 Andrea Schuessler
Andrea Schuessler David G. Walker2
David G. Walker2 Rajiv Khanna
Rajiv Khanna- 1QIMR Berghofer Centre for Immunotherapy and Vaccine Development, QIMR Berghofer Medical Research Institute, Brisbane, QLD, Australia
- 2BrizBrain and Spine, Newro Foundation, Wesley Hospital, Brisbane, QLD, Australia
Progress in the treatment of glioblastoma multiforme (GBM) over the last few decades has remained marginal and GBM is still universally fatal with short survival times after initial diagnosis. Much research is focused on finding new therapeutics for GBM and immune-based approaches have shown great promise. The detection of cytomegalovirus (CMV) antigens in malignant cells has suggested that treatment strategies based on immunological intervention, such as adoptive transfer of antiviral T cells or vaccination with viral epitopes, could be exploited as cancer therapy. Here, we review the rationale for using CMV as a therapeutic target and discuss the first clinical evidence for safety and efficacy of CMV-specific cellular immunotherapy for GBM.
Introduction
Temozolomide was the last drug to bring a significant improvement of survival for patients with glioblastoma multiforme (GBM) brain cancer (1). Now, almost a decade later, further major advances in drug development have remained elusive. GBM continues to be the most aggressive human brain cancer with 5 year survival rates below 10% (2). Standard of care treatment includes surgical resection followed by radiation and chemotherapy, however, even with optimal treatment median survival is only 15 months (1). Glioblastomas are incurable and inevitably recur with a median survival of only 6 months (3). There is an urgent need to find novel therapeutic targets and develop new treatment strategies. Immune-based approaches have great potential, in particular, because they have the great advantage of being safer and less toxic than chemotherapy drugs (4). Bevacizumab (brand name Avastin) is the only immunotherapy drug currently approved for the treatment of recurrent GBM. Bevacizumab is a monoclonal antibody that blocks vascular endothelial growth factor (VEGF), thereby reducing angiogenesis. Recently, two large clinical studies evaluating bevacizumab for the treatment of primary GBM indicated a prolonged progression free survival but failed to show a significant benefit for overall survival (5, 6). Glioblastomas are very heterogeneous tumors and it is likely that a single agent will be insufficient to achieve therapeutic benefit in a majority of patients. More than 30 studies are currently underway to assess the potential of combination therapies using bevacizumab together with radiation or other chemotherapy drugs (7). Most cellular immunotherapies for GBM under investigation at the moment focus on using tumor lysate loaded dendritic cells (DCs) or vaccination using tumor peptides [for an overview see recent review in Ref. (4)]. Preliminary results have shown some promise, however, further optimization is needed to improve anti-tumor immune responses.
Cytomegalovirus as a Target for GBM Immunotherapy
The first report of detection of cytomegalovirus (CMV) antigens in histological sections of GBM (8) was received with much controversy. Subsequent studies have disputed (9–11) as well as confirmed (12–14) the original finding. While CMV sequences have been detected in GBM tissues (15, 16), the topic continues to be controversial as recent deep sequencing studies fail to detect CMV in GBM (17, 18). Interestingly, a clinical study evaluating vaccination using DCs pulsed with autologous tumor lysate described a patient that developed enhanced CMV-specific T cell responses after one dose of the vaccine. No such response was found in a patient with a CMV negative tumor enrolled into the same study, which provided the first immunological evidence for the presence of CMV antigens in glioma cells (19). Specific killing of primary GBM cells by autologous CMV-specific T cells was demonstrated recently and further argues for the existence of CMV antigens in brain tumors (20). The