 Rimas J. Orentas1*
Rimas J. Orentas1* Jessica Nordlund2
Jessica Nordlund2 Jianbin He1
Jianbin He1 Sivasish Sindiri1
Sivasish Sindiri1 Crystal Mackall1
Crystal Mackall1 Terry J. Fry1
Terry J. Fry1 Javed Khan1
Javed Khan1
- 1Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
- 2Molecular Medicine, Department of Medical Sciences and Science for Life Laboratory, Uppsala University, Uppsala, Sweden
Pediatric lymphoid leukemia has the highest cure rate of all pediatric malignancies, yet due to its prevalence, still accounts for the majority of childhood cancer deaths and requires long-term highly toxic therapy. The ability to target B-cell ALL with immunoglobulin-like binders, whether anti-CD22 antibody or anti-CD19 CAR-Ts, has impacted treatment options for some patients. The development of new ways to target B-cell antigens continues at rapid pace. T-cell ALL accounts for up to 20% of childhood leukemia but has yet to see a set of high-value immunotherapeutic targets identified. To find new targets for T-ALL immunotherapy, we employed a bioinformatic comparison to broad normal tissue arrays, hematopoietic stem cells (HSC), and mature lymphocytes, then filtered the results for transcripts encoding plasma membrane proteins. T-ALL bears a core T-cell signature and transcripts encoding TCR/CD3 components and canonical markers of T-cell development predominate, especially when comparison was made to normal tissue or HSC. However, when comparison to mature lymphocytes was also undertaken, we identified two antigens that may drive, or be associated with leukemogenesis; TALLA-1 and hedgehog interacting protein. In addition, TCR subfamilies, CD1, activation and adhesion markers, membrane-organizing molecules, and receptors linked to metabolism and inflammation were also identified. Of these, only CD52, CD37, and CD98 are currently being targeted clinically. This work provides a set of targets to be considered for future development of immunotherapies for T-ALL.
Introduction
The origin of leukemia may be best understood through the specific genomic mutations that alter the regulation, growth, and differentiation of the lymphocytic cells that comprise the disease. These mutations both define the biology of the disease at presentation and can also reveal the genetic origins of the leukemic stem cell. The promise of next-generation sequencing technologies to more fully describe these mutations is starting to be fulfilled as exomic or genomic sequencing has begun to supplement and may someday replace traditional methods of leukemia diagnosis and classification based on pathology, immunophenotyping, and molecular cytogenetics (including FISH, fluorescence-based in situ hybridization, and PCR, polymerase chain reaction, for known genetic lesions) (1).
Genomic technology, however, cannot stand on its own as verification of target expression is still required at the protein level. Thus, it is immunophenotyping that ultimately informs the field of immunotherapeutics whether or not a genetic target could serve as therapeutic target for either antibody or T-cells transduced to express chimeric antigen receptors (CAR-Ts). The advent of CD19–CAR-T-cell therapy has impacted the treatment of pre-B-cell ALL for some patients with advanced disease. Indeed, we and others have proposed a number of targets that may be suitable for pediatric B-ALL (2, 3). However, attractive targets for T-cell leukemia have yet to be recognized and exploited. We present here potential targets for treating T-cell ALL with antibody or CAR-Ts, using strategies developed for the analysis of pediatric solid tumors and B-ALL (4).
In 1993, Pui et al. reviewed ontogeny marker expression in T-ALL in light of normal T-cell antigen expression during thymic development (5). T-ALL was considered as either prothymocyte- (expressing CD7), early thymocyte- (expressing CD5, CD2, and CD1), intermediate thymocyte- (CD1, CD4, or CD8), or mature thymocyte-like (CD3 and TCR surface expressed). The CD1 antigen, expressed on cortical thymocytes, Langerhans cells, and a subset of B-cells, is the only one of these developmental antigens to be turned off upon reaching T-cell maturity. Reinherz originally proposed that T-ALL be classified along the lines of CD1 and CD3 expression with stage I (early thymocyte) expressing CD2, CD5, CD7, and no CD1, CD4, CD8, or CD3; stage 2 (intermediate) expressing CD1, CD2, CD5, and CD7 with variable 4 and 8, and weak CD3; and stage 3 (mature) expressing CD2, CD3, CD5, CD7, and CD4 or CD8 (usually only one or the other) (6, 7). In most simplistic terms, mature or medullary T-ALL expresses surface CD3, but not CD1a. Cortical or thymic T-ALL expresses CD1a, but not surface CD3; and early T-cell precursor T-ALL (ETP-ALL, which encompasses Pro-T-ALL and Pre-T-ALL) does not express CD3 or CD1a.
The answer to the challenge of finding T-cell restricted targets (that is a mature T-cell antigen present on ALL that can be safely eliminated, as CD19 for B-cells), or a more T-ALL restricted target (especially for the more immature forms of the disease) may lay in the nature of the progenitor cell itself. As elegantly presented by the St. Jude – Washington University Pediatric Cancer Genome Project, early precursor T-cell ALL shares many similarities to double negative thymocytes that have the potential to differentiate into cells of either T-cell or myeloid lineage (8, 9). This pluripotency makes the antigenic expression profile for T-ALL far more generalized. At the other end of the spectrum, the most mature forms of T-ALL may benefit from new immunotherapeutic approaches that target the T-cell receptor, specifically, different subclasses that have clonally expanded. Although this was once deemed an approach to be of little interest due to the low number of cases, and the need for an almost individualized treatment approach, the success of CAR-T-cell therapy, which is the essence of personalized or individualized medicine, has brought this approach to the fore once more.
Data Interrogation and Results
T-cell ALL (acute lymphocyte/lymphoblastic leukemia) accounts for 15–18% of all childhood leukemias and 25% of ALL in adults (5, 10). However, we have yet to see a set of high-value targets proposed for T-cell ALL as we have for B- or pre-B-ALL. To that end we undertook a bioinformatics approach to describe the cell surface proteins expressed on T-ALL. Although we have begun to accumulate and analyze T-ALL cases at the Pediatric Oncology Branch of the NCI, a large data set was recently made available at Gene Expression Omnibus (GEO) using the Affymetrix Human Genome U133 Plus 2.0 platform. In this submission, GSE47051, over 100 cases of pediatric T-ALL and pre-B-ALL are available for analysis. The samples were of high quality and derived from bone marrow or peripheral blood aspirates of pediatric ALL patients enrolled in two co-operative group trials (NOPHO, Nordic Society for Pediatric Hematology-Oncology), fully described in the original publications for this sample set (11, 12). Samples were obtained under informed consent, approved by the Regional Ethical Board in Uppsala, Sweden, and in accordance with the guidelines set forth in the Declaration of Helsinki.
By navigating to the GEO home page, the CEL files for these annotated samples were accessed and analyzed. To test the accuracy of the pathological categorization of this sample set, we carried out hierarchical clustering using the freely available ArrayMiningtool (Online Microarray Data Mining Tool)1 (13). In Figure 1 we demonstrated the partitioning of the pre-B-ALL and T-ALL samples, confirming their original pathological classification. Moreover, the genes driving this classification are unmistakably well-recognized components of ALL, Table 1. The T-ALL set contains CD3D, CD7, and CD28; while in the pre-B-ALL set we find CD19, CD79B, and CD22. In Table 1 we present the top 10 cell surface genes with regard to Pearson correlation (gene vs. outcome), and in Figure 2, we present the top four overall scores, as expressed by their respective sample sets. Not surprisingly, the gene expression values that drove this classification were highly divergent from each other, such as CD3D for T-ALL and CD19 and EBF1 (early B-cell factor 1) for pre-B-ALL. Having confidence in the submitted sample classification, we then analyzed the T-ALL and pre-B-ALL samples for differences in average gene expression in comparison to broad normal tissue arrays. For each cell surface membrane transcript, an average expression level was calculated, and then compared to the average expression of the transcript in normal tissue, thus defining a potential space for immunotherapeutic intervention. Methods for this analysis were published previously (4). To confirm our analysis, we also analyzed normal and disease gene expression files using the Partek® Genomics Suite™ v6.5. The GEO database was used to download individual CEL files into the software suite, and data analyzed for quality, differential expression, and with statistical packages for differential gene expression (14–16).
