 Laura Amo1†
Laura Amo1† Estíbaliz Tamayo-Orbegozo1†
Estíbaliz Tamayo-Orbegozo1† Natalia Maruri1
Natalia Maruri1 Cristina Eguizabal2
Cristina Eguizabal2 Olatz Zenarruzabeitia3
Olatz Zenarruzabeitia3 Marta Riñón1Arantza Arrieta1Silvia Santos2
Marta Riñón1Arantza Arrieta1Silvia Santos2 Jorge Monge2
Jorge Monge2 Miguel Angel Vesga2
Miguel Angel Vesga2 Francisco Borrego3,4
Francisco Borrego3,4 Susana Larrucea1*
Susana Larrucea1*- 1Regulation of the Immune System Group, BioCruces Health Research Institute, Hospital Universitario Cruces, Barakaldo, Spain
- 2Basque Center for Transfusion and Human Tissues, Galdakao, Spain
- 3Immunopathology Group, BioCruces Health Research Institute, Hospital Universitario Cruces, Barakaldo, Spain
- 4Ikerbasque, Basque Foundation for Science, Bilbao, Spain
Besides their essential role in hemostasis and thrombosis, platelets are involved in the onset of cancer metastasis by interacting with tumor cells. Platelets release secretory factors that promote tumor growth, angiogenesis, and metastasis. Furthermore, the formation of platelet–tumor cell aggregates in the bloodstream provides cancer cells with an immune escape mechanism by protecting circulating malignant cells from immune-mediated lysis by natural killer (NK) cells. Platelet–tumor cell interaction is accomplished by specific adhesion molecules, including integrins, selectins, and their ligands. Podocalyxin-like protein 1 (PCLP1) is a selectin-ligand protein in which overexpression has been associated with several aggressive cancers. PCLP1 expression enhances cell adherence to platelets in an integrin-dependent process and through the interaction with P-selectin expressed on activated platelets. However, the involvement of PCLP1-induced tumor–platelet interaction in tumor immune evasion still remains unexplored. The identification of selectin ligands involved in the interaction of platelets with tumor cells may provide help for the development of effective therapies to restrain cancer cell dissemination. This article summarizes the current knowledge on molecules that participate in platelet–tumor cell interaction as well as discusses the potential role of PCLP1 as a molecule implicated in tumor immune evasion.
Introduction
The metastasis to distant organ sites depends on the interaction between tumor cells and the microenvironment of the bloodstream and tissues, including platelets, immune system cells, endothelial cells, stromal cells, and extracellular matrix (1). The participation of platelets and coagulation in tumor metastasis has been observed for a long time (2). Moreover, thrombocytosis and thrombosis have been associated with poor prognosis in various tumor types (3). These observations and the finding that suppression of platelet–tumor cell aggregates with antiplatelet agents inhibits experimental metastasis point to an important role of platelets in the development of tumor metastasis (4). During platelet–tumor cell interaction, the communication between both cell types is bilateral, as tumor cells express factors that trigger platelet activation and coagulation, and activated platelets promote tumor growth, metastasis, and angiogenesis (5).
The contribution of selectins and its ligands to the communication between tumor cells and platelets has been confirmed by several preclinical models (6). Here, we describe the molecules involved in platelet–tumor interaction, specially focusing on the mechanisms by which PCLP1, a transmembrane sialomucin with selectin-ligand activity, participates in platelet–tumor aggregate formation. We also hypothesize that PCLP1 might be involved in protecting tumor cell from natural killer (NK) cell-mediated cytotoxicity.
Platelets in Tumor Progression
Metastasis is the main complication and principal cause of death in most types of cancer. The process of metastasis consists of different phases: (1) detachment of cancer cells from the primary tumor, (2) intravasation into the vascular system and transport through the bloodstream, (3) extravasation, and (4) proliferation in a distant tissue (7, 8).
During circulation through the bloodstream and vascular adhesion at distant sites, tumor cells are prone to death induced by shear stress and attack of immune cells. However, once tumor cells intravasate into blood, tumor-derived platelet agonists, such as adenosine diphosphate (ADP) and thrombin, induce platelet activation, followed by the formation of heterotypic aggregates that protect tumor cells from immune attack and physical damage (9, 10). Tissue factor expressed by tumor cells also leads to thrombin generation through the activation of the coagulation cascade that ultimately results in fibrin formation and platelet activation (11). These processes, among others, promote the development of venous thromboembolism in solid tumor and hematological malignancies, including lymphomas and myeloma, and disseminated intravascular coagulation with severe bleeding, which is frequently associated with acute leukemia (12). The relevance of tumor-induced platelet aggregation in cancer progression is highlighted by the correlation between the ability of tumor cells to form platelet aggregates and their metastatic potential.
Platelets promote tumor cell arrest within the vasculature, endothelium cell retraction, and subsequent extravasation and tissue invasion through the release of chemokines, proteolytic enzymes such as gelatinase and heparanase, various metalloproteinases, and microparticules (10). Moreover, platelets contain proangiogenic and antiangiogenic molecules within α-granules, which are selectively released depending on the stimuli and are the most important source of vascular endothelial growth factor (VEGF). The expression of the proangiogenic molecule VEGF increases substantially in patients with cancer, promoting the formation of new vessels (13, 14). Platelets also release soluble factors that increase tumor growth at sites of extravasation. Thrombin and lysophosphatidic acid (LPA) released by platelets enhance tumor cell growth and metastasis through the activation of thrombin receptor PAR1 and LPA receptor, respectively, both expressed on tumor cells (1).
Platelets in Tumor Immune Evasion
Platelets also contribute to metastasis by protecting disseminating tumor cells from NK cell cytolytic activity as supported by the observation that the antimetastatic effect of platelet removal is abolished by simultaneous depletion of NK cells (15–17). NK cells are potent cytolytic lymphocytes of the innate immune system that play a critical role in the elimination of tumor cells. These lymphocytes recognize target cells without prior sensitization by means of signals received from activating and inhibitory receptors, being the balance between these two opposing signals, which determines the activation state of NK cells (18). Nevertheless, tumor cells have developed different mechanisms to evade NK immunosurveillance, including the modulation of NK receptor–ligand expression patterns and the secretion of immunoregulatory molecules like IDO and PGE2 or immunosuppressive cytokines like IL-10 and tumor growth factor-β (TGF-β) (19–22).