precise role of CMV in GBM remains unclear. In contrast to other members of the herpes virus family like Epstein–Barr virus, CMV is not an oncogenic virus. However, CMV encodes many genes that may enable hallmarks of cancer such as pro-angiogenic signaling, immune evasion, and deregulation of the cell cycle (21–26). From a therapeutic perspective, the presence of CMV antigens provides a unique opportunity to exploit pre-existing antiviral immunity for immune-based GBM treatment. Further support for this rationale stems from the finding that lower levels of CMV antigens in GBM sections are associated with prolonged survival (27, 28). Therapeutic use of antiviral drug valganciclovir for GBM patients has been explored without major side effects, but further studies are needed to assess efficacy (29–31). In the context of immunotherapy, a previous study of CMV-specific T cell responses in GBM patients has detected functional impairment of antiviral T cells. Despite similar frequencies in GBM patients and healthy individuals, CMV-specific cytotoxic T cells in GBM patients showed limited ability to produce multiple cytokines (macrophage inflammatory protein MIP-1β, tumor necrosis factor TNF, interferon IFNγ) and to mobilize CD107a in response to CMV epitopes (32). Importantly, polyfunctionality of CMV-specific T cells isolated from GBM patients could be restored by in vitro stimulation with CMV antigens and γC cytokines (32). This suggests that adoptive transfer of in vitro expanded T cells could improve CMV-specific immune responses in GBM patients. Indeed, this preliminary study has shown that immunotherapy using CMV-specific T cells was coincident with prolonged survival in one patient (32). CMV-specific immunity is characterized by high frequencies of CMV-specific cytotoxic T cells in seropositive individuals. While tumor associated antigens are usually poorly immunogenic, viral antigens provide a strong stimulus that makes expansion of high-frequency antiviral T cell cultures comparatively easy. Consequently, several laboratories have established efficient protocols for the in vitro expansion of CMV-specific cytotoxic T cells from GBM patients for the purpose of immunotherapy (20, 32, 33). Efficient killing of autologous primary tumor cells by CMV-specific T cells was demonstrated in vitro, thus, providing direct evidence that CMV-specific cytotoxic T cells can be applied for GBM therapy (20).
Current Investigations of Cytomegalovirus Specific GBM Immunotherapy
Two clinical studies using CMV-specific immunotherapy are currently recruiting participants and two further studies using CMV-specific autologous lymphocyte transfer and DC vaccination have completed enrollment (Table 1). Of the currently recruiting studies, one study is testing genetically modified CMV-specific cytotoxic cells for the treatment of recurrent GBM (clinical trials identifier NCT01109095). CMV-specific T cells are engineered to express a chimeric antigen receptor (CAR) recognizing human epidermal growth factor receptor 2 (HER-2) coupled to CD28 (7). HER-2 antigen is expressed on a majority of GBM cells and CD28 promotes sustained T cell activity. While the primary objective of the study is safety, it might provide some insight if the targeting of glioma cells using T cells specific for CMV and HER-2 can provide a survival benefit. The second CMV-specific GBM immunotherapy under investigation is based on a combination of DC vaccination and a monoclonal antibody directed against CD25, which blocks interleukin IL-2 signaling (clinical trials identifier NCT00626483). Autologous DCs are loaded with CMV pp65-LAMP mRNA and administered with different doses of CD25 antibody, which is expected to inhibit regulatory T cells (7, 34). In this approach, alleviating the suppressive effect of regulatory T cells might lead to more efficient CMV-specific T cell activity. Results from these studies are expected by 2016 and 2015, respectively.
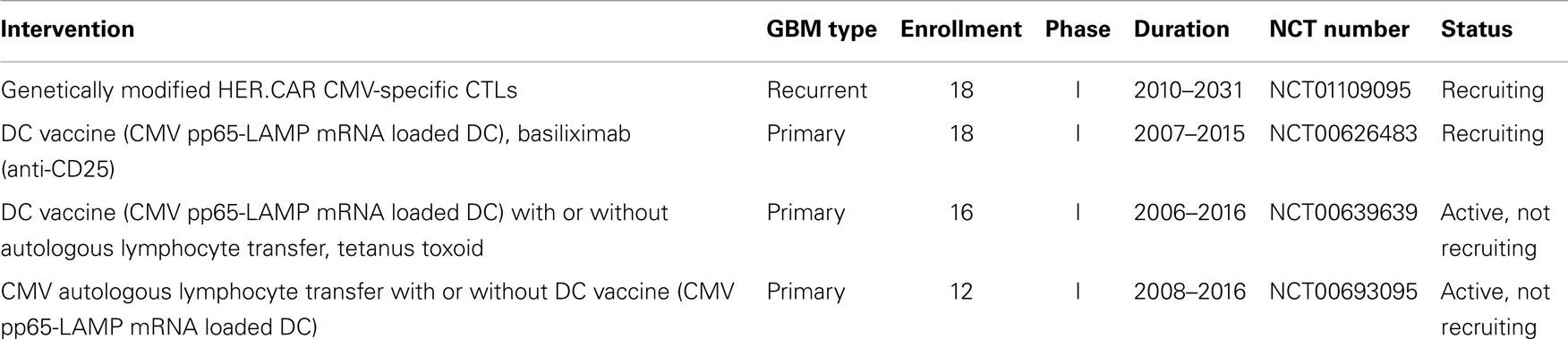
Table 1. Current clinical trials evaluating CMV-specific immunotherapy for GBM.
