noteworthy that when mitochondria are uncoupled, shRNA-mediated caspase-9 depletion cannot protect the cells against cell death, suggesting modulation of the mode of the caspase cascade. It has been shown that alterations in mitochondrial function, such as OXPHOS, affect the response of tumor cells to apoptosis induced by death ligands including generation (68, 69), while death ligands affect mitochondrial metabolism and function. Thus, mitochondrial function and death ligand-induced apoptosis are intimately associated with one another. In this functional relationship, loss of ΔΨm, i.e., mitochondrial membrane depolarization (MMD), may play a central role, because it is not only a major cause of mROS generation but also provokes membrane integrity disruption and caspase cascade activation. There is accumulating evidence to suggest that mROS play a key role in MPT induction by affecting the mPTP conformation. First, ROS are byproducts of OXPHOS and excessive ROS generation is potentially deleterious to mitochondrial and cellular functions. Second, ANT has three cysteine residues whose oxidation is critical for mPTP open–closed transitions and Ca2+ release from the mitochondrial matrix, and mPTPs are believed to be particularly vulnerable to ROS (30–32). Consequently, the MPT can be triggered by excessive mROS generation and/or disruption of the mitochondrial redox homeostasis (70–73). Third, within mitochondria, cytochrome c is bound to the outer surface of the IMM by its association with the mitochondrial phospholipid cardiolipin, and oxidation of cardiolipin is thought to decrease this contact (74). Thus, oxidation of cardiolipin may also be required to liberate sufficient cytochrome c to trigger caspase activation and induce apoptosis. Consequently, oxidation of cardiolipin may serve as a biochemical hallmark of mitochondrial oxidative stress and apoptosis. As the fluorescent dye 10-N-acridine orange (NAO) binds to the non-oxidized, but not oxidized, form of cardiolipin independently of ΔΨm, measurements of NAO fluorescence enable monitoring of the oxidation of cardiolipin in mitochondria (75). It has been shown that in parallel with the increase in MitoSOX signals, TRAIL treatment results in oxidation of cardiolipin in human leukemia and melanoma cells (20, 21). Agonistic antibodies against DR4 and DR5, which trigger the formation of multimeric complexes containing only specific TRAIL-Rs (76, 77) also induce robust cardiolipin oxidation in a dose-dependent manner, indicating that this oxidation is mediated by DR4/DR5 (21). The MPT also results in dissipation of ΔΨm and enhances mROS production via disintegration of the ETC, thereby progressively shutting down OXPHOS and impairing energetic metabolism (78). Hence, the MPT is a rate-limiting and self-amplifying process for apoptosis in which ROS and MMD play key roles. Indeed, MMD is the most common event observed concomitantly with ROS generation in cell death induced by diverse death-inducing stimuli (Tables 1 and 2).
Crosstalk between Membrane Depolarization and ROS
Several lines of evidence suggest that membrane depolarization and ROS are intimately associated with one another. First, membrane depolarization controls the generation of ROS via NADPH oxidase activation. In endothelial cells (ECs), it has been shown that loss of fluid shear stress or ischemia results in the generation of ROS associated with the activation of EC NADPH oxidase (79, 80). It has been suggested that this activation of ROS generation is caused by the activation and assembly of NOX2 through phosphoinositide-3-kinase (PI3K)/serine threonine kinase Akt (81). It is noted that the activation of ROS generation is triggered by EC membrane depolarization caused by inactivation of KATP channels. It is widely accepted that cancer cells express various NOX family members (NOX 1–5, Duox 1, 2) and that these NADPH oxidases play important roles in cancer cell proliferation, death, function, and tumorigenesis (82). For instance, it has been shown that binding of SDF-1α to the chemokine receptor, cysteine (C)-X-C receptor-4, which contributes to the enhanced metastatic functions in prostate cancer cells, promotes ROS generation via NOX2 activation through the PI3K/Akt signaling pathway (83). VEGF that plays a critical role in vascular pathophysiology and induces ROS generation in B1647 cells, a human leukemia cell line, through activation of NOX2 and NOX4, and the NOX-generated ROS are required to sustain cell viability and proliferation, and prevent apoptosis (84). Second, ROS were shown to regulate the sustained membrane depolarization during tumor cell apoptosis. As mentioned above, inactivation of Na+–K+-ATPase is responsible for the sustained membrane depolarization during Fas-induced apoptosis (14, 15). This Na+–K+-ATPase inactivation was shown to be associated with cleavage of the 42-kDa β subunit and decreased levels of the 110-kDa α subunit (15). Fas ligation also triggers the internalization of plasma membrane Na+–K+-ATPase in tumor and normal cells, which is induced by intracellular glutathione depletion and H2O2 generation (85). A molecular mechanism for the H2O2-induced internalization of Na+–K+-ATPase is considered to be serine phosphorylation of the α1 subunit and regulation by mitochondrial H2O2 and the anti-apoptotic protein Bcl-2 (15, 85). Collectively, these observations suggest that ROS can control depolarization in malignant cells through the modification, degradation, and inactivation of Na+–K+-ATPase. Recently, another crosstalk between depolarization and mROS in cancer cells was suggested. Membrane-depolarizing agents by themselves increase mROS and potentiate TRAIL-induced mROS generation, indicating that depolarization controls mROS (21). It is notable that depolarization increases the surface expression of DR5, the triggering of which increases mROS. Since depolarization potentiates TRAIL-induced activation of the transcription factor XBP-1, which is engaged in the regulation of surface DR5 expression (26), it is possible to speculate that the upregulation of surface DR5 expression results in increased mROS accumulation. However, scavenging of mROS by the antioxidant MnTBaP reduces depolarization, while mROS accumulation caused by mitochondrial metabolic dysfunction potentiates the depolarization (21). Collectively, these observations suggest that depolarization and mROS mutually regulate one another. Figure 1 shows the current model for the potentiation of TRAIL-induced apoptosis in cancer cells by membrane depolarization in connection with ROS.
