 Demin Li
Demin Li Massimo Masiero1
Massimo Masiero1 Alison H. Banham
Alison H. Banham- 1Radcliffe Department of Medicine, Nuffield Division of Clinical Laboratory Sciences, Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, UK
- 2Cancer Research UK Molecular Oncology Laboratories, Department of Oncology, Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, UK
The Notch pathway is increasingly attracting attention as a source of therapeutic targets for cancer. Ligand-induced Notch signaling has been implicated in various aspects of cancer biology; as a consequence, pan-Notch inhibitors and therapeutic antibodies targeting one or more of the Notch receptors have been investigated for cancer therapy. Alternatively, Notch ligands provide attractive options for therapy in cancer treatment due to their more restricted expression and better-defined functions, as well as their low rate of mutations in cancer. One of the Notch ligands, Jagged1 (JAG1), is overexpressed in many cancer types, and plays an important role in several aspects of tumor biology. In fact, JAG1-stimulated Notch activation is directly implicated in tumor growth through maintaining cancer stem cell populations, promoting cell survival, inhibiting apoptosis, and driving cell proliferation and metastasis. In addition, JAG1 can indirectly affect cancer by influencing tumor microenvironment components such as tumor vasculature and immune cell infiltration. This article gives an overview of JAG1 and its role in tumor biology, and its potential as a therapeutic target.
Jagged1 (JAG1) is one of the five canonical ligands for Notch receptors expressed by mammalian cells. Along with Jagged2 (JAG2), it belongs to the Serrate/Jagged family, as opposed to the Delta/Delta-like family of ligands (DLL1, DLL3, and DLL4) (1, 2). JAG1, like the other canonical ligands, binds to Notch receptors and triggers activation, an interaction involving the Delta/Serrate/Lag-2 (DSL) domain of the JAG1 extracellular region (3, 4) (Figure 1). The JAG1 DSL domain is also responsible for its binding to the CD46 complement regulator, and this interaction is implicated in the functionality of T-helper cells (5). Of note, the binding and activating ability of JAG1 is tightly regulated by Notch receptor glycosylation (6–8). Recently, binding of the N-terminal C2 domain to phospholipid bilayers has been identified as a novel mechanism of modulating Notch activation induced by JAG1 (9). In addition, a number of publications indicate that the JAG1 intracellular domain can be released via γ-secretase-mediated cleavage, and the processed fragment, containing a PDZ-ligand motif at its C-terminus, has been reported to induce intrinsic reverse signaling within the ligand expressing cell (10–13). A soluble JAG1 extracellular domain, generated by ADAM17-mediated proteolytic cleavage, has also been implicated in mediating paracrine Notch signaling between endothelial cells and tumor cells (14), thus enabling Notch activation in more distant cells.
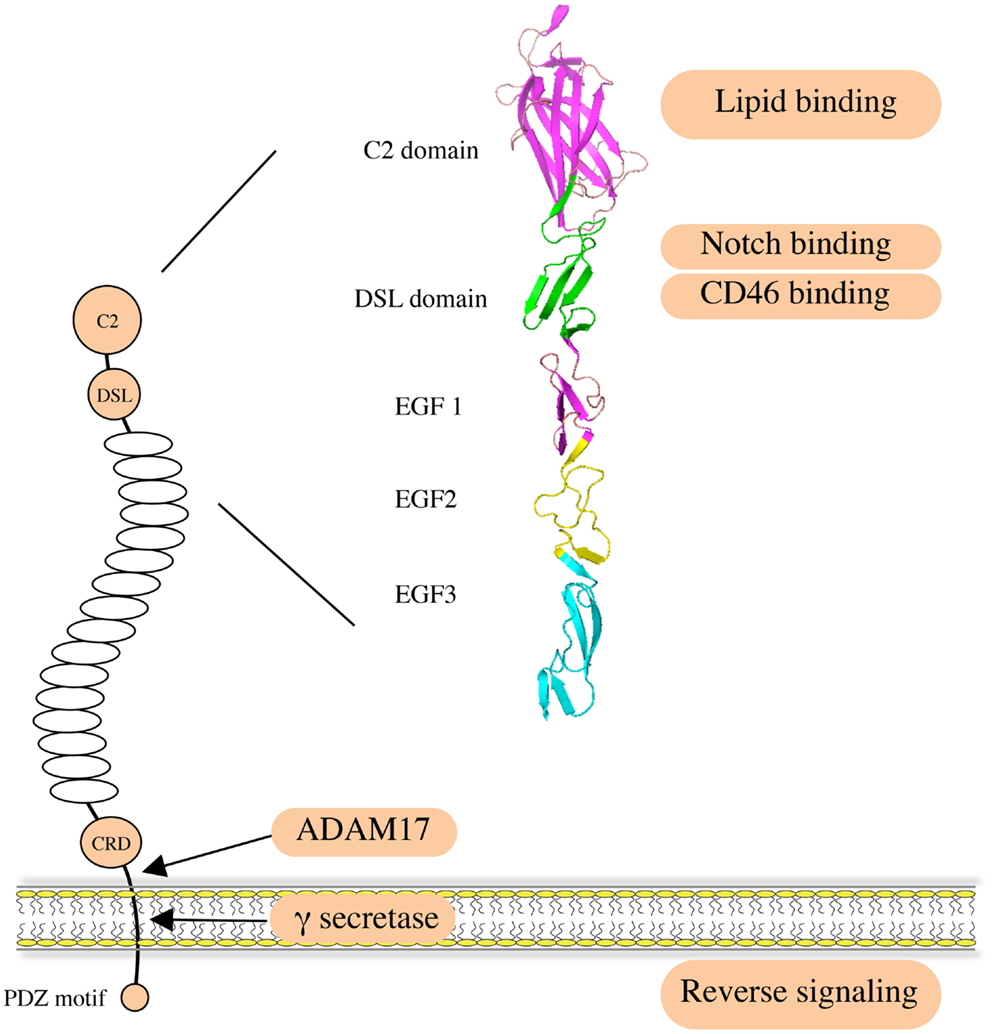
Figure 1. Structure and function of human JAG1. JAG1 is a type I transmembrane protein with an extracellular region featuring 16 epidermal growth factor (EGF) repeats followed by a cysteine-rich domain (CRD). Notch receptors bind to the DSL domain that is located N-terminal to the EGF repeats. A C2 phospholipid recognition domain at the very N-terminus reduces Notch activation upon binding to phospholipid bilayers. The CD46 recognition site largely overlaps with the Notch binding site. JAG1 can be cleaved by ADAM17 metalloprotease to release soluble protein, mediating paracrine Notch signaling on neighboring cells. It can also be processed intramembranously by γ-secretase to release the intracellular domain, within which the C-terminal PDZ-ligand motif is responsible for the intrinsic reverse signaling induced by JAG1.
Being a key component of the Notch signaling pathway, JAG1 plays an important role in both physiological and pathological conditions, including embryonic development and cancer. Among its physiological functions, it is worth noting that Jag1 gene knockout in mice causes severe vascular defects that are lethal in early embryogenesis (15), and that JAG1 mutations in human beings are responsible for Alagille syndrome, an inherited multi-organ developmental disorder (16). In this review, we summarize what has been discovered about the contribution of JAG1 to tumor biology to date, and discuss the evidence supporting JAG1 as a valid target for cancer therapy.
JAG1 Involvement in Cancer
Besides its role in Notch signaling in general (17), JAG1 has also been proven to play roles in multiple aspects of cancer biology, including tumor angiogenesis, neoplastic cell growth, cancer stem cells (CSCs), epithelial–mesenchymal transition (EMT), the metastatic process, and resistance to therapy in several types of cancer. Interestingly, JAG1 has been reported not only to be expressed and to play a role in cancer cells but its expression and activity have also been described in other cell types present in the tumor microenvironment such as mesothelial (18) and endothelial cells (14, 19), astrocytes (20), and osteoblasts (21). Importantly, JAG1 expression can be induced by other signaling pathways that are important in cancer such as TGF-β, WNT/β-catenin, IL-6, and NF-κB, as well as by the Notch pathway itself (22–26). We will first present and describe the mechanisms by which JAG1 exerts its functions in tumor biology (Figure 2), and then discuss its role in selected tumor types for which function and/or clinical relevance have been most extensively reported.
