 Johanna Pflaum
Johanna Pflaum Sophie Schlosser
Sophie Schlosser Martina Müller
Martina Müller- Department of Internal Medicine I, University Hospital Regensburg, Regensburg, Germany
p53 is an important tumor suppressor gene, which is stimulated by cellular stress like ionizing radiation, hypoxia, carcinogens, and oxidative stress. Upon activation, p53 leads to cell-cycle arrest and promotes DNA repair or induces apoptosis via several pathways. p63 and p73 are structural homologs of p53 that can act similarly to the protein and also hold functions distinct from p53. Today more than 40 different isoforms of the p53 family members are known. They result from transcription via different promoters and alternative splicing. Some isoforms have carcinogenic properties and mediate resistance to chemotherapy. Therefore, expression patterns of the p53 family genes can offer prognostic information in several malignant tumors. Furthermore, the p53 family constitutes a potential target for cancer therapy. Small molecules (e.g., Nutlins, RITA, PRIMA-1, and MIRA-1 among others) have been objects of intense research interest in recent years. They restore pro-apoptotic wild-type p53 function and were shown to break chemotherapeutic resistance. Due to p53 family interactions small molecules also influence p63 and p73 activity. Thus, the members of the p53 family are key players in the cellular stress response in cancer and are expected to grow in importance as therapeutic targets.
Introduction
Human cells are constantly exposed to external and internal stressors, which cause damage to the integrity of the cell and to its genome. In order to guarantee the survival of the organism, cells have developed numerous strategies to adapt to stressors. In this review, we would like to discuss the influence of cellular stress on tumor development as well as strategies in cancer therapy targeting pathways involved in cell-cycle control and apoptosis. Special emphasis is put on the members of the p53 family.
Cellular Stress Response in Cancer Development
The development of cancer is a multistep process that involves a series of mutations in the progenitor cell (1). It enables clonal proliferation, uncontrolled growth, and finally invasion (2, 3). Cellular stress can be caused by a multitude of external or internal influences such as ultraviolet radiation (4–6), ionizing radiation (7), hypoxia (8), carcinogens (e.g., aflatoxin) (9, 10), cigarette smoke (11), oxidative stress (12–14), and oncogene activation (15). This can lead to DNA damage and, in consequence, to malignant transformation of the cell. In order to restore its integrity, the cell disposes of a number of damage control mechanisms. These mechanisms are older than the human species and can already be found 1 billion years ago in descendants of choanoflagellates and the early metazoan sea anemone (16). Human tumor protein p53, often described as the “guardian of the genome,” and its target genes play key roles in cell-cycle control and induction of apoptosis. In its capacity as tumor suppressor protein, p53 is not only able to act as transcription factor for genes of pro-apoptotic effector proteins but it is also involved in transcription-independent cellular signaling leading directly to cell death via pathways originating from the mitochondria or the cytosol (17–19). Furthermore, p53 induces transcription of DNA repair enzymes, thereby promoting cell survival (20–22). This shows the functional dichotomy of p53. To date, the exact mechanisms deciding about death or survival of the damaged cell still remain to be elucidated. Under physiological conditions, cellular p53 levels are low and the protein has a relatively short-half-life of 20 min. Upon DNA damage, p53 levels rise primarily through stabilization of the protein (23).
While p53 has been known for more than three decades, two further members of the p53 family, p63 and p73, have been discovered more recently. The three genes exhibit a high degree of homology and there is increasing evidence that they have risen from the triplication of a common ancestral gene (24, 25). All three genes consist of important structural elements including a DNA-binding domain (DBD), an oligomerization domain (OD), and a transactivation domain (TAD) (26). p63 (27, 28) and p73 (29) have been shown to induce apoptosis similarly to p53 via activation of several of its downstream target genes (30–32). Yet, both family members also exhibit functions distinct from p53 (Figure 1).
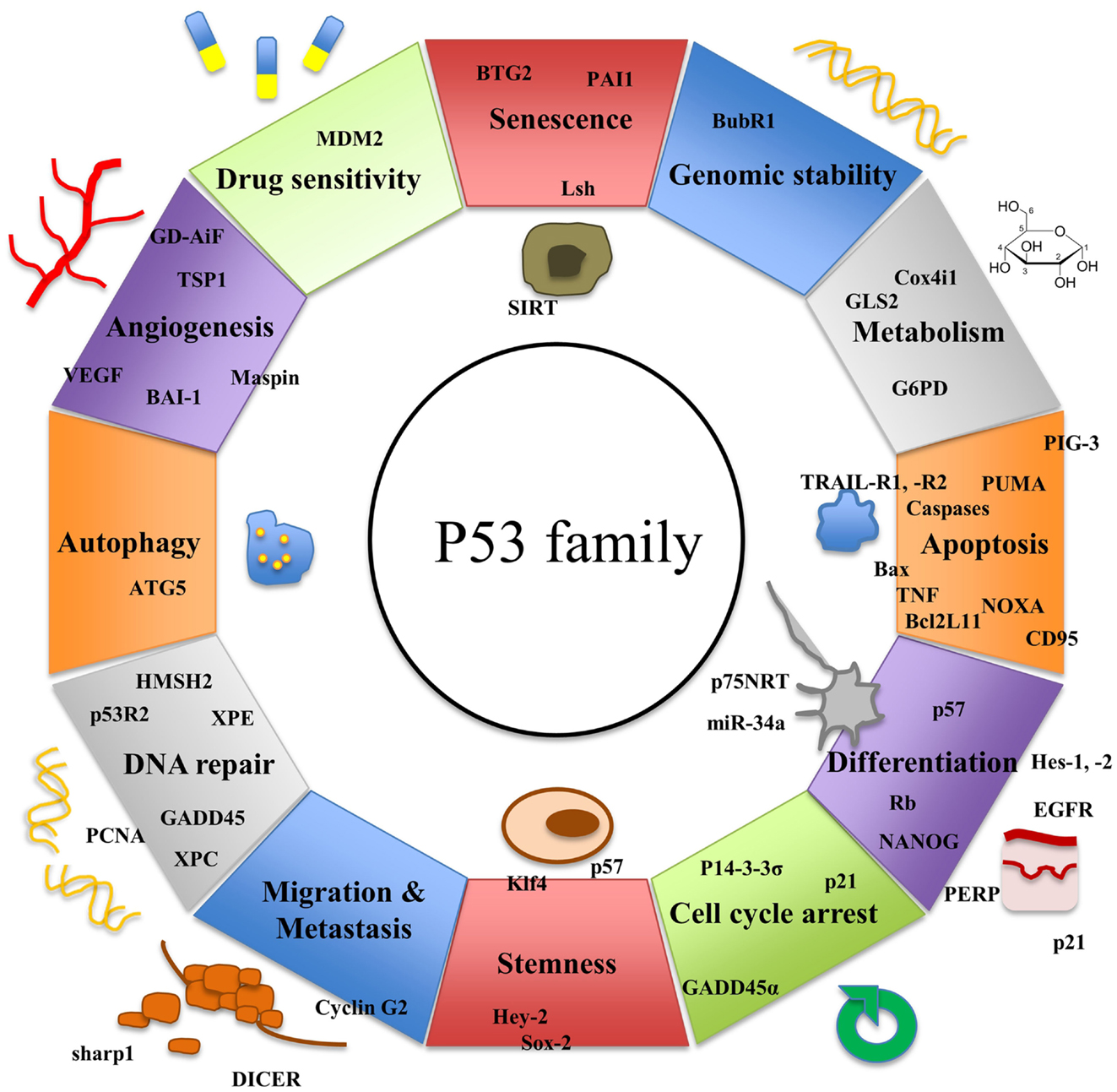
Figure 1. Functions of p53 and its homologs p63 and p73 and their target genes.