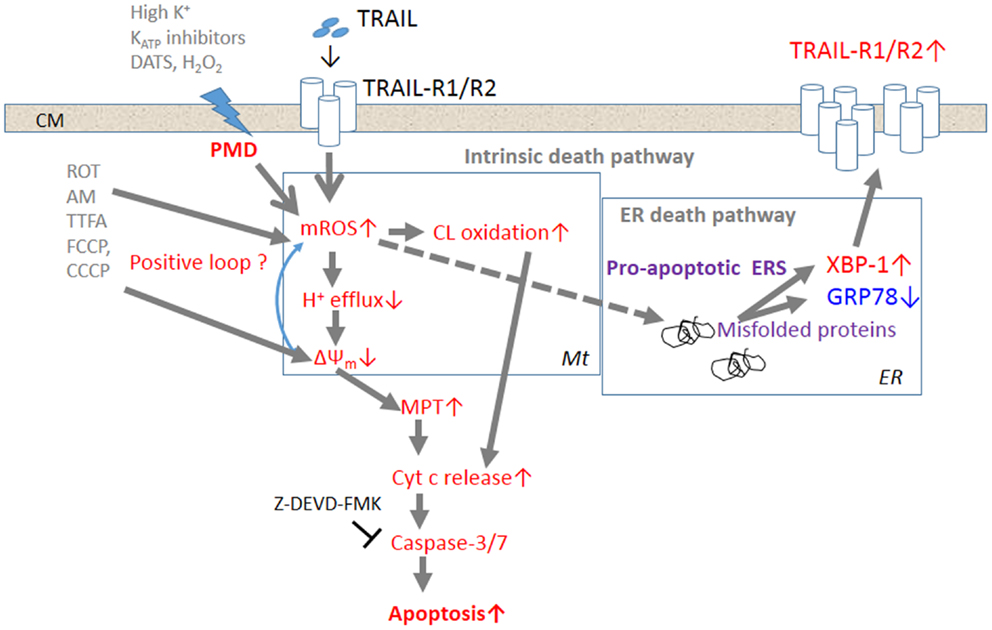
Figure 1. The current model for the potentiation of TRAIL-induced apoptosis in cancer cells by depolarization. Triggering of TRAIL-R1 (DR4)/TRAIL-R2 (DR5) induces the generation and accumulation of mROS, leading to impairment of the ETC function and cardiolipin oxidation. Impairment of the ETC complex I/III function decreases H+ efflux, thereby causing ΔΨm dissipation, i.e., MMD, and additional mROS generation and cardiolipin oxidation, thereby forming a positive loop. Cardiolipin oxidation and ΔΨm dissipation cooperatively promote the MPT and liberation of sufficient cytochrome c to trigger caspase activation and induce apoptosis. TRAIL-resistant cancer cells appear to gain considerable tolerance for oxidative stress-mediated activation of the intrinsic death pathway. Accumulation of mROS can also promote the formation of unfolded or misfolded proteins, thereby provoking ERS responses such as activation of the transcriptional factor XBP-1. XBP-1 activation leads to upregulation of the surface TRAIL-R2 expression level, thereby enhancing the death signaling. Activation of this alternative death pathway may contribute to commit TRAIL-resistant cancer cells to apoptosis.
Several TRAIL-resistant cancer cell types, such as malignant melanoma cells, appear to gain considerable tolerance for oxidative stress-mediated activation of the intrinsic death pathway. However, accumulation of mROS can also promote the formation of unfolded or misfolded proteins, thereby provoking ERS responses such as activation of the transcriptional factor XBP-1. XBP-1 activation leads to upregulation of the surface TRAIL-R2 expression level, thereby enhancing the death signaling. When used together with TRAIL, membrane-depolarizing agents can potentiate mROS accumulation sufficiently to activate not only the intrinsic death pathway, but also the ERS-mediated death pathway, thereby committing drug-resistant cancer cells to apoptosis. Although the precise biochemical and biological consequences between membrane depolarization and mROS generation remain to be elucidated, these two events occur in parallel, indicating that they are intimately associated. Since TRAIL signaling alone can induce sustained depolarization, it is possible that the ROS increase causes this response by inactivating plasma membrane Na+–K+-ATPase and/or KATP channels.