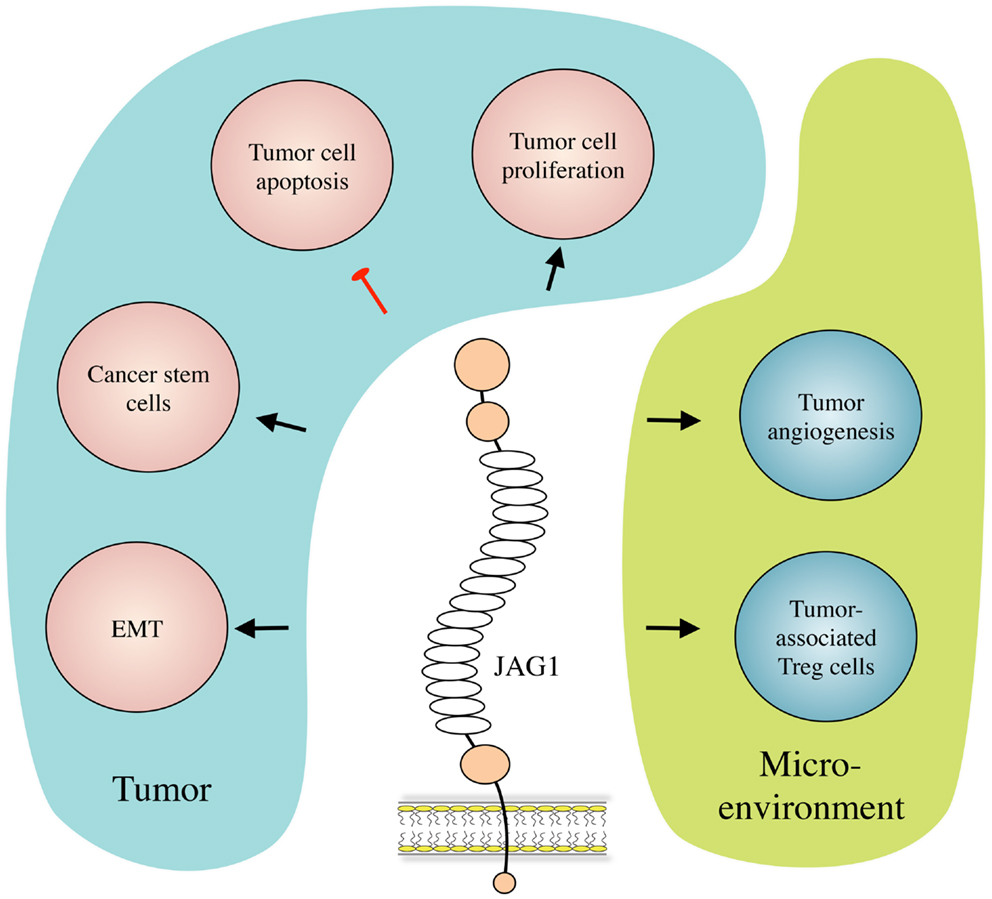
Figure 2. JAG1 in cancer biology. JAG1 expressed by cancer and/or stromal cells induces tumor cell growth and inhibits their apoptosis. JAG1 also induces and helps maintaining the cancer stem cell population, and enhances metastasis formation by inducing EMT. Meanwhile, in the tumor microenvironment, JAG1 promotes tumor-associated angiogenesis, and inhibits tumor-specific immunity by inducing regulatory T (Treg) cells.
Tumor Angiogenesis
Angiogenesis refers to the growth of new blood vessels from existing ones, which is important in normal physiological processes such as embryonic development and wound healing. Angiogenesis also plays a key role in cancer biology, and it is recognized as one of the hallmarks of cancer (27, 28). Sprouting angiogenesis, the main mechanistic variant of this process, is initiated with endothelial tip cell invasion, followed by a series of maturation steps including lumen formation, and recruitment of perivascular cells. Notch ligands expressed on endothelial cells, and their cognate receptors on both endothelial and perivascular cells, smooth muscle cells, and pericytes, are involved in multiple stages of blood vessel formation from initial sprouting until vessel maturation (29, 30). DLL4 expressed by endothelial tip cells suppresses the tip phenotype in neighboring stalk cells, thus maintaining a sufficient number of endothelial cells for vascular integrity and adequate tissue perfusion (7). In some models, JAG1 has been proven to have the opposite effect in that it promotes endothelial cell proliferation and sprouting, and inhibits DLL4-induced Notch signaling in endothelial cells (7). Thus, JAG1 deletion inhibits sprouting angiogenesis, and JAG1 overexpression opposes DLL4 to promote sprouting.
JAG1 is also indispensable for vascular smooth muscle cell coverage of newly formed vessels, as well as in maintaining the interaction between endothelial cells and the perivascular cells. Endothelium-specific Jag1 deletion causes deficits in vascular smooth muscle and fatal vascular defects (31). Endothelium-expressed JAG1 induces αvβ3 integrin expression, which in turn binds to VWF enriched on the basement membrane of the endothelial cells, facilitating smooth muscle adhesion, leading to vessel maturation. Genetic or pharmacologic disruption of JAG1, Notch, αvβ3, or VWF suppresses smooth muscle coverage of nascent vessels and arterial maturation during vascular development (32). The attachment of perivascular cells, such as smooth muscle cells, in turn regulates JAG1 expression and signaling through their surface-expressed Notch receptors. Perivascular cell-expressed Notch3 can also be induced by endothelial JAG1 and subsequently upregulates JAG1 expression on perivascular cells to form an autoregulatory loop that promotes both their maturation as pericytes and angiogenesis (33). Blockade of Notch signaling by knocking down Notch3 or JAG1 expression abrogates angiogenesis. This role during both the early and late stages of angiogenesis makes JAG1 the only Notch ligand for which such a broad function has been identified.
JAG1 has been reported to be strongly expressed by tumor-associated blood vessels, for example, in brain and ovarian cancer (34, 35), where it can trigger Notch signaling in tumor cells (angiocrine function) to promote tumor growth (19, 36, 37). Functionally, head and neck squamous cell carcinoma (HNSCC) tumors have been reported to secrete growth factors, such as HGF and TGF-α, that act in an autocrine/juxtacrine fashion to upregulate JAG1 expression through the MAKP pathway. This tumor-expressed JAG1 then stimulates endothelial cell sprouting, promoting angiogenesis and tumor growth in a mouse xenograft model (38). Furthermore, JAG1 expression levels also correlate with microvessel formation in human HNSCC tissues (38). A JAG1 pro-angiogenic role has also been reported in ovarian cancer models, where it was proven that Jag1 stromal silencing drastically reduced tumor microvascular density and neoplastic growth (39). All these points suggest that JAG1-targeted therapies could be of benefit to cancer patients even in the absence of tumoral JAG1 expression due to its role in tumor angiogenesis.
Cancer Stem Cells
The Notch pathway plays an important role in normal stem cell biology, and this resulted in unacceptable levels of gastrointestinal toxicity when pan-Notch ablation was attempted using γ-secretase inhibitors (40, 41). Interestingly, unlike the Dll1/4 ligands, Jag1 was found to be dispensable for the homeostasis of normal intestinal stem cells (42), suggesting that JAG1 targeting is likely to have less side effects.
In the context of cancer, a small, distinct subpopulation of cells within tumors termed “cancer stem cells” (CSCs), tumorigenic cells, or cancer-initiating cells has been identified in several tumor types (43), including breast cancer (CD44+CD24−/lowlineage− and Aldefluor+ cell fractions), colon cancer (CD133+ cells), and cervical cancer (CD66+ cells). These cells are characterized by their self-renewal, high clonogenic potential, and asymmetric division producing daughter stem and differentiated cancer cells, enabling them to regenerate a tumor even when injected in very low numbers (44–51). CSCs are deemed to have increased invasive potential and resistance to several anti-cancer treatments and are often thought to be responsible for patient relapse and metastasis (52, 53). Therefore, therapies specifically targeting CSCs hold great potential for improving cancer treatment and outcome.
Notch signaling is important for both CSC maintenance and self-renewal (54, 55). Notch1 and Notch4 have been reported to have higher activity in the enriched breast CSC population, and inhibition of Notch signaling reduced stem cell activity in vitro and tumor formation in vivo (54, 56, 57). Similarly, Notch activity is 10- to 30-fold higher in the colon CSC population, where it has an important role in preventing apoptosis (58).
Several studies have functionally linked JAG1 to “stemness” in cancer, and it appears to be the main ligand driving CSC Notch signaling. In breast cancer, high levels of JAG1 promote stem cell self-renewal and potentiate mammosphere formation in vitro (23). In this context, JAG1 seems to play a central role in linking various pathways, involving well-established cancer-related molecules such as Notch3, interleukin-6 (IL-6), carbonic anhydrase IX (CAIX), and NF-κB (23, 26, 59, 60). JAG1 involvement in breast CSC has also been confirmed by mouse models in which mammary-specific deletion of Lfng, an N-acetylglucosamine transferase that prevents Notch activation by Jagged ligands, induces basal-like breast cancer with higher Jag1 activity and enhanced CSC proliferation (61). Likewise, JAG1 has also been involved in CSC biology in other tumor types. For example, JAG1 expressed by both tumor and endothelial cells plays an important role in glioma/glioblastoma-initiating cells (36, 37). Tumor-associated endothelial cell-expressed JAG1 has been also described to mediate lymphoma (19) and colon CSC maintenance. In the latter, soluble JAG1 produced by tumor-associated endothelial cells promoted the CSC phenotype in human colorectal cancer cells (14). Overall, these data indicate JAG1 as an important inducer of the stem cell phenotype in different cancer types and, importantly, demonstrates that both tumor and stromal JAG1 expression are relevant targets for CSCs.
