 Sascha Venturelli1Tobias W. Sinnberg2Alexander Berger1Seema Noor2Mitchell Paul Levesque3Alexander Böcker4
Sascha Venturelli1Tobias W. Sinnberg2Alexander Berger1Seema Noor2Mitchell Paul Levesque3Alexander Böcker4 Heike Niessner2Ulrich M. Lauer1
Heike Niessner2Ulrich M. Lauer1 Michael Bitzer1Claus Garbe2
Michael Bitzer1Claus Garbe2 Christian Busch2*
Christian Busch2*- 1Department of Internal Medicine I, Medical University Hospital, Tuebingen, Germany
- 2Division of Dermatologic Oncology, Department of Dermatology and Allergology, University of Tuebingen, Tuebingen, Germany
- 3Department of Dermatology, University Hospital Zurich, Zurich, Switzerland
- 4Evotec AG, Hamburg, Germany
In recent years, increasing evidence has emerged demonstrating that high-dose ascorbate bears cytotoxic effects on cancer cells in vitro and in vivo, making ascorbate a pro-oxidative drug that catalyzes hydrogen peroxide production in tissues instead of acting as a radical scavenger. This anticancer effect of ascorbate is hypoxia-inducible factor-1α- and O2-dependent. However, whether the intracellular mechanisms governing this effect are modulated by epigenetic phenomena remains unknown. We treated human melanoma cells with physiological (200 μM) or pharmacological (8 mM) ascorbate for 1 h to record the impact on DNA methyltransferase (DNMT)-activity, histone deacetylases (HDACs), and microRNA (miRNA) expression after 12 h. The results were analyzed with the MIRUMIR online tool that estimates the power of miRNA to serve as potential biomarkers to predict survival of cancer patients. FACS cell-cycle analyses showed that 8 mM ascorbate shifted BLM melanoma cells toward the sub-G1 fraction starting at 12 h after an initial primary G2/M arrest, indicative for secondary apoptosis induction. In pharmacological doses, ascorbate inhibited the DNMT activity in nuclear extracts of MeWo and BLM melanoma cells, but did not inhibit human HDAC enzymes of classes I, II, and IV. The expression of 151 miRNAs was altered 12 h after ascorbate treatment of BLM cells in physiological or pharmacological doses. Pharmacological doses up-regulated 32 miRNAs (≥4-fold) mainly involved in tumor suppression and drug resistance in our preliminary miRNA screening array. The most prominently up-regulated miRNAs correlated with a significantly increased overall survival of breast cancer or nasopharyngeal carcinoma patients of the MIRUMIR database with high expression of the respective miRNA. Our results suggest a possible epigenetic signature of pharmacological doses of ascorbate in human melanoma cells and support further pre-clinical and possibly even clinical evaluation of ascorbate for melanoma therapy.
Introduction
In recent years, a large number of studies demonstrated that in pharmacological doses, ascorbic acid (ascorbate, vitamin C) in the low micromolar-range exerts cytotoxic effects on cancer cells in vitro and in vivo (1–3) via pro-oxidative mechanisms (4). This cytotoxicity is conducted by ascorbyl radicals and H2O2 being catalyzed by serum components (5). Hypoxic conditions and hypoxia-inducible factor-1α (HIF-1α)-signaling, both present in cancer metastases, confer resistance to the cancer cells toward ascorbate-induced cytotoxicity (5), while ascorbate inhibits HIF-1 with mechanisms of iron competition (6). This bears a strong clinical implication, since increased tumor ascorbate is associated with extended disease-free survival and decreased HIF-1 activation in human colorectal cancer (7). Likewise, low ascorbate levels are associated with increased HIF-1 activity and an aggressive tumor phenotype in endometrial cancer (8). Interestingly, ascorbate has a preferential toxicity toward melanoma cells (9). In B16, melanoma-bearing mice spontaneous lung metastasis is inhibited by sodium ascorbate supplementation in drinking water in mice fed a restricted diet (low in tyrosine and phenylalanine) (10). In vitro, the induction of a pro-oxidant state by ascorbate and a subsequent reduction in mitochondrial membrane potential are involved in a caspase-8-independent apoptotic pathway of B16F10 melanoma cells (11). Further, oral ascorbate supplementation modulates B16FO melanoma growth, metastasis, and inflammatory cytokine secretion as well as enhanced encapsulation of tumors in scorbutic (l-gulono-gamma lactone oxidase −/−) mice (12, 13).
In this respect, we recently demonstrated that patients afflicted with metastatic melanoma (stage IV) have lower plasma ascorbate levels compared to healthy controls and that polychemotherapy or immunotherapy further decreases plasma ascorbate levels in stage IV melanoma patients (14). However, the ascorbate concentration required for cytotoxicity in cancer cells can only be achieved via intravenous (i.v.) administration (15); up to 49 mM ascorbate blood peak concentrations are thus achievable by administration of 70 g/m2 (16). Yet, in recent phase I clinical trials, ascorbate failed to demonstrate a significant anticancer activity (16–19), although it enhanced chemosensitivity of ovarian cancer cells and reduced toxicity of chemotherapy (20). This obvious discrepancy between impressive anticancer efficacy in various pre-clinical models and lack of a reproducible anticancer activity in cancer patients clearly demonstrates that crucial (co-)factors executing the anticancer efficacy and an appropriate clinical treatment regimen remain to be deciphered. Due to the broad concentration range of ascorbate in humans and its numerous biochemical functions and effects, which seem to differ in somatic and malignantly transformed human cells (1), further research is needed for the understanding of the precise cytotoxic molecular impacts of ascorbate in cancer cells.
Many naturally occurring compounds and nutrients exert beneficial anticancer effects (e.g., suppression of tumor growth or induction of apoptosis), some of which are linked to modulation of epigenetic mechanisms (21–23). In general, epigenetic modifications influence gene expression without altering the DNA sequence and are therefore potentially reversible. Several epigenetic changes were distinguished, including histone acetylation and DNA methylation, and are currently investigated as potential targets for anticancer therapy (24). Both of the latter regulate the expression of microRNAs (miRNAs) and at the same time, are in part controlled by miRNAs via a regulatory circuit (25, 26). miRNAs correlate with clinical outcome in cancer patients in clinical studies (27). To test the possible relation between the expression of any given miRNA and the clinical outcome of cancer patients, the free online MIRUMIR tool, which performs survival analyses and draws Kaplan–Meier plots for any miRNA across several available data sets, was recently established (28).
In the present study, we provide novel evidence that in human metastatic melanoma cells only pharmacological doses of ascorbate induce substantial epigenetic changes. For 8 mM ascorbate, we detected a moderate inhibition of cellular histone deacetylase (HDAC) enzymes and a prominent DNA methyltransferase (DNMT) inhibition. Only pharmacological doses of ascorbate seemed to alter the miRNA expression profile by up-regulating 32 miRNAs mainly involved in tumor suppression and drug resistance, as demonstrated by preliminary miRNA chip expression analyses. Together, our results suggest that high doses of ascorbate only achievable in patients by i.v. administration might have epigenetic impacts on melanoma cells that might be beneficial in combination with classical or novel therapeutic anticancer approaches.
Materials and Methods
Cell Lines and Chemicals
Metastatic melanoma cell lines [MeWo: derived from a lymph node metastasis of a 78 years Caucasian donor; mutation status: BRAF wild-type, NRAS wild-type (29), BLM: subline of BRO melanoma cells isolated from lung metastases after subcutaneous inoculation of nude mice with BRO cells; mutation status: BRAF wild-type, NRAS mutated (30)] were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin and streptomycin, and 1% l-glutamine. All cell culture experiments were performed at 37°C and 5% CO2. The following chemical was used: injectable vitamin C solution (Pascorbin®, 150 mg ascorbate/1 ml injection solution, pH 7.0; Pascoe pharmazeutische Praeparate GmbH, Giessen, Germany).
Cell-Cycle Analysis
BLM cells were incubated with ascorbate at 8 mM. After 0–24 h (in 2 h intervals) the cells (1 × 106) were harvested, washed with cold PBS, fixed with 75% ethanol, and incubated at 4°C for at least 1 h. Cells were then centrifuged and washed twice in cold PBS. Intracellular DNA was labeled with propidium iodide solution [propidium iodide 40 mg/ml (Sigma) and RNase 100 mg/ml (Thermo Scientific) in PBS] and incubated at 4°C for 30 min in the dark. Cell cycle was analyzed using flow cytometry and FACSDiva software (BD Biosciences, Heidelberg, Germany).
In silico (Docking-) Analysis of Histone Deacetylase Inhibition
Ligand preparation
For this study, docking was performed into human HDACs 2, 4, 7, and 8 with trichostatin A (TSA) and the two major resonance structures of ascorbic acid (Figure 1). All ligands were prepared using the molecular operation environment (MOE, version 2007.09, Chemical Computing Group, Inc., Montreal, QC, Canada). 3D representations of the ligands were obtained by energy minimization (Rebuild3D function with preservation of existing chiral centers) using MM94x force field and a Born Solvation model without cutoff constraints. All other parameters were left at default.