First Clinical Outcomes of Cytomegalovirus Specific Therapy for GBM
The first formal clinical assessment of CMV-specific adoptive T cell immunotherapy was completed recently (35). Ten CMV seropositive patients with recurrent GBM received three or four infusions of autologous CMV-specific cytotoxic T cells generated following in vitro stimulation with synthetic CMV epitopes. The treatment was shown to be safe and only mild side effects were recorded. While the patient cohort was too small to draw definite conclusions about efficacy and effects on overall survival, it is notable that 4 out of 10 patients remained completely disease free during the study period. At present, the follow up time for these patients since initiation of T cell therapy ranges from 10 months to more than 4 years, which extends well beyond the expected survival median time of 6 months after tumor recurrence. Immunological and molecular analysis revealed a number of important insights. Analysis of a range of immunological parameters on peripheral blood mononuclear cells (PBMC) before and after T cell therapy failed to reveal major changes. In contrast, molecular analysis of the T cell product used for adoptive therapy showed a signature of seven genes (EOMES, IFNG, BCL6, XAF1, CCL5, CTLA-4, FOXP3) that distinguished individuals with long-term progression free survival from patients that progressed more rapidly. This signature was consistent with T cell activation (i.e., upregulation of T cell transcription factor Eomes and effector molecule IFNγ, downregulation of inhibitory receptor CTLA-4) suggesting that more functional CMV-specific T cells are more efficient in controlling cancer relapse. One patient had a tumor recurrence after therapy and isolation of T cells from the resected tissue revealed the first evidence that CMV-specific T cells are present in tumor tissues. In addition, intratumoral T cells were found to express higher levels of immune inhibitory molecules, indicating that local immunosuppression might be an important factor in the development of effective therapies.
Immune Regulatory Mechanisms to Consider
Immunosuppression is one of the classic hallmarks of human cancers (36). In the context of GBM, this principle applies locally in the tumor microenvironment as well as systemically (4). Immune inhibitory receptors expressed on T cells such as PD-1, CTLA-4, TIM-3, or BTLA play an important role for modulation of T cell responses. Expression of PD-L1, the ligand for PD-1, has been detected in glioma samples (37) and a recent study found high-PD-L1 levels in tumor tissue to be associated with poor survival (38). Blockade of such checkpoint inhibitors might therefore be an attractive treatment option to boost intrinsic tumor defenses. Interference with the PD-1/PD-L1 interaction by monoclonal antibodies directed against PD-1 is currently under clinical investigation for the treatment of recurrent GBM (clinical trials identifier NCT01952769). Similarly, a monoclonal antibody blocking CTLA-4 (ipilimumab) that is currently approved for the treatment of metastatic melanoma might be effective in enhancing anti glioma immune responses. A combination therapy of anti-VEGF and anti-PD-1 or anti-CTLA-4 is in phase II testing for recurrent GBM (clinical trials identifier NCT02017717). In the context of CMV-specific immunotherapy for GBM, analysis of intratumoral CMV-specific T cells in a patient that relapsed after T cell therapy indicated that local immunosuppression might have contributed to treatment failure (35). While further testing is needed to confirm this finding in a larger patient cohort, it is tempting to speculate that combination of CMV-specific T cells with blockade of inhibitory receptors might boost efficacy of adoptive immunotherapy.