Abnormal Increases in ROS in Cancer Cells – A Target for Selective Killing
The steady-state levels of endogenous ROS including and H2O2 are elevated in cancer cells (86, 87). The increase in ROS is thought to contribute to maintenance of the cancer cell phenotype through their effects on cell growth, proliferation, and genetic instability (88, 89). Increased ROS generation is common in cancer cells with active metabolism and genetic instability under the impact of various oncogenes such as Bcr/Abl, Ras, c-Myc, and FLT3, FMS-like tyrosine kinase (90–95). The imbalance between the prooxidant system and the antioxidant system is a consequence of increased ROS generation and/or decreased antioxidants in cancer cells. Such abnormal increases in ROS makes cancer cells more vulnerable than normal cells to cell damage induced by exogenous ROS-inducing agents and can be exploited to selectively kill cancer cells (96–98) (Figure 2). In this respect, it is noted that diverse natural products and synthetic chemicals that exhibit selective killing effects are capable of inducing intracellular ROS generation (Tables 1 and 2). For instance, β-phenylethyl isothiocyanate (PEITC) is a natural product found in cruciferous vegetables with chemopreventive activity, and has been shown to increase ROS generation and induce apoptosis (99, 100). Moreover, recent work has shown the selective killing of oncogenically transformed cells by PEITC (101–103), supporting the emerging view that an intrinsic excess oxidative stress under the control of oncogenic transformation can be exploited as a cancer-selective target.
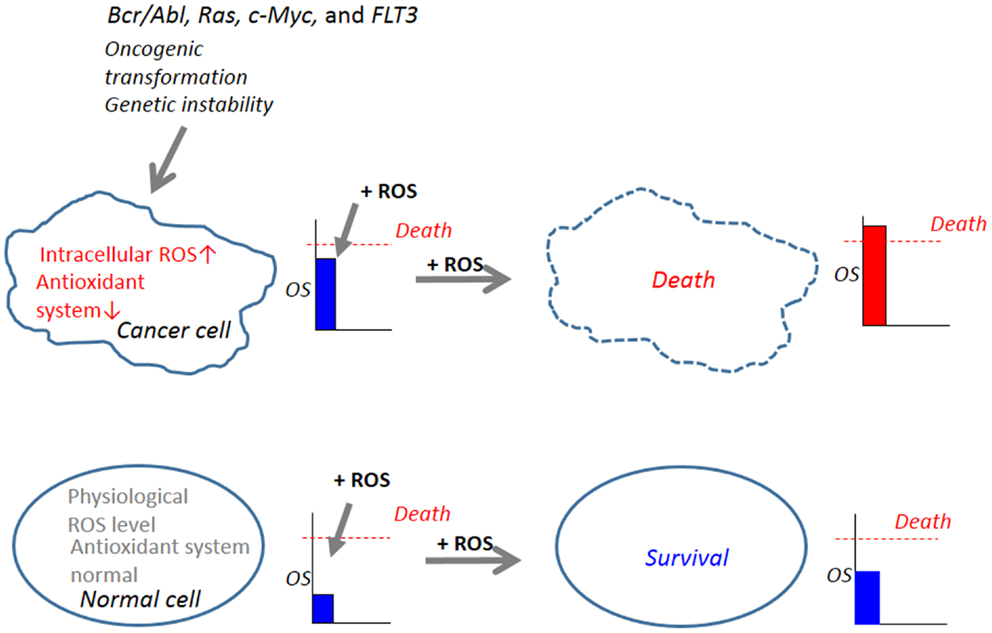
Figure 2. Abnormal increases in ROS in cancer cells serves as a target for tumor-selective killing. Cellular oxidative stress (OS) level is regulated by the balance between the machinery for ROS generation (prooxidant system) and the machinery for ROS scavenging (antioxidant system). In normal cells, the antioxidant system is normal and low physiological levels of ROS, which can function as second messengers in intracellular signaling and are required for normal cell function, are generated. Owing to their active metabolism and genetic instability under the control of oncogenic transformation such as Bcr/Abl, Ras, c-Myc, and FLT3, which causes increased ROS generation and decreased antioxidant systems, cancer cells harbor an excess OS over normal cells. When equivalent levels of OS are added by the administration of exogenous ROS-inducing agents (+ROS), OS levels in cancer cells can readily over the threshold of cell death, while OS levels in normal cells do not. Hence, cancer cells are expected to be more vulnerable than normal cells to cell damage induced by ROS-generating agents and this vulnerability can be exploited to selectively kill these cells.
Conclusion
Recent work has revealed that membrane depolarization is an early and prerequisite event during death ligand-induced apoptosis in malignant cells. Moreover, it has been shown that persistent membrane depolarization can facilitate tumor-selective killing and TRAIL-sensitizing effects by promoting mitochondrial and ER dysfunctions. It is noted that membrane depolarization and ROS regulate one another although the precise mechanisms underlying the mutual regulation remain to be elucidated. The emerging concept is that owing to their active metabolism and genetic instability under the control of oncogenic transformation, cancer cells harbor an excess oxidative stress over normal cells and that this intrinsic oxidative stress can be exploited to selectively kill malignant cells. Thus, the crosstalk is one possible rationale why diverse membrane-depolarizing agents can exhibit tumor-selective killing and TRAIL-sensitizing effects in cancer cells. Since depolarization is intimately linked to not only mitochondrial integrity disruption but also ER dysfunction, and impacts cancer cells selectively, further studies on its role in cancer cell death may afford a novel approach for tumor-selective killing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to their collaborators. This work was supported in part by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology (KAKENHI 23591631; to Yoshihiro Suzuki-Karasaki) and a Grant-in-Aid from Nihon University (to Yoshihiro Suzuki-Karasaki and Mayumi Uchida).
