 Stephen E. Langabeer
Stephen E. Langabeer Karl Haslam
Karl Haslam Eibhlin Conneally2
Eibhlin Conneally2
- 1Cancer Molecular Diagnostics, St. James’s Hospital, Dublin, Ireland
- 2Department of Haematology, St. James’s Hospital, Dublin, Ireland
The Philadelphia chromosome-negative myeloproliferative neoplasms (MPN) are a group of clonal hematopoietic diseases characterized by bone marrow proliferation of one or more of the myeloid cell lineages with no marked alterations in cellular maturation. MPN classically comprise the clinically and pathologically related polycythemia vera (PV), essential thromobocythemia (ET), and primary myelofibrosis (PMF). Identification of the JAK2 V617F mutation has revolutionized the molecular diagnosis of MPN as this mutation is present in >95% of patients with PV and in 50% of patients with ET and PMF. In PV and ET, the potential exists for the disease to transform to a myelofibrotic stage and, together with PMF, transformation to acute myeloid leukemia (AML). The introduction of small molecule inhibitors that abrogate both normal and mutant JAK protein intracellular signaling in PMF has undoubtedly been a major advance in the treatment of these types of malignancies (1). Nevertheless, the only potentially curative option for these diseases, particularly PMF, is allogeneic stem cell transplantation (ASCT). Though ASCT was previously considered only for those patients with advanced or transformed disease, improvements in candidate patient selection and stratification, timing of transplantation, and conditioning regimens have significantly reduced the transplant related morbidity and increased the overall survival for MPN patients undergoing this procedure (2). However, as relapse is a major cause of treatment failure post-ASCT with salvage options limited and subsequent outcome relatively poor, identification of those patients at high-risk of relapse would be highly desirable, potentially enabling therapeutic intervention before overt relapse.
Conventionally, donor chimerism status is used to assess engraftment post-ASCT for hematological malignancies. Comparison of donor and recipient profiles can be achieved by either short tandem repeat (STR) analysis or quantitative polymerase chain reaction (qPCR) possessing sensitivities of 1–2%, with post-ASCT surveillance performed at one to three-monthly intervals. Post-ASCT monitoring utilizing additional patient-specific markers is most likely to provide a more beneficial, personalized profile with this approach already applied to many MPN patients undergoing ASCT. Early studies demonstrated the achievement of a molecular remission in JAK2 V617F-positive MPN post-ASCT using qualitative PCR (3). Development of more sensitive, JAK2 V617F-specific qPCR methodologies capable of detecting one mutant allele in 104 wild type copies has subsequently been shown to provide information on the rate of disease eradication and the identification of patients, at defined time points post-ASCT, at an increased risk of relapse (4–6). These sensitive JAK2 V617F qPCR assays have also been shown to be of value in triggering adoptive immunotherapies such as donor lymphocyte infusions that are able to elicit a graft-versus-tumor effect both preemptively and for salvage, post-relapse (7, 8). Even so, such qPCR methodologies require optimization across platforms and rigorous attention to reliability and sensitivity to ensure continued clinical utility (9). Mutations of MPL that encodes the receptor for thrombopoietin are also recurrent in ET and PMF but at a much lower frequency than the JAK2 V617F. In those reported MPL-mutation positive MPN who have undergone ASCT, rapid clearance of the MPL W515L mutation correlated well with peripheral blood counts and donor chimerism status (10).
More recently, whole exome sequencing has identified insertion and/or deletion mutation in CALR, a gene that encodes the endoplasmic reticulum-associated, calcium binding protein calreticulin. These mutations, which occur exclusively in CALR exon nine, appear not to be found in PV, and are present in up to 80% of ET and PMF patients who are JAK2 V617F-and MPL-negative (11, 12). As CALR mutations are likely initiating events in MPN pathogenesis, the possibility arises to assess these mutations as markers of residual disease in MPN patients post-ASCT. An initial assessment of CALR mutant allele burden, using semi-quantitative PCR fragment analysis, has shown that eradication and persistence of CALR mutations mirror donor chimerism status providing initial validation for future prospective serial monitoring of CALR mutations post-ASCT (13).
Apart from the common JAK2 V617F, MPL, and CALR mutations, several other recurrent mutations are observed in MPN but are also present in the myelodysplastic syndromes (MDS), MDS/MPN syndromes, and AML limiting their diagnostic utility but affording the potential for their use as markers of residual disease. These include mutations of TET2, ASXLI, CBL, NRAS, and EZH2. Rarely, mutations within some of these genes have been demonstrated in normal individuals (14) advocating caution when employed as markers of residual disease. Next generation sequencing (NGS) technologies now afford the opportunity to identify pathogenic mutations in multiple amplicons from these and many other genes simultaneously. A recent proof of principle study has utilized NGS to identify molecular mutations in MDS/MPN patients pre-ASCT with detection of the corresponding mutation in the post-ASCT period predictive of relapse (15). Whereas qPCR techniques provide relative quantitation of target amplicons, the emerging technology of digital PCR (dPCR) allows the absolute quantitation of mutant alleles with very small fold changes quantifiable. This approach has been applied to detect and monitor extremely low levels of BCR-ABL1 transcripts in chronic myeloid leukemia and acute lymphoblastic leukemia patients who have achieved deep molecular responses with tyrosine kinase inhibitor therapy (16, 17).