In early phases of the metastatic process, platelets may surround tumor cells and, in conjunction with fibrinogen, form a dense coat around tumor cells, providing protection from NK cell recognition and destruction (23, 24). Besides, both TGF-β and platelet-derived growth factor (PDGF) secreted by activated platelets inhibit NK cell effector functions. Particularly, it has been reported that TGF-β down-regulates the expression of the activating receptor NKG2D on NK cells after interaction with tumor cells and, to a lesser extent, NKp30, NKp44, NKp46, and NKp80 expression (25). Furthermore, TGF-β reduces IFN-γ production by NK cells and promotes epithelial–mesenchymal-like transition in various cancer cell lines (26). PDGF diminishes the interaction of NK with target cells and NK cell cytotoxic activity (27–29). Recently, it has been reported that glucocorticoid-induced TNF-related ligand (GITRL) up-regulated upon platelet activation inhibits NK cell cytotoxicity and IFN-γ production via GITR expressed on NK cells (30).
Molecules Involved in Platelet–Tumor Interaction
Tumor cell interaction with platelets, leukocytes, and endothelium is mediated mainly by integrins and their ligands and by the binding of P-selectin with selectin ligands expressed on tumor cells.
Integrins
Integrins are transmembrane glycoproteins composed of an α and a β subunits forming a non-covalent heterodimer that promote cell–cell and cell–extracellular matrix adhesion, and modulates multiple cellular processes such as cell migration, proliferation, and survival. Integrins become activated through a process involving an “inside-out” signal from other receptors that results in a change in the integrin structure from a low affinity conformation to a high affinity form that enables ligand binding (31).
During the hematogenous phase of metastasis, binding of soluble platelet agonists to their receptors elicit various signaling transduction events that ultimately induce inside-out signaling processes that culminate in activation of the ligand-binding function of integrin αIIbβ3 (GPIIb/IIIa) and granule secretion (10). The homotypic interaction between platelets occurs via cross-linking of integrins, primarily αIIbβ3, by adhesive ligands that serve as bridging proteins present in plasma or released from activated platelets, including fibrinogen, fibronectin, von Willebrand factor (vWF), and other ligands containing the arginine–glycine–aspartate (RGD) integrin-binding domain (32). Ligand binding to integrin αIIbβ3 then mediates platelet aggregation and triggers “outside-in” signaling that amplifies platelet response, resulting in the stabilization of platelet adhesion and aggregation, and clot retraction.
Platelet–tumor cell aggregates result from cross-linking of platelet integrins, primarily αIIbβ3, with integrins expressed on tumor cells, such as αvβ3, by the above mentioned adhesive ligands (33). The relevance of αIIbβ3 and αvβ3 interaction in tumor metastasis has been demonstrated in in vivo models, which showed a decrease of pulmonary metastasis following inhibition of αvβ3 with a specific monoclonal antibody, an effect that was significantly reduced after platelet depletion (34). αIIbβ3 and αvβ3 integrins also support the arrest of tumor cells to the endothelium of metastatic sites. Other integrins such as α5β1 and α3β1 as well as the adhesive ligands vitronectin and laminin have been implicated in platelet–tumor interaction, tumor adhesion, and metastasis (35) (Figure 1).
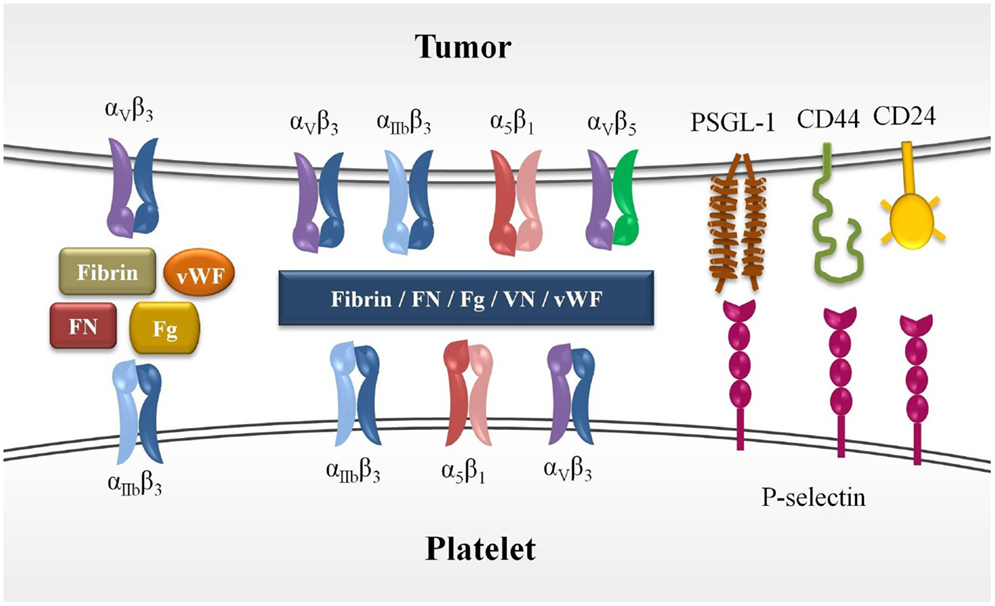
Figure 1. Molecules involved in platelet–tumor cell interaction. Platelet–tumor cell aggregates are formed (1) by cross-linking of platelet integrins, primarily αIIbβ3 integrins, with αvβ3 integrin expressed on tumor cells through their ligands, which act as bringing molecules (Fg, FN, fibrin, and vWF), (2) by interaction of platelet P-selectin with its selectin-ligands expressed on tumor cells (PSGL-1, CD44, CD24). The ectopic expression of megakaryocytic genes in various tumor cells leads to the expression of functional αIIbβ3, and therefore, the heterotypic interaction between platelets and tumor cells may take place through cross-linking of this integrin. Other integrins has also been suggested to participate in platelet–tumor aggregates formation. FN, fibronectin; Fg, fibrinogen; vWF, von Willebrand factor; VN, vitronectin.