Epithelial–Mesenchymal Transition, Invasion, and Metastasis
The ability of tumor cells to invade the surrounding tissues and the ability of tumor cells to colonize distant organs (metastatic process) are both key features of aggressive cancers of pivotal clinical relevance. In order to escape their local environment, epithelial cells can exploit a reversible developmental program called EMT, during which loss of epithelial features (e.g., E-cadherin expression and cell-to-cell adhesion) and the acquisition of mesenchymal traits enables tumor cells to invade, resist apoptosis, disseminate and, as more recently observed, acquire stem cell features (28, 62–64). Notch signaling has been extensively studied in this context (55), and several reports have described JAG1 involvement in EMT, invasive potential, and metastasis particularly but not exclusively, in breast cancer. JAG1-induced signaling in breast cancer inhibits the epithelial phenotype via upregulation of the EMT master-regulator SLUG, and promotes tumor growth and metastasis (65). This is also reported to be important for TGFβ-induced EMT in mammary gland cells (22). Furthermore, JAG1 can increase tumor migratory and invasive behavior by inducing the urokinase-type plasminogen activator (uPA), a well-known marker of recurrence and metastasis (66). Finally, but not least, JAG1 has also been shown to be involved in the tissue specificity of breast cancer dissemination since it has been described to have significant roles in metastasis to the bone and brain (20, 21).
A role in EMT has also been described in cervical cancer, where JAG1 expression correlates with the rapid induction of phosphoinositol-3-kinase (PI3K)-mediated EMT (67); in hepatocellular carcinoma where it is repressed by the tumor-suppressor RUNX3 (68); and in treatment-resistant pancreatic cancer cells, where the JAG1-Notch2 axis controls several EMT transcription factors such as SNAIL, SLUG, and ZEB1 (69). Pro-invasive, migratory, and metastatic function has been also demonstrated for prostate cancer, where high JAG1 expression has been clinically linked to metastasis development and regulation of migration/invasion via NF-κB (70, 71), and for colon cancer, where it mediates APEX1 pro-tumorigenic functions and induces the metastasis markers MMP-2 and MMP-9 (72, 73). Overall, JAG1-induced Notch signaling appears to be implicated in different steps of the invasion/metastatic process. This, in conjunction with the relevance demonstrated in different tumor types, suggests JAG1 as an interesting therapeutic target to inhibit tumor cell invasiveness/metastasis.
Tumor Cell Proliferation, Cell Cycle Regulation, Drug Resistance, and Survival
As for generalized Notch signaling, JAG1 can also directly affect more basic cellular functions such as cell cycle progression/proliferation and apoptosis/cell survival. In several cancer types, JAG1 induces tumor cell growth and promotes cell cycle progression. For example, it can directly regulate the cell cycle and induce proliferation by inducing cyclin D1 in breast cancer (74), cyclin D1, cyclin E, and c-Myc in colon cancer (73), or by promoting CDK2 kinase activity while repressing the p27 cell cycle suppressor in prostate cancer cells (75). Similar pro-proliferative functions have also been reported for glioma, myeloma, and ovarian cancer (18, 39, 76, 77). As anticipated, JAG1 can also affect cell death and there are several studies indicating that this Notch ligand exerts anti-apoptotic functions, although a mechanism remains to be fully elucidated. Interestingly, JAG1 can prevent both spontaneous apoptosis, for example, in glioma and prostate cancer cells (71, 77), as well as chemotherapy-induced cell death. Examples of the latter include lymphoma cells with respect to doxorubicin (19), ovarian cancer and taxanes (39), and pancreatic cancer and gemcitabine (69). While a role in spontaneous apoptosis prevention informs us regarding a physiological role played by JAG1, its role in chemo-protection represents a key finding as it highlights the opportunity to treat unresponsive tumor cases via combination chemotherapy treatments that include JAG1-Notch signaling blockade.
T-Cell Regulation
A favorable tumor microenvironment is vital for cancer growth and survival. Various immune cells are found within the proximity of tumors, including T-cells, dendritic cells, macrophages, neutrophils, etc., but rarely the natural killer (NK) cells that are highly efficient in killing MHC downregulated tumor cells (78). Importantly, in many cancer types, there is an enrichment of T regulatory (Treg) cells capable of inhibiting tumor-specific immune responses and thus helping tumor cells evade immunosurveillance (79). Treg cells, in combination with other factors, such as myeloid-derived suppressor cells and cytokines, foster an immunosuppressive tumor microenvironment that supports tumor growth. Notably, inhibitors to immune checkpoints such as PD-1 and CTLA-4 can activate the tumor microenvironment and are showing exciting promise in the clinic in a variety of cancer types (78).
The induction and expansion of Treg cells in the tumor microenvironment involves crosstalk between tumor cells and dendritic cells, in which Notch signaling, and in particular JAG1-induced Notch activation, plays an important role. JAG1-Notch3 signaling has been reported to be essential for Treg induction and expansion stimulated by OX40L (80), whilst JAG1 expression in antigen presenting cells induces antigen-specific Tregs (81, 82). Furthermore, maturation of dendritic cells via JAG1 promotes survival and proliferation of Tregs (83).
JAG1-induced Notch signaling may also have impact on CD4+ T helper (Th) cell activation through a recently identified interaction between JAG1 and CD46. CD46 (MCP) is a ubiquitously expressed human type I transmembrane glycoprotein that was originally discovered as a complement regulatory protein and then a cell-entry receptor enabling viral infection. Activation of CD46 on CD4+ T-cells was shown to regulate the expression of Notch and its ligands, and, furthermore, JAG1 was identified as an additional physiological ligand for CD46 (5). The JAG1 binding site of CD46 overlaps with that of Notch receptors (5), and therefore, crosstalk between the complement and Notch systems may also be significant in cancer patients.
JAG1 in Individual Cancer Types
There is a significant body of literature describing JAG1 functionality in a variety of tumor types, which is summarized in Table 1. The cancer types that have been most extensively studied are discussed in more detail below.
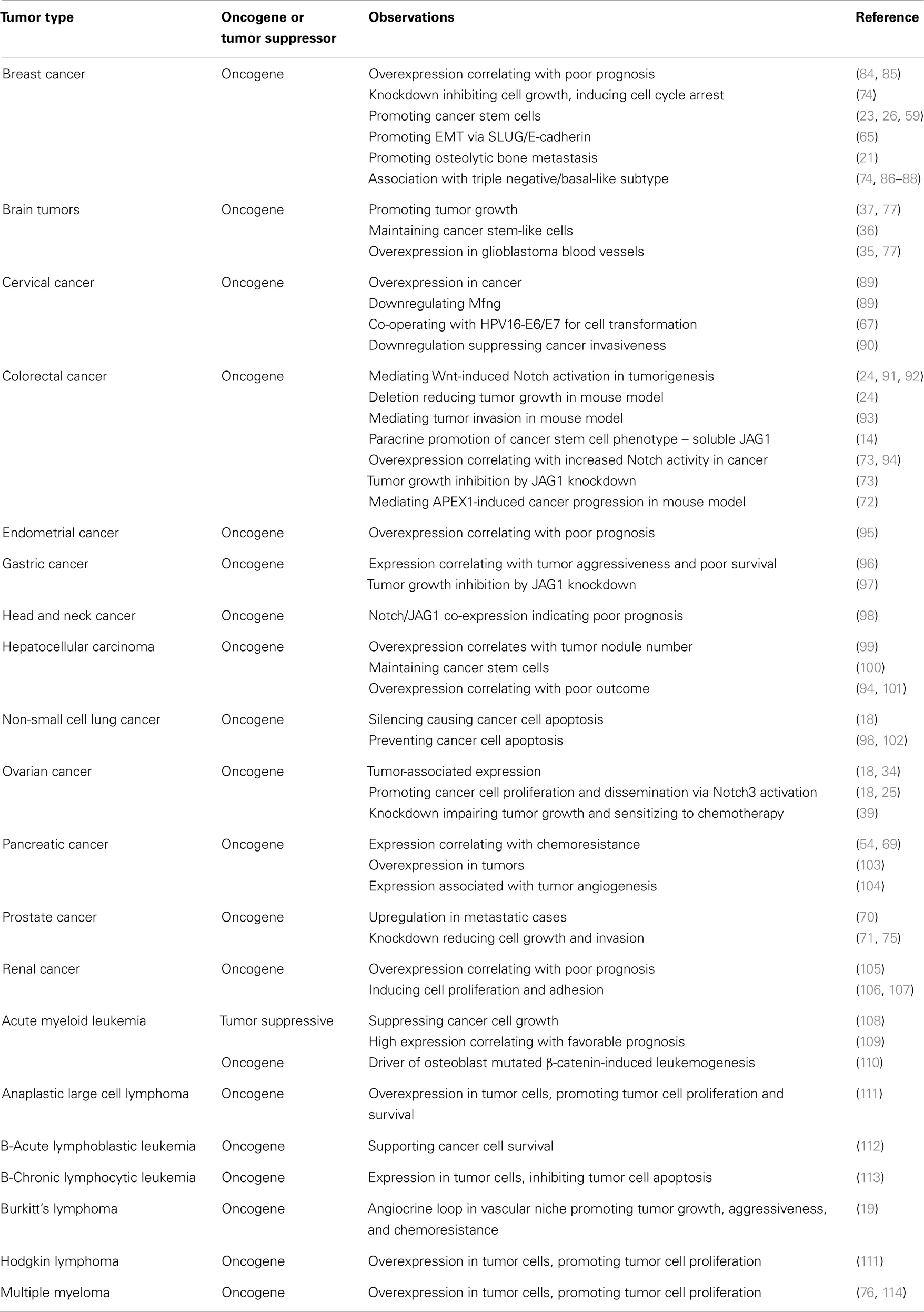
Table 1. JAG1 involvement in individual cancer types.