Abbreviations
AIF, apoptosis-inducing factor; ANT, adenine nucleotide translocase; Apaf-1, apoptotic peptidase activating factor 1; ATF6, activating transcription factor 6; DADS, diallyl disulfide; DAS, diallyl sulfide; DATS, diallyl trisulfide DCFH-DA, 2′,7′-dichlorofluorescein diacetate; DcR, decoy receptor; DHE, dihydroethidine; DR, death receptor; ΔΨm, mitochondrial membrane potential; EC, endothelial cell; eIF2, eukaryotic initiation factor-2; ER, endoplasmic reticulum; ERK, extracellular signal-regulated protein kinase; ERS, endoplasmic reticulum stress; ETC, electron transport chain; FasL, Fas ligand; GRP78, glucose-related protein 78; IMM, inner mitochondrial membrane; IP3, inositol-1,4,5-triphosphate; IRE1, inositol requiring enzyme 1; JNK, Jun-N-terminal kinase; KATP, ATP-sensitive K+; KCa, calcium-dependent K+; Kv, voltage-dependent K+; mAbs, monoclonal antibodies; MHP, mitochondrial hyperpolarization, MMD, mitochondrial membrane depolarization; MnTBaP, Mn(III)tetrakis(4-benzoic acid)porphyrin chloride; MPT, mitochondrial permeability transition; mPTP, mitochondrial permeability transition pore; mROS, mitochondrial reactive oxygen species; NAC, N-acetyl-l-cysteine; NAO, 10-N-acrydine orange; NSCLC, non-small cell lung cancer; OMM, outer mitochondrial membrane; OXPHOS, oxidative phosphorylation; PARAs, pro-apoptotic receptor agonists; PEITC, β-phenyethyl isothiocyanate; PI3K, phosphoinositide-3-kinase; PMD, plasma membrane depolarization; rh, recombinant human; ROS, reactive oxygen species; siRNA, small interfering RNA; SMAC, second mitochondrial activator of caspases; SOD, superoxide dismutase; SUR, sulfonylurea receptor; TEA, tetraethylammonium; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; TRAIL-R, TNF-related apoptosis-inducing ligand receptor; TRPV1, transient receptor potential vanilloid 1; UPR, unfolded protein response; VDAC, voltage-dependent anion channel; VEGF, vascular endothelial growth factor; XBP-1, X-box-binding protein-1; XIAP, X-linked inhibitor of apoptosis protein.
References
1. LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy. Cell Death Differ (2003) 10:66–75. doi: 10.1038/sj.cdd.4401187
2. Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer (2008) 8:782–98. doi:10.1038/nrc2465
3. Wang S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene (2008) 27:6207–15. doi:10.1038/onc.2008.298
4. Sayers TJ. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother (2011) 60:1173–80. doi:10.1007/s00262-011-1008-4
5. Brost S, Zimmermann A, Koschny R, Sykora J, Stremmel W, Schirmacher P, et al. Hepatocyte expression of TRAIL pathway regulators correlates with histopathological and clinical parameters in chronic HCV infection. Pathol Res Pract (2014) 210:83–91. doi:10.1016/j.prp.2013.10.005
6. Badmann A, Langsch S, Keogh A, Brunner T, Kaufmann T, Corazza N. TRAIL enhances paracetamol-induced liver sinusoidal endothelial cell death in a Bim- and Bid-dependent manner. Cell Death Dis (2012) 20(3):e447. doi:10.1038/cddis.2012.185
7. Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem (2011) 286:39336–48. doi:10.1074/jbc.M111.280420
8. Corazza N, Jakob S, Schaer C, Frese S, Keogh A, Stroka D, et al. TRAIL receptor-mediated JNK activation and Bim phosphorylation critically regulate Fas-mediated liver damage and lethality. J Clin Invest (2006) 116:2493–9. doi:10.1172/JCI27726
9. Förster A, Preussner LM, Seeger JM, Rabenhorst A, Kashkar H, Mrowietz U, et al. Dimethylfumarate induces apoptosis in human mast cells. Exp Dermatol (2013) 22:719–24. doi:10.1111/exd.12247
10. Dyer MJ, MacFarlane M, Cohen GM. Barriers to effective TRAIL-targeted therapy of malignancy. J Clin Oncol (2007) 25(28):4505–6.
11. Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene (2013) 32:1341–50. doi:10.1038/onc.2012.164
12. McCarthy JV, Cotter TG. Cell shrinkage and apoptosis: a role for potassium and sodium ion efflux. Cell Death Differ (1997) 4:756–70. doi:10.1038/sj.cdd.4400296
13. Lang F, Föller M, Lang K, Lang P, Ritter M, Vereninov A, et al. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol (2007) 428:209–25. doi:10.1016/S0076-6879(07)28011-5
14. Bortner CD, Gomez-Angelats M, Cidlowski JA. Plasma membrane depolarization without repolarization is an early molecular event in anti-Fas-induced apoptosis. J Biol Chem (2001) 276:4304–14. doi:10.1074/jbc.M005171200
15. Yin W, Li X, Feng S, Cheng W, Tang B, Shi YL, et al. Plasma membrane depolarization and Na,K-ATPase impairment induced by mitochondrial toxins augment leukemia cell apoptosis via a novel mitochondrial amplification mechanism. Biochem Pharmacol (2009) 78:191–202. doi:10.1016/j.bcp.2009.03.025
16. Nolte F, Friedrich O, Rojewski M, Fink RH, Schrezenmeier H, Körper S. Depolarisation of the plasma membrane in the arsenic trioxide (As2O3)-and anti-CD95-induced apoptosis in myeloid cells. FEBS Lett (2004) 578:85–9. doi:10.1016/j.febslet.2004.10.075
17. Ghoumari AM, Piochon C, Tomkiewicz C, Eychenne B, Levenes C, Dusart I, et al. Neuroprotective effect of mifepristone involves neuron depolarization. FASEB J (2006) 20:1377–86. doi:10.1096/fj.05-5832com
18. Frese S, Frese-Schaper M, Andres AC, Miescher D, Zumkehr B, Schmid RA. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res (2006) 66:5867–74. doi:10.1158/0008-5472.CAN-05-3544
19. Suzuki Y, Inoue T, Murai M, Suzuki-Karasaki M, Ochiai T, Ra C. Depolarization potentiates TRAIL-induced apoptosis in human melanoma cells: role for ATP-sensitive K+ channels and endoplasmic reticulum stress. Int J Oncol (2012) 41:465–75. doi:10.3892/ijo.2012.1483
20. Inoue T, Suzuki-Karasaki Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic Biol Med (2013) 61:273–84. doi:10.1016/j.freeradbiomed.2013.04.020
21. Suzuki-Karasaki M, Ochiai T, Suzuki-Karasaki Y. Crosstalk between mitochondrial ROS and depolarization in the potentiation of TRAIL-induced apoptosis in human tumor cells. Int J Oncol (2014) 44:616–28. doi:10.3892/ijo.2013.2215
22. Murai M, Inoue T, Suzuki-Karasaki M, Ochiai T, Ra C, Nishida S, et al. Diallyl trisulfide sensitizes human melanoma cells to TRAIL-induced cell death by promoting endoplasmic reticulum-mediated apoptosis. Int J Oncol (2012) 41:2029–37. doi:10.3892/ijo.2012.1656
23. Tochigi M, Inoue T, Suzuki-Karasaki M, Ochiai T, Ra C, Suzuki-Karasaki Y. Hydrogen peroxide induces cell death in human TRAIL-resistant melanoma through intracellular superoxide generation. Int J Oncol (2013) 42:863–72. doi:10.3892/ijo.2013.1769
24. Chen LH, Jiang CC, Kiejda KA, Wang YF, Thorne RF, Zhang XD, et al. Thapsigargin sensitizes human melanoma cells to TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the unfolded protein response. Carcinogenesis (2007) 28:2328–36. doi:10.1093/carcin/bgm173
25. Jiang CC, Chen LH, Gillespie S, Kiejda KA, Mhaidat N, Wang YF, et al. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res (2007) 67:5880–8. doi:10.1158/0008-5472.CAN-07-0213
26. Liu H, Jiang CC, Lavis CJ, Croft A, Dong L, Tseng HY, et al. 2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human melanoma cells through XBP-1-mediated up-regulation of TRAIL-R2. Mol Cancer (2009) 8:122. doi:10.1186/1476-4598-8-122
27. Hail N Jr. Mitochondria: a novel target for the chemoprevention of cancer. Apoptosis (2005) 10:687–705. doi:10.1007/s10495-005-0792-8
28. Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene (2006) 25:4744–56. doi:10.1038/sj.onc.1209609
29. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell (2004) 116:205–19. doi:10.1016/S0092-8674(04)00046-7
30. Halestrap AP, Brennerb C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem (2003) 10:1507–25. doi:10.2174/0929867033457278
31. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta (2009) 1787:1395–401. doi:10.1016/j.bbabio.2009.06.009
32. Zhivotovsky B, Galluzzi L, Kepp O, Kroemer G. Adenine nucleotide translocase: a component of the phylogenetically conserved cell death machinery. Cell Death Differ (2009) 16:1419–25. doi:10.1038/cdd.2009.118
33. Dalla Via L, García-Argáez AN, Martínez-Vázquez M, Grancara S, Martinis P, Toninello A. Mitochondrial permeability transition as target of anticancer drugs. Curr Pharm Des (2014) 20:223–44.
35. Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ (2006) 13:363–73. doi:10.1038/sj.cdd.4401817
36. Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci (2003) 1010:186–94. doi:10.1196/annals.1299.032
37. Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol (2004) 14:20–8. doi:10.1016/j.tcb.2003.11.001