Selectins
Selectins are cell-surface adhesion molecules with a carbohydrate-binding domain that bind with low affinity to sialylated and fucosylated glycan structures present on selectin ligands and induce integrin activation. Several studies have shown that selectins may transduce outside-in signals upon interaction with their ligands (36, 37). In cancer cell interactions, selectins expressed on platelets, leukocytes, and endothelium bind to selectin ligands present on tumor cells, leading to the formation of platelet–tumor-leukocyte aggregates and tumor cell arrest in the microvasculature (38).
The selectin family consists of three molecules with selective cell distribution. P-selectin is stored in the alpha and dense granules of platelets and in the Weibel–Palade bodies of endothelial cells and translocated to surface upon cellular activation by agonists. P-selectin binds to a variety of human cancer cells, such as colon, lung, and breast cancer, as well as melanoma and neuroblastoma (39). Platelets from P-selectin-deficient mice exhibit a reduced interaction with tumor cells, resulting in a marked decrease of metastasis and reflecting the importance of this protein in tumor progression (40, 41). L-selectin, a molecule constitutively expressed on the majority of leukocytes, enables leukocyte homing to lymphoid organs and extravasation into inflamed tissues. This molecule facilitates tumor metastasis and acts synergistically with P-selectin (42). Although E-selectin, expressed on endothelial cells, has not been implicated in platelet–tumor interaction, it participates in the homing of metastatic cancer cells to distant organs (43).
Selectin Ligands
The tetrasaccharide sialyl-Lewisx (sLex) and its isomer sialyl-Lewisa (sLea) recognized by selectins are located in terminal chains of glycolipids and N-/O-glycoproteins displayed on selectin ligands. High cell-surface expression of sLex and sLea or altered glycosylation on tumor cells has been associated with tumor progression and metastasis (44). Selectin ligands are mainly sialylated, fucosylated, sulfated glycans localized on tumor cell mucins, that is, heavily glycosylated proteins with O-linked oligosaccharides.
Several mucin-like molecules with P-selectin ligand activity have been identified. P-selectin glycoprotein ligand-1 (PSGL-1) is a sialylated mucin-type disulfide-linked homodimer expressed on most leukocytes, which presents a high-affinity binding to P-selectin and is essential for the homing of leukocytes to tissue. PSGL-1 has also been described as a P-selectin ligand on lung cancer and myeloma cells, and an E-selectin ligand on prostate tumor cells (45–47). CD24 modified by sLex serves as the major P-selectin-reactive ligand on the surface of breast cancer cells and its enhanced expression is related to cancer progression and poor prognosis (48, 49). CD44 variant isoforms (CD44v) act as E-/L-/P-selectin ligands on colon cancer cells and as E-selectin ligand on breast cancer cells (50, 51). Podocalyxin-like protein 1 (PCLP1), a cell-surface sialomucin expressed in a wide range of normal cell as well as in various types of cancer, has been also associated with metastasis (52).
PCLP1, a Selectin Ligand Involved in Platelet–Tumor Interaction
Podocalyxin-like protein 1 is a cell-surface glycoprotein that belongs to the CD34 family of sialomucins (53). Initially identified on kidney podocytes, PCLP1 is widely expressed on vascular endothelium, mesothelial cells, hematopoietic stem and progenitor cells, a subset of neuronal cells, and platelets (54–58). PCLP1 is attached to the actin cytoskeleton through its association with ezrin, a protein that regulates cell adhesion, motility, and survival (59). In renal glomerular podocytes, PCLP1 functions as an anti-adhesive molecule, which plays a vital role in maintaining opened the filtration slits due to its highly negative charge, as evidenced by the observation of severe renal malformation and anuria in PCLP1-deficient mice (60). On the contrary, in high endothelial venules, PCLP1 serves as a pro-adhesive molecule participating in the tethering and rolling of lymphocytes via interaction with L-selectin (53). During embryonic development, PCLP1 expression by mesothelial cells is required for the retraction of the gut from the umbilical cord (60). In the hematopoietic system, PCLP1 participates in cell migration to distant hematopoietic tissues (56). PCLP1 is also involved in neuronal development and synapse formation (61).
The overexpression of PCLP1 has been associated with a more aggressive phenotype and poor prognosis in numerous types of cancer, including breast, colorectal, prostate, bladder, ovarian cancer, renal carcinoma, and oral squamous cell carcinoma, which suggests an important role of this protein in cancer progression and metastasis, although the underlying mechanisms are poorly understood (62–69). PCLP1 is also overexpressed in blasts of acute leukemia, testicular tumor, hepatic carcinoma, astrocytic tumor, and small cell lung carcinoma (70–74). PCLP1 increases in vitro migration and invasion of tumor cells through its interaction with ezrin in breast cancer cells (75) and promotes invadopodia formation and metastasis by activating Rac1/Cdc42/cortactin signaling cascade (76). Recently, it has been demonstrated that PCLP1 expression in A549 human lung carcinoma cell line is increased during TGF-β-induced epithelial–mesenchymal transition and regulates the molecular changes associated with this process (77).
Although data support a correlation between PCLP1 expression and enhanced binding of metastatic tumor cells to selectins, evidences showing a functional role of PCLP1 as a selectin-ligand in cancer are limited. Recent reports show that PCLP1 expressed on colon carcinoma or pancreatic cells display E- and L-, but not P-, selectin-mediated adhesion under flow conditions (78, 79). The first study was conducted by perfusing E- and P-selectin-expressing Chinese Ovarian (CHO) cells and L-selectin-expressing human peripheral blood lymphocytes over purified PCLP1 protein derived from colon carcinoma cells and by cell-free flow-based adhesion assays using PCLP1-coated microspheres (78). Interestingly, the results proved that carbohydrate determinants involved in binding of PCLP1 expressed in colon carcinoma cells to L- and E-selectins differ from that displayed by PCLP1 expressed in high endothelial venules. The second report used PCLP1-knockdown pancreatic cells perfused over immobilized E- and L-selectin, demonstrating that PCLP1 is functionally relevant in tumor cell-selectin interaction (79).