Breast Cancer
Multiple lines of evidence suggest that Notch signaling is involved in breast cancer development, maintenance and metastasis, and overexpression of Notch-1, -3, and -4 activated intracellular domains in mice causes aggressive and metastatic mammary tumors (115–119). Active forms of the Notch1 and Notch4 receptors also transform both normal human and murine mammary epithelial cells (116, 120, 121). When primary breast cancer samples are examined, accumulation of activated Notch1 and Notch3 is frequently observed in tumor cells (121, 122). Conversely, loss of a Notch negative regulator, Numb, is found to be associated with higher grade and worse prognosis in primary breast cancer (121, 123), and suppression of Notch activity by Notch3 and CSL knockdown promotes cancer cell apoptosis and inhibits tumor cell growth (122).
Unlike Notch pathway activation in T-cell acute lymphoblastic leukemia (T-ALL), which is mostly caused by Notch1 gene mutations, the induction of Notch signaling in breast cancer (and other carcinomas) is predominantly associated with ligand-dependent mechanisms of activation. Studies indicate that JAG1 is the most prominent ligand involved in this aberrant Notch activation in breast cancer. JAG1 mRNA and protein are overexpressed in this tumor type, with high expression levels correlating with poor prognosis (84, 85). Functionally, in vitro JAG1 knockdown inhibits tumor cell growth inducing cell cycle arrest (74). JAG1-stimulated Notch signaling induces uPA, which is a validated marker of recurrence, high metastasic risk, and death from breast malignancy (66). JAG1 has a proven role in regulating breast CSC numbers (23, 26), EMT (65), and the metastatic process (21).
Two overlapping subtypes, triple negative (TN) breast cancer lacking estrogen receptor (ER), progesterone receptor (PR), and Her2 receptors, and basal-like breast cancer, which is generally associated with BRCA1 activation, are normally more aggressive and have poorer prognoses (124). These subtypes of breast cancers generally have higher levels of JAG1 expression, which correlate with reduced disease free survival (DFS). In contrast, the less aggressive luminal subtype, which is more associated with BRCA2 mutations, has lower JAG1 expression (74, 87, 88). In addition, a rare aggressive cell population in luminal-like cancers, lumino-basal cells, shares a gene signature with basal-like cancer, and their responsiveness to hormone therapy can be enhanced by blocking Notch signaling. Although a specific role for JAG1 was not investigated, these cells do have higher JAG1 expression than typical luminal-like cancers (125). These observations have also been confirmed by functional studies in breast cancer mouse models in which Jag1 played a key role in inducing a basal-like phenotype (61).
Cervical Cancer
Cervical cancer is the second most common cancer in women. Infection by high-risk human papillomavirus (HPV) such as HPV-16 and HPV-18, and the continued expression of viral oncoprotein E6 and E7 is linked to its development and progression (126).
The first indication for the involvement of Notch signaling in cervical cancer was the consistent pathway activation observed in this tumor type and in cervical metaplastic tissues but not in normal specimens (127). Furthermore, Notch1 expression correlates with disease progression, with little or no protein expression in normal cervical epithelium and high expression in precancerous and cancer tissues (128). Importantly, Notch1 protein is detected at high levels in the nucleus, indicating that activated Notch signaling may contribute to the progression of HPV-associated cervical neoplasia. Notch activation seems to contribute to cervical cancer development through its co-operation with HPV-16 E6 and E7 oncoproteins. Notch activation synergizes with HPV proteins in the transformation of immortalized human keratinocytes and primary keratinocytes (129, 130), and co-expression of the activated intracellular form of Notch1, along with HPV E6 and E7, can also support tumor growth in vivo (131).
Cervical cancer cells do not seem to present Notch-activating mutations, as sequencing of Notch1 alleles failed to detect any of the mutations previously associated with T-ALL development (132). However, increased expression of the Notch ligand JAG1 was observed in cervical cancer cell lines and primary samples. JAG1 activity was indispensable for tumor maintenance as dominant negative JAG1 and RNA interference reduced cell line tumorigenicity in vitro (89). Overexpression of JAG1 in cervical cancer samples was coupled with the downregulation of Mfng, a negative regulator of the Jagged-Notch1 interaction (89). JAG1 also co-operates with HPV16-E6 and E7 oncoproteins in cell transformation and during in vivo tumor growth (67). The involvement of other Notch ligands in cervical cancer is unclear, but DLL1 expression was not upregulated in primary samples (89).
Targeting JAG1-induced Notch1 activation, by Notch1 RNA interference or by a γ-secretase inhibitor, suppressed cervical cancer invasiveness. Expression of microRNA-34a, which downregulates both Notch1 and JAG1, inhibits abnormal cell growth and suppresses cervical cancer invasiveness by repressing Notch-JAG1 signaling (90).
However, not all evidence supports a tumor-promoting role of Notch signaling in cervical cancer. Talora et al. found that Notch1 activity downregulation was required for sustained HPV-E6/E7 expression and subsequent steps of malignant transformation. Consequently, Notch1 activity was absent in their cohort of invasive cervical cancer samples (133). Notch1 activation has also been reported to induce the cervical cancer cell line HeLa to undergo apoptosis, growth arrest, and tumor growth suppression in vivo (134–136), although the role of JAG1 has not specifically been investigated within this tumor-suppressor context. The discrepancies between these results and the others could be due to the differences in sample cohorts, cell lines, and/or experimental reagents, and further studies are needed to reconcile the differences and to validate the role of Notch signaling in cervical cancer pathogenesis.
Colon Cancer
Colon cancer is one of the leading causes of cancer death worldwide, and the genetic causes of colon cancer involve mutations of oncogenes, suppressor genes, and multiple developmental pathways, including Wnt, Notch, Hedgehog, and BMP pathways. Mutations in the Wnt pathway cause colon cancer through constitutive activation of the β-catenin/TCF transcription factor complex (137). It has been demonstrated that pharmacological blockade of Notch activity using a γ-secretase inhibitor impairs intestinal homeostasis, and suppresses adenoma cell proliferation and induces differentiation, suggesting that tumorigenesis in this model requires a concerted activation of both Notch and Wnt (138). Further investigation proved that Notch works downstream of Wnt, as this pathway transcriptionally induces the Notch ligand JAG1 to trigger Notch activity (24, 91). Accordingly, deletion of the Wnt signaling inhibitor progastrin decreased JAG1 expression and Notch activation, and subsequently promoted the differentiation of colon cancer cells (92). Furthermore, deletion of JAG1 reduced tumor growth in the ApcMin/+ mouse model, confirming JAG1 as a pathological link between Wnt and Notch pathways in colon cancer (24). Stromal JAG1-induced Notch activation also mediated the tumor invasion and intravasation caused by the deletion of metastasis-suppressor gene Aes (Grg5) in ApcΔ716 intestinal polyposis mice, suggesting that JAG1-induced Notch signaling can be a promising target for prevention and treatment of colon cancer metastasis (93). Similarly, JAG1 expressed by endothelial cells has been implicated in fostering colorectal CSCs (14).
In human beings, Notch signaling was shown to be strongly activated in primary human colorectal cancers, and has an important role in cancer initiation and progression through the regulation of the main cellular functions associated with tumorigenesis, such as apoptosis, proliferation, angiogenesis, and cell migration. Microarray analysis discovered that the expression levels of Notch1, and its target Hes1, increased with increasing tumor grade (139). In situ hybridization on 130 colorectal cancer samples found that Notch signaling is constantly activated as measured by Hes1 expression (86). Immunohistochemistry on a colon cancer tissue microarray confirmed that Hes1 is overexpressed in primary colon cancer tissues (140).
The cause of Notch overactivation in colon cancer appears to be ligand-dependent and to correlate with elevated JAG1 expression levels (73, 94). Accordingly, JAG1 knockdown leads to reduced Notch signaling activity that is accompanied by cell growth inhibition, cell cycle arrest, migration, and invasion inhibition, as well as tumor growth suppression (73). Apurinic-apyrimidinic endonuclease-1 (APEX1), a multiple-functional DNA repair enzyme, promotes colon cancer progression through activating the JAG1/Notch signaling pathway. APEX1-induced JAG1 expression in colon cancer cells, which subsequently activated Notch signaling to promote tumorigenicity, migration, invasion, angiogenesis, tumor formation, and metastasis in mouse xenograft models (72).