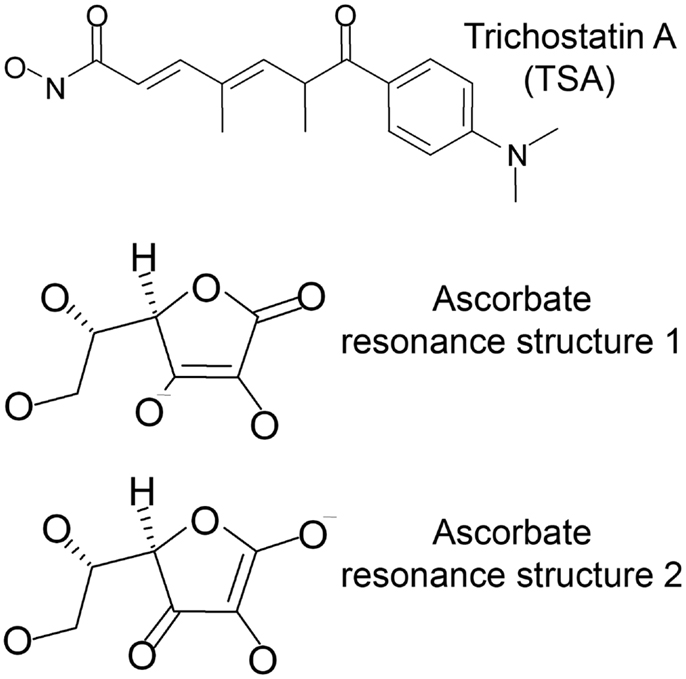
Figure 1. Ligands used for docking into the crystal structures of HDAC-2, -4, -7, and -8.
Protein preparation
Crystal structures of HDAC2 (PDB code: 3max), HDAC4 (PDB code: 2vqm), HDAC7 (PDB code: 3c10), and HDAC8 (PDB code: 1t64) were retrieved from the protein data bank1 (PDB) and loaded into MOE. The Protonate3D functionality was applied to assign the correct ionization state and geometries to the protein atoms and to add hydrogen atoms (31). For the final docking, water molecules were discarded.
Docking
Docking was performed using GOLD (version 4.1.2, The Cambridge Crystallographic Data Centre, Cambridge, UK). No additional protein preparation was applied. Binding sites were defined by all residues within 5 Å distance from the corresponding ligands in the crystal structure. Docking was performed using GoldScore as scoring function. All other parameters were left at default. Docking poses were analyzed in MOE. To optimize the ligand–receptor interactions energy minimizations were applied using MM94x force field and a Born Solvation model without cutoff constraints.
HDAC-Inhibitor Screening Assay
Determination of a possible HDAC-inhibitor activity of ascorbate was done by using the HDAC assay kit (Active Motif, Rixensart, Belgium). Ascorbate was diluted in assay buffer to the final concentrations of 5, 10, 20, 50, 100, 200 μM, and 8 mM. Assay was performed according to manufacturer’s protocol. Briefly, ascorbate was incubated with HeLa nuclear extract as a source of human HDACs for 2 h at 37°C and the developing time was set to 10 min. Each experiment was performed in triplicates and repeated three times.
HDAC-Inhibitor Profiling Assay
The HDAC profiling assay was performed on basis of fluorometric measurement by Scottish Biomedical (Scottish Biomedical, Glasgow, UK). The percentage inhibition values of 50 μM and 8 mM ascorbate against human HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, HDAC6, HDAC7, HDAC8, HDAC9, HDAC10, and HDAC11 was determined. Both concentrations of ascorbate were tested in two experiments, each in duplicates. TSA is used as a standard inhibitor by Scottish Biomedical for this assay and was deployed according to the information of the manufacturer in the following concentrations; HDAC1, HDAC2, HDAC3, HDAC6, HDAC10, and HDAC11 were tested at 10 nM TSA, HDAC8 at 100 nM and HDAC4, HDAC5, HDAC7, and HDAC9 were tested at 10 μM TSA.
Measurement of DNA Methyltransferase Activity
Nuclear extracts were prepared from BLM and MeWo melanoma cells (in triplicates) 12 h after 1 h treatment with ascorbate (untreated, 200 μM and 8 mM) by using the Nuclear Extract Kit (Active Motif) according to the procedure described by the manufacturer. DNMT activity was analyzed in the nuclear extracts with the DNMT activity/inhibition assay (Active Motif) according to the procedure described by the manufacturer.
miRNA Expression Analysis
microRNA was isolated from BLM cells using the miRNeasy kit (Qiagen, Hilden, Germany) according to the procedure described by the manufacturer. miRNA expression analysis was performed on BLM melanoma cells (five groups: untreated; 200 μM, 8 mM ascorbate treated, 4 and 12 h after the 1 h treatment, all in triplicates) using the human miRNA Microarray Release 14.0, 8x15K (Agilent, Waldbronn, Germany) based on Sanger miRbase (release 14.0). Two hundred nanograms of RNA were used per sample. The miRNA expression analysis was kindly performed at the Genomic Core Facility of the European Molecular Biology Laboratory (EMBL, Heidelberg, Germany) according to the supplier’s instructions. Evaluation of raw data generated at the Genomic Core Facility of the EMBL was performed as described previously (32).
MIRUMIR miRNA Analysis
Five highly up-regulated miRNA 12 h after 8 mM ascorbate treatment (miR-596, miR-630, miR-490, miR-375, and miR-708) were analyzed using the free online MIRUMIR database (28), which is incorporated into BioProfiling.de, an analytical portal for high-throughput cell biology2. The MIRUMIR database draws Kaplan–Meier plots for the submitted miRNAs after an implemented statistical procedure to account for multiple testing; P-values are generated automatically.
Statistical Analysis
Statistical analysis was performed with One-way ANOVA Dunnett’s multiple comparison test using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA, USA). According to One-way ANOVA Dunnett’s multiple comparison test, all ascorbate treatment groups were compared vs. vehicle/control. All values of P > 0.05 were defined as statistically not significant. The miRNA chip array was analyzed as described previously in detail (32).
Results
Pharmacological Ascorbate Induces Apoptosis in Human Metastatic BLM Melanoma Cells in a Time-Dependent Manner
As shown previously (5), pharmacological doses of ascorbate in the low millimolar-range induce cell death in human cancer cells. However, since in the latter publication we only performed end-point analyses 24 h after incubation of the cells with ascorbate, for the current project we exposed BLM cells to pharmacological 8 mM ascorbate for 1 h, and the cells were ethanol-fixed every 2 h for 24 h to closely monitor cell-cycle alterations at 2 h intervals over 24 h. The cell cycle was analyzed with FACS after staining of the cells with propidium iodide. We observed that the G2/M fraction of cells initially steadily increased starting at 2 h after ascorbate exposure, while 12 h post-treatment a subsequent increase of the sub-G1 fraction of DNA fragmented cells was evident (indicative for apoptotic cells). At 20 h post-treatment, the cell cycle was already completely shifted toward the sub-G1 fraction (Figure 2). At 12 h post ascorbate exposure only a small percentage of the BLM cells were shifted toward the sub-G1 fraction of cells. Therefore, the 12 h time point after ascorbate exposure was chosen for the following experiments.