Whether NGS technologies will be of value in routine practice in the post-ASCT setting remains to be proven. Currently, limits of detection require improvement in order to approach those achieved by qPCR necessary for clinical application (Figure 1). Furthermore, issues such as intra- and inter-laboratory will require addressing for both NGS and dPCR; preliminary studies with NGS have demonstrated technical feasibility and concordance in the diagnostic setting (18). These emerging methodologies might soon be applied to identify and monitor mutational events enabling a distinct, personalized profile in those MPN patients undergoing ASCT. The goal would be the prompt recognition of those patients with an increased risk of relapse in which early therapeutic intervention is warranted, ultimately leading to gains in long-term disease-free and overall survival.
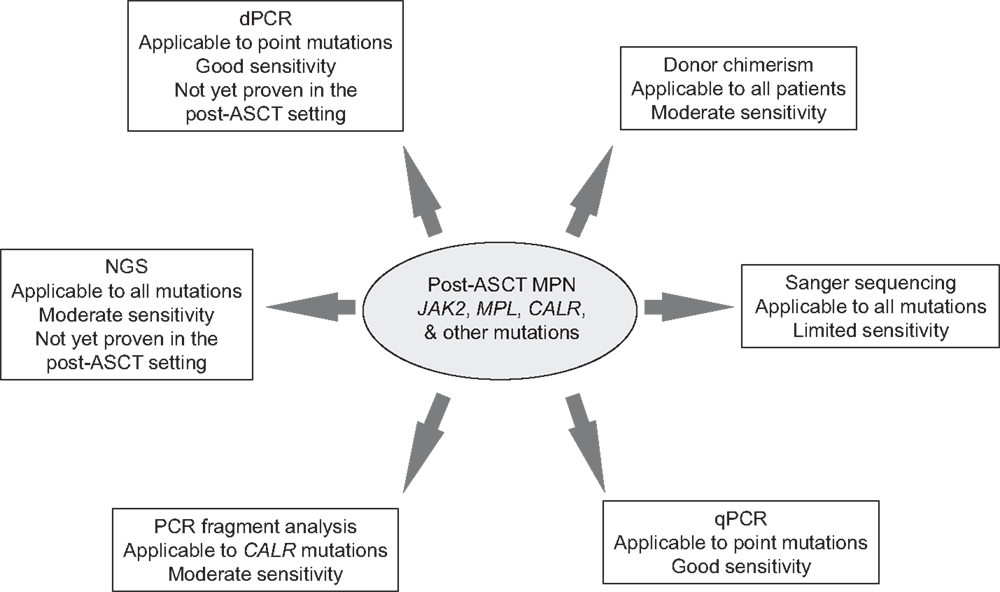
Figure 1. Summary of methodologies for monitoring MPN-specific mutations post-allogeneic stem cell transplantation. ASCT, allogeneic stem cell transplantation; MPN, myeloproliferative neoplasm; qPCR, quantitative polymerase chain reaction; NGS, next generation sequencing; and dPCR, digital polymerase chain reaction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Santos FP, Verstovsek S. Therapy with JAK2 inhibitors for myeloproliferative neoplasms. Hematol Oncol Clin North Am (2012) 26:1083–99. doi: 10.1016/j.hoc2012.07.008
2. Fleischman AG, Maziarz RT. Hematopoietic stem cell transplantation for myelofibrosis: where are we now? Curr Opin Hematol (2013) 20:130–6. doi:10.1097/MOH.0b013e32835dd862
3. Steckel NK, Koldehoff M, Ditschkowski M, Beelen DW, Elmaagacli AH. Use of the activating gene mutation (Val617Phe) JAK2 as a minimal residual disease marker in patients with myelofibrosis and myeloid metaplasia after allogeneic stem cell transplantation. Transplantation (2007) 83:1518–20. doi:10.1097/01.tp.0000263393.65764.f4
4. Alchalby H, Badbaran A, Zabelina T, Kobbe G, Hahn J, Wolff D, et al. Impact of JAK2 V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood (2010) 116:3572–81. doi:10.1182/blood-2009-12-260588
5. Lange T, Edelmann A, Siebolts U, Krahl R, Nehring C, Jäkel N, et al. JAK2 p.V617F allele burden in myeloproliferative neoplasms one month after allogeneic stem cell transplantation significantly predicts outcome and risk of relapse. Haematologica (2013) 98:722–8. doi:10.3324/haematol.2012.076901
6. Haslam K, Molloy KM, Conneally E, Langabeer SE. Evaluation of a JAK2 V617F quantitative PCR to monitor residual disease post-allogeneic hematopoietic stem cell transplantation for myeloproliferative neoplasms. Clin Chem Lab Med (2014) 52:e29–31. doi:10.1515/cclm-2013-0768
7. Benjamini O, Koren-Michowitz M, Amariglio N, Kroger N, Nagler A, Shimoni A. Relapse of post-polycythemia myelofibrosis after allogeneic stem cell transplantation in polycythemic phase: successful treatment with donor lymphocyte infusion directed by quantitative PCR test for V617F-JAK2 mutation. Leukemia (2008) 22:1961–3. doi:10.1038/leu.2008.215
8. Kröger N, Alchalby H, Klyuchnikov E, Badbaran A, Hidebrandt Y, Ayuk F, et al. JAK2 V617F-triggered pre-emptive and salvage adoptive immunotherapy with donor lymphocyte infusion in patients with myelofibrosis after allogeneic stem cell transplantation. Blood (2009) 113:1866–8. doi:10.1182/blood-2008-11-190975