Ovarian Cancer
JAG1 is the main Notch ligand expressed by ovarian cancer cells; it is also strongly expressed by peritoneal mesothelial cells (18) and tumor-associated endothelial cells (34). JAG1 activates primarily Notch3 in ovarian cancer and promotes proliferation and dissemination within the intraperitoneal cavity (18). Its expression was induced by Notch3 activity itself, and was repressed by the WNT/β-catenin pathway (25). JAG1 gene silencing in tumor cells reduced viability and sensitized them to taxane treatment both in vitro and in vivo, where it drastically reduced tumor growth. Silencing of stromal-expressed Jag1 also impaired tumor growth, without affecting tumor cell proliferation, by reducing the microvascular density. Combined tumor and stromal silencing proved to be synergistic, indicating the importance of targeting both the tumor and its microenvironment (39).
Hematological Cancers
The first oncogenic role for Notch signaling was described in T-ALL as a consequence of the identification of a high frequency of activating Notch1 mutations (141–143). Notch1 is also one of the most frequently mutated genes in chronic lymphocytic leukemia (CLL) (144, 145). In contrast to solid tumors, aberrations in hematological malignancies generally involve Notch receptors or pathway regulator mutations. While some mutations may enable ligand-independent pathway activation, others that stabilize the Notch intracellular domain (e.g., by targeting PEST motifs) still initially require their ligand-induced activation (143, 146).
Despite the predominance of genetic alterations as mechanisms of pathway alteration, Notch ligands such as JAG1 do play a part in some hematological cancers as they do in normal hematopoiesis (147, 148). Non-mutated Notch1 is highly expressed in Hodgkin lymphoma (HL) and anaplastic large cell lymphoma (ALCL) cells, and tumor-associated JAG1, overexpressed by bystander cells as well as by neighboring tumor cells, induces Notch1 activation and promotes tumor cell proliferation and survival (111). Similarly, in multiple myelomas, Notch1, Notch2, and JAG1 were highly expressed in primary tumor samples, and JAG1-induced Notch activation drove myeloma cell proliferation (76, 114).
Ligand-induced Notch overactivation was also observed in a subpopulation of primary B-CLL cells that are protected from spontaneous apoptosis as a result of JAG1 stimulation in ex vivo cultures (113). These cells overexpressed Notch ligands such as JAG1 and JAG2, and soluble JAG1 could stimulate Notch activation and increase B-CLL survival through the NF-κB pathway (113). Although this study was purely based on ex vivo systems, the results provide a good indication that JAG1 may play an important role in sustaining B-CLL cell survival.
Apart from inducing Notch activation from cancer cells themselves, JAG1 expressed by stromal cell also plays an important role in supporting cancer cell survival and tumor growth in hematological malignancies. B-ALL stroma express JAG1, JAG2, and DLL1, and these ligands are responsible for the synergistic activation of cancer cells that expressed Notch3 and Notch4, which ultimately support B-ALL cell survival (112). Interestingly, B-cell lymphomas have been reported to produce FGF4, which upregulates Jag1 on adjacent endothelial cells, that in turn induces Notch2 regulation of Hey1 in the lymphoma cells (19). This angiocrine FGF4-FGFR1/Jag1-Notch2 loop contributed to extranodal invasion and chemoresistance, thus extending the clinical relevance of Jag1 targeting into lymphomas that lack Notch receptor mutations or JAG1 expression.
Interestingly, Notch pathway activation does not always promote the pathogenesis of hematological malignancies. For example, Notch activation in acute myeloid leukemia (AML) suppressed, rather than promoted, cancer growth, whilst whole-genome expression analysis discovered that Notch signaling was silenced in AML (149, 150). In vivo and ex vivo activation of tumoral Notch, using genetic modification, induced cell cycle arrest, differentiation, and apoptosis in AML-initiating cells, and suppressed AML growth. JAG1 stimulation was also found to suppress AML cell line growth (108), and high JAG1 surface levels in leukemia cells was proposed as an independent favorable prognostic factor in AML patients (109). These results suggest that promoting JAG1-stimulated Notch activity could be a potential route for novel therapy in AML. However, further studies are required; first, as the expression level of the Notch1 ICD did not correlate with AML outcome (109), and second, a separate study reported activating mutations of β-catenin in mouse osteoblasts that induced AML development through Jag1-induced Notch activation (110). This group reported that a large proportion (38%) of patients with myelodysplastic syndromes or AML had osteoblast associated β-catenin activation-induced Notch signaling (110).
Therapeutic Potential and Implications
It is well established that the Notch pathway (17), and in particular JAG1-induced Notch activation, plays important roles in tumor biology, affecting both cancer cells and multiple components of the neoplastic microenvironment (e.g., vasculature and immune cells). This, in addition to the facts that JAG1 is often upregulated in tumor cells (although generally not mutated in cancer), and that ligand-induced activation is required even in the presence of some Notch receptor mutations, makes it a particularly attractive target for therapy (143).
Generally, the main concern in targeting the Notch pathway by pan-Notch inhibitors was the resulting gastrointestinal toxicity (41). Antibodies targeting individual Notch receptors (151, 152) seem to avoid toxicity but, in some situations, tumors may have high levels of more than one receptor (e.g., Notch1 and Notch2 in CLL, Notch1 and Notch4 in breast CSCs), or potentially, the roles of individual Notch receptors may contribute differently in the tumor versus its microenvironment, which adds further complications. For example, while Notch1 is often oncogenic when expressed by cancer cells, Notch3 expressed by perivascular cells plays a major role in JAG1-mediated vascular function (33), and it is also important in Treg induction and expansion in the tumor microenvironment (80).
JAG1 has a number of advantages as a target for anti-cancer therapy over other Notch ligands. DLL4 dysfunction exhibited haploinsufficiency (153), while that of JAG1 did not (15). Thus, targeting JAG1 may provide a greater therapeutic window in which to reduce Notch activity without causing severe adverse effects. In addition, while in the context of cancer, DLL4 mainly functions in the vasculature, JAG1 has roles in vasculature, immunosuppressive Treg cells, and the tumor/stem cells. Thus, targeting JAG1 on both stroma and tumor cells could induce synergistic effects as demonstrated in an ovarian cancer model (39). The fact that chronic blockade of DLL4 causes severe pathological changes in animal models (154) makes alternative targets such as JAG1 even more appealing.
Crosstalk between Notch signaling and other pathways, such as Wnt, Hedgehog, and vascular endothelial growth factor (VEGF) pathways, as well as with the immune system, make Notch and its ligands attractive approaches for combination therapy. Bevacizumab treatment targeting VEGF-induced angiogenesis has made a profound impact on various cancer types, but resistance to treatment is frequently observed in both preclinical and clinical settings (155). Increased pericyte coverage of the tumor vasculature that maintains its integrity is thought to be one of the main mechanisms that cause resistance to anti-angiogenic treatments (156). Therefore, the role of endothelium-expressed JAG1 in vascular smooth muscle biology (32) suggests that targeting JAG1 may have beneficial synergistic effects with anti-angiogenic approaches.
Furthermore, due to its anti-apoptotic and pro-“stemness” functions, JAG1 blockade represents an attractive option also for combination therapy approaches with standard chemotherapy as demonstrated in preclinical models of ovarian and pancreatic cancer and lymphoma (19, 39, 69).
In conclusion, targeting JAG1 will provide a new approach to act on multiple aspects of tumor biology, and represents a promising new strategy in developing novel anti-tumor therapies.
Conflict of Interest Statement
The authors are inventors and contributors of a patent on Jagged1 therapeutic antibody development.
Acknowledgments
This work is supported by a Cancer Research UK program grant A10702. The authors thank Dr. Amanda Anderson for critical reading of the manuscript.