38. Groenendyk J, Michalak M. Endoplasmic reticulum quality control and apoptosis. Acta Biochim Pol (2005) 52:381–95.
39. Martin S, Hill DS, Paton JC, Paton AW, Birch-Machin MA, Lovat PE, et al. Targeting GRP78 to enhance melanoma cell death. Pigment Cell Melanoma Res (2010) 23:675–82. doi:10.1111/j.1755-148X.2010.00731.x
40. Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J (2006) 20:1589–98. doi:10.1096/fj.05-5603rev
41. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med (2010) 48:749–62. doi:10.1016/j.freeradbiomed.2009.12.022
42. Suzuki Y, Ono Y, Hirabayashi Y. Rapid and specific reactive oxygen species generation via NADPH oxidase activation during Fas-mediated apoptosis. FEBS Lett (1998) 425:209–12. doi:10.1016/S0014-5793(98)00228-2
43. Miñana JB, Gómez-Cambronero L, Lloret A, Pallardó FV, Del Olmo J, Escudero A, et al. Mitochondrial oxidative stress and CD95 ligand: a dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology (2002) 35:1205–14. doi:10.1053/jhep.2002.32969
44. Sato T, Machida T, Takahashi S, Iyama S, Sato Y, Kuribayashi K, et al. Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J Immunol (2004) 173:285–96. doi:10.4049/jimmunol.173.1.285
45. Reinehr R, Becker S, Eberle A, Grether-Beck S, Häussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem (2005) 280:27179–94. doi:10.1074/jbc.M414361200
46. Goossens V, Stangé G, Moens K, Pipeleers D, Grooten J. Regulation of tumor necrosis factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron flux through the electron transport chain complex I. Antioxid Redox Signal (1999) 1:285–95. doi:10.1089/ars.1999.1.3-285
47. Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell (2007) 26:675–87. doi:10.1016/j.molcel.2007.04.021
48. Mellier G, Pervaiz S. The three Rs along the TRAIL: resistance, re-sensitization and reactive oxygen species (ROS). Free Radic Res (2012) 46:996–1003. doi:10.3109/10715762.2012.690514
49. Shenoy K, Wu Y, Pervaiz S. LY303511 enhances TRAIL sensitivity of SHEP-1 neuroblastoma cells via hydrogen peroxide-mediated mitogen-activated protein kinase activation and up-regulation of death receptors. Cancer Res (2009) 69:1941–50. doi:10.1158/0008-5472.CAN-08-1996
50. Lee DH, Rhee JG, Lee YJ. Reactive oxygen species up-regulate p53 and Puma; a possible mechanism for apoptosis during combined treatment with TRAIL and wogonin. Br J Pharmacol (2009) 157:1189–202. doi:10.1111/j.1476-5381.2009.00245.x
51. Das A, Banik NL, Ray SK. Garlic compounds generate reactive oxygen species leading to activation of stress kinases and cysteine proteases for apoptosis in human glioblastoma T98G and U87MG cells. Cancer (2007) 110:1083–95. doi:10.1002/cncr.22888
52. Powolny AA, Singh SV. Multitargeted prevention and therapy of cancer by diallyl trisulfide and related Allium vegetable-derived organosulfur compounds. Cancer Lett (2008) 269:305–14. doi:10.1016/j.canlet.2008.05.027
53. Pallepati P, Averill-Bates DA. Activation of ER stress and apoptosis by hydrogen peroxide in HeLa cells: protective role of mild heat preconditioning at 40°C. Biochim Biophys Acta (2011) 1813:1987–99. doi:10.1016/j.bbamcr.2011.07.021
54. Delhalle S, Deregowski V, Benoit V, Merville MP, Bours V. NF-kappaB-dependent MnSOD expression protects adenocarcinoma cells from TNF-alpha-induced apoptosis. Oncogene (2002) 21:3917–24. doi:10.1038/sj.onc.1205489
55. Perez-Cruz I, Carcamo JM, Golde DW. Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood (2003) 102:336–43. doi:10.1182/blood-2002-11-3559
56. Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ (2009) 16:1040–52. doi:10.1038/cdd.2009.49
57. Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy (2012) 8:1462–76. doi:10.4161/auto.21211
58. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res (2014) 74:647–51. doi:10.1158/0008-5472.CAN-13-2966
59. Gewirtz DA. Cytoprotective and nonprotective autophagy in cancer therapy. Autophagy (2013) 9:1263–5. doi:10.4161/auto.25233
60. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J (2012) 441:523–40. doi:10.1042/BJ20111451
61. Chaabane W, User SD, El-Gazzah M, Jaksik R, Sajjadi E, Rzeszowska-Wolny J, et al. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp (2013) 61:43–58. doi:10.1007/s00005-012-0205-y
62. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195:59–70. doi:10.1084/jem.20010659
63. Carter WO, Narayanan PK, Robinson JP. Intracellular hydrogen peroxide and superoxide anion detection in endothelial cells. J Leukoc Biol (1994) 55:253–8.
64. Suzuki Y, Yoshimaru T, Inoue T, Ra C. Discrete generations of intracellular hydrogen peroxide and superoxide in antigen-stimulated mast cells: reciprocal regulation of store-operated Ca2+ channel activity. Mol Immunol (2009) 46:2200–9. doi:10.1016/j.molimm.2009.04.013
65. Mukhopadhyay P, Rajesh M, Yoshihiro K, Haskó G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun (2007) 358:203–8. doi:10.1016/j.bbrc.2007.04.106
66. Batinic-Haberle I, Cuzzocrea S, Rebouças JS, Ferrer-Sueta G, Mazzon E, Di Paola R, et al. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med (2009) 46:192–201. doi:10.1016/j.freeradbiomed.2008.09.042
67. Izeradjene K, Douglas L, Tillman DM, Delaney AB, Houghton JA. Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res (2005) 65:7436–45. doi:10.1158/0008-5472.CAN-04-2628
68. Jia L, Kelsey SM, Grahn MF, Jiang XR, Newland AC. Increased activity and sensitivity of mitochondrial respiratory enzymes to tumor necrosis factor alpha-mediated inhibition is associated with increased cytotoxicity in drug-resistant leukemic cell lines. Blood (1996) 87:2401–10.
69. Guidarelli A, Cerioni L, Cantoni O. Inhibition of complex III promotes loss of Ca2+ dependence for mitochondrial superoxide formation and permeability transition evoked by peroxynitrite. J Cell Sci (2007) 120:1908–14. doi:10.1242/jcs.003228
70. Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett (1996) 397:7–10. doi:10.1016/0014-5793(96)00989-1
71. Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev (2007) 39:443–55. doi:10.1080/03602530701468516
72. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis (2007) 12:913–22. doi:10.1007/s10495-007-0756-2
73. Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Moreno-Sánchez R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol Aspects Med (2010) 31:29–59. doi:10.1016/j.mam.2009.12.006
74. Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ (2007) 14:1243–7. doi:10.1038/sj.cdd.4402135
75. Petit JM, Maftah A, Ratinaud MH, Julien R. 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur J Biochem (1992) 209:267–73. doi:10.1111/j.1432-1033.1992.tb17285.x
76. Griffith TS, Rauch CT, Smolak PJ, Waugh JY, Boiani N, Lynch DH, et al. Functional analysis of TRAIL receptors using monoclonal antibodies. J Immunol (1999) 162:2597–605.