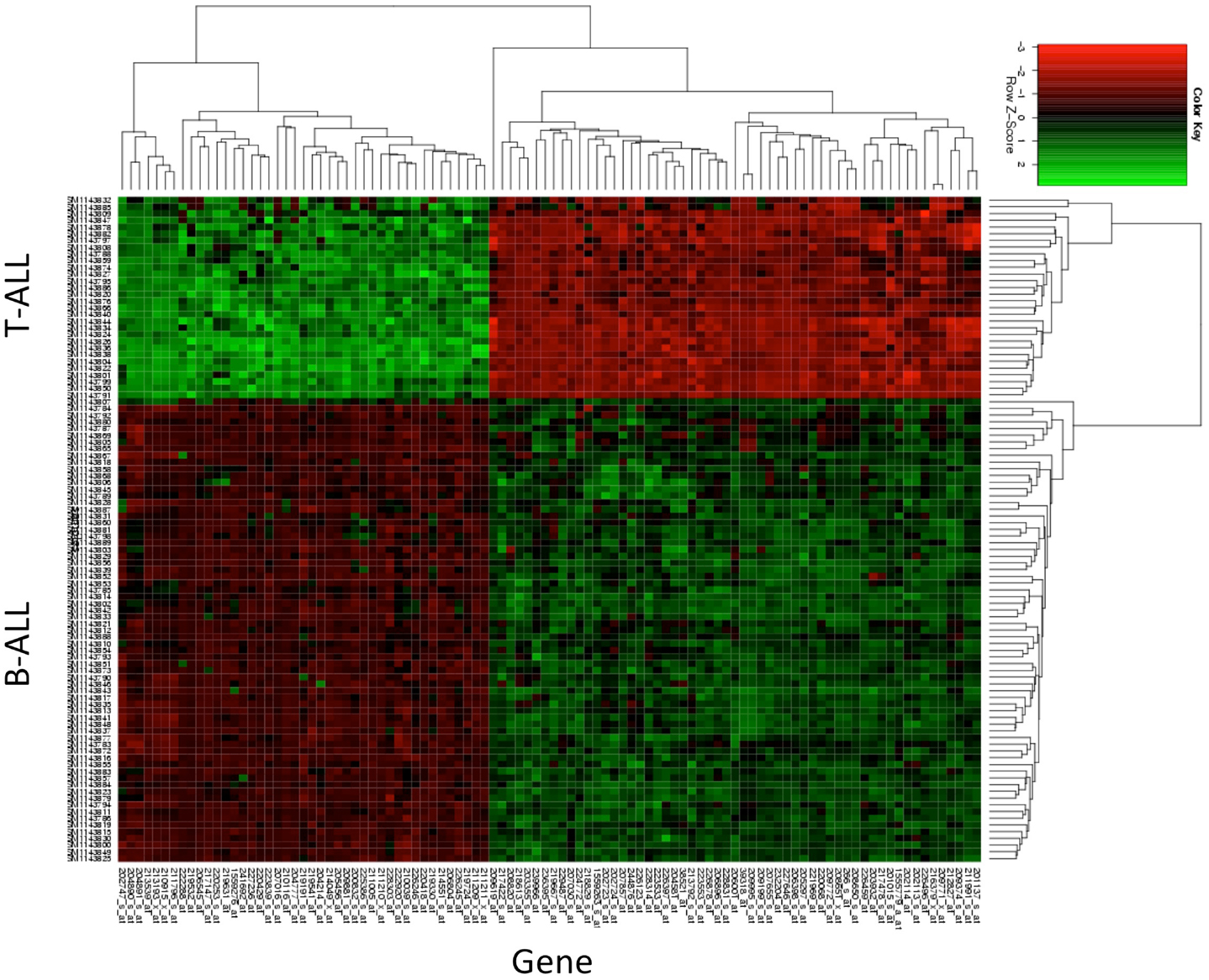
Figure 1. Hierarchical clustering of T-ALL and pre-B-ALL. Samples from GEO data set GSE47051 were downloaded and then clustered without reference to their diagnostic category (T-ALL or B-ALL). The top genes driving the subsequent clustering are listed in the x-axis, and the disease categories of those samples on the y-axis. Gene expression (log2cs) was normalized by z score (x-mean/std.dev.) across all leukemia samples for purposes of comparison.
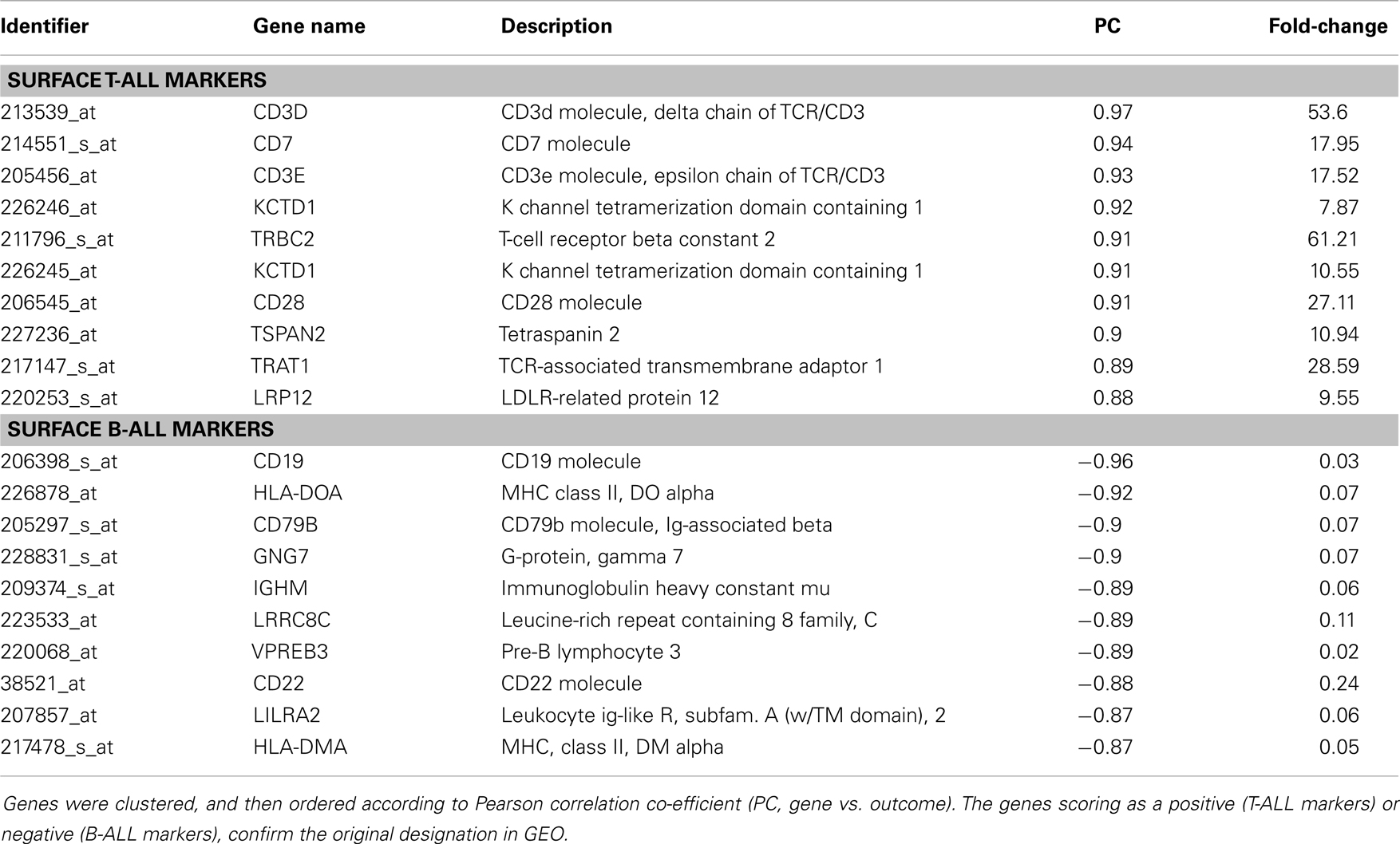
Table 1. Top 10 cell surface genes driving classification.