References
1. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol (2006) 7:678–89. doi:10.1038/nrm2009
2. Fiuza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol (2007) 194:459–74. doi:10.1677/JOE-07-0242
3. Cordle J, Johnson S, Tay JZ, Roversi P, Wilkin MB, De Madrid BH, et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat Struct Mol Biol (2008) 15:849–57. doi:10.1038/nsmb.1457
4. Whiteman P, De Madrid BH, Taylor P, Li D, Heslop R, Viticheep N, et al. Molecular basis for Jagged-1/Serrate ligand recognition by the Notch receptor. J Biol Chem (2013) 288:7305–12. doi:10.1074/jbc.M112.428854
5. Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA, Laing A, et al. The CD46-Jagged1 interaction is critical for human T(H)1 immunity. Nat Immunol (2012) 13(12):1213–21. doi:10.1038/ni.2454
6. Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell (2005) 16:927–42. doi:10.1091/mbc.E04-07-0614
7. Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell (2009) 137:1124–35. doi:10.1016/j.cell.2009.03.025
8. Taylor P, Takeuchi H, Sheppard D, Chillakuri C, Lea SM, Haltiwanger RS, et al. Fringe-mediated extension of O-linked fucose in the ligand-binding region of Notch1 increases binding to mammalian Notch ligands. Proc Natl Acad Sci U S A (2014) 111:7290–5. doi:10.1073/pnas.1319683111
9. Chillakuri CR, Sheppard D, Ilagan MX, Holt LR, Abbott F, Liang S, et al. Structural analysis uncovers lipid-binding properties of Notch ligands. Cell Rep (2013) 5:861–7. doi:10.1016/j.celrep.2013.10.029
10. Hock B, Bohme B, Karn T, Yamamoto T, Kaibuchi K, Holtrich U, et al. PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proc Natl Acad Sci U S A (1998) 95:9779–84. doi:10.1073/pnas.95.17.9779
11. Ascano JM, Beverly LJ, Capobianco AJ. The C-terminal PDZ-ligand of JAGGED1 is essential for cellular transformation. J Biol Chem (2003) 278:8771–9. doi:10.1074/jbc.M211427200
12. LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem (2003) 278:34427–37. doi:10.1074/jbc.M302659200
13. Duryagina R, Thieme S, Anastassiadis K, Werner C, Schneider S, Wobus M, et al. Overexpression of Jagged-1 and its intracellular domain in human mesenchymal stromal cells differentially affect the interaction with hematopoietic stem and progenitor cells. Stem Cells Dev (2013) 22(20):2736–50. doi:10.1089/scd.2012.0638
14. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell (2013) 23:171–85. doi:10.1016/j.ccr.2012.12.021
15. Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet (1999) 8:723–30. doi:10.1093/hmg/8.5.723
16. Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in Alagille syndrome. Hum Mutat (2001) 17:18–33. doi:10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T
17. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell (2014) 25:318–34. doi:10.1016/j.ccr.2014.02.018
18. Choi JH, Park JT, Davidson B, Morin PJ, Shih IeM, Wang TL. Jagged-1 and Notch3 juxtacrine loop regulates ovarian tumor growth and adhesion. Cancer Res (2008) 68:5716–23. doi:10.1158/0008-5472.CAN-08-0001
19. Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell (2014) 25:350–65. doi:10.1016/j.ccr.2014.02.005
20. Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med (2013) 5:384–96. doi:10.1002/emmm.201201623
21. Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell (2011) 19:192–205. doi:10.1016/j.ccr.2010.12.022
22. Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J (2004) 23:1155–65. doi:10.1038/sj.emboj.7600069
23. Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest (2007) 117:3988–4002. doi:10.1172/JCI32533
24. Rodilla V, Villanueva A, Obrador-Hevia A, Robert-Moreno A, Fernandez-Majada V, Grilli A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A (2009) 106:6315–20. doi:10.1073/pnas.0813221106
25. Chen X, Stoeck A, Lee SJ, Shih IeM, Wang MM, Wang TL. Jagged1 expression regulated by Notch3 and Wnt/beta-catenin signaling pathways in ovarian cancer. Oncotarget (2010) 1:210–8.
26. Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun (2013) 4:2299. doi:10.1038/ncomms3299
27. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi:10.1016/S0092-8674(00)81683-9
28. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
29. Sainson RC, Harris AL. Regulation of angiogenesis by homotypic and heterotypic notch signalling in endothelial cells and pericytes: from basic research to potential therapies. Angiogenesis (2008) 11:41–51. doi:10.1007/s10456-008-9098-0
30. Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell (2009) 16:196–208. doi:10.1016/j.devcel.2009.01.015
31. High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A (2008) 105:1955–9. doi:10.1073/pnas.0709663105
32. Scheppke L, Murphy EA, Zarpellon A, Hofmann JJ, Merkulova A, Shields DJ, et al. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood (2012) 119:2149–58. doi:10.1182/blood-2011-04-348706
33. Liu H, Kennard S, Lilly B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ Res (2009) 104:466–75. doi:10.1161/CIRCRESAHA.108.184846
34. Lu C, Bonome T, Li Y, Kamat AA, Han LY, Schmandt R, et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res (2007) 67:1757–68. doi:10.1158/0008-5472.CAN-06-3700
35. Jubb AM, Browning L, Campo L, Turley H, Steers G, Thurston G, et al. Expression of vascular Notch ligands Delta-like 4 and Jagged-1 in glioblastoma. Histopathology (2012) 60:740–7. doi:10.1111/j.1365-2559.2011.04138.x
36. Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res (2011) 71:6061–72. doi:10.1158/0008-5472.CAN-10-4269
37. Jeon HM, Kim SH, Jin X, Park JB, Kim SH, Joshi K, et al. Crosstalk between glioma-initiating cells and endothelial cells drives tumor progression. Cancer Res (2014) 74(16):4482–92. doi:10.1158/0008-5472.CAN-13-1597
38. Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell (2005) 8:13–23. doi:10.1016/j.ccr.2005.06.004
39. Steg AD, Katre AA, Goodman B, Han HD, Nick AM, Stone RL, et al. Targeting the notch ligand JAGGED1 in both tumor cells and stroma in ovarian cancer. Clin Cancer Res (2011) 17:5674–85. doi:10.1158/1078-0432.CCR-11-0432
40. Deangelo DJ, Stone RM, Silverman LB, Stock W, Attar EC, Fearen I, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol ASCO Ann Meet Proc Part I (2006) 24:6585.
41. Imbimbo BP. Therapeutic potential of gamma-secretase inhibitors and modulators. Curr Top Med Chem (2008) 8:54–61. doi:10.2174/156802608783334015
42. Pellegrinet L, Rodilla V, Liu Z, Chen S, Koch U, Espinosa L, et al. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology (2011) 140(1230–1240):e1231–7. doi:10.1053/j.gastro.2011.01.005
43. Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs (2007) 21:299–310. doi:10.2165/00063030-200721050-00002
44. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100:3983–8. doi:10.1073/pnas.0530291100
45. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res (2005) 65:5506–11. doi:10.1158/0008-5472.CAN-05-0626
46. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1:555–67. doi:10.1016/j.stem.2007.08.014
47. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature (2007) 445:106–10. doi:10.1038/nature05372
48. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature (2007) 445:111–5. doi:10.1038/nature05384
49. Bajaj J, Maliekal TT, Vivien E, Pattabiraman C, Srivastava S, Krishnamurthy H, et al. Notch signaling in CD66+ cells drives the progression of human cervical cancers. Cancer Res (2011) 71:4888–97. doi:10.1158/0008-5472.CAN-11-0543
50. Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G, et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia (2012) 17:111–7. doi:10.1007/s10911-012-9255-3
51. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports (2014) 2:78–91. doi:10.1016/j.stemcr.2013.11.009
52. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A (2009) 106:13820–5. doi:10.1073/pnas.0905718106
53. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene (2010) 29:4741–51. doi:10.1038/onc.2010.215
54. Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett (2009) 279:8–12. doi:10.1016/j.canlet.2008.09.030
55. Espinoza I, Pochampally R, Xing F, Watabe K, Miele L. Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther (2013) 6:1249–59. doi:10.2147/OTT.S36162
56. Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res (2005) 65:6207–19. doi:10.1158/0008-5472.CAN-05-0592
57. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res (2010) 70:709–18. doi:10.1158/0008-5472.CAN-09-1681
58. Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, et al. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res (2010) 70:1469–78. doi:10.1158/0008-5472.CAN-09-2557
59. Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, et al. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells (2007) 25:807–15. doi:10.1634/stemcells.2006-0442
60. Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C, et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene (2012) 32(44):5210–9. doi:10.1038/onc.2012.550
61. Xu K, Usary J, Kousis PC, Prat A, Wang DY, Adams JR, et al. Lunatic fringe deficiency cooperates with the Met/caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell (2012) 21:626–41. doi:10.1016/j.ccr.2012.03.041
62. Kong D, Li Y, Wang Z, Sarkar FH. Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: are they cousins or twins? Cancers (Basel) (2011) 3:716–29. doi:10.3390/cancers30100716
63. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15:178–96. doi:10.1038/nrm3758
64. Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene (2014) 33:1755–63. doi:10.1038/onc.2013.128
65. Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med (2007) 204:2935–48. doi:10.1084/jem.20071082
66. Shimizu M, Cohen B, Goldvasser P, Berman H, Virtanen C, Reedijk M. Plasminogen activator uPA is a direct transcriptional target of the JAG1-Notch receptor signaling pathway in breast cancer. Cancer Res (2011) 71:277–86. doi:10.1158/0008-5472.CAN-10-2523