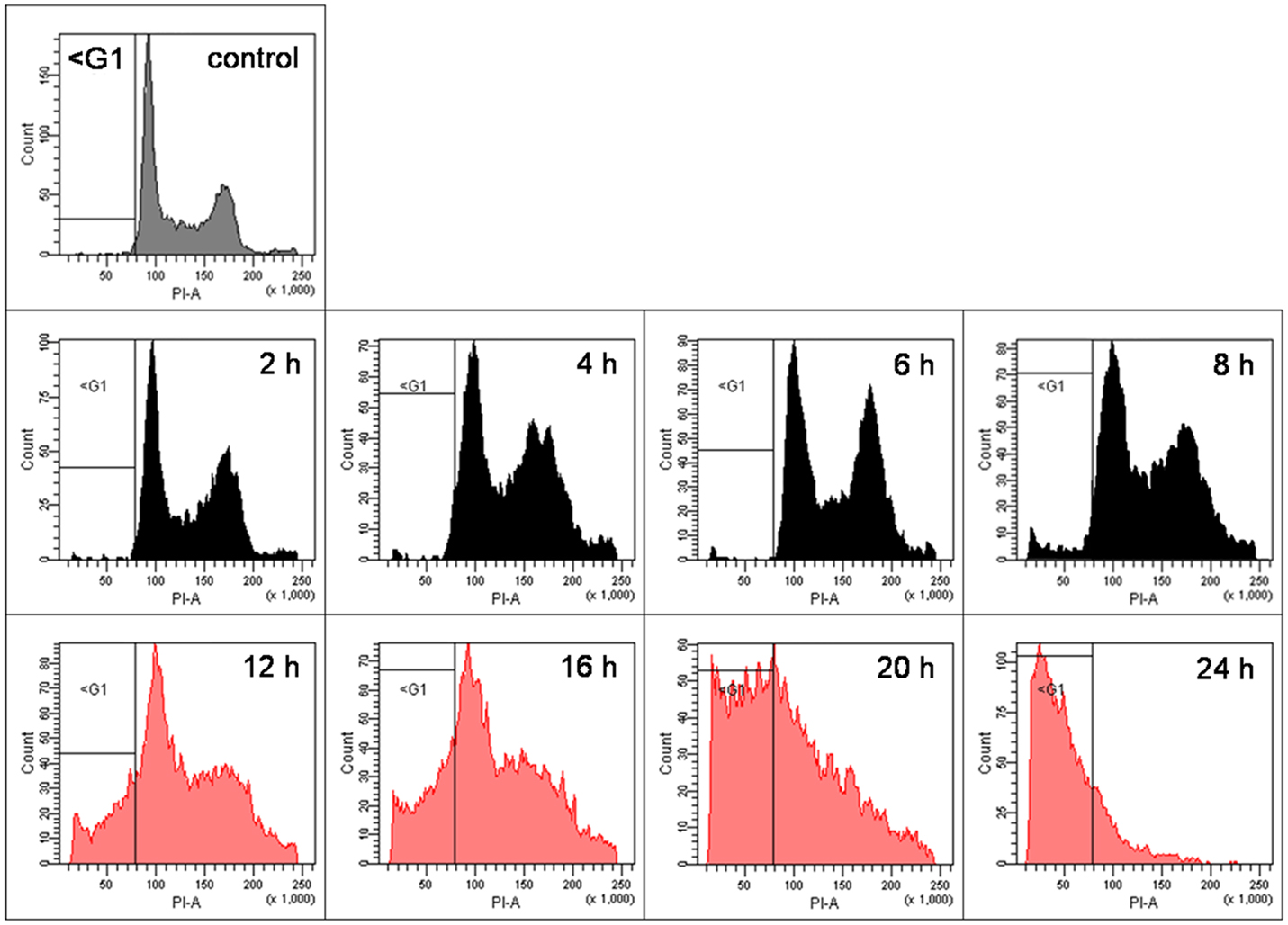
Figure 2. Pharmacological ascorbate induces apoptosis in human metastatic BLM melanoma cells in a time-dependent manner. BLM cells were treated with 8 mM ascorbate for 1 h. Cells were ethanol-fixed every 2 h for 24 h, stained with propidium iodide, and cell cycle was analyzed with FACS. Depicted are the FACS plots for the untreated control cells and for 2, 4, 6, 8, 12, 16, 20, and 24 h after ascorbate exposure. The G2/M fraction of cells steadily increased from 2 to 8 h, while 12 h post-treatment a prominent increase of the sub-G1 fraction of DNA fragmented cells started (indicative for apoptosis induction). At 20 h post-treatment, the cell cycle was already completely shifted toward the sub-G1 fraction. Gray colored graphs indicate the control cells; black colored graphs (2–8 h) the initial shift into G2/M phase; red colored graphs (12–24 h) the subsequent shift into sub-G1 fraction.
Pharmacological Ascorbate Moderately Inhibits Histone Deacetylases
Epigenetic modifications such as histone acetylation or DNA methylation play an important role in cancer development and progression (24). Whether ascorbate has epigenetic effects on cancer cells has not been investigated yet. However, since reactive oxygen species (ROS) induce hypermethylation of the E-cadherin promoter regions in hepatoma cells (33), we hypothesized that ascorbate, a pro-oxidative radical-inducing drug in pharmacological concentrations (2, 5), might bear similar epigenetic effects on melanoma cells. To verify our hypothesis, we therefore analyzed two major epigenetic mechanisms, inhibition of DNMTs and HDACs.
First we analyzed, if ascorbate possessed an HDAC-inhibitory activity. To this end, an in silico docking analysis was performed. The in silico analysis revealed that ascorbate was able to penetrate into the binding pocket of class I and II HDACs and to interact with the zinc ion, two issues that are important for HDAC inhibitors (Figures 3A,B). Calculated GoldScores representing the binding affinity, supported these first data, leading us to the assumption that ascorbate, in a given setup, could be a similar strong binding partner to the binding pockets as the well-known HDAC-inhibitor TSA (Figure 3C) (22). Due to the positive results obtained by the docking experiments, we next performed a cell free HDAC-inhibitor assay. In this assay, nuclear extract of the well-characterized human HeLa cell line was used as HDAC enzyme source. The results showed that in contrast to the in silico docking data, in vitro only a marginal inhibitory activity of ascorbate on HDACs could be detected (Figure 4A). The latter data could be verified by a profiling of all known HDAC enzymes of class I, II, and IV. As before, TSA was used as reference HDAC inhibitor in this experimental setting. In line with the HDAC-inhibitor assay above, neither the physiological 50 μM nor the pharmacologic 8 mM ascorbate showed a significant inhibition of the 11 conserved human HDACs tested when compared to the potent inhibition mediated by TSA (Figure 4B).
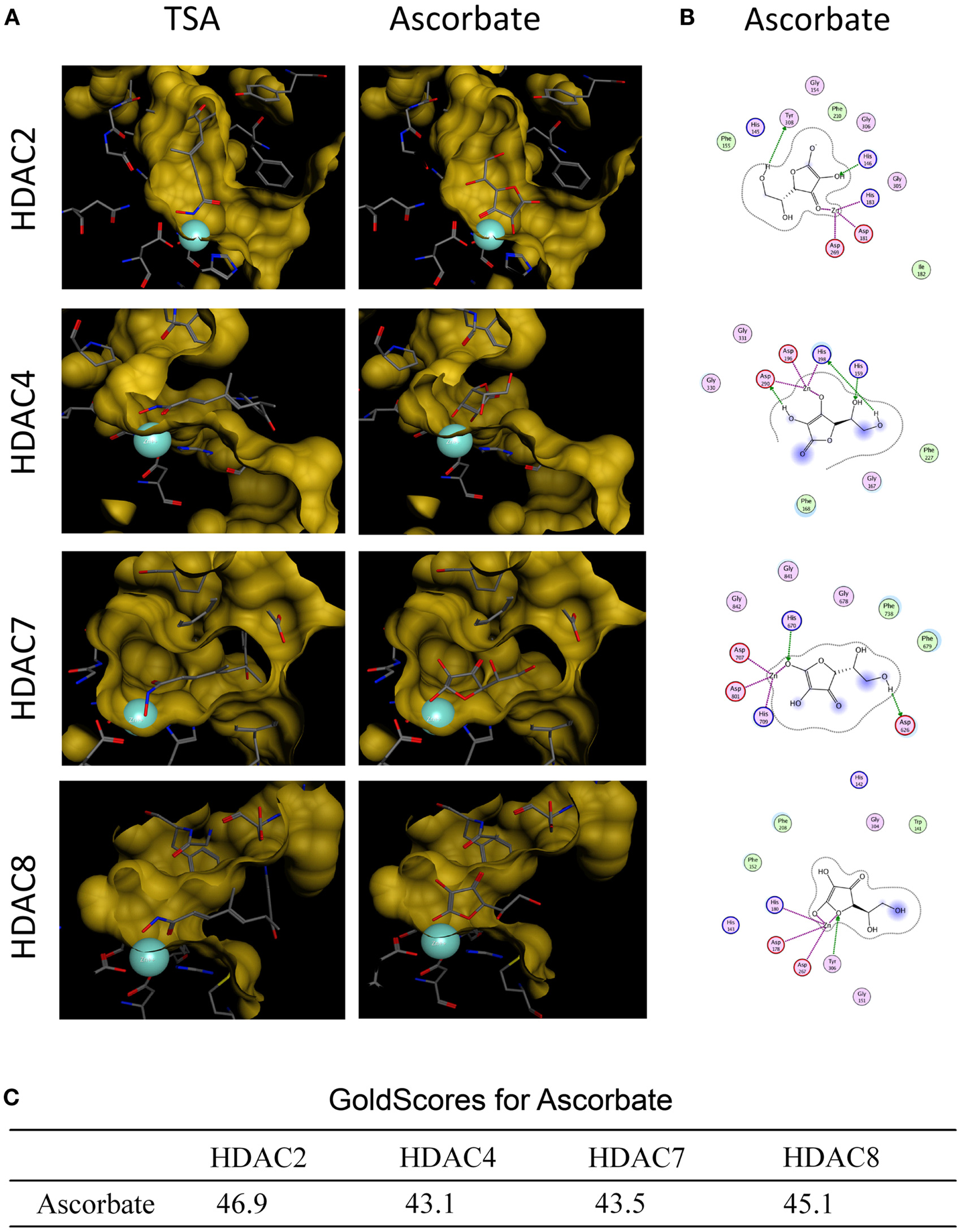
Figure 3. In silico docking analysis of ascorbate and HDACs. (A) In silico docking analyses of ascorbate and HDAC2, HDAC4, HDAC7, and HDAC8. Trichostatin A (TSA) served as positive control. The analysis demonstrates the fitting of ascorbate into each HDAC binding pocket and the ability to interact with the HDAC-derived zinc ion (turquoise sphere) of the catalytic center. (B) 2D depiction of ligand is shown along with interacting amino acids. Green circles represent greasy, purple circles polar, red circles acidic, and blue circles basic amino acids. HDAC contacts are depicted by a blue half moon around the amino acids. Blue arrows represent backbone acceptors, green ones depict side chain acceptors and side chain donors. Green benzoyl rings with a “+” describe an arene–cation binding, two benzoyl rings an arene–arene binding. Areas with a blue background are exposed to the ligand. The purple dotted lines represent metal contact. (C) Docking analysis of ascorbate in the individual HDAC binding pockets were performed using GOLD software (version 4.1.2) and MOE.