While p63 is crucially involved in craniofacial, limb, and skin development (33), p73 plays an important role during neurogenesis (34). Multiple isoforms of the p53 family members are generated using different promoters and alternative splicing. They can carry out contrary functions. Whereas some isoforms have oncogenic potential, others can act as tumor suppressors (35). However, many isoforms seem to have both capacities depending on the entity of the cell they are expressed in and the tissue context. To date, regulation and interactions of the three members of the p53 family are still under investigation.
Apoptosis
Malignant tumors often exhibit defects in apoptosis signaling pathways, resulting in tumor cell survival. Therefore, understanding the exact mechanisms of apoptosis can provide new strategies for the development of anti-cancer treatments. The extrinsic apoptosis signaling pathway is initiated by ligands such as TNFα, CD95L, and TRAIL binding to death receptors (36–38). The best characterized members of the death receptor family are TNFR1, CD95, DR3, TRAIL-R1 (CD4), TRAIL-R2 (CD5), and DR6 (39, 40).
Death receptor signaling leads to activation of caspases. Caspases are cysteinyl aspartate proteinases, which are synthesized as inactive zymogens and, upon stimulation, are initialized by autolytic cleavage (41). Initiator caspases, such as caspase 8 und 9, form signaling complexes, which activate downstream effector caspases, including caspase 3 and 7, through proteolytic cleavage (41, 42). Effector caspases cannot self-activate but process a multitude of cellular substrates during cell death (43). The intrinsic apoptosis signaling pathway originates in the mitochondria and is part of the cellular stress response. It is regulated by proteins of the Bcl-2 family. Pro-apoptotic members of the protein family include Bax, Bak, and their subclass of BH-3 only proteins such as BAD, BID, BIM, Hrk, PUMA, BMF, and Noxa, whereas A1, Bcl-2, Bcl-w, Bcl-XL, and Mcl-1 are among the anti-apoptotic members (44). The anti-apoptotic Bcl-2 proteins exert their function by stabilizing the outer mitochondrial membrane (45). Upon cellular stress, Bid and Bim mediate homo-oligomerization of Bax and Bak, which leads to the release of cytochrome c from the mitochondrial intermembrane space (46). By binding Bcl-2 proteins Bad, Noxa, and PUMA lead to inhibition of the proteins (44). Being released into the cytosol, cytochrome c forms a complex with APAF-1 and pro-caspase 9. After cleavage, caspase 9 activates effector caspase 3 (44).
p53 and Its Isoforms
p53 is encoded by the TP53 gene on the short arm of chromosome 17 and has a molecular mass of 43.7 kDa (25). It spans 19,200 bp including 11 exons (Figure 2). There are three known promoters within the p53 gene: two sites upstream of exon 1 producing full-length p53 and one internal site within intron 4 leading to transcription of amino-terminally truncated Δ133p53 (47). Δ40p53 isoforms, which have lost a part of the N-terminal TAD, can be obtained by alternative splicing of exon 2 and alternative initiation of translation at ATG40 (24), while Δ160p53 isoforms, which lack the first 159 residues, arise from translational initiation at ATG160 (48). Alternative splicing of intron 9 generates additional three isoforms, full-length p53, p53β, and p53γ (24). Both 53β and p53γ lack the OD (24). To date, a total of 12 p53 isoforms have been described: p53, p53β, p53γ, Δ40p53α, Δ40p53β, Δ40p53γ, Δ133p53α, Δ133p53β, Δ133p53γ, Δ160p53α, Δ160p53β, and Δ160p53γ (49, 50). While some p53 isoforms exert functions similar to full-length p53, others have antagonizing properties. Δ133p53, for example, inhibits p53-mediated apoptosis and causes cell-cycle arrest at the G2/M checkpoint (47, 50). Δ40p53 isoforms control the development of pluripotent embryonic stem cells into differentiated somatic cells by modulating IGF-1-R levels (51). Very little is known about the clinical role of p53 isoforms and further investigation is needed to determine if they could prove valuable as targets for anti-cancer therapy.
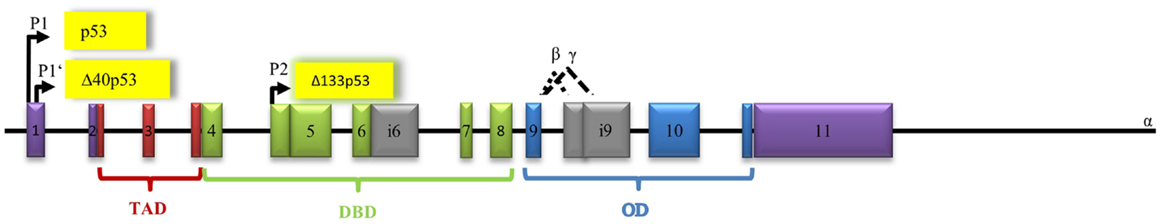
Figure 2. Architecture of the human p53 gene structure: alternative splicing (α, β, γ), alternative promoters (P1, P1′, P2), transactivation domain (TAD), DNA-binding domain (DBD), and oligomerization domain (OD) are indicated. The P1 promoter generates full-length-proteins with a transactivation domain (TAD), whereas the P1′- and P2 promoters generate proteins lacking the TAD.