67. Veeraraghavalu K, Subbaiah VK, Srivastava S, Chakrabarti O, Syal R, Krishna S. Complementation of human papillomavirus type 16 E6 and E7 by Jagged1-specific Notch1-phosphatidylinositol 3-kinase signaling involves pleiotropic oncogenic functions independent of CBF1;Su(H);Lag-1 activation. J Virol (2005) 79:7889–98. doi:10.1128/JVI.79.12.7889-7898.2005
68. Tanaka S, Shiraha H, Nakanishi Y, Nishina S, Matsubara M, Horiguchi S, et al. Runt-related transcription factor 3 reverses epithelial-mesenchymal transition in hepatocellular carcinoma. Int J Cancer (2012) 131:2537–46. doi:10.1002/ijc.27575
69. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res (2009) 69:2400–7. doi:10.1158/0008-5472.CAN-08-4312
70. Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res (2004) 64:6854–7. doi:10.1158/0008-5472.CAN-04-2500
71. Wang Z, Li Y, Banerjee S, Kong D, Ahmad A, Nogueira V, et al. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J Cell Biochem (2010) 109:726–36. doi:10.1002/jcb.22451
72. Kim MH, Kim HB, Yoon SP, Lim SC, Cha MJ, Jeon YJ, et al. Colon cancer progression is driven by APEX1-mediated upregulation of Jagged. J Clin Invest (2013) 123:3211–30. doi:10.1172/JCI65521
73. Dai Y, Wilson G, Huang B, Peng M, Teng G, Zhang D, et al. Silencing of Jagged1 inhibits cell growth and invasion in colorectal cancer. Cell Death Dis (2014) 5:e1170. doi:10.1038/cddis.2014.137
74. Cohen B, Shimizu M, Izrailit J, Ng NF, Buchman Y, Pan JG, et al. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res Treat (2010) 123:113–24. doi:10.1007/s10549-009-0621-9
75. Zhang Y, Wang Z, Ahmed F, Banerjee S, Li Y, Sarkar FH. Down-regulation of Jagged-1 induces cell growth inhibition and S phase arrest in prostate cancer cells. Int J Cancer (2006) 119:2071–7. doi:10.1002/ijc.22077
76. Jundt F, Probsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, et al. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood (2004) 103:3511–5. doi:10.1182/blood-2003-07-2254
77. Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res (2005) 65:2353–63. doi:10.1158/0008-5472.CAN-04-1890
78. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene (2008) 27:5904–12. doi:10.1038/onc.2008.271
79. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res (2007) 13:4345–54. doi:10.1158/1078-0432.CCR-07-0472
80. Gopisetty A, Bhattacharya P, Haddad C, Bruno JC Jr, Vasu C, Miele L, et al. OX40L/Jagged1 cosignaling by GM-CSF-induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol (2013) 190:5516–25. doi:10.4049/jimmunol.1202298
81. Vigouroux S, Yvon E, Wagner HJ, Biagi E, Dotti G, Sili U, et al. Induction of antigen-specific regulatory T cells following overexpression of a Notch ligand by human B lymphocytes. J Virol (2003) 77:10872–80. doi:10.1128/JVI.77.20.10872-10880.2003
82. Yvon ES, Vigouroux S, Rousseau RF, Biagi E, Amrolia P, Dotti G, et al. Overexpression of the Notch ligand, Jagged-1, induces alloantigen-specific human regulatory T cells. Blood (2003) 102:3815–21. doi:10.1182/blood-2002-12-3826
83. Bugeon L, Gardner LM, Rose A, Gentle M, Dallman MJ. Cutting edge: Notch signaling induces a distinct cytokine profile in dendritic cells that supports T cell-mediated regulation and IL-2-dependent IL-17 production. J Immunol (2008) 181:8189–93. doi:10.4049/jimmunol.181.12.8189
84. Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res (2005) 65:8530–7. doi:10.1158/0008-5472.CAN-05-1069
85. Dickson BC, Mulligan AM, Zhang H, Lockwood G, O’Malley FP, Egan SE, et al. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol (2007) 20:685–93. doi:10.1038/modpathol.3800785
86. Reedijk M, Odorcic S, Zhang H, Chetty R, Tennert C, Dickson BC, et al. Activation of Notch signaling in human colon adenocarcinoma. Int J Oncol (2008) 33:1223–9. doi:10.3892/ijo_00000112
87. Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, et al. JAG1 expression is associated with a basal phenotype and recurrence in lymph node-negative breast cancer. Breast Cancer Res Treat (2008) 111:439–48. doi:10.1007/s10549-007-9805-3
88. Bane AL, Pinnaduwage D, Colby S, Reedijk M, Egan SE, Bull SB, et al. Expression profiling of familial breast cancers demonstrates higher expression of FGFR2 in BRCA2-associated tumors. Breast Cancer Res Treat (2009) 117:183–91. doi:10.1007/s10549-008-0087-1
89. Veeraraghavalu K, Pett M, Kumar RV, Nair P, Rangarajan A, Stanley MA, et al. Papillomavirus-mediated neoplastic progression is associated with reciprocal changes in JAGGED1 and manic fringe expression linked to notch activation. J Virol (2004) 78:8687–700. doi:10.1128/JVI.78.16.8687-8700.2004
90. Pang RT, Leung CO, Ye TM, Liu W, Chiu PC, Lam KK, et al. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis (2010) 31:1037–44. doi:10.1093/carcin/bgq066
91. Guilmeau S, Flandez M, Mariadason JM, Augenlicht LH. Heterogeneity of Jagged1 expression in human and mouse intestinal tumors: implications for targeting Notch signaling. Oncogene (2010) 29:992–1002. doi:10.1038/onc.2009.393
92. Pannequin J, Bonnans C, Delaunay N, Ryan J, Bourgaux JF, Joubert D, et al. The wnt target jagged-1 mediates the activation of notch signaling by progastrin in human colorectal cancer cells. Cancer Res (2009) 69:6065–73. doi:10.1158/0008-5472.CAN-08-2409
93. Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y, et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell (2011) 19:125–37. doi:10.1016/j.ccr.2010.11.008
94. Gao J, Liu J, Fan D, Xu H, Xiong Y, Wang Y, et al. Up-regulated expression of Notch1 and Jagged1 in human colon adenocarcinoma. Pathol Biol (Paris) (2011) 59:298–302. doi:10.1016/j.patbio.2010.11.001
95. Mitsuhashi Y, Horiuchi A, Miyamoto T, Kashima H, Suzuki A, Shiozawa T. Prognostic significance of Notch signalling molecules and their involvement in the invasiveness of endometrial carcinoma cells. Histopathology (2012) 60:826–37. doi:10.1111/j.1365-2559.2011.04158.x
96. Yeh TS, Wu CW, Hsu KW, Liao WJ, Yang MC, Li AF, et al. The activated Notch1 signal pathway is associated with gastric cancer progression through cyclooxygenase-2. Cancer Res (2009) 69:5039–48. doi:10.1158/0008-5472.can-08-4021
97. Sun HW, Wu C, Tan HY, Wang QS. Combination DLL4 with Jagged1-siRNA can enhance inhibition of the proliferation and invasiveness activity of human gastric carcinoma by Notch1/VEGF pathway. Hepatogastroenterology (2012) 59:924–9. doi:10.5754/hge11484
98. Lin JT, Chen MK, Yeh KT, Chang CS, Chang TH, Lin CY, et al. Association of high levels of Jagged-1 and Notch-1 expression with poor prognosis in head and neck cancer. Ann Surg Oncol (2010) 17:2976–83. doi:10.1245/s10434-010-1118-9
99. Wang XQ, Zhang W, Lui EL, Zhu Y, Lu P, Yu X, et al. Notch1-Snail1-E-cadherin pathway in metastatic hepatocellular carcinoma. Int J Cancer (2012) 131:E163–72. doi:10.1002/ijc.27336
100. Nishina S, Shiraha H, Nakanishi Y, Tanaka S, Matsubara M, Takaoka N, et al. Restored expression of the tumor suppressor gene RUNX3 reduces cancer stem cells in hepatocellular carcinoma by suppressing Jagged1-Notch signaling. Oncol Rep (2011) 26:523–31. doi:10.3892/or.2011.1336
101. Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K, et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology (2013) 144:1530–42.e12. doi:10.1053/j.gastro.2013.02.009
102. Konishi J, Kawaguchi KS, Vo H, Haruki N, Gonzalez A, Carbone DP, et al. Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res (2007) 67:8051–7. doi:10.1158/0008-5472.can-07-1022
103. Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, Dang TP, et al. Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer (2011) 10:138. doi:10.1186/1476-4598-10-138
104. Buchler P, Gazdhar A, Schubert M, Giese N, Reber HA, Hines OJ, et al. The Notch signaling pathway is related to neurovascular progression of pancreatic cancer. Ann Surg (2005) 242:791–800; discussion 800–1. doi:10.1097/01.sla.0000189115.94847.f1
105. Wu K, Xu L, Zhang L, Lin Z, Hou J. High Jagged1 expression predicts poor outcome in clear cell renal cell carcinoma. Jpn J Clin Oncol (2011) 41:411–6. doi:10.1093/jjco/hyq205
106. Ai Q, Ma X, Huang Q, Liu S, Shi T, Zhang C, et al. High-level expression of Notch1 increased the risk of metastasis in T1 stage clear cell renal cell carcinoma. PLoS One (2012). 7:e35022. doi:10.1371/journal.pone.0035022
107. Wu K, Zhang L, Lin Y, Yang K, Cheng Y. Inhibition of gammasecretase induces G2/M arrest and triggers apoptosis in renal cell carcinoma. Oncol Lett (2014) 8:55–61. doi:10.3892/ol.2014.2078
108. Murata-Ohsawa M, Tohda S, Nara N. Cellular analysis of growth suppression induced by the Notch ligands, Delta-1 and Jagged-1 in two myeloid leukemia cell lines. Int J Mol Med (2004) 14:223–6. doi:10.3892/ijmm.14.2.223
109. Czemerska M, Pluta A, Szmigielska-Kaplon A, Wawrzyniak E, Cebula-Obrzut B, Medra A, et al. Jagged-1 a new promising factor associated with favorable prognosis in AML patients. Leuk Lymphoma (2014). doi:10.3109/10428194.2014.917638
110. Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature (2014) 506:240–4. doi:10.1038/nature12883
111. Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H, Dorken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood (2002) 99:3398–403. doi:10.1182/blood.V99.9.3398
112. Nwabo Kamdje AH, Mosna F, Bifari F, Lisi V, Bassi G, Malpeli G, et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood (2011) 118:380–9. doi:10.1182/blood-2010-12-326694
113. Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood (2009) 113:856–65. doi:10.1182/blood-2008-02-139725
114. Skrtic A, Korac P, Kristo DR, Ajdukovic Stojisavljevic R, Ivankovic D, Dominis M. Immunohistochemical analysis of NOTCH1 and JAGGED1 expression in multiple myeloma and monoclonal gammopathy of undetermined significance. Hum Pathol (2010) 41:1702–10. doi:10.1016/j.humpath.2010.05.002
115. Gallahan D, Jhappan C, Robinson G, Hennighausen L, Sharp R, Kordon E, et al. Expression of a truncated Int3 gene in developing secretory mammary epithelium specifically retards lobular differentiation resulting in tumorigenesis. Cancer Res (1996) 56:1775–85.