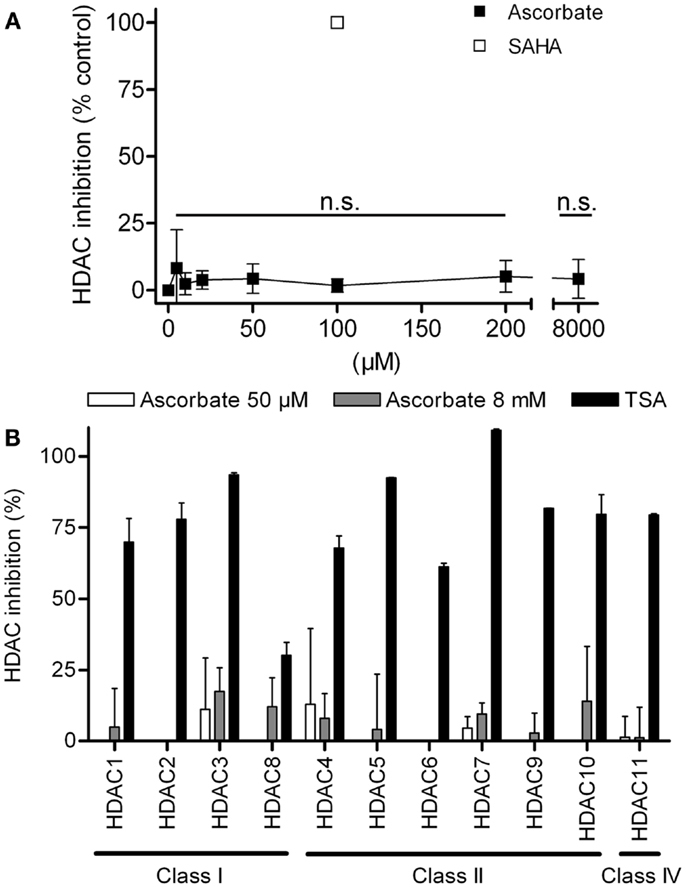
Figure 4. Ascorbate does not act as histone deacytelase inhibitor (HDACi). (A) Overall HDAC inhibition in cellular extracts of the human cell line HeLa by increasing concentrations of ascorbate (5–8000 μM). As reference inhibitor 100 μM suberoylanilide hydroxamic acid (SAHA) was used. Every concentration was tested three times in triplicates. (B) Specific fluorometric profiling assay using recombinant human HDACs of classes I, II, and IV. Specific inhibition values were generated for the treatment with 50 μM and 8 mM ascorbate. Inhibition values for every HDAC were yielded by two experiments, each performed in duplicates. Shown are mean ± SD. One-way ANOVA Dunnett’s multiple comparison test, n.s. indicates not significant.
Pharmacological Ascorbate Inhibits DNA Methyltransferases
Due to the negative results of the HDAC inhibition assays, we next investigated if ascorbate had a DNMT inhibitory activity in the human metastatic MeWo and BLM melanoma cells. Twelve hours after treatment of the respective melanoma cells with either physiological 200 μM or pharmacological 8 mM ascorbate for 1 h, a nuclear extract was prepared and the amount of methylated DNA was measured. The experiments showed that the physiological concentration of ascorbate (200 μM) increased the DNMT activity in a moderate fashion in both cell lines. Interestingly, the pharmacological concentration of 8 mM ascorbate clearly inhibited DNMTs in both cell lines by up to 40% (Figure 5).
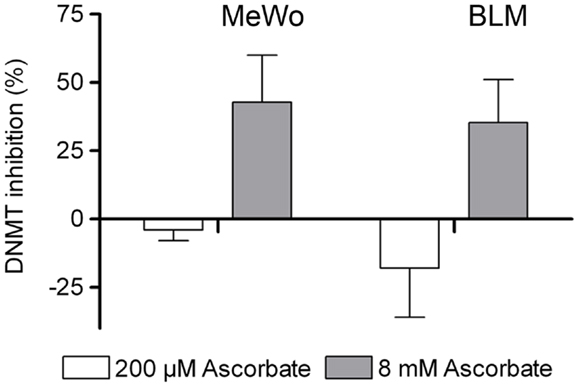
Figure 5. Ascorbate inhibits DNA methyltransferase (DNMT) activity in melanoma cells. Global DNMT activity was determined in nuclear extracts of MeWo and BLM cells 12 h after 1 h treatment with 200 μM or 8 mM ascorbate. Treatment with 200 μM ascorbate increases DNMT activity in MeWo and BLM cells by 4 and 18%, respectively, and 8 mM ascorbate inhibits DNMT activity by 43 and 35%, respectively.
Pharmacological Ascorbate Strongly Modifies miRNA Expression
The novel classification of pharmacological ascorbate as DNMT inhibitor rose the question if the inhibition of DNMTs within the melanoma cells subsequently had an impact on the cellular miRNA expression profile. To answer this question, we performed miRNA expression chip analysis. The investigation revealed that the expression of 151 miRNAs was significantly altered when comparing ascorbate treatment for 1 h at 8 mM with treatment at 200 μM (Figure 6). The IC50 of ascorbate is in the low millimolar range in melanoma cells (5). Since in this study, we mainly investigated the role of epigenetic mechanisms accompanying ascorbate-induced cytotoxicity, we did not focus on miRNA expression changes induced by physiological ascorbate (200 μM), which bears no cytotoxic effect on melanoma cells. Comparing the impact of the pharmacological dose of ascorbate (8 mM) with the impact of ascorbate at the maximum physiological plasma condition of 200 μM, after 12 h a significant up-regulation of 32 miRNAs (4- to 38-fold) could be stated. Interestingly, 14 of these miRNAs (miR-596, miR-630, miR-422a, miR-490-5p, miR-375, miR-708, miR-345, miR-125b-2, miR-516a-3p, miR-135a, miR-1228, miR-1915, miR-134, and miR-663) have established roles in tumor suppression and drug resistance, while 5 miRNAs (miR-630, miR-375, miR-345, miR-1228, and miR-134) are known to inhibit epithelial–mesenchymal transition and invasion in cancer cells. Eleven of the up-regulated miRNAs (miR-887, miR-583, miR-662, miR-1973, miR-718, miR-1268, miR-2117, miR-614, miR-617, miR-1972, and miR-1181) have no reported functions in cancer cells yet. A detailed list of the 32 up-regulated miRNAs, their reported expression profile in cancer, functions and predicted RNA-targets are given in Table 1. To further analyze a possible clinical significance of the up-regulated miRNA upon ascorbate administration, we screened the free MIRUMIR online database (28), which tests any given miRNA as biomarker to predict survival in available clinical data sets that cover more than 800 cancer patients. We were able to find a strong correlation of high expression of miR-596, miR-630, miR-490, miR-375, and miR-708 with overall long-term survival in breast cancer or nasopharyngeal carcinoma patients when compared to low expression of the respective miRNA in the same cohorts of patients. The Kaplan–Meier plots as depicted in Figure 7 were automatically generated by MIUMIR upon submission of the respective miRNAs (miR-596, miR-630, miR-490, miR-375, and miR-708).