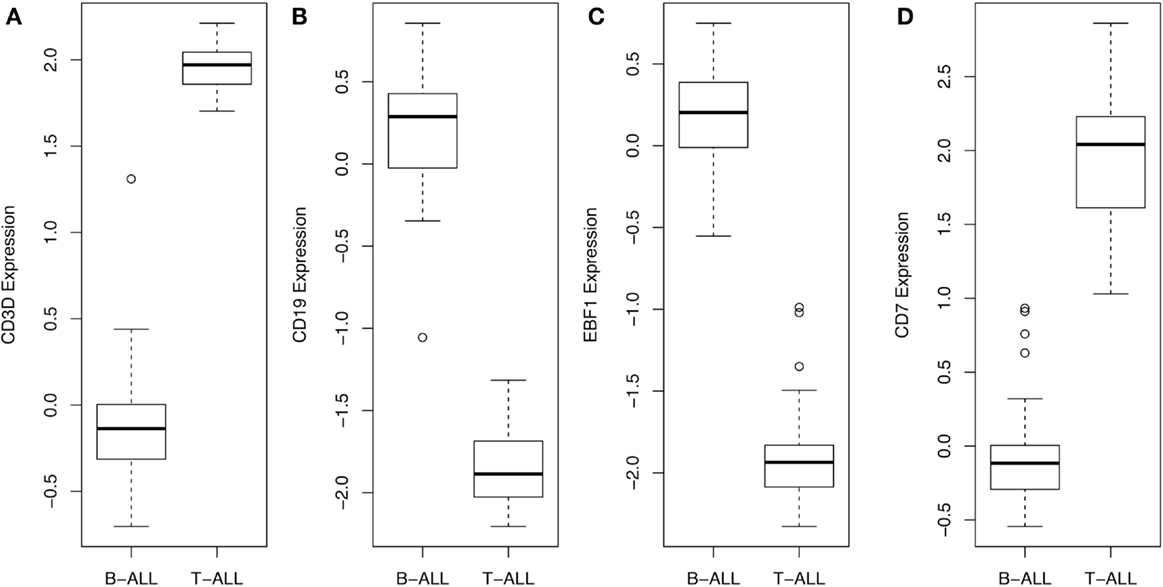
Figure 2. Gene expression values, across all samples, for top scoring hits. The average and range of values of the top four genes are presented in this box and whisker plot, demonstrating that CD3D and CD7 for T-ALL and CD19 and EBF1 for BCP-ALL (B-ALL) can be used to readily distinguish and verify the original pathological classification of these disease samples.
Table 2 lists the top 40 genes for each disease category, as a function of the ratio of an individual transcript’s expression level in ALL vs. a set of 117 normal tissues, our broad normal tissue array. A third of the transcripts (15 of the 46 transcripts, 33%) were present in both pre-B-ALL and T-ALL. These transcripts are by definition shared more between the two leukemias than with normal tissue. They are (bold italics in the table): PTGER4, ITGA4, CD37, CD52, CD62L (L-selectin), CXCR4, CD69, EVI2B (CD361), SLC39A8, MICB, LRRC70, CLELC2B, HMHA1, LST1, and CMTM6 (CKLFSF6).
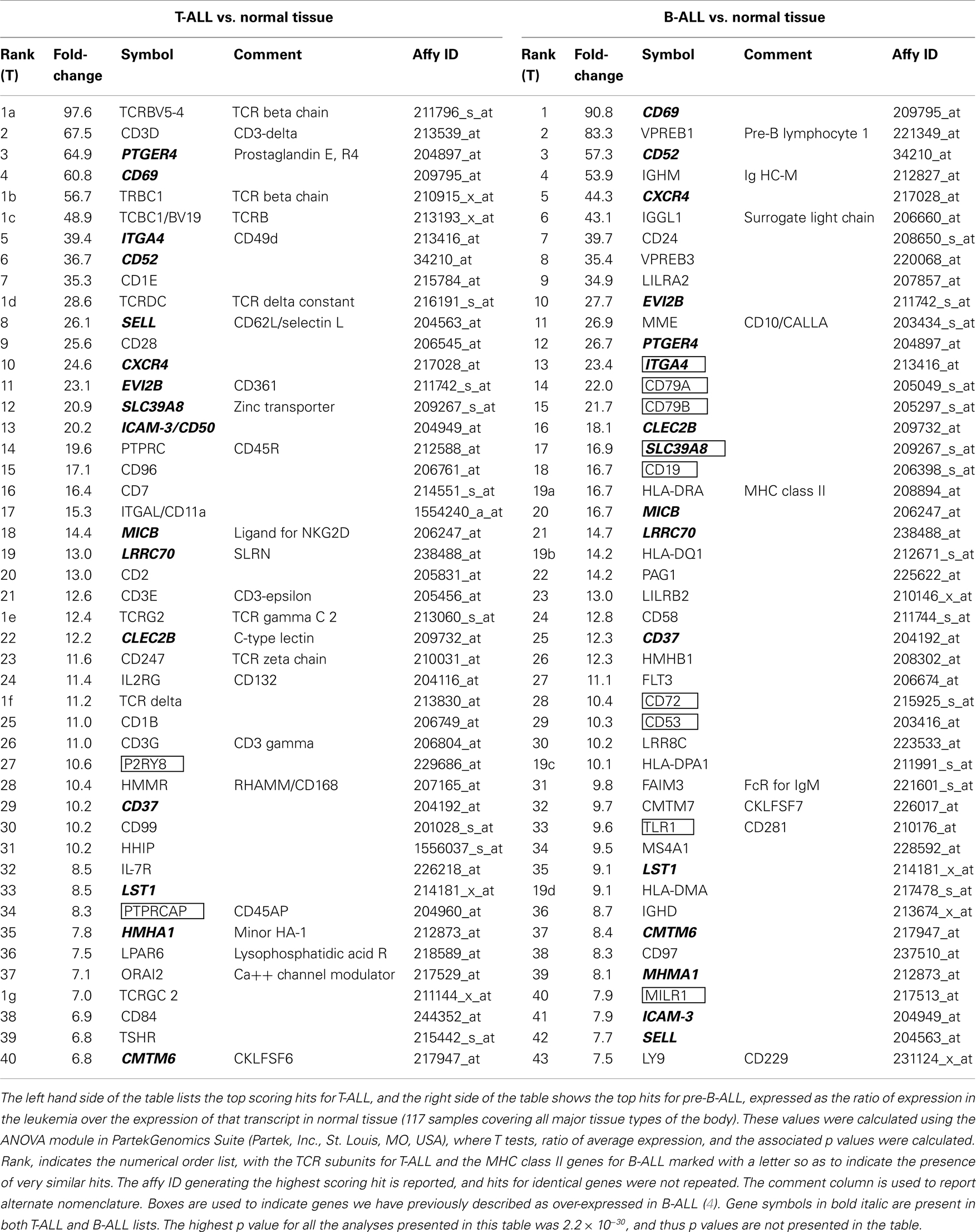
Table 2. T-ALL and B-ALL cell surface immunotherapy targets.
Shared pre-B-ALL and T-ALL Genes
SLC39A8 is the easiest to classify as this zinc transporter is a solute carrier protein expressed during inflammation. In short, it is a response to metabolic demand (17). The next group of markers that naturally fall together has to do with core homing and adhesion functions of lymphocytes, specifically CD37, a tetraspanin that associates with integrins; CD49d, an integrin expressed on activated lymphocytes; CD62L, a homing receptor; CD52, an adhesion receptor; CXCR4, whose natural ligand is SDF-1 and mediates homing to the bone marrow. Tetraspanins organize plasma membrane domains in order to co-ordinate signaling and cellular functions. In B-cells, the CD81/CD21/CD19 co-receptor complex interacts with CD82 and lipid rafts, while in T-cells the CD151/CD81/CD37/Tssc-6 complex is thought to perform a similar structural-signaling role (18, 19). This linkage to the tetraspanin CD37 is significant, as our data now link the expression of CD37 with known adhesion receptors of the integrin family in ALL. Anti-CD37 antibody is currently being evaluated in adult B-cell malignancies (NHL, CLL, MM, ALL) as a drug conjugate with the maytansine derivative DM1 (20).