The role for PCLP1 in promoting cell–platelet interactions is supported by a study performed in Tera-1 cell line, derived from an embryonal carcinoma of the testis expressing high levels of PCLP1, and in CHO cells ectopically expressing PCLP1 (80). This report provided evidence that PCLP1 increases the adhesion of cells to fibrinogen-immobilized platelets in a sialylation-dependent manner, further enhanced by activation of platelets by agonist. PCLP1-induced adhesion is partially inhibited by blockers of P-selectin and by an inhibitor of integrins αvβ3/αvβ5, and almost totally restrained by RGD peptide, a potent inhibitor of many integrin–ligand interactions, indicating the contribution of these adhesion molecules to this process. Besides, PCLP1 colocalizes with P-selectin expressed on activated platelets at cell contact sites. Another report demonstrated that PCLP1 increases cell adherence to human vascular endothelial cells, an effect that is amplified after stimulation of these cells with thrombin or histamine (81). Furthermore, PCLP1 enhances cell adhesion to immobilized fibronectin as well as cell spreading and migration. PCLP1-mediated cell adhesion to fibronectin is also dependent on the activity of integrins, as it is prevented by RGD peptides or by inhibitors of αvβ3 integrin, whereas cell spreading is controlled by α5β1 integrin.
Fibronectin, a protein that forms part of the extracellular matrix, is also abundant in plasma and mediates platelet aggregation by binding β3 integrins and tethering adjacent platelets (82, 83). It has been reported that incorporation of plasma fibronectin into fibrin clots enhances metastasis to the lungs (84). Thus, the ability of PCLP1 to potentiate cell binding to fibronectin through the activation of integrins could increase the interaction of platelets with tumor cells, fibronectin acting as a bridge molecule between αvβ3 or αvβ5 expressed on tumor cells and αIIbβ3, αvβ3, and α5β1 integrins expressed on platelets. Subsequently, the interaction between fibronectin and integrins might trigger outside-in signals that would potentiate the activation state of platelets, thereby stabilizing the aggregates. Other adhesive molecules such as fibrinogen, vitronectin, fibrin, and vWF could also serve as bridge proteins in this process (Figure 2).
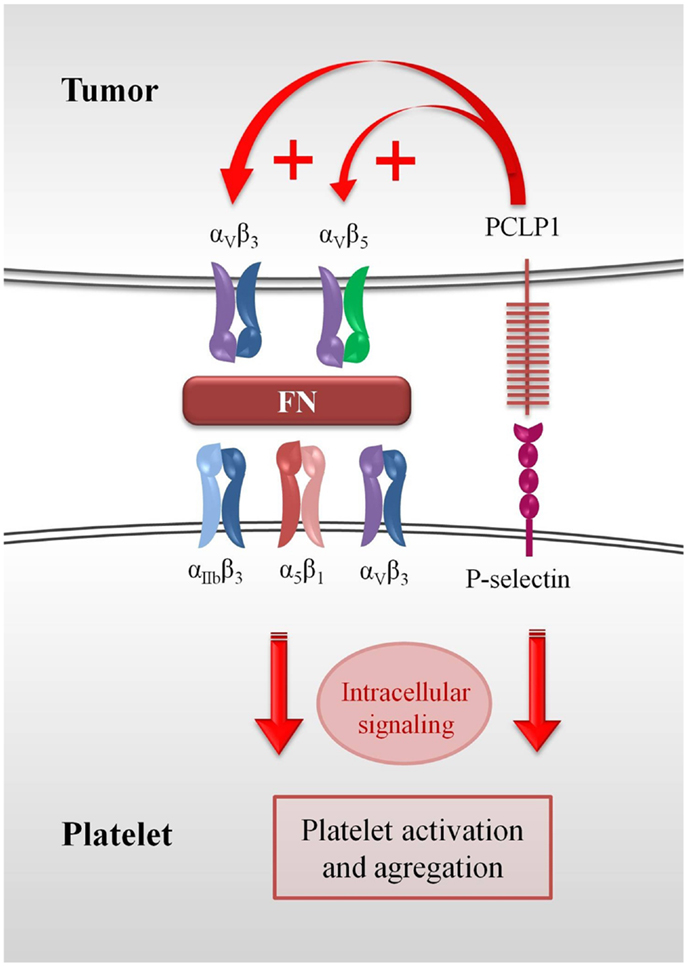
Figure 2. Role of PCLP1 in platelet–tumor cell interaction. The ability of PCLP1 to potentiate cell binding to fibronectin through the activation of integrins could increase the interaction of platelets with tumor cells. Fibronectin would act as a bridge molecule between αvβ3 or αvβ5 expressed on tumor cells and αIIbβ3, αvβ3, and α5β1 integrins expressed on platelets. This interaction might trigger outside-in signals that would potentiate the activation state of platelets, eventually leading to the stabilization of the hetero-aggregates. Moreover, PCLP1 binding to P-selectin could also result in the generation of intracellular signaling that would reinforce the hetero-aggregates.
Although the signaling triggered in platelet by the interaction of P-selectin with PCLP1 has not been explored so far, it is known that selectins may transduce outside-in signals delivered by selectin ligands through their cytoplasmic tail (36). In that way, binding of PSGL-1 to P-selectin expressed on platelets induces calcium influx and a signaling cascade that triggers αIIbβ3 activation, microaggregates generation, and thrombus formation (85, 86). Platelet activation is followed by the phosphorylation of tyrosine, serine, threonine, and histidine residues on the cytoplasmic tail of P-selectin (87–89). Similarly, the interaction of PCLP1 with P-selectin might induce outside-in signals that could ultimately lead to enhanced expression of adhesion molecules on platelets that would reinforce platelet–tumor cell–leukocytes aggregates, creating a positive feedback loop.