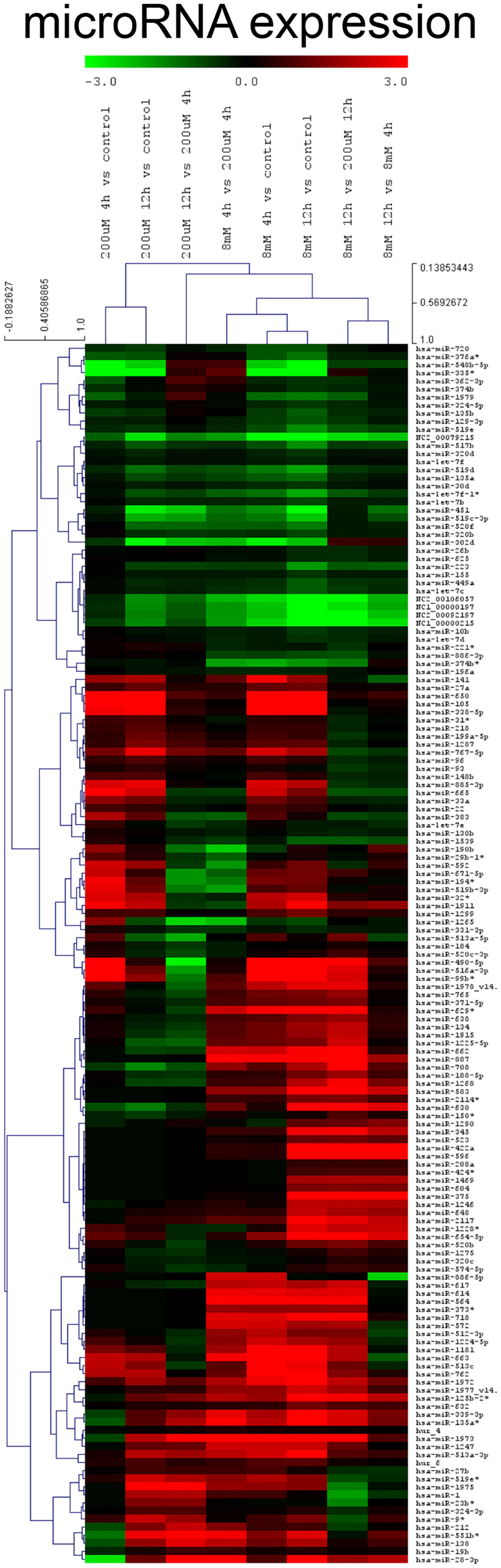
Figure 6. Ascorbate alters the expression of miRNA in melanoma cells. A miRNA expression chip analysis was performed on human metastatic BLM melanoma cells (5 groups: untreated, 200 μM ascorbate, 8 mM ascorbate, both 4 and 12 h after ascorbate exposure; all in triplicates) using the human miRNA Microarray Release 14.0, 8x15K (Agilent Technologies) based on Sanger miRbase (release 14.0). A total of 151 miRNAs were differentially expressed in response to ascorbate. Incubation of BLM cells with 8 mM ascorbate for 1 h up-regulated 32 miRNAs (4- to 38-fold) involved in tumor suppression and drug resistance compared to physiological (200 μM) ascorbate after 12 h.

Table 1. miRNA expression profile of BLM melanoma cells after ascorbate treatment.
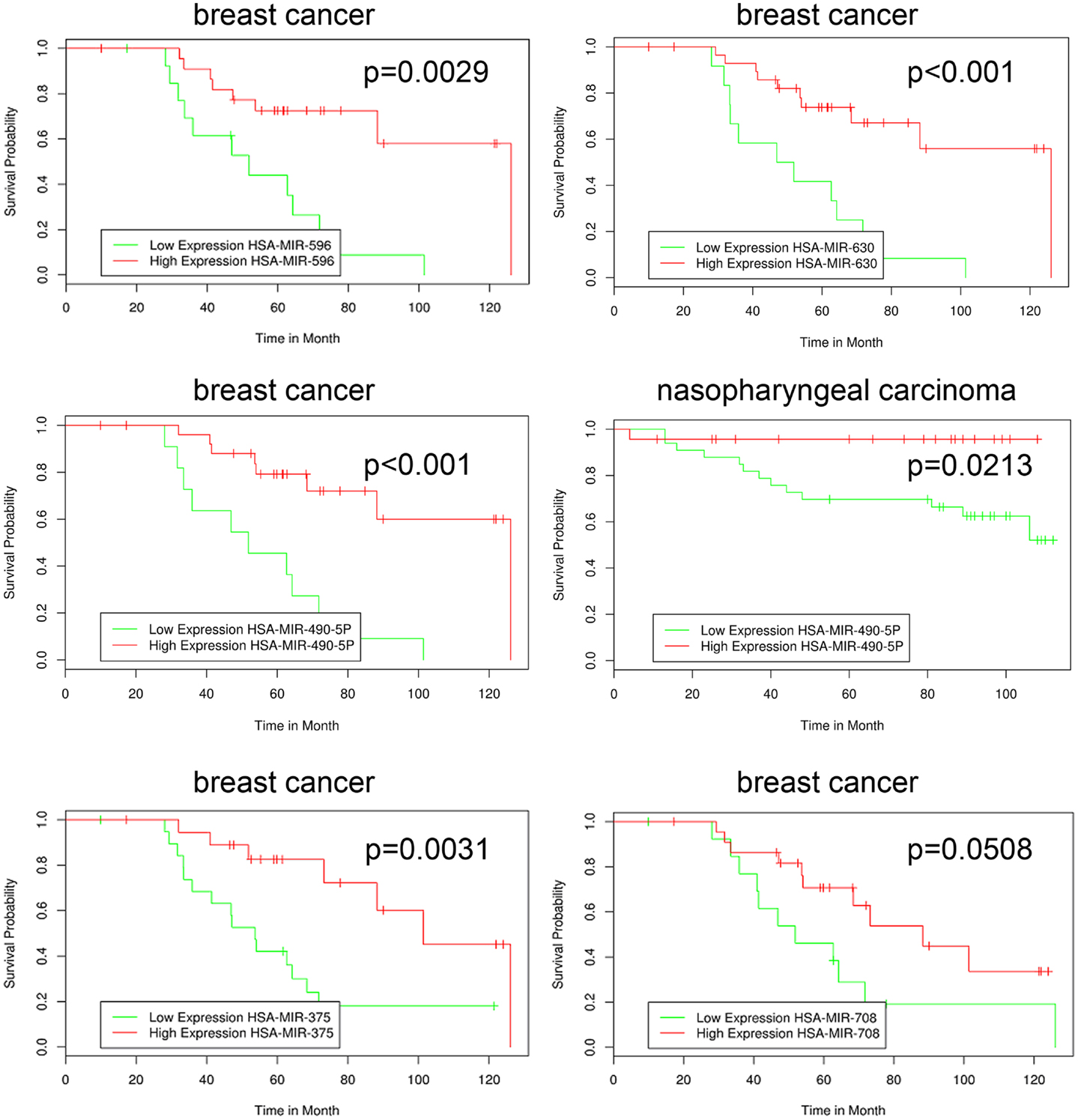
Figure 7. High expression of miRNA up-regulated by ascorbate in melanoma cells correlates with increased overall survival in cancer patients. The MIRUMIR online database was screened to detect an impact of the up-regulated miRNA (compare Figure 6) on cancer patient survival. The depicted Kaplan–Meier plots are automatically drawn by MIRUMIR upon submission of the respective miRNAs. A significant correlation of high expression of miR-596, miR-630, miR-490, miR-375, and miR-708 with overall long-term survival in breast cancer (GEO dataset IDs: GSE37405 and GSE37405) or nasopharyngeal carcinoma patients (GEO dataset ID: GSE36682) was observed when compared to low expression of the respective miRNA.
Discussion
Cutaneous melanoma is an aggressive malignancy with increasing incidence. Up to now curative therapies for stage IV patients, which have an overall survival of 9–14 months, are lacking (81–83). Therefore, in spite of the recently approved targeted drugs (BRAF- or MEK-inhibitors) or available immunotherapies (anti-CTLA-4- or anti-PD1-antibodies) novel therapeutic strategies are still urgently needed. Numerous alternative treatment approaches are therefore currently investigated in the context of cancer therapy. A highly controversial and also emotionally discussed approach, the application of ascorbate in pharmacological doses, was first proposed and described by Pauling and Cameron in the 1970s (84, 85). Although their original hypothesis concerning the mode of action was incorrect (encapsulation of tumors by collagen induction), in the mean time it became evident that the anticancer effects of ascorbate are principally mediated by induction of radicals (1, 4). Furthermore, the crucial need for i.v. administration instead of oral supplementation to assure a sufficient, cytotoxic drug concentration has widely been acknowledged (86). This partly explains why the clinical observations of Pauling and Cameron could not be reproduced in clinical trials conducted in the 1980s. We recently observed functional effects of ascorbate on survival of melanoma cells (5). In the latter paper, we showed the cytotoxic effect of ascorbate on all 60 cancer cell lines of the NCI60 panel of cancer cells, which includes 9 human melanoma cell lines. The IC50 of ascorbate was 3.1 mM for all melanoma cell lines (0.2–8.5 mM); the overall IC50 of all 60 cancer cell lines was 4.5 mM. 8 mM ascorbate generated a high amount of intracellular peroxide radicals in LOX-IMVI melanoma cells leading to an increased percentage of sub-G1 (apoptotic) cells determined by FACS. In the melanoma cell lines, ascorbate treatment at the individual IC50 concentrations decreased GLUT-1 expression (pro-survival HIF-1α downstream target). In line, in the present paper, we observed a similar time-dependent prominent increase of BLM melanoma cells in the sub-G1 fraction, beginning at 12 h after incubation of the cells with 8 mM ascorbate.
Surprisingly, despite the abundance of scientific reports elucidating the mechanistic background of pharmacological ascorbate-induced cancer cell cytotoxicity, independent of the cellular mutation status, the successful transfer into the clinics has failed so far. The most likely explanations for this discrepancy are (i) the observation of induced ascorbate resistance by exogenous factors such as hypoxia present in metastatic tissue of cancer patients, which clinically has not been taken into consideration yet (5), and (ii) the existence of possible additional endogenous mechanistic features driven by ascorbate. Such additional effects might severely influence its cytotoxic efficacy for the treatment of cancer.