Human p53 protein consists of several domains. The central DNA-binding domain (DBD) (core domain) is shared by most p53 isoforms and binds to response elements of target genes. A large number of p53 mutations occur within this region of the gene (52). The N-terminal transcription–activation domain (TA) is the binding-site for positive (e.g., p300/CBP, TAFII40/60) or negative regulators (e.g., MDM2 and MDMX) of p53 gene transcription (53). The C-terminal oligomerization (CTD) domain is subject to alternative splicing and post-translational modification. The CTD has been shown to influence DNA binding and transcriptional activity of the p53 family members (54).
p53 Regulates Cell-Cycle, Induces Apoptosis, and Promotes Cell Differentiation
p53 controls a large number of genes mediating G2/M and G1 cell-cycle arrest, DNA damage recognition, DNA repair, apoptosis, and senescence (25) (Figure 1). Absence of one parental copy of p53 through germline mutation of TP53, a condition called Li–Fraumeni syndrome, leads to development of several tumors, particularly sarcomas and cancers of the breast, brain, and adrenal glands (55, 56). Even in young individuals suffering from this condition multiple malignant tumors may develop. p53 knock-out mice have been shown to be prone to development of various types of malignancies demonstrating the important role of p53 in cancer biology (57). When initiated during the cellular stress response, p53 activates transcription of p21, a cyclin-dependent kinase inhibitor. p21 blocks CDK-1 and -2 leading to cell-cycle arrest at G1 and S phase (58). Since p53 counteracts cell growth and development, it is crucial that p53 function is strictly regulated. The E3 ubiquitin ligase MDM2 blocks p53’s transcriptional activity by binding to the N-terminal TA domain of the protein (59, 60). MDM2 is also capable of inducing the ubiquitin-mediated proteasomal degradation of the tumor suppressor protein (61, 62). In return, p53 positively regulates expression of MDM2. Thereby, it creates an auto-regulatory loop that controls the level of active p53 in the cell (63–65). During the cellular stress response, MDM2 is inhibited by different regulator proteins leading to accumulation of p53 in the cell (66).
Another important upstream regulator of p53 activity is p14ARF, a protein transcribed from an alternate reading frame of the CDKN2A gene locus that also encodes for the tumor suppressor p16INK4a (67, 68). p14ARF is part of the cell’s response to oncogenic activation (69–73). It acts as an inhibitor of MDM2-medited degradation of p53 (74). Therefore, ARF-deficient mice are prone to developing tumors of various entities (75). In a negative feedback loop, ARF promotes degradation of its activator E2F-1 and is suppressed by its downstream target p53 (76, 77).
Primarily, p53 is a transcription factor. It is involved in the intrinsic and extrinsic apoptosis signaling pathways by initiating transcription of functional proteins such as PUMA, Bax, Bid, CD95, and TRAIL-R2 (78). Yet, transcription-independent functions have been described. In the cytosol, p53 induces cell death by forming inhibitory complexes with Bcl-XL and Bcl-2, which leads to the permeabilization of the mitochondrial membrane and cytochrome c release (79, 80). Furthermore, cytosolic p53 can activate pro-apoptotic proteins such as Bax and Bak through direct protein–protein interaction (18, 81, 82).
Recently, it was observed that p53 also plays an important role in stem cell biology. In embryonic stem cells, p53 guarantees genetic stability via induction of differentiation (83) while limiting generation of induced pluripotent stem cells and tightly controls reprogramming (84). The cancer stem cell (CSC) hypothesis suggests that every tumor holds a pool of CSCs capable of renewal. They are essential for sustenance and growth of the tumor and respond poorly to conventional chemotherapy (85). CSCs result from either dedifferentiation of somatic cells or mutations in existing stem or progenitor cells (84). Targeting CSCs via activation of p53-linked pathways could trigger cell differentiation. In consequence, malignant cells would be more susceptible to DNA damaging agents and their capacity of self-renewal would be reduced.
In 1997, the cloning of p73 as a new p53 family member was reported, this was followed by the discovery of p63 – the third member of the p53 family (54, 86–89). The protein architecture is highly conserved among the three members of the p53 family (30). The highest degree of sequence homology has been described for the DNA-binding core domain (30). In contrast, the C-terminal domains are diverse and subject to alternative splicing and post-translational modification. Sauer et al. demonstrated that the C-terminal domains influence DNA binding and transcriptional activity (54) and suggested that the diversity of the C-terminal domains of the p53 family influences cell fate decisions and cellular responses that are regulated by the p53 family members (90).
p63 and Its Isoforms
The p53 homolog p63 contains three promoters that are known to encode three types of isoforms (91). The first promoter has only recently been discovered by Beyer et al. In response to DNA damage, it leads to activation of human male germ-cell-encoded TAp63 protein, which is specifically expressed in testes and protects the genomic integrity of the male germline (91, 92). The second promoter mediates transcription of TA isoforms, which contain a N-terminal TAD (22% identical with the TAD of p53) followed by a DBD (60% identical with the DBD of p53), an OD (38% identical with the OD of p53), and the sterile alpha motif (SAM) (30). In contrast, there is no SAM in the p53 gene. The third promoter is located between exon 3 and 4. Loss of exons 2 and 3 and incorporation of exon 3′ through the third promoter results in different ΔN isoforms (93). Additionally, alternative splicing at the 3′-terminus leads to the generation of five isoforms (α, β, γ, δ, and ε) and contributes to the variety of proteins (93) Premature transcriptional termination in exon 10 generates isoform ε (94) (Figure 3).
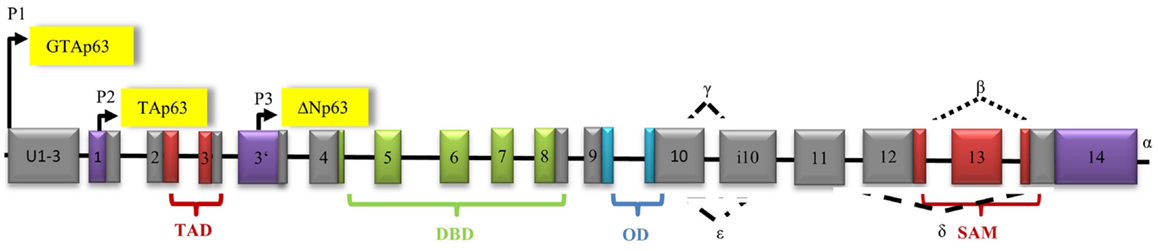
Figure 3. Architecture of the human p63 gene structure: alternative splicing (α, β, γ, δ, ε), alternative promoters (P1, P2, P3), transactivation domain (TAD), DNA-binding domain (DBD), oligomerization domain (OD), and sterile alpha motif domain (SAM) are indicated.