CXCR4 is over-expressed in more than 20 different cancers, and expression in normal tissues (other than in the CD34 stem cell niche in bone marrow) is measurably lower (21–23). CD49d (alpha 4, beta 1/beta 7 integrin) is an adhesion receptor that mediates both rolling and firm adhesion to endothelium (24). CD49D expression in tandem with CXCR4 has been studied in pediatric and adult ALL. In children with ALL, no difference in expression levels was correlated to outcome, while adults with high CXCR4 and low CD49D (VLA-4) did worse (25).
CD62L is the canonical homing receptor for lymphocytes to enter secondary lymphoid tissues via high endothelial venules. Recently, targeting CD62L has been proposed for CLL, and thus it may be a viable target in pediatric ALL (26). CD52 is the well-characterized target of the anti-CAMPATH-1 antibody alemtuzumab, and is expressed on activated T-cells. This represents a second opportunity to more fully explore in pediatric ALL. CLEC2B (C-type lectin domain family 2B), is encoded in the NK cell killer gene complex on chromosome 12, and is also known as activation induced C-type lectin of T-cells, called AICL (27). AICL/CLELC2B map adjacent to CD69 whose C-type lectin domain is similar in sequence. CD69, a well-described activation marker for T-cells has also been found to be valid marker for prognosis in CLL, reflecting its ability to be up-regulated in either cell lineage (28). MICB (MHC class I polypeptide-related sequence B), is a ligand for NKG2D, and it thus activates T or NK cells. The shedding of the MICB protein is thought to help tumors evade NK-mediated immunosurveillance (29). Although not expressed on pre-B-ALL, CD96 is also an immunoglobulin superfamily member, and we discuss it here because it is also a ligand for activated T or NK cells. CD96 has also been described as a leukemia stem cell marker in AML (30). Since the expression of CD96 is low or absent on normal hematopoietic stem cells (HSC), Staudinger et al., have proposed using anti-CD96 antibody to purge bone marrow during hematopoietic stem cell transplantation (HSCT) for the disease (31).
CD361 (EVI2B) is a recently described antigen that was classified in 10th human leukocyte differentiation antigens (HLDA) conference as part of the B-cell panel, showing specific staining of ALL (32). This gene is found in an intron of the NF1 (neurofibromatosis type 1) gene, along with EVI2A and OMpg, being transcribed in the opposite direction from NF1 (33). Little is known of its function, and it may be over-expressed simply because of its genomic location. The CMTM6 (CKLF-like MARVEL transmembrane containing 6, CKLFSF6) is a chemokine-like gene that also contains a domain that bears sequence similar to tetraspanins. Thus, it appears to play both a signaling and a membrane-organizing role. The biological function is still being investigated.
LRRC70 (leucine-rich repeat containing 70/synleurin), is also not well-described and may play a role in cell adhesion or may bind a growth factor (34). Few functional studies have been carried out on the LRR protein family. PTGER4, is a prostaglandin receptor. This G-protein-coupled receptor is known to activate T-cells. Lymphocytic colitis was recently shown to be associated with increased TNFA, IFNG, and PTGER4, and a role for prostaglandin proposed in activating pathogenic lymphocytes (35). LST1 (leukocyte specific transcript 1) is expressed normally in cells of myeloid lineage, serving as transmembrane adaptor protein, interacting with the SHP-1 and SHP-2 phosphatases (36). There is no described association with LST1 and ALL to this point.
Mutis et al., generated cytotoxic T-cells restricted to the non-self HLA molecule HA-1/MMHA1 (histocompatibility, minor, HA-1), and proposed using this reactivity to actively target residual disease during HSCT (37, 38). The restriction of this antigen to the hematopoietic system highlights the fact that HSCT, which is often part of ALL therapy, could be augmented by targeting leukemia-expressed HA-1. In clinical trials, HA-1 and HA-2 peptide vaccines are being tested for the induction of increased graft vs. leukemia effect following allogeneic HSCT (NCT00943293). In reviewing this list of genes over-expressed in both T-ALL and pre-B-ALL, in comparison to normal tissue, a general picture of immune activation arises. While some of the proteins encoded by the transcripts expressed by both T-ALL and B-ALL are not as well-characterized, they still fit this theme and are also likely to be expressed throughout the immune system. Obvious therapeutic correlation can be made for CD52 and CD37. Antibody to CD96 is used for purging marrow, because it is too broadly expressed in normal tissue to be targeted by an active agent. The ability of MICB and CD96 to activate T-cells or NK may also be an important insight, as adoptive immunotherapy with T-cells engineered to express NK ligands has been demonstrated and their presence may therefore be associated with an activated mature phenotype (39). Also of importance to immunotherapy is the presence of minor antigen (HA-1) in ALL that may be exploited during HSCT. We now turn our attention to the transcripts expressed in the disease most in need of new approaches, pediatric T-ALL.
T-ALL Genes
Among the T-ALL transcripts listed as being over-expressed in Table 2, 7 of 46 are TCR chains (TCRBV5-4, TCRBC1, TCBC1/TCRBV, TCRDC, TCRG2, TCR delta, and TCRGC2) and 4 are members of the CD3 complex (CD3-delta, -epsilon, -gamma, and -zeta), making 11 of 46 (23%) of the T-ALL hits in Table 2 directly related to clonotypic T-cell marker expression. This analysis does not inform us directly if these molecules are on the surface, as the presence of the surface protein CD3-epsilon is the key diagnostic criterion for classification of T-ALL as being mature/medullary type, but many of these are re-arranged TCR chains indicating the T-ALL data set we are analyzing is weighted toward a more mature phenotype. Thinking of these re-arranged proteins as immunotherapeutic targets will be discussed later.
Transcripts for many canonical normal T-cell markers are also seen in T-ALL: such as, CD2, CD7, CD11a, CD28, CD45R, CD45AP, CD84, CD99, IL2RG, IL-7R, and the CD1 family markers CD1E and CD1B. If we consider this set of transcripts as an aggregate diagnostic of a single case, the presence of CD2 and CD1 transcripts, along with CD7 would classify this sample as a cortical or thymic T-ALL, which is in keeping with the predominance of re-arranged TCR transcripts detected (as opposed to an early T-cell precursor type ALL). CD84 is in the SLAM (signaling lymphocyte activation molecule) subset of the CD2 family of proteins, and mediates T-cell activation through homotypic adhesion (40). CD28 is the well-characterized second signaling molecule, CD11A (LFA-1 alpha chain) is a well-recognized lymphocyte integrin, and CD45 is LCA (leukocyte common antigen). CD45AP is known to positively regulate CD45 function. The transcripts for IL2 and IL7 receptor chains are also in keeping with the T-cell transcriptome of T-ALL. CD99 is a well-characterized marker of T-ALL and in pediatric disease its high level of expression has been proposed as a marker for detecting minimal residual disease along with CD3, CD7, and/or TdT (41). Hence, this set of transcripts is largely a picture of a normal activated immune cell.
The expression of CD1A is a key diagnostic for T-ALL, representing the key transition, along with high expression of CD2, to a more mature phenotype. In our analysis, we detected transcripts for CD1B and CD1E. CD1A is thought to be the primary cell surface CD1 family member while CD1B interacts with intracellular lipids and CD1E functions as a lipid chaperone, reviewed in Ref. (42). Nevertheless, CD1B clearly presents mycobacterial antigens to T-cells and is on the surface (43). In studying LEF1 mutations in pediatric T-ALL, Gutierrez et al. demonstrated that this subset of pediatric T-ALL was arrested at the cortical stage and they demonstrated the presence of CD1B and CD1E by gene expression profiling (44). We can infer from this that the CD1 family is induced as a group, and also hope that unique subsets of lipid antigens may be present on T-ALL. None have yet been reported.