116. Dievart A, Beaulieu N, Jolicoeur P. Involvement of Notch1 in the development of mouse mammary tumors. Oncogene (1999) 18:5973–81. doi:10.1038/sj.onc.1202991
117. Kiaris H, Politi K, Grimm LM, Szabolcs M, Fisher P, Efstratiadis A, et al. Modulation of notch signaling elicits signature tumors and inhibits hras1-induced oncogenesis in the mouse mammary epithelium. Am J Pathol (2004) 165:695–705. doi:10.1016/S0002-9440(10)63333-0
118. Raafat A, Bargo S, Anver MR, Callahan R. Mammary development and tumorigenesis in mice expressing a truncated human Notch4/Int3 intracellular domain (h-Int3sh). Oncogene (2004) 23:9401–7. doi:10.1038/sj.onc.1208187
119. Hu C, Dievart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of activated murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am J Pathol (2006) 168:973–90. doi:10.2353/ajpath.2006.050416
120. Imatani A, Callahan R. Identification of a novel NOTCH-4/INT-3 RNA species encoding an activated gene product in certain human tumor cell lines. Oncogene (2000) 19:223–31. doi:10.1038/sj.onc.1203295
121. Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res (2006) 66:1517–25. doi:10.1158/0008-5472.CAN-05-3054
122. Yamaguchi N, Oyama T, Ito E, Satoh H, Azuma S, Hayashi M, et al. NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer Res (2008) 68:1881–8. doi:10.1158/0008-5472.CAN-07-1597
123. Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol (2004) 167:215–21. doi:10.1083/jcb.200406140
124. Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res (2007) 9:R6. doi:10.1186/bcr1639
125. Haughian JM, Pinto MP, Harrell JC, Bliesner BS, Joensuu KM, Dye WW, et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc Natl Acad Sci U S A (2012) 109:2742–7. doi:10.1073/pnas.1106509108
126. Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet (2007) 370:890–907. doi:10.1016/S0140-6736(07)61416-0
127. Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci U S A (1995) 92:6414–8. doi:10.1073/pnas.92.14.6414
128. Daniel B, Rangarajan A, Mukherjee G, Vallikad E, Krishna S. The link between integration and expression of human papillomavirus type 16 genomes and cellular changes in the evolution of cervical intraepithelial neoplastic lesions. J Gen Virol (1997) 78(Pt 5):1095–101.
129. Rangarajan A, Syal R, Selvarajah S, Chakrabarti O, Sarin A, Krishna S. Activated Notch1 signaling cooperates with papillomavirus oncogenes in transformation and generates resistance to apoptosis on matrix withdrawal through PKB/Akt. Virology (2001) 286:23–30. doi:10.1006/viro.2001.0867
130. Lathion S, Schaper J, Beard P, Raj K. Notch1 can contribute to viral-induced transformation of primary human keratinocytes. Cancer Res (2003) 63:8687–94.
131. Chakrabarti O, Veeraraghavalu K, Tergaonkar V, Liu Y, Androphy EJ, Stanley MA, et al. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J Virol (2004) 78:5934–45. doi:10.1128/JVI.78.11.5934-5945.2004
132. Maliekal TT, Bajaj J, Giri V, Subramanyam D, Krishna S. The role of Notch signaling in human cervical cancer: implications for solid tumors. Oncogene (2008) 27:5110–4. doi:10.1038/onc.2008.224
133. Talora C, Sgroi DC, Crum CP, Dotto GP. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev (2002) 16:2252–63. doi:10.1101/gad.988902
134. Wang L, Qin H, Chen B, Xin X, Li J, Han H. Overexpressed active Notch1 induces cell growth arrest of HeLa cervical carcinoma cells. Int J Gynecol Cancer (2007) 17:1283–92. doi:10.1111/j.1525-1438.2007.00927.x
135. Yao J, Duan L, Fan M, Yuan J, Wu X. Notch1 induces cell cycle arrest and apoptosis in human cervical cancer cells: involvement of nuclear factor kappa B inhibition. Int J Gynecol Cancer (2007) 17:502–10. doi:10.1111/j.1525-1438.2007.00872.x
136. Franko-Tobin LG, Mackey LV, Huang W, Song X, Jin B, Luo J, et al. Notch1-mediated tumor suppression in cervical cancer with the involvement of SST signaling and its application in enhanced SSTR-targeted therapeutics. Oncologist (2012) 17:220–32. doi:10.1634/theoncologist.2011-0269
137. Bertrand FE, Angus CW, Partis WJ, Sigounas G. Developmental pathways in colon cancer: crosstalk between WNT, BMP, Hedgehog and Notch. Cell Cycle (2012) 11:4344–51. doi:10.4161/cc.22134
138. van Es JH, Van Gijn ME, Riccio O, Van Den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature (2005) 435:959–63. doi:10.1038/nature03659
139. Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, et al. gamma-secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res (2009) 69:573–82. doi:10.1158/0008-5472.CAN-08-2088
140. Veenendaal LM, Kranenburg O, Smakman N, Klomp A, Borel Rinkes IH, Van Diest PJ. Differential Notch and TGFbeta signaling in primary colorectal tumors and their corresponding metastases. Cell Oncol (2008) 30:1–11.
141. Reynolds TC, Smith SD, Sklar J. Analysis of DNA surrounding the breakpoints of chromosomal translocations involving the beta T cell receptor gene in human lymphoblastic neoplasms. Cell (1987) 50:107–17. doi:10.1016/0092-8674(87)90667-2
142. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell (1991) 66:649–61. doi:10.1016/0092-8674(91)90111-B
143. Weng AP, Ferrando AA, Lee W, Morris JPT, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306:269–71. doi:10.1126/science.1102160
144. Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med (2011) 208:1389–401. doi:10.1084/jem.20110921
145. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475:101–5. doi:10.1038/nature10113
146. Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature (2011) 473:234–8. doi:10.1038/nature09917
147. Robert-Moreno A, Guiu J, Ruiz-Herguido C, Lopez ME, Ingles-Esteve J, Riera L, et al. Impaired embryonic haematopoiesis yet normal arterial development in the absence of the Notch ligand Jagged1. EMBO J (2008) 27:1886–95. doi:10.1038/emboj.2008.113
148. Poulos MG, Guo P, Kofler NM, Pinho S, Gutkin MC, Tikhonova A, et al. Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep (2013) 4:1022–34. doi:10.1016/j.celrep.2013.07.048
149. Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM, et al. Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med (2013) 210:321–37. doi:10.1084/jem.20121527
150. Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, Zhu N, et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med (2013) 210:301–19. doi:10.1084/jem.20121484
151. Aste-Amezaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One (2010) 5:e9094. doi:10.1371/journal.pone.0009094
152. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, De Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature (2010) 464:1052–7. doi:10.1038/nature08878
153. Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev (2004) 18:2469–73. doi:10.1101/gad.1239204
154. Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature (2010) 463:E6–7. doi:10.1038/nature08751
155. Kerr DJ, Young AM. Targeted therapies: bevacizumab-has it reached its final resting place? Nat Rev Clin Oncol (2011) 8:195–6. doi:10.1038/nrclinonc.2011.32
Keywords: Jagged1, Notch pathway, cancer therapy, cancer stem cells, angiogenesis
Citation: Li D, Masiero M, Banham AH and Harris AL (2014) The Notch ligand Jagged1 as a target for anti-tumor therapy. Front. Oncol. 4:254. doi: 10.3389/fonc.2014.00254
Received: 13 August 2014; Accepted: 04 September 2014;
Published online: 25 September 2014.
Edited by:
Aleksandra Filipovic, Imperial College London, UKReviewed by:
Hongying Wang, Chinese Academy of Medical Sciences, ChinaMaria Felice Brizzi, University of Turin, Italy
Copyright: © 2014 Li, Masiero, Banham and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian L. Harris, Molecular Oncology Laboratories, Oxford University Department of Oncology, Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford OX3 9DS, UK e-mail:YWRyaWFuLmhhcnJpc0BvbmNvbG9neS5veC5hYy51aw==