TAp63 is predominantly expressed in oocytes, although it has also been identified in other tissues like epidermis. In TAp63 knock-out mice, a phenotype with ulcers, hair defects, and reduced wound healing can be observed (95).
When first discovered, ΔN isoforms were thought to exclusively repress transcription. But, ΔN isoforms gain their transcriptional activity from two additional TADs within the residue, one located between the OD and the SAM domain and another located in proximity to the proline-rich domain (96, 97). Therefore, they do not only repress functions of the TA isoforms by inhibiting transcription of TA dependent genes but also transactivate their own target genes (98). ΔN63 is found in epidermal cells, in particular (99). Knock-out mice with down-regulated ΔNp63 show severe skin wounds as well as delayed wound healing (100). ΔNp63 expression can be found in multiple tumors, particularly in those with unfavorable prognosis (101). Of importance for clinical use is the fact that ΔNp63α expression is a prognostic marker for poor response to cisplatin chemotherapy in HNSCC (102). However, categorizing ΔNp63 isoforms as proto-oncogenes and TAp63 isoforms as tumor suppressors would be far too simple (103). For instance, diffuse large human B-cell lymphomas do not show enhanced expression of ΔNp63 protein, but overexpression of TAp63 (104, 105).
p63 function is regulated by post-translational modifications that influence p63 protein stability. For example, E3 ligases like Pirh2 and ITCH lead to polyubiquitination and subsequent proteasomal degradation of the protein (106). RNA-binding proteins such as RNPC1, HuR, or PCB1 control stability of p63 by binding AU-, CU-, or U-rich elements in 5′ or 3′ UTRs of p63 mRNA (107–109).
p63 and p53 have common and distinct downstream target genes (110), thereby sharing functions in cell-cycle control and apoptosis (Figure 1). TAp63 causes G1 cell-cycle arrest through transcriptional up-regulation of p21 and p57/Kip2 (111). Furthermore, p63 induces apoptosis via the extrinsic and the intrinsic apoptosis signaling pathway by enhanced expression of Bax, RAD9, DAP3, APAF-1, CD95, TNF-R, or TRAIL-R death receptors (27).
In addition, p63 assumes defined functions within the cell distinct from those of p53. In oocytes, DNA damage directly induces phosphorylation of p63, which leads to oocyte death (112, 113). p63 knock-out mice show a phenotype that is lethal soon after birth. They suffer from significant epithelial abnormalities, concerning skin, glands, teeth, and hair follicles (114). Their limbs are truncated and craniofacial anomalies are characteristic (93, 115). Human heterozygous mutations of p63 result in dysplasia of hair, teeth, digits, sweat glands, and nails (93). Therefore, p63 is essential for epithelial development. Furthermore, in a recent study, D’Aguanno et al. suggested that p63 might be involved in cancer cell metabolism. Colon CSCs showed a higher glycolytic activity when expressing TAp63 instead of ΔNp63 (116). Consistent with these observations, Giacobbe et al. reported that TAp63 isoforms can enhance expression of the mitochondrial glutaminase 2 (GLS2) gene, both in primary cells and in tumor cell lines (117).
Loss of function mutations of p63 are extremely rare in malignancies in contrast to p53 mutations (30) and controversial phenotypes have been described. Development of spontaneous tumors could be found as well as no increase in tumor disposition (111, 118–120). However, alterations in p63 expression patterns play an important role in tumorigenesis (121). In addition, mice heterozygous for mutations in both p53 and p63 (p53+/−; p63+/−) show higher tumor burden in comparison to mice heterozygous for p53 only (118). Knock-down of p63 (p63−/−) can lead to loss of p53 and thereby to cancer development (118). In fact, mice lacking p53 and p63 show increased Ras-mediated sarcoma development (111) and are prone to malignant transformations of embryonic fibroblasts (122). Furthermore, TAp63 has been shown to play an important role in tumor dissemination. Interactions of TGFβ, Ras, and mutant p53 induce formation of a ternary complex of mutant p53, Smads, and the p63 protein, which opposes the anti-metastatic function of p53 (123, 124). TAp63 leads to overexpression of metastasis suppressor genes or microRNAs like DICER1, mir-130b, and integrin recycling genes (116). Mutant p53 can reduce Dicer expression via inhibition of TAp63, thus enabling tumor metastasis (125). The p63 gene controls transcription of the miR-200 family, which regulate CSCs and epithelial–mesenchymal transition (126). ΔNp63α induces miR-205 transcription and regulates epithelial–mesenchymal transition in human bladder cancer cells (127). Therefore, controlling p63 could be a promising approach to control or prevent metastasis in cancer.
p73 and Its Isoforms
The p73 gene consists of 15 exons and is located on chromosome 1p36. Like p63, p73 has several TA isoforms containing a specific TAD and ΔN isoforms lacking it (Figure 4). The first promoter, located on exon 1, can induce transcription of several truncated ΔNp73 isoforms. They are either lacking exon 2 or exon 2 and exon 3 (ΔEx2p73 and ΔEx2/3p73). In variant ΔN’p73, exon 3 is substituted by exon 3′. The TAD of p73 is 30% identical to p53. The consecutive p73 DBD shares 63% and the OD 38% identity with p53 (30). The OD is followed by the SAM domain, which is crucial for activating the molecule via tetramerization. At least seven different 3′ terminal splicing variants are known (α, β, γ, δ, ε, ζ, η) (128). Different cell types just express a selection of p73 isoforms (129). Splice variants α and β are rarely expressed in malignant cells (130). Expression of γ, δ, ∈, and θ isoforms has been described in acute myeloid leukemia (AML) and in chronic myeloid leukemia (CML) (131).