Three immune cell adhesion receptors are over-expressed in T-ALL. ICAM-3 (CD50) is constitutively expressed on most B-cell malignancies, except for Hodgkin lymphoma, and myeloid cells, but appears to be absent from other tissues, with the possible exception of tumor-associated vasculature (45, 46). We report here that it is likely to be found on pediatric T-ALL as well. HMMR (hyaluronan-mediated motility receptor, RHAMM, or CD168), is found in a complex containing BRCA1 in breast tissue where it may govern apicobasal polarity (47). TCRs recognizing RHAMM were generated in a model system and found to inhibit AML growth in a xenogeneic system (48). However, these T-cells also recognized CD34+ HSC in an HLA-A2 restricted manner. Thus, an active T-cell population targeting HMMR could only be envisioned for clinical use in an HLA mismatch setting.
LPAR6, lysophosphatidic acid receptor 6, is another interesting transcript that may be expressed either for its contribution to cellular transformation or it may be part of an active genomic locus, as it is transcribed from an intron of the RB gene (49). On its own, this G-coupled receptor is known to stimulate cell activation upon encountering its ligand, lysophosphatidic acid, and may play a role in tumor cell tissue invasion (50). Like the zinc transporter, another T-cell metabolic protein, ORAI2 (ORAI calcium release-activated calcium modulator 2) is over-expressed in T-ALL. It is likely to function in a manner similar to ORAI1 where STIM1 activates the translocation of this calcium channel to the surface of the T-cell upon depletion of intracellular calcium stores (51).
Three transcripts expressed by T-ALL are a little more enigmatic, but reflect the fact that we are dealing with a transformed cell population. HHIP (Hedgehog interacting protein) interacts with all forms of sonic hedgehog (SHH), a secreted factor that guides tissue formation during development, and is a key mediator of its function (52). HHIP function on T-ALL has not been explored, but an AML study has described the ability of AML-derived stromal cells to express HHIP and to interact with AML stem cells that now re-express SHH and its receptor, smoothened (SMO) (53). TSHR (thyroid stimulating hormone receptor) is not normally considered a T-cell antigen, but mutations of this receptor have been associated with increased vasculature and the development of pituitary adenomas (54, 55). Clinical studies with TSHR primarily focus on Grave’s disease. P2RY8 (purinergic receptor P2Y, G-protein-coupled 8), is best known as supplying the promoter that drives CRLF/TSLPR overexpression, upon deletion of an intervening pseudoautosomal region, in certain cases of B-ALL (56, 57). In a fascinating study, Fujiwara et al., screened for transforming genes in the rare biphenotypic acute leukemia (BAL), which bears both lymphoid and myeloid markers (58). They found P2RY8 expression to activate a number of cellular activation pathways, including those mediated by CERB and Elk-1. Thus, P2RY8 may be over-expressed simply by residing in a very active genomic region or it may play a functional role in transforming leukemia cell clones.
In order to further analyze T-ALL gene expression signatures, we sought to determine if the normal gene sets we used for comparison had an impact on the type of transcripts identified as being over-expressed. We ran two subsequent analyses using CD34+ bone marrow-derived HSC, and normal peripheral blood mononuclear cells, as the normal tissue comparators, Table 3. HSC CEL files were submitted by Dehashis Sahoo to GEO as GSE32719 by the Weissman Lab at Stanford University, USA (59). We selected CD34+ HSC that was from normal donors aged 19–31 years old (n = 14). For normal PBMC, we used control patients from the GEO Submission GSE21942 by Kemppinen and Saarela at the University of Cambridge, UK (n = 15) (60). In the top half of Table 3, where comparison is made to HSC, a profile similar to the normal tissue array of Table 2 is seen, where T-cells transcripts predominate. In fact 60 and 70% of the transcripts (ranked by T score and fold-change, respectively), were shared. However, when comparison was made to PBMC, the percentage of shared transcripts dropped to 10 and 15% for T score and fold-change, respectively. Only three transcripts were shared between the lists featured in the top and bottom half (HSC vs. PBMC) Table 3 shows CD3D (the delta chain of the TCR complex), HHIP, and TALLA-1 (TSPAN7). The presence of T-cell associated like transcripts has been discussed, but HHIP and TALLA-1 warrant special attention.
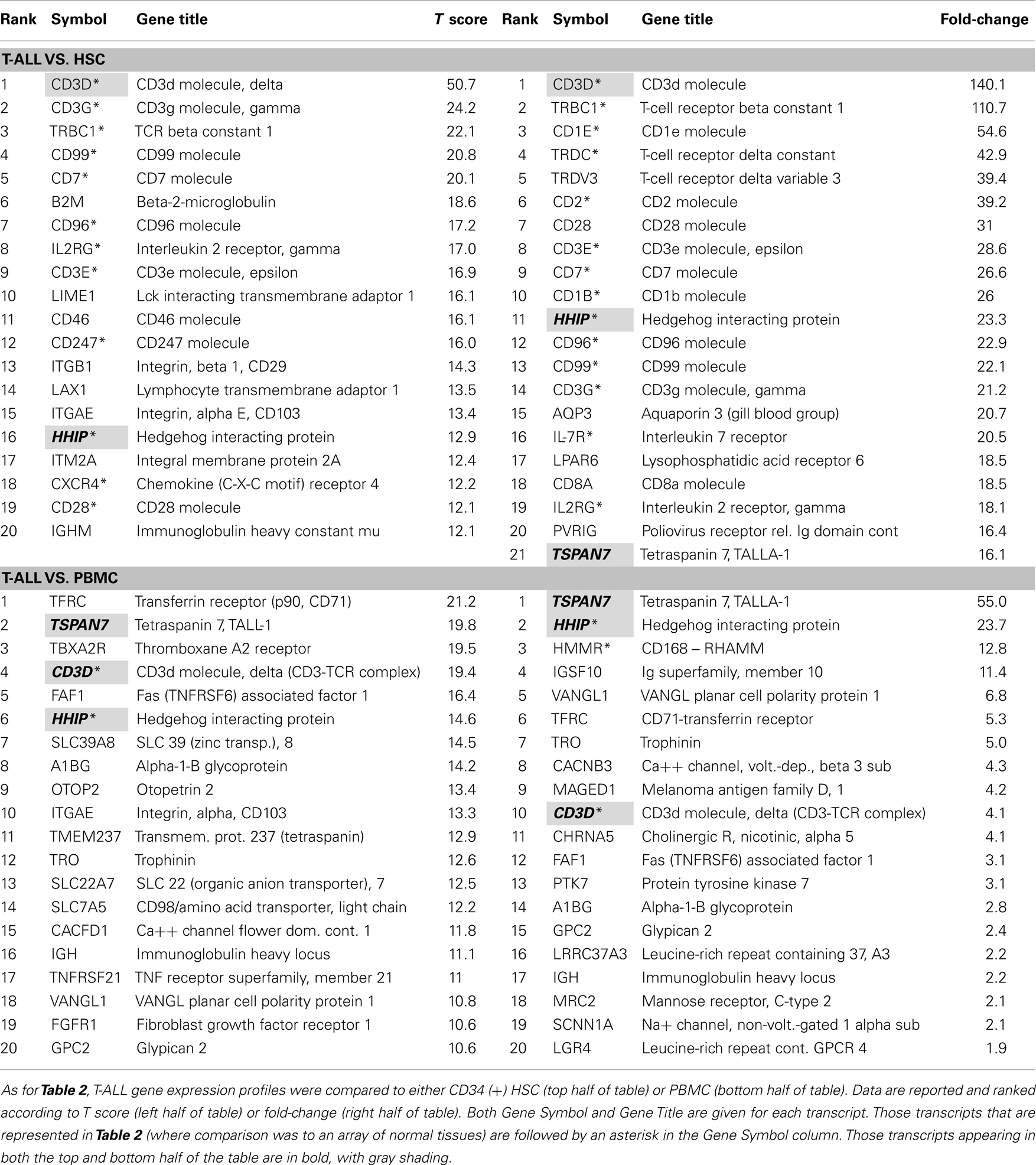
Table 3. Comparison of T-ALL to HSC and PBMC normal gene expression profiles.