9. Jovanovic JV, Ivey A, Vannucchi AM, Lippert E, Oppliger Leibundgut E, Cassinat B, et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2 V617F-associated myeloproliferative neoplasms: a joint European LeukemiaNet/MPN & MPNr-EuroNet (COST action BM0902) study. Leukemia (2013) 27:2032–9. doi:10.1038/leu.2013.219
10. Alchalby H, Badbaran A, Bock O, Fehse B, Bacher U, Zander AR, et al. Screening and monitoring of MPL W515L mutation with real-time PCR in patients with myelofibrosis undergoing allogeneic-SCT. Bone Marrow Transplant (2010) 45:1404–7. doi:10.1038/bmt.2009.367
11. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JB, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369:2379–90. doi:10.1056/NEJMoa1311347
12. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med (2013) 369:2391–405. doi:10.1056/NEJMoa1312542
13. Haslam K, Langabeer SE, Molloy K, McMullin MF, Conneally E. Assessment of CALR mutations in myelofibrosis patients, post-allogeneic stem cell transplantation. Br J Haematol (2014). [Epub ahead of print] doi:10.1111/bjh.12904
14. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet (2012) 44:1179–81. doi:10.1038/ng.2413
15. Fu Y, Schroeder T, Zabelina T, Badbaran A, Bacher U, Kobbe G, et al. Post-allogeneic monitoring with molecular markers detected by pre-transplant next-generation or Sanger sequencing predicts clinical relapse in patients with myelodysplastic/myeloproliferative neoplasms. Eur J Haematol (2014) 92:189–94. doi:10.1111/ejh.12223
16. Jennings LJ, George D, Czech J, Yu M, Joseph L. Detection and quantification of BCR-ABL1 fusion transcripts by droplet digital PCR. J Mol Diagn (2014) 16:174–9. doi:10.1016/j.jmoldx.2013.10.007
17. Iacobucci I, Lonetti A, Venturi C, Ferrari A, Papayannidis C, Ottaviani E, et al. Use of a high sensitive nanofluidic array for the detection of rare copies of BCR-ABL1 transcript in patients with Philadelphia-positive acute lymphoblastic leukemia in complete response. Leuk Res (2014) 38:581–5. doi:10.1016/j.leukres.2014.02.005
18. Kohlmann A, Klein HU, Weissman S, Bresolin S, Chaplin T, Cuppens H, et al. The Interlaboratory Robustness of Next-generation sequencing (IRON) study: a deep sequencing investigation of TET2, CBL and KRAS mutations by an international consortium involving 10 laboratories. Leukemia (2011) 25:1840–8. doi:10.1038/leu.2011.155
Keywords: myeloproliferative neoplasms, allogeneic stem cell transplantation, residual disease, JAK2 V617F, CALR, next generation sequencing, digital PCR
Citation: Langabeer SE, Haslam K and Conneally E (2014) Monitoring residual disease in the Ph-negative myeloproliferative neoplasms post-allogeneic stem cell transplantation: more mutations and more methodologies. Front. Oncol. 4:212. doi: 10.3389/fonc.2014.00212
Received: 30 May 2014; Accepted: 24 July 2014;
Published online: 08 August 2014.
Edited by:
David Grimwade, King’s College London School of Medicine, UKReviewed by:
David Grimwade, King’s College London School of Medicine, UKChristian Thiede, Technische Universität Dresden, Germany
Susanne Schnittger, MLL Munich Leukemia Laboratory, Germany
Donal McLornan, Guy’s and St Thomas’ NHS Foundation Trust, UK
Copyright: © 2014 Langabeer, Haslam and Conneally. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:c2xhbmdhYmVlckBzdGphbWVzLmll