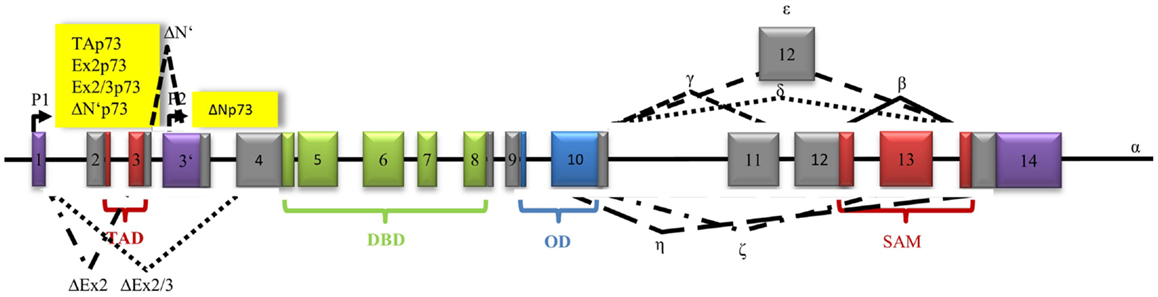
Figure 4. Architecture of the human p73 gene structure: alternative splicing (α, β, γ, δ, ε, ζ, η), alternative promoters (P1, P2), transactivation domain (TAD), DNA-binding domain (DBD), oligomerization domain (OD), and sterile alpha motif domain (SAM) are indicated. The P1 promoter generates full-length-proteins with a transactivation domain (TAD), whereas the P2 promoter generates proteins lacking the TAD. Alternative splicing of exon 2 produces Ex2p73 proteins that contain part of the TAD, alternative splicing of exon 2 and 3 produces Ex2/3p73 proteins that have completely lost the TAD. Alternative splicing of exon 3′ generates ΔN′p73.
There are several molecular mechanisms that regulate p73 function on transcriptional, post-translational, and protein level (32). Enhancers of p73 transcription are p300 (132), E2F-1 (133), CREB-binding protein (CBP) (134), YAP (135), and MM1 (my modulator 1) (136), while MDM2 (137) and c-myc (136) inhibit p73 transcriptional activity. On the post-translational level, p73 activity is reduced by sumoylation by PIAS-1 (138), deacetylation by SIRT (139), threonine phosphorylation by CDK2/CDK-1 (140), neddylation by NEDD8 (141), and conjugation and ubiquitination by Itch (142). In contrast, acetylation by p300 and pCAF (143) or phosphorylation by c-Abl (144), p38MAPk or PKCδ (145) stimulate p73 activity. The RING finger E3 ubiquitin ligase PIR2 selectively ubiquitinates ΔNp73 variants (146). ASPP proteins are also able to regulate p73 function via their poly-C-binding domain (147).
Functions of p73 are diverse. Similarly to its family members p73 plays an important role at different regulatory checkpoints of the cell-cycle. TAp73 induces G1 cell-cycle arrest via enhanced expression of p21 and p57/Kip2 (148). Furthermore, TAp73 represses genes relevant in G2/M-phase like CDC25B and CDC25C (149), Cyclin B1 (150), and Cyclin B2 (149). p73 binds to FLASH and leads to cell-cycle arrest in S-phase (151). As known from p53, DNA damage stimulates p73 to induce apoptosis involving endoplasmic reticulum (ER) stress (152).
Neuronal differentiation is regarded as innate p73 function that is not shared with p53. Phenotype studies of genetically modified mice support this thesis. Most p73 knock-out mice die within the first 4 weeks after birth. They show hippocampal dysgenesis, hydrocephalus ex vacuo, atypical social and reproductive behavior, and often suffer from chronic infections (34). Heterozygous mice develop an Alzheimer’s disease-like phenotype with impaired motor and cognitive functions (153, 154). Autopsy revealed accumulation of phosphor-tau positive filaments in the brain and in atrophic neurons (153). TAp73 knock-out mice develop a less severe phenotype characterized by malformations of the hippocampal dentate gyrus (155), whereas ΔNp73 knock-out mice present with reduced neuronal density in the motor cortex, loss of vomeronasal neurons, and Cajal–Retzius cells, as well as choroid plexus atrophy (156, 157). Latest research revealed that TAp73 is a transcriptional activator of the p75 neurotrophin receptor (p75NTR), which plays an important role during neurogenesis. TAp73 knock-out mice show reduced levels of p75NTR and suffer from peripheral nerve defect, including myelin thickness and thermal sensitivity (158).
Similarly to p63, p73 executes a set of important functions in tumor metabolism. TAp73 induces the expression of glucose-6-phosphate dehydrogenase (G6PD), which is essential for the oxidative pentose phosphate pathway (159). Cox4il is another p73 target gene relevant in metabolism. Deletion of TAp73 leads to impairment of oxidative phosphorylation via Cox4il. As a result, levels of reactive oxygen species in cells accumulate (160).
p73 is rarely mutated in human cancer (<1%), but overexpression of p73 can be found in several malignancies, for example, in hepatocellular carcinoma (29, 161, 162), neuroblastoma (163), lung cancer (164), prostate cancer (165, 166), urothelial cancer (167), colorectal carcinoma (168), and breast cancer (>40%) (169). Seventy percent of TAp73 knock-out mice or mice heterozygous for p73 suffer from malignant tumors. Colorectal and breast cancer predominantly show an increase in ΔNp73 (170). Overexpression of both, TA and ΔN isoforms, has been detected in thyroid cancer and in chronic B-cell leukemia (171), whereas diminished p73 expression has been reported for pancreatic malignancies (172). p73 heterozygous mice (p73+/−) have an increased probability for the development of spontaneous tumors such as lung adenocarcinoma, lymphomas of the thyme, and hemangiosarcoma (118). Mice heterozygous for mutations in both p53 and p73 (p53+/−; p73+/−) develop a severe disease pattern due to a severe tumor burden and more aggressive tumor dissemination (118).
p53 Family as a Target of Small Molecules
Large-scale genome sequencing has shown that over half of human malignancies exhibit point mutations in the p53 gene impairing p53 function. Most p53 mutations are missense point mutations located within the DBD. Many of them lead to destabilization of folding of the domain at physiological temperatures and interfere with its DNA-binding ability (173). Certain mutations lead to a gain-of-function of p53 and result in oncogenicity (52, 174, 175). In many other tumors p53, though intact, is inactive following enhanced degradation or reduced activation (176). Loss of wild-type p53 function or gain-of-function is often associated with aggressive tumor growth, poor prognosis, and resistance to chemotherapy. Restoration of p53 function in mice suffering from lymphomas or sarcomas has been shown to induce tumor regression (177, 178). Therefore, restoring wild-type function of p53 holds great promise as a future strategy for cancer treatment.