In a series of papers in the 1980s, Seon et al. described a unique human T-cell leukemia antigen identified by a monoclonal antibody, designated it TALLA (61, 62). Immunotoxin-conjugated antibody to TALLA was found to control the growth of leukemic xenografts in nude mouse models (63). The antigen, a tetraspanin, is now referred to as TSPAN7, is expressed from the X chromosome, and has been most recently been explored with regard to its role in differentiating glutamatergic neurons (64). As discussed above, little is known about HHIP and leukemia. As HHIP is likely to regulate stem cell-like characteristics of either leukemia cells, or stromal cells that support them it should be considered a high-value target (53). Also of interest was our detection of SLC7A5, which is also known as CD98, in the T score but not fold-change column of Table 3, where comparison is made to PBMC. A humanized monoclonal antibody to CD98, IGN523, is currently being tested in refractory or relapsed AML (NCT02040506). CD98 is composed of two chains, and has the ability to signal though both mTOR and AKT pathways (65).
All of the gene expression data are presented in our fully annotated Oncogenomics database2.
Discussion
In seeking to analyze a series of pediatric malignancies for potential immunotherapeutic targets, we did not have a data set sufficiently robust to analyze pediatric T-ALL. In the data presented here, we now can add T-ALL to our list of pediatric malignancies that have been analyzed at the transcript level. Future studies will be enhanced by our ability to look at next-generation sequencing data, and to perhaps identify differential expression between tumor and normal at even finer levels, such as comparing alternative splicing profiles. Although we did not discuss our data for pre-B-ALL in a transcript by transcript level, our results were consistent with our earlier work looking at a more restricted set of normal tissues (4). The antigens CD19, CD79A, CD79B, CD49d, CD53, CD72, TLR1, and MILR1 were all reported previously. Thus, our pre-B-ALL analysis sets are consistent. New and potentially interesting hits that arose in this new analysis for B-ALL include CD52, CD24, and CD10/CALLA. Therapeutic antibodies have been developed for all of them.
The analysis of pediatric T-ALL is novel, and seems to reflect the perplexing nature of this disease in that the majority of the over-expressed transcripts are likely to be broadly expressed throughout the immune system. Thus CD2, CD69, and CD99 are all very risky targets that may require more sophisticated approaches than a single antibody or CAR-T. This holds true for CD96 as well, which is being used to purge bone marrow, but is too broadly expressed to be targeted systemically. We do have some reasonable antigens to target, but the data may be telling us something else. It may be time to consider more fully the use of clonotypic antibodies or targeted CAR-Ts to permanently eliminate specific T-cell subsets.
In the clinic, minimal residual disease can be detected in >95% of ALL patients by PCR detection of patient-specific rearrangements of the TCR or the immunoglobulin locus. This argues that at a genetic level the TCR is a valid target for therapeutic development (66). Campana et al. looked carefully at the expression of intracellular vs. membrane-expressed CD3 and of the TCR in T-ALL (67). In a series of ALL where patients ranged in age from 6 to 65 (median age, 20), 11 of 40 showed CD3 and either TCRAB (9 of 11) or TCRGD (2/11) on the membrane. Cytoplasmic TCR was detected in 12/40; while 17/40 had neither CD3 or TCR surface expression. In a study of adult T-ALL associated with HTLV-1 infection in Japan, all samples expressed surface TCR, albeit with a low fluorescence intensity (68). In this study, 7/12 patient samples expressed TCR on the surface on >95% of their blasts. Thus, the disease association with differentiation status holds, and we can state that a third of all patients with T-ALL are likely to have surface TCR. Unfortunately, our acuity in diagnosis of T-ALL according to the degree of cellular progression along the T-cell developmental pathway has not resulted in any useful differentiation of outcome status, with the possible exception of a slightly better outcome for CD10+ disease (69).
We propose a second method for targeting certain sub-types of T-ALL that express a surface T-cell receptor. We hypothesize that deletion of a specific family or sub-family of TCR-bearing ALL, for example the TCRBV5-4 family that ranked the highest of all differentially expressed genes in Table 2, would effectively eliminate that leukemic clone. Using CART-Ts specific for a single TCR family would result in only a partial depletion of normal T-cells, and the physiological consequences with respect to induced immunosuppression may be minimal. This is in contrast to current CD19–CAR-T therapy for B-ALL, where the entire healthy B-cell compartment is ablated. Alternatively, the generation of target lists, such as we present here may be exploited in the future when a new generation of immunotherapeutic agents where more than one set of antigens is required to fully activate an effector T-cell-can be developed. The creation of a treatment strategy for prostate cancer that requires recognition of both PMSA and PSCA to fully trigger a dual-engineered T-cell population may be one such approach (70). Our next step will be to validate surface expression of the transcripts described here by flow cytometry, and thus complement gene expression data with the demonstrated presence of protein that could be targeted on the surface of pediatric T-ALL.
Finally, we also explored the effect of altering the normal tissue comparator from a collection of normal tissues throughout the body, to two more restricted normal tissue sample sets. The first of these was the CD34+ HSC compartment in bone marrow. As this is the tissue from which the leukemia arises, one might anticipate that a more tumor-like set of transcript would be identified. However, the transcripts remained very much T lymphocyte specific, and up to 70% of the transcripts were shared when comparison was made between T-ALL and normal HSCs (Table 2 vs. Table 3). We did see a noticeably different set of transcripts identified when peripheral blood lymphocytes were used as the normal tissue comparison. The T lymphocyte specific transcripts were lost and a more generic tumor-like set of transcripts were noted. Interestingly, the CD3-delta chain of the TCR complex remained highly over-expressed in all comparisons, as did TALLA-1 (in 2 of 3) and HHIP (in 3 of 3). TALLA-1 has been described previously as a T-cell leukemia specific antigen and our analysis confirms that this is a high priority target worthy of further exploration (61). Shared between all comparisons was HHIP. HHIP has been elegantly described as a modulator of SHH signaling, adding another level of regulation to the patched (PTC1) and SMO pathway that modulates expression of Hedgehog (Hh)-regulated genes (52). A report demonstrating participation of this pathway in regulating myeloid leukemia supports further research into the role of HHIP in lymphocytic leukemia (53). In conclusion, there may be no single “correct” normal tissue to use as a comparator for gene expression profiles from leukemia samples. Instead, we should view each comparison as a means to identifying unique aspects of leukemia-associated gene expression patterns. These approaches highlight that general tissue comparisons yield results that reflect the tissue of origin of the malignancy, as seen in Table 2, while comparison to similar tissues types (in this case mature lymphocytes) reveals transcripts that are biased toward those that are associated with tumorigenesis, Table 3. However, both types of targets are of value, especially in the context of immunotherapy, where finding tumor-specific antigens that spare the host from fatal toxicity is the goal. If we restricted ourselves to the HSC or lymphocytic comparators in studying B-ALL, we would have missed antigens like CD19 and CD22 that are currently being targeted in experimental trials. When an antigen is found in all comparator scenarios, such as HHIP, it certainly warrants further investigation. This is also the case for TALLA-1, although it may not differ sufficiently from normal tissue. The antigens identified by using a broad tissue sample are certainly valuable, but as discussed, must be evaluated for the impact of targeting the normal tissues that may share these antigens, such as mature T-cell subsets.
In the Pediatric Cancer Genomes project, a database of prevalent mutations found in pediatric cancers, including T-ALL is being assembled3. Currently, this database is very useful for identifying mutations, which differs from our analysis, focusing on gene expression levels relative to normal tissue data sets. The largest collection of T-ALL samples at the Pediatric Cancer Genomes database is derived from ETP-ALL (9), and when this and other datasets are explored more closely it is apparent that only a very small minority of the genes identified are cell surface membrane proteins, with one exception, IL-7R. We also identified in IL-7R in T-ALL when HSC was used as a comparator in the fold-change analysis (Table 3), and as hit number 32 in the comparison run with our broad normal tissue panel (Table 2). This highlights again that different targets of interest will be identified when different normal comparators are used. This also encourages mining the “normal” T-cell developmental antigens we have identified for potentially harboring oncogenic mutations as well. The entire data in our fully annotated (including membranous protein assignment) gene expression database where users can search specific genes and compare gene expression between T-ALL, B-ALL, HSC, PBMC, and a range of normal tissue are available at: http://home.ccr.cancer.gov/oncology/oncogenomics/.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, Center for Cancer Research (CCR), NCI. Research was also supported by a Stand Up To Cancer – St. Baldrick’s – NCI Pediatric Dream Team Translational Cancer Research Grant and by the Hyundai Hope On Wheels, Hope Grant Program.