As mentioned previously, PCLP1 is also expressed on activated platelets. Mice overexpressing human PCLP1 on platelets exhibit a phenotype characterized by decreased bleeding time and enhanced platelet aggregation after agonist stimulation (90). Inversely, the deletion of pclp1 gene in mouse platelets produce a prolonged bleeding time, decreased platelet aggregation, decreased in vivo thrombosis, and reduced adherence of platelets to fibrinogen under flow, all this pointing to a role of PCLP1 in the control of hemostasis (91). Thereby, PCLP1 expressed on platelet could strengthen the heterotypic conjugates formation during hematogenous metastasis and promote their retention to the vessel wall of target organs by enhancing cell adhesion to fibronectin localized on subendothelial matrix, which is exposed following endothelial cell retraction at metastatic sites.
Considering the aforementioned observations, we hypothesize that PCLP1 could have a role in the escape of tumor cells from the immune surveillance by inducing platelet–tumor interaction. PCLP1 might participate in tumor cell evasion by fostering the formation of a physical barrier to NK cell-mediated cytotoxicity comprised of platelets and adhesive proteins through the mechanisms described above. Moreover, PCLP1 contribution to platelet–tumor interaction with vascular endothelial cells may result in a decreased permanence of hetero-aggregates in the bloodstream, thereby diminishing their exposure to NK cells.
Future Perspectives
The evidence provided by the literature suggests that an anti-PCLP1 antibody aimed at disrupting platelet–tumor aggregation could be useful as a therapeutic drug for the treatment of PCLP1-expressing cancers. As PCLP1 expression has also been reported in normal tissues, including neuron, podocytes, and hematopoietic stem cells, antibodies that display cross-reactivity with normal cells may not be appropriate as therapeutic agents. Given that selectin-binding determinants exhibited on PCLP1 expressed in tumor cells are different from those described in normal cells (78), an antibody recognizing specific glycan epitopes displayed on tumor cells might avoid undesirable side effects. In addition to antibodies, alternative antagonists may be used such as peptides that block PCLP1–selectin interaction, specific aptamers, which are oligonucleotides displaying high affinity and specificity for target molecules, and glycomimetic compounds, synthetic oligosaccharide analogs that mimic native carbohydrates.
A systematic study of PCLP1 contribution to the interaction of platelet with different tumor types has not been addressed to date. The functional properties of PCLP1 as an adhesive protein that promotes platelet–tumor aggregates presented herein may encourage researchers to reveal yet hidden aspects of this molecule.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by EITB Maratoia-BIOEF (Basque Foundation for Health Innovation and Research) (BIO11/CM/007).
References
1. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer (2011) 11(2):123–34. doi: 10.1038/nrc3004
2. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood (2007) 110(6):1723–9. doi:10.1182/blood-2006-10-053736
3. Buergy D, Wenz F, Groden C, Brockmann MA. Tumor-platelet interaction in solid tumors. Int J Cancer (2012) 130(12):2747–60. doi:10.1002/ijc.27441
4. Gil-Bernabe AM, Lucotti S, Muschel RJ. Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol (2013) 162(4):433–41. doi:10.1111/bjh.12381
5. Goubran HA, Stakiw J, Radosevic M, Burnouf T. Platelet-cancer interactions. Semin Thromb Hemost (2014) 40(3):296–305. doi:10.1055/s-0034-1370767
6. Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol (2012) 2012:676731. doi:10.1155/2012/676731
7. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med (2006) 12(8):895–904. doi:10.1038/nm1469
8. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov (2012) 2(12):1091–9. doi:10.1158/2159-8290.CD-12-0329
9. Boukerche H, Berthier-Vergnes O, Penin F, Tabone E, Lizard G, Bailly M, et al. Human melanoma cell lines differ in their capacity to release ADP and aggregate platelets. Br J Haematol (1994) 87(4):763–72. doi:10.1111/j.1365-2141.1994.tb06736.x
10. Bambace NM, Holmes CE. The platelet contribution to cancer progression. J Thromb Haemost (2011) 9(2):237–49. doi:10.1111/j.1538-7836.2010.04131.x
11. Milsom C, Rak J. Tissue factor and cancer. Pathophysiol Haemost Thromb (2008) 36(3–4):160–76. doi:10.1159/000175154
12. Falanga A, Russo L, Milesi V. The coagulopathy of cancer. Curr Opin Hematol (2014) 21(5):423–9. doi:10.1097/MOH.0000000000000072
13. Italiano JE Jr., Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood (2008) 111(3):1227–33. doi:10.1182/blood-2007-09-113837
14. Peterson JE, Zurakowski D, Italiano JE Jr., Michel LV, Fox L, Klement GL, et al. Normal ranges of angiogenesis regulatory proteins in human platelets. Am J Hematol (2010) 85(7):487–93. doi:10.1002/ajh.21732
15. Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res (1999) 59(6):1295–300.
16. Gorelik E, Wiltrout RH, Okumura K, Habu S, Herberman RB. Role of NK cells in the control of metastatic spread and growth of tumor cells in mice. Int J Cancer (1982) 30(1):107–12. doi:10.1002/ijc.2910300118
17. Hanna N. The role of natural killer cells in the control of tumor growth and metastasis. Biochim Biophys Acta (1985) 780(3):213–26.
18. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
19. Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite l-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood (2006) 108(13):4118–25. doi:10.1182/blood-2006-03-006700
20. Yakar I, Melamed R, Shakhar G, Shakhar K, Rosenne E, Abudarham N, et al. Prostaglandin e(2) suppresses NK activity in vivo and promotes postoperative tumor metastasis in rats. Ann Surg Oncol (2003) 10(4):469–79. doi:10.1245/ASO.2003.08.017
21. Sabry M, Lowdell MW. Tumor-primed NK cells: waiting for the green light. Front Immunol (2013) 4:408. doi:10.3389/fimmu.2013.00408
22. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100(7):4120–5. doi:10.1073/pnas.0730640100
23. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood (2005) 105(1):178–85. doi:10.1182/blood-2004-06-2272
24. Zheng S, Shen J, Jiao Y, Liu Y, Zhang C, Wei M, et al. Platelets and fibrinogen facilitate each other in protecting tumor cells from natural killer cytotoxicity. Cancer Sci (2009) 100(5):859–65. doi:10.1111/j.1349-7006.2009.01115.x
25. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res (2009) 69(19):7775–83. doi:10.1158/0008-5472.CAN-09-2123
26. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res (2009) 19(2):156–72. doi:10.1038/cr.2009.5
27. Gersuk GM, Westermark B, Mohabeer AJ, Challita PM, Pattamakom S, Pattengale PK. Inhibition of human natural killer cell activity by platelet-derived growth factor (PDGF). III. Membrane binding studies and differential biological effect of recombinant PDGF isoforms. Scand J Immunol (1991) 33(5):521–32. doi:10.1111/j.1365-3083.1991.tb02522.x
28. Gersuk GM, Chang WC, Pattengale PK. Inhibition of human natural killer cell activity by platelet-derived growth factor. II. Membrane binding studies, effects of recombinant IFN-alpha and IL-2, and lack of effect on T cell and antibody-dependent cellular cytotoxicity. J Immunol (1988) 141(11):4031–8.
29. Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med (2013) 19(8):460–73. doi:10.1016/j.molmed.2013.05.002
30. Placke T, Salih HR, Kopp HG. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J Immunol (2012) 189(1):154–60. doi:10.4049/jimmunol.1103194
31. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110(6):673–87. doi:10.1016/S0092-8674(02)00971-6
32. Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res (2007) 100(12):1673–85. doi:10.1161/01.RES.0000267878.97021.ab
33. Felding-Habermann B, Habermann R, Saldivar E, Ruggeri ZM. Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J Biol Chem (1996) 271(10):5892–900. doi:10.1074/jbc.271.10.5892
34. Lonsdorf AS, Kramer BF, Fahrleitner M, Schonberger T, Gnerlich S, Ring S, et al. Engagement of alphaIIbbeta3 (GPIIb/IIIa) with alphanubeta3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. J Biol Chem (2012) 287(3):2168–78. doi:10.1074/jbc.M111.269811
35. McNicol A, Israels SJ. Beyond hemostasis: the role of platelets in inflammation, malignancy and infection. Cardiovasc Hematol Disord Drug Targets (2008) 8(2):99–117. doi:10.2174/187152908784533739
36. Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, et al. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol (1998) 142(5):1381–91. doi:10.1083/jcb.142.5.1381
37. Simon SI, Cherapanov V, Nadra I, Waddell TK, Seo SM, Wang Q, et al. Signaling functions of L-selectin in neutrophils: alterations in the cytoskeleton and colocalization with CD18. J Immunol (1999) 163(5):2891–901.
38. Laubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol (2010) 20(3):169–77. doi:10.1016/j.semcancer.2010.04.005
39. Chen M, Geng JG. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp (Warsz) (2006) 54(2):75–84. doi:10.1007/s00005-006-0010-6
40. Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci U S A (1998) 95(16):9325–30. doi:10.1073/pnas.95.16.9325
41. Qi CL, Wei B, Ye J, Yang Y, Li B, Zhang QQ, et al. P-selectin-mediated platelet adhesion promotes the metastasis of murine melanoma cells. PLoS One (2014) 9(3):e91320. doi:10.1371/journal.pone.0091320
42. Borsig L, Wong R, Hynes RO, Varki NM, Varki A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc Natl Acad Sci U S A (2002) 99(4):2193–8. doi:10.1073/pnas.261704098
43. Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer (1997) 71(4):612–9. doi:10.1002/(SICI)1097-0215(19970516)71:4<612::AID-IJC17>3.3.CO;2-1
44. Hauselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol (2014) 4:28. doi:10.3389/fonc.2014.00028
45. Gong L, Cai Y, Zhou X, Yang H. Activated platelets interact with lung cancer cells through P-selectin glycoprotein ligand-1. Pathol Oncol Res (2012) 18(4):989–96. doi:10.1007/s12253-012-9531-y
46. Azab AK, Quang P, Azab F, Pitsillides C, Thompson B, Chonghaile T, et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood (2012) 119(6):1468–78. doi:10.1182/blood-2011-07-368050
47. Dimitroff CJ, Descheny L, Trujillo N, Kim R, Nguyen V, Huang W, et al. Identification of leukocyte E-selectin ligands, P-selectin glycoprotein ligand-1 and E-selectin ligand-1, on human metastatic prostate tumor cells. Cancer Res (2005) 65(13):5750–60. doi:10.1158/0008-5472.CAN-04-4653
48. Aigner S, Ramos CL, Hafezi-Moghadam A, Lawrence MB, Friederichs J, Altevogt P, et al. CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J (1998) 12(12):1241–51.