77. Georgakis GV, Li Y, Humphreys R, Andreeff M, O’Brien S, Younes M, et al. Activity of selective fully human agonistic antibodies to the TRAIL death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured lymphoma cells: induction of apoptosis and enhancement of doxorubicin- and bortezomib-induced cell death. Br J Haematol (2005) 130:501–10. doi:10.1111/j.1365-2141.2005.05656.x
78. Kroemer G, Petit P, Zamzami N, Vayssière JL, Mignotte B. The biochemistry of programmed cell death. FASEB J (1995) 9:1277–87.
79. Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, et al. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res (1998) 83:730–7. doi:10.1161/01.RES.83.7.730
80. Zhang Q, Matsuzaki I, Chatterjee S, Fisher AB. Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am J Physiol Lung Cell Mol Physiol (2005) 289:L954–61. doi:10.1152/ajplung.00210.2005
81. Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, et al. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol (2012) 302:H105–14. doi:10.1152/ajpheart.00298.2011
82. Bonner MY, Arbiser JL. Targeting NADPH oxidases for the treatment of cancer and inflammation. Cell Mol Life Sci (2012) 69:2435–42. doi:10.1007/s00018-012-1017-2
83. Jones KJ, Chetram MA, Bethea DA, Bryant LK, Odero-Marah V, Hinton CV. Cysteine (C)-X-C receptor 4 regulates NADPH oxidase-2 during oxidative stress in prostate cancer cells. Cancer Microenviron (2013) 6:277–88. doi:10.1007/s12307-013-0136-0
84. Maraldi T, Prata C, Caliceti C, Vieceli Dalla Sega F, Zambonin L, Fiorentini D, et al. VEGF-induced ROS generation from NAD(P)H oxidases protects human leukemic cells from apoptosis. Int J Oncol (2010) 36:1581–9.
85. Yin W, Cheng W, Shen W, Shu L, Zhao J, Zhang J, et al. Impairment of Na(+),K(+)-ATPase in CD95(APO-1)-induced human T-cell leukemia cell apoptosis mediated by glutathione depletion and generation of hydrogen peroxide. Leukemia (2007) 21:1669–78. doi:10.1038/sj.leu.2404791
86. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res (1991) 51:794–8.
87. Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J (2009) 418:29–37. doi:10.1042/BJ20081258
88. Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem (2005) 280:39485–92. doi:10.1074/jbc.M503296200
89. Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature (2005) 436:123–7. doi:10.1038/nature03688
90. Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science (1997) 275:1649–52. doi:10.1126/science.275.5306.1649
91. Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem (2000) 275:24273–8. doi:10.1074/jbc.M002094200
92. Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell (2002) 9:1031–44. doi:10.1016/S1097-2765(02)00520-8
93. Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans (2003) 31:1441–4. doi:10.1042/BST0311441
94. Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood (2005) 105:1717–23. doi:10.1182/blood-2004-03-0849
95. Naughton R, Quiney C, Turner SD, Cotter TG. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia (2009) 23:1432–40. doi:10.1038/leu.2009.49
96. Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature (2000) 407:390–5. doi:10.1038/35030140
97. Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood (2003) 101:4098–104. doi:10.1182/blood-2002-08-2512
98. Yu R, Mandlekar S, Harvey KJ, Ucker DS, Kong AN. Chemopreventive isothiocyanates induce apoptosis and caspase-3-like protease activity. Cancer Res (1998) 58:402–8.
99. Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol Cancer Ther (2003) 2:1045–52.