Small Molecules Targeting Wild-Type p53
To date, a number of small molecules have been identified, which are able to restore wild-type p53 function to cancer cells (Figure 5). The first small molecule inhibitors, which target p53/MDM2-interaction, are Nutlins. Nutlins are a family of three (Nutlin-1, Nutlin-2, Nutlin-3) cis-imidazoline analogs. They occupy the deep hydrophobic pocket of MDM2 that mediates p53 interaction (179). Hence, Nutlins prevent p53 degradation and lead to p53 accumulation and stabilization. There is evidence that Nutlins do not only enhance p53 function but also upregulate p73 in different in vitro and in vivo settings (180). Nutlin-3a has even proven effective at inducing apoptosis in p53-deficient colorectal carcinoma cells and hepatocellular carcinoma cell lines via activation of p73 (181, 182). A number of preclinical studies, mostly using Nutlin-3 as a therapeutic agent, have been carried out focusing especially on hematological malignancies like AML (183, 184), ALL (185), and B-CLL (186, 187). However, Nutlins are also able to induce apoptosis in other cell lines including ovarian cancer (188), sarcoma (189, 190), as well as glioblastoma (191). Yet, effectiveness of Nutlin therapy ultimately presumes the presence of wild-type p53 and latest findings suggest that it strongly depends on the epigenetic profile of p53 target genes (190, 192). Moreover, Michaelis et al. and Aziz et al. reported on several different cancer cell lines that developed de novo p53 mutations and became resistant toward Nutlin-3 mediated apoptosis (193, 194).
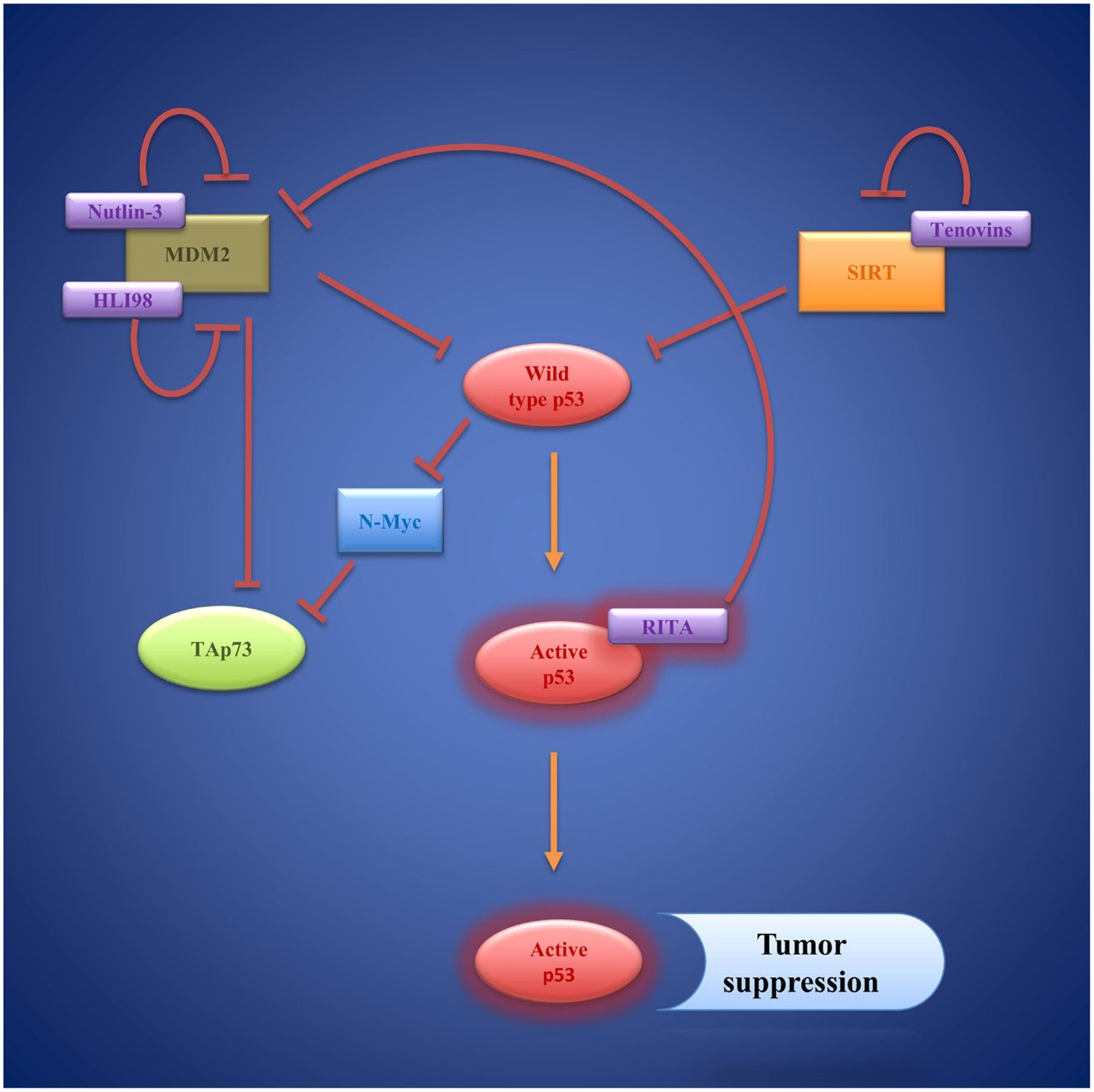
Figure 5. Wild-type p53 as a target of small molecules: Nutlins, HLI98, and RITA compensate MDM2 inhibition of p53 via inhibition of MDM2. Tenovins have been identified as SIRT 1 and SIRT 2 inhibitors that indirectly activate p53. Activated p53 induces transcription of genes regulating cell-cycle arrest and apoptosis, resulting in tumor suppression.
Another small molecule that inhibits p53/MDM2 interaction is RITA (reactivation of p53 and induction of tumor cell apoptosis). RITA binds p53 and thereby induces conformational changes within the molecule that prevent MDM2 association (195, 196). In a human head and neck cancer cell line (HNC), RITA was able to restore p53 function contributing to cytotoxicity of cisplatin therapy and leading to apoptosis in vitro and in vivo (197). The anti-tumoral effect of RITA was also observed in neuroblastoma cell lines (198).