49. Fang X, Zheng P, Tang J, Liu Y. CD24: from A to Z. Cell Mol Immunol (2010) 7(2):100–3. doi:10.1038/cmi.2009.119
50. Alves CS, Burdick MM, Thomas SN, Pawar P, Konstantopoulos K. The dual role of CD44 as a functional P-selectin ligand and fibrin receptor in colon carcinoma cell adhesion. Am J Physiol Cell Physiol (2008) 294(4):C907–16. doi:10.1152/ajpcell.00463.2007
51. Burdick MM, Chu JT, Godar S, Sackstein R. HCELL is the major E- and L-selectin ligand expressed on LS174T colon carcinoma cells. J Biol Chem (2006) 281(20):13899–905. doi:10.1074/jbc.M513617200
52. Nielsen JS, McNagny KM. The role of podocalyxin in health and disease. J Am Soc Nephrol (2009) 20(8):1669–76. doi:10.1681/ASN.2008070782
53. Sassetti C, Tangemann K, Singer MS, Kershaw DB, Rosen SD. Identification of podocalyxin-like protein as a high endothelial venule ligand for L-selectin: parallels to CD34. J Exp Med (1998) 187(12):1965–75. doi:10.1084/jem.187.12.1965
54. Kerjaschki D, Sharkey DJ, Farquhar MG. Identification and characterization of podocalyxin – the major sialoprotein of the renal glomerular epithelial cell. J Cell Biol (1984) 98(4):1591–6. doi:10.1083/jcb.98.4.1591
55. Horvat R, Hovorka A, Dekan G, Poczewski H, Kerjaschki D. Endothelial cell membranes contain podocalyxin – the major sialoprotein of visceral glomerular epithelial cells. J Cell Biol (1986) 102(2):484–91. doi:10.1083/jcb.102.2.484
56. Doyonnas R, Nielsen JS, Chelliah S, Drew E, Hara T, Miyajima A, et al. Podocalyxin is a CD34-related marker of murine hematopoietic stem cells and embryonic erythroid cells. Blood (2005) 105(11):4170–8. doi:10.1182/blood-2004-10-4077
57. Vitureira N, McNagny K, Soriano E, Burgaya F. Pattern of expression of the podocalyxin gene in the mouse brain during development. Gene Expr Patterns (2005) 5(3):349–54. doi:10.1016/j.modgep.2004.10.002
58. Miettinen A, Solin ML, Reivinen J, Juvonen E, Vaisanen R, Holthofer H. Podocalyxin in rat platelets and megakaryocytes. Am J Pathol (1999) 154(3):813–22. doi:10.1016/S0002-9440(10)65328-X
59. Orlando RA, Takeda T, Zak B, Schmieder S, Benoit VM, McQuistan T, et al. The glomerular epithelial cell anti-adhesin podocalyxin associates with the actin cytoskeleton through interactions with ezrin. J Am Soc Nephrol (2001) 12(8):1589–98.
60. Doyonnas R, Kershaw DB, Duhme C, Merkens H, Chelliah S, Graf T, et al. Anuria, omphalocele, and perinatal lethality in mice lacking the CD34-related protein podocalyxin. J Exp Med (2001) 194(1):13–27. doi:10.1084/jem.194.1.13
61. Vitureira N, Andres R, Perez-Martinez E, Martinez A, Bribian A, Blasi J, et al. Podocalyxin is a novel polysialylated neural adhesion protein with multiple roles in neural development and synapse formation. PLoS One (2010) 5(8):e12003. doi:10.1371/journal.pone.0012003
62. Somasiri A, Nielsen JS, Makretsov N, McCoy ML, Prentice L, Gilks CB, et al. Overexpression of the anti-adhesin podocalyxin is an independent predictor of breast cancer progression. Cancer Res (2004) 64(15):5068–73. doi:10.1158/0008-5472.CAN-04-0240
63. Forse CL, Yilmaz YE, Pinnaduwage D, O’Malley FP, Mulligan AM, Bull SB, et al. Elevated expression of podocalyxin is associated with lymphatic invasion, basal-like phenotype, and clinical outcome in axillary lymph node-negative breast cancer. Breast Cancer Res Treat (2013) 137(3):709–19. doi:10.1007/s10549-012-2392-y
64. Larsson A, Johansson ME, Wangefjord S, Gaber A, Nodin B, Kucharzewska P, et al. Overexpression of podocalyxin-like protein is an independent factor of poor prognosis in colorectal cancer. Br J Cancer (2011) 105(5):666–72. doi:10.1038/bjc.2011.295
65. Casey G, Neville PJ, Liu X, Plummer SJ, Cicek MS, Krumroy LM, et al. Podocalyxin variants and risk of prostate cancer and tumor aggressiveness. Hum Mol Genet (2006) 15(5):735–41. doi:10.1093/hmg/ddi487
66. Boman K, Larsson AH, Segersten U, Kuteeva E, Johannesson H, Nodin B, et al. Membranous expression of podocalyxin-like protein is an independent factor of poor prognosis in urothelial bladder cancer. Br J Cancer (2013) 108(11):2321–8. doi:10.1038/bjc.2013.215
67. Cipollone JA, Graves ML, Kobel M, Kalloger SE, Poon T, Gilks CB, et al. The anti-adhesive mucin podocalyxin may help initiate the transperitoneal metastasis of high grade serous ovarian carcinoma. Clin Exp Metastasis (2012) 29(3):239–52. doi:10.1007/s10585-011-9446-0
68. Hsu YH, Lin WL, Hou YT, Pu YS, Shun CT, Chen CL, et al. Podocalyxin EBP50 ezrin molecular complex enhances the metastatic potential of renal cell carcinoma through recruiting Rac1 guanine nucleotide exchange factor ARHGEF7. Am J Pathol (2010) 176(6):3050–61. doi:10.2353/ajpath.2010.090539
69. Lin CW, Sun MS, Wu HC. Podocalyxin-like 1 is associated with tumor aggressiveness and metastatic gene expression in human oral squamous cell carcinoma. Int J Oncol (2014) 45(2):710–8. doi:10.3892/ijo.2014.2427
70. Kelley TW, Huntsman D, McNagny KM, Roskelley CD, Hsi ED. Podocalyxin: a marker of blasts in acute leukemia. Am J Clin Pathol (2005) 124(1):134–42. doi:10.1309/7BHLAHHU0N4MHT7Q
71. Schopperle WM, Kershaw DB, DeWolf WC. Human embryonal carcinoma tumor antigen, Gp200/GCTM-2, is podocalyxin. Biochem Biophys Res Commun (2003) 300(2):285–90. doi:10.1016/S0006-291X(02)02844-9