100. Wu SJ, Ng LT, Lin CC. Effects of antioxidants and caspase-3 inhibitor on the phenylethyl isothiocyanate-induced apoptotic signaling pathways in human PLC/PRF/5 cells. Eur J Pharmacol (2005) 518:96–106. doi:10.1016/j.ejphar.2005.06.021
101. Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell (2006) 10:241–52. doi:10.1016/j.ccr.2006.08.009
102. Wu XJ, Hua X. Targeting ROS: selective killing of cancer cells by a cruciferous vegetable derived pro-oxidant compound. Cancer Biol Ther (2007) 6:646–7. doi:10.4161/cbt.6.5.4092
103. Baumann S, Fas SC, Giaisi M, Müller WW, Merling A, Gülow K, et al. Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCgamma1- and Ca2+-dependent apoptosis. Blood (2008) 111:2354–63. doi:10.1182/blood-2007-06-096198
104. Wei L, Lu N, Dai Q, Rong J, Chen Y, Li Z, et al. Different apoptotic effects of wogonin via induction of H2O2 generation and Ca2+ overload in malignant hepatoma and normal hepatic cells. J Cell Biochem (2010) 111:1629–41. doi:10.1002/jcb.22898
105. Tsai CF, Yeh WL, Huang SM, Tan TW, Lu DY. Wogonin induces reactive oxygen species production and cell apoptosis in human glioma cancer cells. Int J Mol Sci (2012) 13:9877–92. doi:10.3390/ijms13089877
106. Yang WH, Fong YC, Lee CY, Jin TR, Tzen JT, Li TM, et al. Epigallocatechin-3-gallate induces cell apoptosis of human chondrosarcoma cells through apoptosis signal-regulating kinase 1 pathway. J Cell Biochem (2011) 112:1601–11. doi:10.1002/jcb.23072
107. Macho A, Calzado MA, Muñoz-Blanco J, Gómez-Díaz C, Gajate C, Mollinedo F, et al. Selective induction of apoptosis by capsaicin in transformed cells: the role of reactive oxygen species and calcium. Cell Death Differ (1999) 6:155–65. doi:10.1038/sj.cdd.4400465
108. Pramanik KC, Boreddy SR, Srivastava SK. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS One (2011) 6:e20151. doi:10.1371/journal.pone.0020151
109. McLachlan A, Kekre N, McNulty J, Pandey S. Pancratistatin: a natural anti-cancer compound that targets mitochondria specifically in cancer cells to induce apoptosis. Apoptosis (2005) 10:619–30. doi:10.1007/s10495-005-1896-x
110. Griffin C, McNulty J, Pandey S. Pancratistatin induces apoptosis and autophagy in metastatic prostate cancer cells. Int J Oncol (2011) 38:1549–56. doi:10.3892/ijo.2011.977
111. Griffin C, Karnik A, McNulty J, Pandey S. Pancratistatin selectively targets cancer cell mitochondria and reduces growth of human colon tumor xenografts. Mol Cancer Ther (2011) 10:57–68. doi:10.1158/1535-7163.MCT-10-0735
112. Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature (2011) 475:231–4. doi:10.1038/nature10167
113. Karmakar S, Banik NL, Patel SJ, Ray SK. Garlic compounds induced calpain and intrinsic caspase cascade for apoptosis in human malignant neuroblastoma SH-SY5Y cells. Apoptosis (2007) 12:671–84. doi:10.1007/s10495-006-0024-x
114. Gosslau A, Chen M, Ho CT, Chen KY. A methoxy derivative of resveratrol analogue selectively induced activation of the mitochondrial apoptotic pathway in transformed fibroblasts. Br J Cancer (2005) 92:513–21.
115. Chen V, Staub RE, Fong S, Tagliaferri M, Cohen I, Shtivelman E. Bezielle selectively targets mitochondria of cancer cells to inhibit glycolysis and OXPHOS. PLoS One (2012) 7:e30300. doi:10.1371/journal.pone.0030300
116. Basile V, Belluti S, Ferrari E, Gozzoli C, Ganassi S, Quaglino D, et al. bis-Dehydroxy-Curcumin triggers mitochondrial-associated cell death in human colon cancer cells through ER-stress induced autophagy. PLoS One (2013) 8:e53664. doi:10.1371/journal.pone.0053664
117. Pérez-Ortiz JM, Tranque P, Burgos M, Vaquero CF, Llopis J. Glitazones induce astroglioma cell death by releasing reactive oxygen species from mitochondria: modulation of cytotoxicity by nitric oxide. Mol Pharmacol (2007) 72:407–17. doi:10.1124/mol.106.032458
118. Jangamreddy JR, Ghavami S, Grabarek J, Kratz G, Wiechec E, Fredriksson BA, et al. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: differences between primary and cancer cells. Biochim Biophys Acta (2013) 1833:2057–69. doi:10.1016/j.bbamcr.2013.04.011
119. Pérez-Ortiz JM, Tranque P, Vaquero CF, Domingo B, Molina F, Calvo S, et al. Glitazones differentially regulate primary astrocyte and glioma cell survival. Involvement of reactive oxygen species and peroxisome proliferator-activated receptor-gamma. J Biol Chem (2004) 279:8976–85. doi:10.1074/jbc.M308518200
120. Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci (2007) 120:4155–66. doi:10.1242/jcs.011163
121. Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ (2008) 15:171–82. doi:10.1038/sj.cdd.4402233
122. Lee ZW, Zhou J, Chen CS, Zhao Y, Tan CH, Li L, et al. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS One (2011) 6:e21077. doi:10.1371/journal.pone.0021077
Keywords: depolarization, ROS, TRAIL, tumor-selective killing, sensitization, apoptosis, oxidative stress, endoplasmic reticulum stress
Citation: Suzuki-Karasaki Y, Suzuki-Karasaki M, Uchida M and Ochiai T (2014) Depolarization controls TRAIL-sensitization and tumor-selective killing of cancer cells: crosstalk with ROS. Front. Oncol. 4:128. doi: 10.3389/fonc.2014.00128
Received: 26 March 2014; Accepted: 14 May 2014;
Published online: 30 May 2014.
Edited by:
Evzen Krepela, Charles University in Prague, Czech RepublicReviewed by:
Thomi Brunner, University of Konstanz, GermanyOlivier Micheau, Institut National de la Santé et de la Recherche Médicale, France
Copyright: © 2014 Suzuki-Karasaki, Suzuki-Karasaki, Uchida and Ochiai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshihiro Suzuki-Karasaki, Division of Physiology, Department of Biomedical Sciences, Nihon University School of Medicine, 30-1 Oyaguchikami-machi, Itabashi-ku, Tokyo 173-8610, Japan e-mail:c3V6dWtpLnlvc2hpaGlyb0BuaWhvbi11LmFjLmpw