Rational design led to construction of the spiro-oxindole MI-219, which is a highly specific small molecule inhibitor of p53/HDM2-interaction (199). Later, it was discovered that MI-219 does not only induce dissociation of the two molecules but also leads to auto-ubiquitination and degradation of HDM2 (200). MI-219 has been shown to activate p53-dependent pathways, which initiated cell-cycle arrest and apoptosis in a number of cancer cell lines, whereas primary cells remained unaffected by these p53-mediated effects (199). In a preclinical trial, the pharmacological properties of MI-219 were tested and dosages were predicted for use in phase I clinical studies (201).
As an alternative to interfering with p53/MDM2-interaction, degradation of p53 can be prevented by inhibiting the E3 ligase activity of MDM2, and therefore, preventing ubiquitination of p53 (202). A series of 5-deazaflavin derivatives, named HDM2 ligase inhibitor 98 class (HLI98), which bind the C-terminal RING-domain of MDM2, were identified (203–205). Later, it was shown that the nitro group of the molecules is not needed to convey inhibitory function, which led to the synthesis of novel 5-deazaflavin derivatives named MDP compounds (206). While HLI98 and MDP compounds demonstrate an interesting proof of concept, there are still obstacles to overcome in terms of chemical properties such as solubility as well as selectivity for MDM2 (206). Another important question, which needs further attention, is whether inhibition of MDM2 function leads to induction of MDM2 formation via the p53 feedback loop.
The tryptamine JNJ-26854165 (Serdemetan) effectively prevents p53/HDM2 from binding to the proteasome, thereby inhibiting degradation of p53 (207). In acute myeloid and lymphoid leukemia cells, JNJ-26854165 induces apoptosis via p53 by transcription-dependent and -independent pathways (207). A phase I clinical trial assessing safety and dosage of Serdemetan in advance stage and refractory solid tumors showed good bioavailability of the substance and p53 levels in skin biopsies increased. Forty percent of patients showed stable disease, yet in some patients QTc prolongation was observed as an adverse effect (208). However, increased MDM2 levels could render substances like Nutlins, RITA, MDP compounds, and JNJ-26854165 less efficient (209).
SIRT1, a nicotinamide adenine dinucleotide-dependent class III histone deacetylase, deacetylates p53 at Lys382, thereby reducing its activity (210). Hence, blocking SIRT function is a new strategy of restoring p53 function independent of MDM2 (211). Two small molecules, tenovin 1 and the more water-soluble tenovin 6, which block SIRT1 and SIRT2 function efficiently, were discovered by Lain et al. (212). Tenovin 1 was shown to induce apoptosis in cutaneous T-cell lymphoma cells (213). Interestingly, following tenovin 6 treatments cell death was observed in five different colon cancer cell lines independent of their p53 status (214). Also, tenovin 6 activated autophagy-lysosomal pathway genes in chronic lymphocytic leukemia cells without affecting p53 pathways (215). Both findings point toward additional cellular mechanisms mediating the anti-tumor effect of tenovins.
Small Molecules Targeting Mutant p53
In tumors that harbor p53 mutations, which often lead to loss of its DNA-binding function, targets for small molecules other than MDM2 are needed. An increasing number of p53 mutations have been described so far. Nevertheless, most mutations cause unfolding of the DBD rendering it unable to bind to target genes for transactivation (216, 217). Therefore, a number of small molecules aiming at restoring and stabilizing the original DBD conformation have been developed (Figure 6). Bykov et al. identified two small molecules by screening a library of low-molecular-weight compounds for substances, which are able to restore wild-type function of mutant p53: PRIMA-1 and MIRA-1 (218, 219). PRIMA-1 (p53 reactivation and induction of massive apoptosis) is a pro-drug (220). The molecule effectively induces apoptosis in bladder cancer cell lines (221). Later, PRIMA-1MET (APR-246), a compound that bears great structural similarities to PRIMA-1, but has higher activity than its predecessor, was discovered (222). Interestingly, PRIMA-1MET can not only restore the pro-apoptotic function of p53 but also of mutant TAp63γ and of TAp73β, while exerting little effect on TAp73α (223). Furthermore, PRIMA-1MET is involved in activating downstream target genes of the p53 family (223–225).
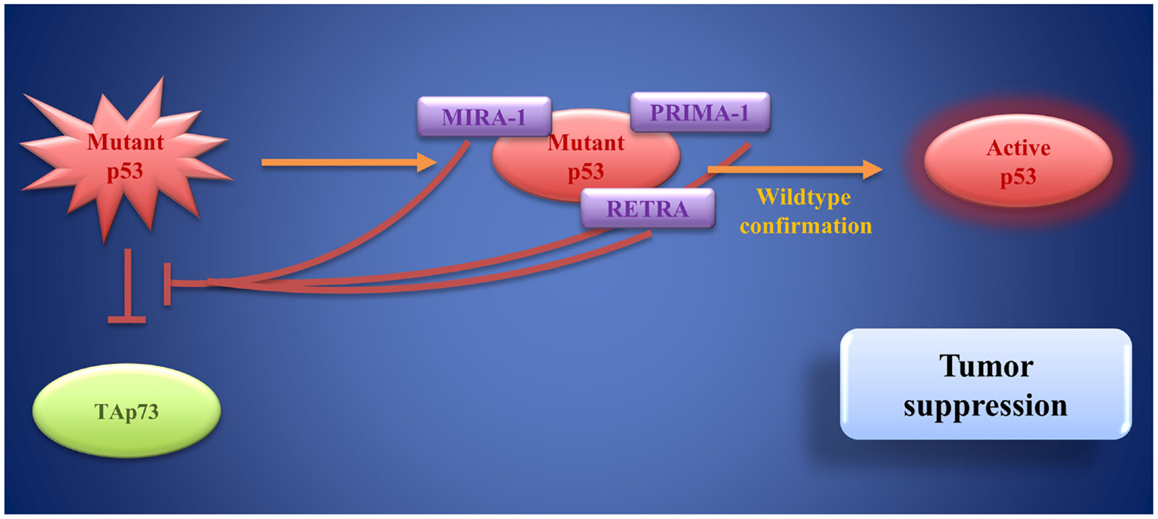
Figure 6. Mutant p53 as a target of small molecules: PRIMA-1, MIRA-1, and RETRA bind to mutant p53 and restore wild-type p53 function. Moreover, they block mutant p53-induced inhibition of TAp73. These activities result in tumor suppression.