In the last few years, increasing evidence demonstrated that natural products and edibles harbor epigenetic activities, which might be beneficial for cancer therapy (24, 87). Epigenetic alterations that induce multiple changes in gene expression profiles are substantial features of cancer (88–90). Several epigenetic mechanisms have been described so far and the investigation of this complex molecular machinery is ongoing. Two principal mechanisms that cause a silencing of control genes and mediate tumor formation as well as tumor progression are the modulation of HDACs and the regulation of DNMTs. Therefore, in the present study, we investigated possible epigenetic impacts of ascorbate on melanoma cells to gain a more profound understanding of this alternative therapeutic approach widely used in complimentary medicine (91).
In particular, our in silico findings showed that ascorbate fits into the catalytic pocket of human HDACs and interacts with the zinc ion as well as other residues of the active site making it an interesting candidate that could act as a histone deacytelase inhibitor (HDACi). The obtained GoldScore values even assigned ascorbate to be a similar potent inhibitor as the well-known HDACi TSA. To verify these results, different HDAC inhibition assays were performed. However, neither the inhibition experiments with nuclear extracts nor the extensive profiling study could prove that ascorbate substantially inhibits classical human HDACs in vitro. Our second attempt was to test, if ascorbate can inhibit DNMTs within melanoma cells. Indeed, we could show that physiological levels of ascorbate in the micromolar range have no or a slightly activating activity on DNMTs, whereas pharmacological levels of ascorbate in the millimolar-range (achievable in patients via i.v.-administration) inhibit cellular DNMTs in melanoma cell lines. Based on these results, we conclude that ascorbate bears a novel DNMT inhibitory activity in high concentrations, but no HDAC-inhibitory potential.
Due to the newfound epigenetic activity of pharmacological ascorbate on DNMTs we next analyzed its impact on expression of miRNAs. The results of the chip analysis highlighted that upon stimulation of melanoma cells with either physiological or pharmacological ascorbate, a total of 151 miRNAs were differentially regulated in comparison to the untreated cells. Most interestingly, by comparing the melanoma cells incubated with the maximum physiological dose of 200 μM to cells pre-conditioned with pharmacological 8 mM to specifically analyze the impact of cytotoxicity-inducing drug concentrations, it became obvious that the majority of the up-regulated miRNAs are known to be involved in tumor suppression, cancer cell drug resistance, or inhibition of migration and invasion through inhibition of epithelial mesenchymal transition. The last-mentioned is a typical morphogenetic feature in the developing embryo untimely reappearing in cancer cells in general and in melanoma cells, in particular, due to their neural crest origin (92–94). Since the miRNAs found to be up-regulated upon ascorbate stimulation in the chip analysis were not validated by additional real-time PCR analyses, definite conclusions or a clear clinical significance cannot be drawn of the rather preliminary results yet. However, the up-regulated expression of miRNA due to ascorbate in melanoma cells correlated with an increased overall survival of breast cancer or nasopharyngeal carcinoma patients of the MIRUMIR database (28) with high expression of the respective miRNAs, therefore, suggesting a possible beneficial clinical relevance of the specific miRNA up-regulation by ascorbate. At this point, our results therefore allow us to generate the hypothesis that pharmacological ascorbate might modify the miRNA signature of melanoma cells, which subsequently might be beneficial for overall survival of melanoma patients in analogy to the endogenous miRNA expression profiles of breast cancer and nasopharyngeal carcinoma patients with either short- or long-term survival.
Considering the observed preliminary up-regulation of specific miRNAs by ascorbate (possibly via its novel DNMT inhibitory activity) governing a broad spectrum of tumor-suppressive effects including apoptosis induction, antiproliferative activity, and decrease of cancer cell invasion, this novel epigenetic signature of ascorbate might open the door for the exploration of ascorbate in combination with other classical or even epigenetically active molecules for cancer therapy (90) and therefore warrants further pre-clinical and clinical investigation.
Author Contributions
Tobias W. Sinnberg, Alexander Berger, and Seema Noor: acquisition, analysis, and interpretation of data for the work; revising of the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work. Mitchell Paul Levesque, Alexander Böcker, Heike Niessner, Ulrich M. Lauer, Michael Bitzer, and Claus Garbe: acquisition and analysis of data; revising of the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work. Sascha Venturelli and Christian Busch: design of the work; analysis and interpretation of data; drafting of the manuscript; final approval of the version to be published; agreement to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Eve Holtorf for technical assistance. This work was supported by grants from the DFG SFB 773: “understanding and overcoming drug resistance of solid tumors” to Claus Garbe, Ulrich M. Lauer, Michael Bitzer, and Christian Busch. Sascha Venturelli was supported in part by the DFG (VE671/2-1) and Innovation Grant of the University Tuebingen. Tobias W. Sinnberg and Alexander Berger were supported by the fortune program of the University Clinic Tuebingen (2198-0-0 and 1966-0-0). Christian Busch and Sascha Venturelli received a research grant from the Wissenschatsfoerderung der Deutschen Brauwirtschaft e.V. (B103). We further acknowledge support by DFG and Open Access Publishing Fund of Tuebingen University. The sponsors had no involvement in the study design, in the collection, analysis, and interpretation of data, in the writing of the manuscript, and in the decision to submit the manuscript for publication.
Abbreviations
DNMT, DNA methyltransferase; DNMTi, DNA methyltransferase inhibitor; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; i.v., intravenous; miRNA, microRNA; ROS, reactive oxygen species; SAHA, suberoylanilide hydroxamic acid; TSA, trichostatin A.
Footnotes
References
1. Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A (2005) 102:13604–9. doi:10.1073/pnas.0506390102
2. Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A (2008) 105:11105–9. doi:10.1073/pnas.0804226105
3. Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res (2010) 16:509–20. doi:10.1158/1078-0432.CCR-09-1713
4. Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A (2007) 104:8749–54. doi:10.1073/pnas.0702854104
5. Sinnberg T, Noor S, Venturelli S, Berger A, Schuler P, Garbe C, et al. The ROS-induced cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer cell lines. J Cell Mol Med (2014) 18:530–41. doi:10.1111/jcmm.12207
6. Kuiper C, Dachs GU, Currie MJ, Vissers MC. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic Biol Med (2014) 69:308–17. doi:10.1016/j.freeradbiomed.2014.01.033
7. Kuiper C, Dachs GU, Munn D, Currie MJ, Robinson BA, Pearson JF, et al. Increased tumor ascorbate is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in human colorectal cancer. Front Oncol (2014) 4:10. doi:10.3389/fonc.2014.00010
8. Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res (2010) 70:5749–58. doi:10.1158/0008-5472.CAN-10-0263
9. Bram S, Froussard P, Guichard M, Jasmin C, Augery Y, Sinoussi-Barre F, et al. Vitamin C preferential toxicity for malignant melanoma cells. Nature (1980) 284:629–31. doi:10.1038/284629a0
10. Meadows GG, Pierson HF, Abdallah RM. Ascorbate in the treatment of experimental transplanted melanoma. Am J Clin Nutr (1991) 54:1284S–91S.
11. Kang JS, Cho D, Kim YI, Hahm E, Yang Y, Kim D, et al. L-ascorbic acid (vitamin C) induces the apoptosis of B16 murine melanoma cells via a caspase-8-independent pathway. Cancer Immunol Immunother (2003) 52:693–8. doi:10.1007/s00262-003-0407-6
12. Cha J, Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp Oncol (2011) 33:226–30.
13. Cha J, Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int J Oncol (2013) 42:55–64. doi:10.3892/ijo.2012.1712
14. Schleich T, Rodemeister S, Venturelli S, Sinnberg T, Garbe C, Busch C. Decreased plasma ascorbate levels in stage IV melanoma patients. Metab Nutr Oncol (2013). doi:10.1055/s-0033-1348256
15. Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic Biol Med (2009) 47:32–40. doi:10.1016/j.freeradbiomed.2009.02.016
16. Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol (2013) 72:139–46. doi:10.1007/s00280-013-2179-9
17. Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol (2008) 19:1969–74. doi:10.1093/annonc/mdn377
18. Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol (2013) 71:765–75. doi:10.1007/s00280-013-2070-8
19. Riordan HD, Casciari JJ, González MJ, Riordan NH, Miranda-Massari JR, Taylor P, et al. A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P R Health Sci J (2005) 24:269–76.
20. Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med (2014) 6:222ra18. doi:10.1126/scitranslmed.3007154
21. Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res (2011) 55:999–1009. doi:10.1002/mnfr.201000547
22. Berger A, Venturelli S, Kallnischkies M, Böcker A, Busch C, Weiland T, et al. Kaempferol, a new nutrition-derived pan-inhibitor of human histone deacetylases. J Nutr Biochem (2013) 24:977–85. doi:10.1016/j.jnutbio.2012.07.001
23. Venturelli S, Berger A, Böcker A, Busch C, Weiland T, Noor S, et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone proteins in human-derived hepatoblastoma cells. PLoS One (2013) 8:e73097. doi:10.1371/journal.pone.0073097
24. Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther (2009) 8:1409–20. doi:10.1158/1535-7163.MCT-08-0860
25. Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res (2007) 61:24R–9R. doi:10.1203/pdr.0b013e3180457684
26. Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. microRNAs and epigenetics. FEBS J (2011) 278:1598–609. doi:10.1111/j.1742-4658.2011.08089.x
27. Lovat F, Valeri N, Croce CM. microRNAs in the pathogenesis of cancer. Semin Oncol (2011) 38:724–33. doi:10.1053/j.seminoncol.2011.08.006
28. Antonov AV, Knight RA, Melino G, Barlev NA, Tsvetkov PO. MIRUMIR: an online tool to test microRNAs as biomarkers to predict survival in cancer using multiple clinical data sets. Cell Death Differ (2013) 20:367. doi:10.1038/cdd.2012.137
29. Carey TE, Takahashi T, Resnick LA, Oettgen HF, Old LJ. Cell surface antigens of human malignant melanoma: mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc Natl Acad Sci U S A (1976) 73:3278–82.
30. Lockshin A, Giovanella BC, De Ipolyi PD, Williams LJ Jr, Mendoza JT, Yim SO, et al. Exceptional lethality for nude mice of cells derived from a primary human melanoma. Cancer Res (1985) 45:345–50.
31. Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins (2009) 75:187–205. doi:10.1002/prot.22234
32. Reischauer S. Lgl2 executes its function as a tumor suppressor by regulating ErbB signaling in the zebrafish epidermis. PLoS Genet (2009) 5:e1000720. doi:10.1371/journal.pgen.1000720
33. Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK, et al. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology (2008) 135:2128–40. doi:10.1053/j.gastro.2008.07.027
34. Williams SV, Platt FM, Hurst CD, Aveyard JS, Taylor CF, Pole JC, et al. High-resolution analysis of genomic alteration on chromosome arm 8p in urothelial carcinoma. Genes Chromosomes Cancer (2010) 49(7):642–59. doi:10.1002/gcc.20775
35. Costa FF, Bischof JM, Vanin EF, Lulla RR, Wang M, Sredni ST, et al. Identification of microRNAs as potential prognostic markers in ependymoma. PLoS One (2011) 6(10):e25114. doi:10.1371/journal.pone.0025114
36. Anwar SL, Albat C, Krech T, Hasemeier B, Schipper E, Schweitzer N, et al. Concordant hypermethylation of intergenic microRNA genes in human hepatocellular carcinoma as new diagnostic and prognostic marker. Int J Cancer (2013) 133(3):660–70. doi:10.1002/ijc.28068
37. Endo H, Muramatsu T, Furuta M, Uzawa N, Pimkhaokham A, Amagasa T, et al. Potential of tumor-suppressive miR-596 targeting LGALS3BP as a therapeutic agent in oral cancer. Carcinogenesis (2013) 34(3):560–9. doi:10.1093/carcin/bgs376
38. Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res (2010) 70(5):1793–803. doi:10.1158/0008-5472.CAN-09-3112
39. Kuo TC, Tan CT, Chang YW, Hong CC, Lee WJ, Chen MW, et al. Angiopoietin-like protein 1 suppresses SLUG to inhibit cancer cell motility. J Clin Invest (2013) 123(3):1082–95. doi:10.1172/JCI64044
40. Farhana L, Dawson MI, Murshed F, Das JK, Rishi AK, Fontana JA. Upregulation of miR-150* and miR-630 induces apoptosis in pancreatic cancer cells by targeting IGF-1R. PLoS One (2013) 8(5):e61015. doi:10.1371/journal.pone.0061015
41. Gougelet A, Pissaloux D, Besse A, Perez J, Duc A, Dutour A, et al. miRNA profiles in osteosarcoma as a predictive tool for ifosfamide response. Int J Cancer (2011) 129(3):680–90. doi:10.1002/ijc.25715
42. Han Y, Chen J, Zhao X, Liang C, Wang Y, Sun L, et al. microRNA expression signatures of bladder cancer revealed by deep sequencing. PLoS One (2011) 6(3):e18286. doi:10.1371/journal.pone.0018286
43. Bhatti I, Lee A, James V, Hall RI, Lund JN, Tufarelli C, et al. Knockdown of microRNA-21 inhibits proliferation and increases cell death by targeting programmed cell death 4 (PDCD4) in pancreatic ductal adenocarcinoma. J Gastrointest Surg (2011) 15(1):199–208. doi:10.1007/s11605-010-1381-x
44. Ding L, Xu Y, Zhang W, Deng Y, Si M, Du Y, et al. MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res (2010) 20(7):784–93. doi:10.1038/cr.2010.79
45. Tsukamoto Y, Nakada C, Noguchi T, Tanigawa M, Nguyen LT, Uchida T, et al. microRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res (2010) 70(6):2339–49. doi:10.1158/0008-5472.CAN-09-2777
46. Liu AM, Poon RT, Luk JM. microRNA-375 targets hippo-signaling effector YAP in liver cancer and inhibits tumor properties. Biochem Biophys Res Commun (2010) 394(3):623–7. doi:10.1016/j.bbrc.2010.03.036
47. Hui AB, Lenarduzzi M, Krushel T, Waldron L, Pintilie M, Shi W, et al. Comprehensive microRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res (2010) 16(4):1129–39. doi:10.1158/1078-0432.CCR-09-2166
48. Mathé EA, Nguyen GH, Bowman ED, Zhao Y, Budhu A, Schetter AJ, et al. microRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res (2009) 15(19):6192–200. doi:10.1158/1078-0432.CCR-09-1467
49. Bierkens M, Krijgsman O, Wilting SM, Bosch L, Jaspers A, Meijer GA, et al. Focal aberrations indicate EYA2 and hsa-miR-375 as oncogene and tumor suppressor in cervical carcinogenesis. Genes Chromosomes Cancer (2013) 52(1):56–68. doi:10.1002/gcc.22006
50. Wilting SM, Verlaat W, Jaspers A, Makazaji NA, Agami R, Meijer CJ, et al. Methylation-mediated transcriptional repression of microRNAs during cervical carcinogenesis. Epigenetics (2013) 8(2):220–8. doi:10.4161/epi.23605
51. Necela BM, Carr JM, Asmann YW, Thompson EA. Differential expression of microRNAs in tumors from chronically inflamed or genetic (APC) models of colon cancer. PLoS One (2011) 6(4):e18501. doi:10.1371/journal.pone.0018501
52. Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y, et al. microRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res (2011) 71(19):6208–19. doi:10.1158/0008-5472.CAN-11-0073
53. Saini S, Majid S, Shahryari V, Arora S, Yamamura S, Chang I, et al. miRNA-708 control of CD44(+) prostate cancer-initiating cells. Cancer Res (2012) 72(14):3618–30. doi:10.1158/0008-5472.CAN-12-0540
54. Guo P, Lan J, Ge J, Nie Q, Mao Q, Qiu Y. miR-708 acts as a tumor suppressor in human glioblastoma cells. Oncol Rep (2013) 30(2):870–6. doi:10.3892/or.2013.2526
55. Östling P, Leivonen SK, Aakula A, Kohonen P, Mäkelä R, Hagman Z, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res (2011) 71(5):1956–67. doi:10.1158/0008-5472.CAN-10-2421
56. Navon R, Wang H, Steinfeld I, Tsalenko A, Ben-Dor A, Yakhini Z. Novel rank-based statistical methods reveal microRNAs with differential expression in multiple cancer types. PLoS One (2009) 4(11):e8003. doi:10.1371/journal.pone.0008003
57. Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, et al. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell (2011) 147(6):1233–47. doi:10.1016/j.cell.2011.10.043
58. Rokah OH, Granot G, Ovcharenko A, Modai S, Pasmanik-Chor M, Toren A, et al. Downregulation of miR-31, miR-155, and miR-564 in chronic myeloid leukemia cells. PLoS One (2012) 7(4):e35501. doi:10.1371/journal.pone.0035501
59. Pogribny IP, Filkowski JN, Tryndyak VP, Golubov A, Shpyleva SI, Kovalchuk O. Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int J Cancer (2010) 127(8):1785–94. doi:10.1002/ijc.25191
60. Tang JT, Wang JL, Du W, Hong J, Zhao SL, Wang YC, et al. microRNA 345, a methylation-sensitive microRNA is involved in cell proliferation and invasion in human colorectal cancer. Carcinogenesis (2011) 32(8):1207–15. doi:10.1093/carcin/bgr114
61. Yamada H, Yanagisawa K, Tokumaru S, Taguchi A, Nimura Y, Osada H, et al. Detailed characterization of a homozygously deleted region corresponding to a candidate tumor suppressor locus at 21q11-21 in human lung cancer. Genes Chromosomes Cancer (2008) 47(9):810–8. doi:10.1002/gcc.20582
62. White NM, Chow TF, Mejia-Guerrero S, Diamandis M, Rofael Y, Faragalla H, et al. Three dysregulated miRNAs control kallikrein 10 expression and cell proliferation in ovarian cancer. Br J Cancer (2010) 102(8):1244–53. doi:10.1038/sj.bjc.6605634
63. Ju X, Li D, Shi Q, Hou H, Sun N, Shen B. Differential microRNA expression in childhood B-cell precursor acute lymphoblastic leukemia. Pediatr Hematol Oncol (2009) 26(1):1–10. doi:10.1080/08880010802378338
64. Holleman A, Chung I, Olsen RR, Kwak B, Mizokami A, Saijo N, et al. miR-135a contributes to paclitaxel resistance in tumor cells both in vitro and in vivo. Oncogene (2011) 30(43):4386–98. doi:10.1038/onc.2011.148
65. Zhou L, Qiu T, Xu J, Wang T, Wang J, Zhou X, et al. miR-135a/b modulate cisplatin resistance of human lung cancer cell line by targeting MCL1. Pathol Oncol Res (2013) 19(4):677–83. doi:10.1007/s12253-013-9630-4
66. Jukic DM, Rao UN, Kelly L, Skaf JS, Drogowski LM, Kirkwood JM, et al. microRNA profiling analysis of differences between the melanoma of young adults and older adults. J Transl Med (2010) 8:27. doi:10.1186/1479-5876-8-27
67. Liu SG, Qin XG, Zhao BS, Qi B, Yao WJ, Wang TY, et al. Differential expression of miRNAs in esophageal cancer tissue. Oncol Lett (2013) 5(5):1639–42.