72. Chen X, Higgins J, Cheung ST, Li R, Mason V, Montgomery K, et al. Novel endothelial cell markers in hepatocellular carcinoma. Mod Pathol (2004) 17(10):1198–210. doi:10.1038/modpathol.3800167
73. Hayatsu N, Kaneko MK, Mishima K, Nishikawa R, Matsutani M, Price JE, et al. Podocalyxin expression in malignant astrocytic tumors. Biochem Biophys Res Commun (2008) 374(2):394–8. doi:10.1016/j.bbrc.2008.07.049
74. Koch LK, Zhou H, Ellinger J, Biermann K, Holler T, von Rucker A, et al. Stem cell marker expression in small cell lung carcinoma and developing lung tissue. Hum Pathol (2008) 39(11):1597–605. doi:10.1016/j.humpath.2008.03.008
75. Sizemore S, Cicek M, Sizemore N, Ng KP, Casey G. Podocalyxin increases the aggressive phenotype of breast and prostate cancer cells in vitro through its interaction with ezrin. Cancer Res (2007) 67(13):6183–91. doi:10.1158/0008-5472.CAN-06-3575
76. Lin CW, Sun MS, Liao MY, Chung CH, Chi YH, Chiou LT, et al. Podocalyxin-like 1 promotes invadopodia formation and metastasis through activation of Rac1/Cdc42/cortactin signaling in breast cancer cells. Carcinogenesis (2014). doi:10.1093/carcin/bgu139
77. Meng X, Ezzati P, Wilkins JA. Requirement of podocalyxin in TGF-beta induced epithelial mesenchymal transition. PLoS One (2011) 6(4):e18715. doi:10.1371/journal.pone.0018715
78. Thomas SN, Schnaar RL, Konstantopoulos K. Podocalyxin-like protein is an E-/L-selectin ligand on colon carcinoma cells: comparative biochemical properties of selectin ligands in host and tumor cells. Am J Physiol Cell Physiol (2009) 296(3):C505–13. doi:10.1152/ajpcell.00472.2008
79. Dallas MR, Chen SH, Streppel MM, Sharma S, Maitra A, Konstantopoulos K. Sialofucosylated podocalyxin is a functional E- and L-selectin ligand expressed by metastatic pancreatic cancer cells. Am J Physiol Cell Physiol (2012) 303(6):C616–24. doi:10.1152/ajpcell.00149.2012
80. Larrucea S, Butta N, Rodriguez RB, Alonso-Martin S, Arias-Salgado EG, Ayuso MS, et al. Podocalyxin enhances the adherence of cells to platelets. Cell Mol Life Sci (2007) 64(22):2965–74. doi:10.1007/s00018-007-7374-6
81. Larrucea S, Butta N, Arias-Salgado EG, Alonso-Martin S, Ayuso MS, Parrilla R. Expression of podocalyxin enhances the adherence, migration, and intercellular communication of cells. Exp Cell Res (2008) 314(10):2004–15. doi:10.1016/j.yexcr.2008.03.009
82. Cho J, Degen JL, Coller BS, Mosher DF. Fibrin but not adsorbed fibrinogen supports fibronectin assembly by spread platelets. Effects of the interaction of alphaIIb beta3 with the C terminus of the fibrinogen gamma-chain. J Biol Chem (2005) 280(42):35490–8. doi:10.1074/jbc.M506289200
83. Cho J, Mosher DF. Role of fibronectin assembly in platelet thrombus formation. J Thromb Haemost (2006) 4(7):1461–9. doi:10.1111/j.1538-7836.2006.01943.x
84. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E, et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res (2010) 70(11):4327–34. doi:10.1158/0008-5472.CAN-09-3312
85. Theoret JF, Yacoub D, Hachem A, Gillis MA, Merhi Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb Res (2011) 128(3):243–50. doi:10.1016/j.thromres.2011.04.018
86. Sathish JG, Falati S, Croce K, Crump C, Furie BC, Furie B, et al. Antibody cross-linking of human platelet P-selectin induces calcium entry by a mechanism dependent upon Fcgamma receptor IIA. Thromb Haemost (2004) 92(3):598–605. doi:10.1160/TH03-11-0689
87. Modderman PW, von dem Borne AE, Sonnenberg A. Tyrosine phosphorylation of P-selectin in intact platelets and in a disulphide-linked complex with immunoprecipitated pp60c-src. Biochem J (1994) 299(Pt 3):613–21.
88. Fujimoto T, McEver RP. The cytoplasmic domain of P-selectin is phosphorylated on serine and threonine residues. Blood (1993) 82(6):1758–66.
89. Crovello CS, Furie BC, Furie B. Histidine phosphorylation of P-selectin upon stimulation of human platelets: a novel pathway for activation-dependent signal transduction. Cell (1995) 82(2):279–86. doi:10.1016/0092-8674(95)90315-1
90. Alonso-Martin S, Nowakowski A, Larrucea S, Fernandez D, Vilar-Egea M, Ayuso MS, et al. Overexpression of podocalyxin in megakaryocytes and platelets decreases the bleeding time and enhances the agonist-induced aggregation of platelets. Thromb Res (2010) 125(6):e300–5. doi:10.1016/j.thromres.2010.02.008
Keywords: platelet–tumor interaction, selectin ligands, podocalyxin, immune evasion
Citation: Amo L, Tamayo-Orbegozo E, Maruri N, Eguizabal C, Zenarruzabeitia O, Riñón M, Arrieta A, Santos S, Monge J, Vesga MA, Borrego F and Larrucea S (2014) Involvement of platelet–tumor cell interaction in immune evasion. Potential role of podocalyxin-like protein 1. Front. Oncol. 4:245. doi: 10.3389/fonc.2014.00245
Received: 15 July 2014; Accepted: 26 August 2014;
Published online: 11 September 2014.
Edited by:
Masoud H. Manjili, Virginia Commonwealth University Massey Cancer Center, USAReviewed by:
Mohey Eldin El Shikh, Queen Mary University of London, UKFabian Benencia, Ohio University, USA
Copyright: © 2014 Amo, Tamayo-Orbegozo, Maruri, Eguizabal, Zenarruzabeitia, Riñón, Arrieta, Santos, Monge, Vesga, Borrego and Larrucea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susana Larrucea, Regulation of Immune System Group, BioCruces Health Research Institute, Plaza de Cruces s/n, Barakaldo 48903, Spain e-mail:c2xhcnJ1Y2VhQG9zYWtpZGV0emEubmV0
†Laura Amo and Estíbaliz Tamayo-Orbegozo have contributed equally to this work.