Conclusion and Perspective
Immunotherapies provide a novel approach to complement standard therapies for GBM treatment (Figure 1). CMV antigens provide an attractive target for cellular immunotherapies that could provide more efficient tumor recognition than tumor associated antigens. The first clinical assessment of CMV-specific T cell therapy has been completed and proved to be safe with potential clinical benefit. Further clinical trials using CMV directed immunotherapy for GBM are underway. Cancer associated immunosuppression has to be taken into account as it might limit the effectiveness of antiviral T cells within the tumor tissue. Future studies should therefore focus on multimodal strategies combining cellular immunotherapy with blockage of inhibitory receptors on T cells.
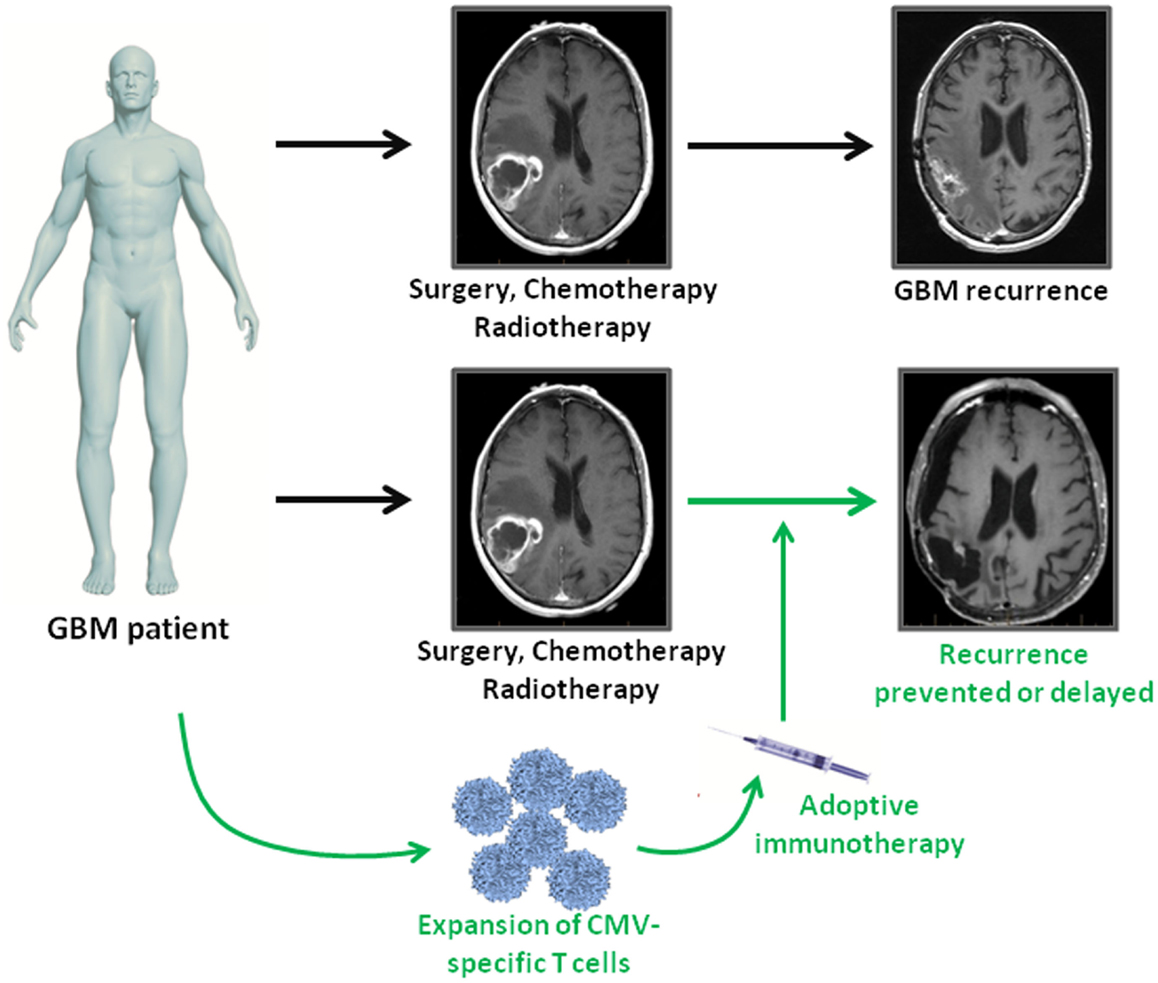
Figure 1. Adoptive immunotherapy as an addition to standard treatment for GBM patients. GBM patients receive standard of care therapy consisting of surgery followed by chemo- and radiotherapy. However, tumor recurrence is inevitable. In GBM patients, who are CMV seropositive, virus-specific T cell immunotherapy can be used as consolidative treatment to enhance intrinsic immune responses against viral antigen expressing tumor cells. If effective, adoptive immunotherapy has the potential to prolong progression free survival or even prevent relapse in GBM patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
2. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi:10.1016/S1470-2045(09)70025-7
3. Reardon DA, Herndon JE II, Peters KB, Desjardins A, Coan A, Lou E, et al. Bevacizumab continuation beyond initial bevacizumab progression among recurrent glioblastoma patients. Br J Cancer (2012) 107(9):1481–7. doi:10.1038/bjc.2012.415
4. Reardon DA, Wucherpfennig KW, Freeman G, Wu CJ, Chiocca EA, Wen PY, et al. An update on vaccine therapy and other immunotherapeutic approaches for glioblastoma. Expert Rev Vaccines (2013) 12(6):597–615. doi:10.1586/erv.13.41
5. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med (2014) 370(8):709–22. doi:10.1056/NEJMoa1308345
6. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med (2014) 370(8):699–708. doi:10.1056/NEJMoa1308573
7. ClinicalTrials.gov. Available from: www.clinicaltrials.gov
8. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res (2002) 62(12):3347–50.
9. Poltermann S, Schlehofer B, Steindorf K, Schnitzler P, Geletneky K, Schlehofer JR. Lack of association of herpesviruses with brain tumors. J Neurovirol (2006) 12(2):90–9. doi:10.1080/13550280600654573
10. Sabatier J, Uro-Coste E, Pommepuy I, Labrousse F, Allart S, Tremoulet M, et al. Detection of human cytomegalovirus genome and gene products in central nervous system tumours. Br J Cancer (2005) 92(4):747–50. doi:10.1038/sj.bjc.6602339