Footnotes
References
1. So CWE. An overview: from discovery of candidate mutations to disease modeling and transformation mechanisms of acute leukemia. In: So CWE, editor. Leukemia: Methods and Protocols. New York: Humana Press (2008). p. 1–5. doi: 10.1007/978-1-59745-418-6_1
2. Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin Cancer Res (2012) 18(10):2780–90. doi:10.1158/1078-0432.CCR-11-1920
3. Orentas RJ, Lee DW, Mackall C. Immunotherapy targets in pediatric cancer. Front Oncol (2012) 2:3. doi:10.3389/fonc.2012.00003
4. Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front Oncol (2012) 2:194. doi:10.3389/fonc.2012.00194
5. Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood (1993) 82(2):343–62.
6. Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF. Discrete stages of human intrathymic differentiation: analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc Natl Acad Sci U S A (1980) 77(3):1588–92. doi:10.1073/pnas.77.3.1588
7. Crist WM, Shuster JJ, Falletta J, Pullen DJ, Berard CW, Vietti TJ, et al. Clinical features and outcome in childhood T-cell leukemia-lymphoma according to stage of thymocyte differentiation: a Pediatric Oncology Group Study. Blood (1988) 72(6):1891–7.
8. Wada H, Masuda K, Satoh R, Kakugawa K, Ikawa T, Katsura Y, et al. Adult T-cell progenitors retain myeloid potential. Nature (2008) 452(7188):768–72. doi:10.1038/nature06839
9. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature (2012) 481(7380):157–63. doi:10.1038/nature10725
10. Copelan EA, McGuire EA. The biology and treatment of acute lymphoblastic leukemia in adults. Blood (1995) 85(5):1151–68.
11. Nordlund J, Backlin CL, Wahlberg P, Busche S, Berglund EC, Eloranta ML, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol (2013) 14(9):r105. doi:10.1186/gb-2013-14-9-r105
12. Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S, et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia (2010) 24(2):345–54. doi:10.1038/leu.2009.251
13. Glaab E, Garibaldi JM, Krasnogor N. ArrayMining: a modular web-application for microarray analysis combining ensemble and consensus methods with cross-study normalization. BMC Bioinformatics (2009) 10:358. doi:10.1186/1471-2105-10-358
14. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics (2003) 19(2):185–93. doi:10.1093/bioinformatics/19.2.185
15. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res (2003) 31(4):e15. doi:10.1093/nar/gng015
16. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (2003) 4(2):249–64. doi:10.1093/biostatistics/4.2.249
17. Liu MJ, Bao S, Galvez-Peralta M, Pyle CJ, Rudawsky AC, Pavlovicz RE, et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep (2013) 3(2):386–400. doi:10.1016/j.celrep.2013.01.009
18. Martin F, Roth DM, Jans DA, Pouton CW, Partridge LJ, Monk PN, et al. Tetraspanins in viral infections: a fundamental role in viral biology? J Virol (2005) 79(17):10839–51. doi:10.1128/JVI.79.17.10839-10851.2005
19. Wright MD, Moseley GW, van Spriel AB. Tetraspanin microdomains in immune cell signalling and malignant disease. Tissue Antigens (2004) 64(5):533–42. doi:10.1111/j.1399-0039.2004.00321.x
20. Palomba ML, Younes A. In the spotlight: a novel CD37 antibody-drug conjugate. Blood (2013) 122(20):3397–8. doi:10.1182/blood-2013-09-526384
21. Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev (2010) 29(4):709–22. doi:10.1007/s10555-010-9256-x
22. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer (2004) 4(7):540–50. doi:10.1038/nrc1388
23. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410(6824):50–6. doi:10.1038/35065016
24. Yue J, Pan Y, Sun L, Zhang K, Liu J, Lu L, et al. The unique disulfide bond-stabilized W1 beta4-beta1 loop in the alpha4 beta-propeller domain regulates integrin alpha4beta7 affinity and signaling. J Biol Chem (2013) 288(20):14228–37. doi:10.1074/jbc.M113.462630
25. Ko SY, Park CJ, Park SH, Cho YU, Jang S, Seo EJ, et al. High CXCR4 and low VLA-4 expression predicts poor survival in adults with acute lymphoblastic leukemia. Leuk Res (2014) 38(1):65–70. doi:10.1016/j.leukres.2013.10.016
26. Burgess M, Gill D, Singhania R, Cheung C, Chambers L, Renyolds BA, et al. CD62L as a therapeutic target in chronic lymphocytic leukemia. Clin Cancer Res (2013) 19(20):5675–85. doi:10.1158/1078-0432.CCR-13-1037
27. Hamann J, Montgomery KT, Lau S, Kucherlapati R, van Lier RA. AICL: a new activation-induced antigen encoded by the human NK gene complex. Immunogenetics (1997) 45(5):295–300. doi:10.1007/s002510050208
28. Del Poeta G, Del Principe MI, Zucchetto A, Luciano F, Buccisano F, Rossi FM, et al. CD69 is independently prognostic in chronic lymphocytic leukemia: a comprehensive clinical and biological profiling study. Haematologica (2012) 97(2):279–87. doi:10.3324/haematol.2011.052829
29. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest (2013) 123(10):4410–22. doi:10.1172/JCI69369
30. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A (2007) 104(26):11008–13. doi:10.1073/pnas.0704271104
31. Staudinger M, Humpe A, Gramatzki M. Strategies for purging CD96 stem cells in vitro and in vivo: new avenues for autologous stem cell transplantation in acute myeloid leukemia. Oncoimmunology (2013) 2(6):e24500. doi:10.4161/onci.24500
32. Faure GC, Amsellem S, Arnoulet C, Bardet V, Campos L, De Carvalho-Bittencourt M, et al. Mutual benefits of B-ALL and HLDA/HCDM HLDA 9th Barcelona 2010. Immunol Lett (2011) 134(2):145–9. doi:10.1016/j.imlet.2010.10.008
33. Kaufmann D, Gruener S, Braun F, Stark M, Griesser J, Hoffmeyer S, et al. EVI2B, a gene lying in an intron of the neurofibromatosis type 1 (NF1) gene, is as the NF1 gene involved in differentiation of melanocytes and keratinocytes and is overexpressed in cells derived from NF1 neurofibromas. DNA Cell Biol (1999) 18(5):345–56. doi:10.1089/104454999315240
34. Wang W, Yang Y, Li L, Shi Y. Synleurin, a novel leucine-rich repeat protein that increases the intensity of pleiotropic cytokine responses. Biochem Biophys Res Commun (2003) 305(4):981–8. doi:10.1016/S0006-291X(03)00876-3
35. Dey I, Beck PL, Chadee K. Lymphocytic colitis is associated with increased pro-inflammatory cytokine profile and up regulation of prostaglandin receptor EP4. PLoS One (2013) 8(4):e61891. doi:10.1371/journal.pone.0061891
36. Draber P, Stepanek O, Hrdinka M, Drobek A, Chmatal L, Mala L, et al. LST1/A is a myeloid leukocyte-specific transmembrane adaptor protein recruiting protein tyrosine phosphatases SHP-1 and SHP-2 to the plasma membrane. J Biol Chem (2012) 287(27):22812–21. doi:10.1074/jbc.M112.339143
37. Mutis T, Blokland E, Kester M, Schrama E, Goulmy E. Generation of minor histocompatibility antigen HA-1-specific cytotoxic T cells restricted by nonself HLA molecules: a potential strategy to treat relapsed leukemia after HLA-mismatched stem cell transplantation. Blood (2002) 100(2):547–52. doi:10.1182/blood-2002-01-0024
38. Marijt WA, Heemskerk MH, Kloosterboer FM, Goulmy E, Kester MG, van der Hoorn MA, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci U S A (2003) 100(5):2742–7. doi:10.1073/pnas.0530192100
39. Lehner M, Gotz G, Proff J, Schaft N, Dorrie J, Full F, et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One (2012) 7(2):e31210. doi:10.1371/journal.pone.0031210
40. Yan Q, Malashkevich VN, Fedorov A, Fedorov E, Cao E, Lary JW, et al. Structure of CD84 provides insight into SLAM family function. Proc Natl Acad Sci U S A (2007) 104(25):10583–8. doi:10.1073/pnas.0703893104
41. Dworzak MN, Froschl G, Printz D, Zen LD, Gaipa G, Ratei R, et al. CD99 expression in T-lineage ALL: implications for flow cytometric detection of minimal residual disease. Leukemia (2004) 18(4):703–8. doi:10.1038/sj.leu.2403303
42. Adams EJ. Lipid presentation by human CD1 molecules and the diverse T cell populations that respond to them. Curr Opin Immunol (2014) 26C:1–6. doi:10.1016/j.coi.2013.09.005
43. Van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol (2013) 14(7):706–13. doi:10.1038/ni.2630
44. Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood (2010) 115(14):2845–51. doi:10.1182/blood-2009-07-234377
45. Molica S, Dattilo A, Mannella A, Levato D. Intercellular adhesion molecules (ICAMs) 2 and 3 are frequently expressed in B cell chronic lymphocytic leukemia. Leukemia (1996) 10(5):907–8.