68. Waltering KK, Porkka KP, Jalava SE, Urbanucci A, Kohonen PJ, Latonen LM, et al. Androgen regulation of micro-RNAs in prostate cancer. Prostate (2011) 71(6):604–14. doi:10.1002/pros.21276
69. Guled M, Lahti L, Lindholm PM, Salmenkivi K, Bagwan I, Nicholson AG, et al. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma-A miRNA microarray analysis. Genes Chromosomes Cancer (2009) 48(7):615–23. doi:10.1002/gcc.20669
70. Jia L, Wu J, Zhang L, Chen J, Zhong D, Xu S, et al. Restoration of miR-1228* expression suppresses epithelial-mesenchymal transition in gastric cancer. PLoS One (2013) 8(3):e58637. doi:10.1371/journal.pone.0058637
71. Li Y, Mine T, Ioannides CG. Short GC-rich RNA similar to miR 1909 and 1915 folds in silico with the 5’-UTR and ORF of Notch and responders: potential for the elimination of cancer stem cells. Oncol Rep (2010) 24(6):1443–53. doi:10.3892/or_00001004
72. Xu K, Liang X, Cui D, Wu Y, Shi W, Liu J. miR-1915 inhibits Bcl-2 to modulate multidrug resistance by increasing drug-sensitivity in human colorectal carcinoma cells. Mol Carcinog (2013) 52(1):70–8. doi:10.1002/mc.21832
73. Guo L, Liu Y, Bai Y, Sun Y, Xiao F, Guo Y. Gene expression profiling of drug-resistant small cell lung cancer cells by combining microRNA and cDNA expression analysis. Eur J Cancer (2010) 46(9):1692–702. doi:10.1016/j.ejca.2010.02.043
74. Li J, Wang Y, Luo J, Fu Z, Ying J, Yu Y, et al. miR-134 inhibits epithelial to mesenchymal transition by targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett (2012) 586(20):3761–5. doi:10.1016/j.febslet.2012.09.016
75. Niu CS, Yang Y, Cheng CD. MiR-134 regulates the proliferation and invasion of glioblastoma cells by reducing nanog expression. Int J Oncol (2013) 42(5):1533–40. doi:10.3892/ijo.2013.1844
76. Yin C, Wang PQ, Xu WP, Yang Y, Zhang Q, Ning BF, et al. Hepatocyte nuclear factor-4α reverses malignancy of hepatocellular carcinoma through regulating miR-134 in the DLK1-DIO3 region. Hepatology (2013) 58(6):1964–76. doi:10.1002/hep.26573
77. Pigati L, Yaddanapudi SC, Iyengar R, Kim DJ, Hearn SA, Danforth D, et al. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS One (2010) 5(10):e13515. doi:10.1371/journal.pone.0013515
78. Tili E, Michaille JJ, Alder H, Volinia S, Delmas D, Latruffe N, et al. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFβ signaling pathway in SW480 cells. Biochem Pharmacol (2010) 80(12):2057–65. doi:10.1016/j.bcp.2010.07.003
79. Pan J, Hu H, Zhou Z, Sun L, Peng L, Yu L, et al. Tumor-suppressive mir-663 gene induces mitotic catastrophe growth arrest in human gastric cancer cells. Oncol Rep (2010) 24(1):105–12. doi:10.3892/or_00000834
80. Sand M, Skrygan M, Sand D, Georgas D, Gambichler T, Hahn SA, et al. Comparative microarray analysis of microRNA expression profiles in primary cutaneous malignant melanoma, cutaneous malignant melanoma metastases, and benign melanocytic nevi. Cell Tissue Res (2013) 351(1):85–98. doi:10.1007/s00441-012-1514-5
81. Eigentler TK, Figl A, Krex D, Mohr P, Mauch C, Rass K, et al. Number of metastases, serum lactate dehydrogenase level, and type of treatment are prognostic factors in patients with brain metastases of malignant melanoma. Cancer (2011) 117:1697–703. doi:10.1002/cncr.25631
82. Garbe C, Peris K, Hauschild A, Saiag P, Middleton M, Spatz A, et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline – update 2012. Eur J Cancer (2012) 48:2375–90. doi:10.1016/j.ejca.2012.06.013
83. Garbe C, Abusaif S, Eigentler TK. Vemurafenib. Recent Results Cancer Res (2014) 201:215–25. doi:10.1007/978-3-642-54490-3_13
84. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A (1976) 73:3685–9. doi:10.1073/pnas.73.10.3685
85. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A (1978) 75:4538–42. doi:10.1073/pnas.75.9.4538
86. Mikirova N, Casciari J, Riordan N, Hunninghake R. Clinical experience with intravenous administration of ascorbic acid: achievable levels in blood for different states of inflammation and disease in cancer patients. J Transl Med (2013) 11:191. doi:10.1186/1479-5876-11-191
87. Meeran SM, Ahmed A, Tollefsbol TO. Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin Epigenetics (2010) 1:101–16. doi:10.1007/s13148-010-0011-5
88. Venturelli S, Berger A, Weiland T, Zimmermann M, Häcker S, Peter C, et al. Dual antitumour effect of 5-azacytidine by inducing a breakdown of resistance-mediating factors and epigenetic modulation. Gut (2011) 60:156–65. doi:10.1136/gut.2010.208041
89. Weiland T, Berger A, Essmann F, Lauer UM, Bitzer M, Venturelli S. Kinetic tracking of therapy-induced senescence using the real-time cell analyzer single plate system. Assay Drug Dev Technol (2012) 10:289–95. doi:10.1089/adt.2011.0402
90. Venturelli S, Berger A, Weiland T, Essmann F, Waibel M, Nuebling T, et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2’-deoxycytidine in solid tumor cells. Mol Cancer Ther (2013) 12:2226–36. doi:10.1158/1535-7163.MCT-13-0137
91. Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS One (2010) 5:e11414. doi:10.1371/journal.pone.0011414
92. Busch C, Krochmann J, Drews U. The chick embryo as an experimental system for melanoma cell invasion. PLoS One (2013) 8:e53970. doi:10.1371/journal.pone.0053970
93. Schriek G, Oppitz M, Busch C, Just L, Drews U. Human SK-Mel 28 melanoma cells resume neural crest cell migration after transplantation into the chick embryo. Melanoma Res (2005) 15:225–34. doi:10.1097/00008390-200508000-00001
Keywords: ascorbate, vitamin C, cancer, melanoma, epigenetics, microRNA, HDAC, DNMT
Citation: Venturelli S, Sinnberg TW, Berger A, Noor S, Levesque MP, Böcker A, Niessner H, Lauer UM, Bitzer M, Garbe C and Busch C (2014) Epigenetic impacts of ascorbate on human metastatic melanoma cells. Front. Oncol. 4:227. doi: 10.3389/fonc.2014.00227
Received: 06 June 2014; Accepted: 06 August 2014;
Published online: 25 August 2014.
Edited by:
Gabi U. Dachs, University of Otago, New ZealandReviewed by:
Giorgio Stassi, University of Palermo, ItalyMassimiliano Agostini, Medical Research Council, UK
Copyright: © 2014 Venturelli, Sinnberg, Berger, Noor, Levesque, Böcker, Niessner, Lauer, Bitzer, Garbe and Busch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Busch, Division of Dermatologic Oncology, Department of Dermatology and Allergology, University Hospital of Tuebingen, Liebermeisterstr. 25, Tuebingen 72076, Germany e-mail:Y2hyaXN0aWFuLmJ1c2NoQG1lZC51bmktdHVlYmluZ2VuLmRl