11. Lau SK, Chen YY, Chen WG, Diamond DJ, Mamelak AN, Zaia JA, et al. Lack of association of cytomegalovirus with human brain tumors. Mod Pathol (2005) 18(6):838–43. doi:10.1038/modpathol.3800352
12. Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol (2011) 103(2):231–8. doi:10.1007/s11060-010-0383-6
13. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol (2008) 10(1):10–8. doi:10.1215/15228517-2007-035
14. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol (2008) 116(1):79–86. doi:10.1007/s00401-008-0359-1
15. Bhattacharjee B, Renzette N, Kowalik TF. Genetic analysis of cytomegalovirus in malignant gliomas. J Virol (2012) 86(12):6815–24. doi:10.1128/JVI.00015-12
16. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J Virol (2012) 86(2):854–64. doi:10.1128/JVI.06097-11
17. Cimino PJ, Zhao G, Wang D, Sehn JK, Lewis JS Jr, Duncavage EJ. Detection of viral pathogens in high grade gliomas from unmapped next-generation sequencing data. Exp Mol Pathol (2014) 96(3):310–5. doi:10.1016/j.yexmp.2014.03.010
18. Cosset E, Petty TJ, Dutoit V, Cordey S, Padioleau I, Otten-Hernandez P, et al. Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral-like type I interferon gene response. Int J Cancer (2013) 135(6):1381–9. doi:10.1002/ijc.28670
19. Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med (2008) 359(5):539–41. doi:10.1056/NEJMc0804818
20. Nair SK, De Leon G, Boczkowski D, Schmittling R, Xie W, Staats J, et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin Cancer Res (2014) 20(10):2684–94. doi:10.1158/1078-0432.CCR-13-3268
21. Noriega V, Redmann V, Gardner T, Tortorella D. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res (2012) 54(1–3):140–51. doi:10.1007/s12026-012-8304-8
22. Smith C, Khanna R. Immune regulation of human herpesviruses and its implications for human transplantation. Am J Transplant (2013) 13(Suppl 3):9–23. doi:10.1111/ajt.12005
23. Maussang D, Langemeijer E, Fitzsimons CP, Stigter-van Walsum M, Dijkman R, Borg MK, et al. The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res (2009) 69(7):2861–9. doi:10.1158/0008-5472.CAN-08-2487
24. Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A, et al. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci Signal (2010) 3(133):ra58. doi:10.1126/scisignal.2001180
25. Soroceanu L, Cobbs CS. Is HCMV a tumor promoter? Virus Res (2011) 157(2):193–203. doi:10.1016/j.virusres.2010.10.026
26. Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M, et al. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res (2011) 71(21):6643–53. doi:10.1158/0008-5472.CAN-11-0744