46. Cordell JL, Pulford K, Turley H, Jones M, Micklem K, Doussis IA, et al. Cellular distribution of human leucocyte adhesion molecule ICAM-3. J Clin Pathol (1994) 47(2):143–7. doi:10.1136/jcp.47.2.143
47. Maxwell CA, Benitez J, Gomez-Baldo L, Osorio A, Bonifaci N, Fernandez-Ramires R, et al. Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol (2011) 9(11):e1001199. doi:10.1371/journal.pbio.1001199
48. Spranger S, Jeremias I, Wilde S, Leisegang M, Starck L, Mosetter B, et al. TCR-transgenic lymphocytes specific for HMMR/Rhamm limit tumor outgrowth in vivo. Blood (2012) 119(15):3440–9. doi:10.1182/blood-2011-06-357939
49. Herzog H, Darby K, Hort YJ, Shine J. Intron 17 of the human retinoblastoma susceptibility gene encodes an actively transcribed G protein-coupled receptor gene. Genome Res (1996) 6(9):858–61. doi:10.1101/gr.6.9.858
50. Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer (2003) 3(8):582–91. doi:10.1038/nrc1143
51. Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell (2009) 136(5):876–90. doi:10.1016/j.cell.2009.02.014
52. Bosanac I, Maun HR, Scales SJ, Wen X, Lingel A, Bazan JF, et al. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat Struct Mol Biol (2009) 16(7):691–7. doi:10.1038/nsmb.1632
53. Kobune M, Iyama S, Kikuchi S, Horiguchi H, Sato T, Murase K, et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J (2012) 2:e87. doi:10.1038/bcj.2012.36
54. Arturi F, Scarpelli D, Coco A, Sacco R, Bruno R, Filetti S, et al. Thyrotropin receptor mutations and thyroid hyperfunctioning adenomas ten years after their first discovery: unresolved questions. Thyroid (2003) 13(4):341–3. doi:10.1089/105072503321669811
55. Celano M, Sponziello M, Tallini G, Maggisano V, Bruno R, Dima M, et al. Increased expression of pro-angiogenic factors and vascularization in thyroid hyperfunctioning adenomas with and without TSH receptor activating mutations. Endocrine (2013) 43(1):147–53. doi:10.1007/s12020-012-9747-3
56. Loh ML, Zhang J, Harvey RC, Roberts K, Payne-Turner D, Kang H, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children’s Oncology Group TARGET Project. Blood (2013) 121(3):485–8. doi:10.1182/blood-2012-04-422691
57. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet (2009) 41(11):1243–6. doi:10.1038/ng.469
58. Fujiwara S, Yamashita Y, Choi YL, Watanabe H, Kurashina K, Soda M, et al. Transforming activity of purinergic receptor P2Y, G protein coupled, 8 revealed by retroviral expression screening. Leuk Lymphoma (2007) 48(5):978–86. doi:10.1080/10428190701225882
59. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A (2011) 108(50):20012–7. doi:10.1073/pnas.1116110108
60. Kemppinen AK, Kaprio J, Palotie A, Saarela J. Systematic review of genome-wide expression studies in multiple sclerosis. BMJ Open (2011) 1(1):e000053. doi:10.1136/bmjopen-2011-000053
61. Seon BK, Negoro S, Barcos MP. Monoclonal antibody that defines a unique human T-cell leukemia antigen. Proc Natl Acad Sci U S A (1983) 80(3):845–9. doi:10.1073/pnas.80.3.845
62. Seon BK, Negoro S, Barcos MP, Tebbi CK, Chervinsky D, Fukukawa T. Monoclonal antibody SN2 defining a human T cell leukemia-associated cell surface glycoprotein. J Immunol (1984) 132(4):2089–95.
63. Hara H, Seon BK. Complete suppression of in vivo growth of human leukemia cells by specific immunotoxins: nude mouse models. Proc Natl Acad Sci U S A (1987) 84(10):3390–4. doi:10.1073/pnas.84.10.3390
64. Bassani S, Cingolani LA, Valnegri P, Folci A, Zapata J, Gianfelice A, et al. The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron (2012) 73(6):1143–58. doi:10.1016/j.neuron.2012.01.021
65. Cantor JM, Ginsberg MH. CD98 at the crossroads of adaptive immunity and cancer. J Cell Sci (2012) 125(Pt 6):1373–82. doi:10.1242/jcs.096040
66. van der Welden VHJ, van Dongen JJM. MRD detection in acute lymphoblastic leukemia patients using Ig/TCR gene rearrangements as targets for real-time quantitative PCR. In: So CWE, editor. Leukemia: Methods and Protocols. New York: Humana Press (2008). p. 115–50. doi:10.1007/978-1-59745-418-6_7
67. Campana D, van Dongen JJ, Mehta A, Coustan-Smith E, Wolvers-Tettero IL, Ganeshaguru K, et al. Stages of T-cell receptor protein expression in T-cell acute lymphoblastic leukemia. Blood (1991) 77(7):1546–54.
68. Suzushima H, Hattori T, Asou N, Wang JX, Nishikawa K, Okubo T, et al. Discordant gene and surface expression of the T-cell receptor/CD3 complex in adult T-cell leukemia cells. Cancer Res (1991) 51(22):6084–8.
69. Campana D, Behm FG. Immunophenotyping of leukemia. J Immunol Methods (2000) 243(1–2):59–75. doi:10.1016/S0022-1759(00)00228-3
Keywords: immunotherapy, chimeric antigen receptors, antibody therapy, adoptive immunotherapy, pediatric leukemia, T-ALL, TALLA-1, HHIP
Citation: Orentas RJ, Nordlund J, He J, Sindiri S, Mackall C, Fry TJ and Khan J (2014) Bioinformatic description of immunotherapy targets for pediatric T-cell leukemia and the impact of normal gene sets used for comparison. Front. Oncol. 4:134. doi: 10.3389/fonc.2014.00134
Received: 15 March 2014; Accepted: 21 May 2014;
Published online: 10 June 2014.
Edited by:
Alan Wayne, Children’s Hospital Los Angeles, USAReviewed by:
Christian Capitini, University of Wisconsin, USADonald Williams Parsons, Texas Children’s Cancer Center, USA
Copyright: © 2014 Orentas, Nordlund, He, Sindiri, Mackall, Fry and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rimas J. Orentas, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 10 Center Dr. 1W3840, Bethesda, MD 20892, USA e-mail:cmltYXMub3JlbnRhc0BuaWguZ292