27. Rahbar A, Orrego A, Peredo I, Dzabic M, Wolmer-Solberg N, Straat K, et al. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J Clin Virol (2013) 57(1):36–42. doi:10.1016/j.jcv.2012.12.018
28. Rahbar A, Stragliotto G, Orrego A, Peredo I, Taher C, Willems J, et al. Low levels of human cytomegalovirus infection in glioblastoma multiforme associates with patient survival – a case-control study. Herpesviridae (2012) 3:3. doi:10.1186/2042-4280-3-3
29. Soderberg-Naucler C, Rahbar A, Stragliotto G. Survival in patients with glioblastoma receiving valganciclovir. N Engl J Med (2013) 369(10):985–6. doi:10.1056/NEJMc1302145
30. Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, et al. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: a randomized, double-blind, hypothesis-generating study. Int J Cancer (2013) 133(5):1204–13. doi:10.1002/ijc.28111
31. Cobbs CS. Does valganciclovir have a role in glioblastoma therapy? Neuro Oncol (2014) 16(3):330–1. doi:10.1093/neuonc/nou009
32. Crough T, Beagley L, Smith C, Jones L, Walker DG, Khanna R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol Cell Biol (2012) 90(9):872–80. doi:10.1038/icb.2012.19
33. Ghazi A, Ashoori A, Hanley PJ, Brawley VS, Shaffer DR, Kew Y, et al. Generation of polyclonal CMV-specific T cells for the adoptive immunotherapy of glioblastoma. J Immunother (2012) 35(2):159–68. doi:10.1097/CJI.0b013e318247642f
34. Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL, et al. Monoclonal antibody blockade of IL-2 receptor alpha during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood (2011) 118(11):3003–12. doi:10.1182/blood-2011-02-334565
35. Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res (2014) 74(13):3466–76. doi:10.1158/0008-5472.CAN-14-0296
36. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
37. Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol (2009) 11(4):394–402. doi:10.1215/15228517-2008-104
Keywords: glioblastoma multiforme, immunotherapy, cytomegalovirus, T cell, adoptive transfer, checkpoint modulators
Citation: Schuessler A, Walker DG and Khanna R (2014) Cytomegalovirus as a novel target for immunotherapy of glioblastoma multiforme. Front. Oncol. 4:275. doi: 10.3389/fonc.2014.00275
Received: 01 August 2014; Accepted: 21 September 2014;
Published online: 07 October 2014.
Edited by:
Charles Stringfellow Cobbs, Swedish Neuroscience Institute, USAReviewed by:
Enrico Lugli, National Institutes of Health, USACecilia Söderberg-Naucler, Karolinska Institutet, Sweden
Copyright: © 2014 Schuessler, Walker and Khanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajiv Khanna, QIMR Berghofer Centre for Immunotherapy and Vaccine Development, Clive Berghofer Cancer Research Centre, 300 Herston Road, Brisbane, QLD 4006, Australia e-mail:cmFqaXYua2hhbm5hQHFpbXJiZXJnaG9mZXIuZWR1LmF1