PRIMA-1MET alone and PRIMA-1MET in combination with chemotherapeutic drugs are effective at inducing tumor cell apoptosis in vivo (221, 222, 225). Also, a phase one clinical trial using PRIMA-1MET (APR-246) in advanced prostate cancer and hematological malignancies, as well as a phase Ib/II clinical trial using this compound in addition to carboplatin in recurrent high-grade serous ovarian cancer are under way and will offer more insight into the effectiveness and practicability of mutant p53 reactivation (National Cancer Institute: Safety Study of APR-246 in patients with refractory hematologic cancer or prostate cancer; p53 suppressor activation in recurrent high-grade serous ovarian cancer, a Phase Ib/II study of systemic carboplatin combination chemotherapy with or without APR-246).
MIRA-1 (mutant p53 reactivation and induction of rapid apoptosis) is a maleimide-derived molecule and has no structural similarity with PRIMA-1, but it is equally able to restore p53 function leading to cell death via apoptosis with even higher potency than PRIMA-1 (219). By reestablishing its DNA-binding capacity and transcriptional transactivation through p53, MIRA-1 leads to programed cell death in multiple myeloma in vitro and in a mouse model (226). To date, little is known about the molecular mechanisms and safety of MIRA-1 treatment and further research is needed before clinical evaluation.
Although PRIMA-1 and MIRA-1 seem to have stabilizing effect on a great variety of p53 mutants, they are not able to restore normal protein configuration to the Phe176 mutant (218). This shows the necessity to test p53 status and to identify the underlying p53 mutations before small molecule treatment (220). In fact, approaches have been made to target distinct mutations. Rational drug design led to the identification of the compound PhiKan083, which stabilizes the Cys 220 p53 mutant and prolongs its half-life, but does not rescue any other p53 mutant (227). PhiKan083 fits into a groove in the defective molecule and induces refolding of the protein (227). In consequence, the melting point of the mutant increases and denaturation is slowed down (227).
CP-31398 was discovered by screening a library of more than 100,000 synthetic compounds for substances that effectively stabilize p53 conformation (228). Initially, CP-31398 was thought to prevent unfolding of wild-type and mutant p53 and increase levels of wild-type p53 by blocking ubiquitination and degradation (229). Yet, further research revealed that it yields a number of p53-independent functions, which mediate its cytotoxic effects (230). In a mouse model of urothelial cancer of the bladder CP-31398 effectively reduced tumor growth and invasion (231).
However, increased p53 activity bares risks for non-cancerous cells that might also be subject to apoptosis and further research is needed to find the adequate dose-response relationship, specific to the compound used (209). In an attempt to identify molecules, which restore p53’s transcriptional activity exclusively in cancer cells holding p53 mutations, reactivation of transcriptional reporter activity (RETRA) was identified by screening compounds from a chemical library (232). Further analysis revealed that RETRA, rather than restoring a functional p53 molecule, leads to an increase in TAp73 levels and to its release from a blocking complex with mutant p53 (232). As mentioned above, p73 can activate various target genes of p53 involved in cell-cycle arrest and apoptosis, thereby mediating tumor cell death (232). In vivo, in a xenograft mouse model, tumor growth could be decelerated by intraperitoneal injection of RETRA (232). Although still in the very early stages of development, RETRA opens up new perspectives for p63- and 73-based cancer treatment options.
Moreover, restoring p53 apoptotic function and modulation of p63 and p73 expression is often essential for sensitivity toward chemotherapeutic drugs or radiation, as lack of p53 and unfavorable expression patterns of p63 and p73 can lead to resistance toward treatment in different malignant tumors (233–235). Reconstitution of p53 function or activation of certain p63 and p73 isoforms might allow reducing the dose of cytotoxic drugs while still maintaining their anti-tumor effects. Simultaneously, this would permit to protect normal tissues from side effects of chemotherapy.
However, restoration of wild-type p53 might not be beneficial in all types of tumors. Jackson et al. showed that doxorubicin lead to cell-cycle arrest and senescence instead of cell death in breast cancer expressing wild-type p53, thereby promoting tumor cell survival and resistance to chemotherapy (236). This shows the necessity to elucidate which p53-dependent pathways are favored in certain malignancies before considering small molecule treatment. Novel treatment approaches could lead to the development of substances that selectively activate p53-mediated apoptosis signaling pathways.
Conclusion
The p53 family plays a central role in cancer development and treatment response. Whereas p53 is often mutated in tumors, p63 and p73 function is preserved, yet altered by different expression patterns of their TA and ΔN isoforms. Increasingly, these expression patterns are evaluated to estimate prognosis and adapt anti-cancer therapy. Nevertheless, the molecular mechanisms regulating the interplay between the different isoforms of the p53 family are only partly understood and are focus of current research. Identifying compounds that interfere with oncogenic signaling induced by certain p63 and p73 isoforms could be a novel approach in anti-cancer therapy.
An increasing number of compounds that re-establish pro-apoptotic p53 function in cancer cells have emerged over the past decade. A variety of small molecules, which aim at increasing p53 function in cancers expressing wild-type p53, have been discovered. Among them are Nutlins, which are already undergoing clinical evaluation, RITA, tenovins, and many others.
In tumors with underlying p53 mutation restoring wild-type activity of p53 has proven more difficult, but nevertheless feasible. PRIMA-1 and MIRA-1 are effective at inducing apoptosis via p53 in tumors that exhibit a great variety of p53 mutations. Yet, there are other small molecules, like PhiKan083, which are more specific and restore wild-type configuration of specific mutants only.
A number of in vivo studies and clinical trials have shown synergistic effects of small molecule treatment and chemotherapeutic drugs in a variety of malignancies. Especially cancer cells, which are resistant to chemotherapy due to impaired p53 function, become more susceptible to treatment.
Taking the approaches of p53 reactivation further, there might be new possibilities of targeting CSCs, which are often insusceptible to chemotherapy. Induction of p53 in these cells could lead to activation of pro-apoptotic pathways via differentiation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ashley DJ. The two “hit” and multiple “hit” theories of carcinogenesis. Br J Cancer (1969) 23(2):313–28. doi: 10.1038/bjc.1969.41
2. Karakosta A, Golias C, Charalabopoulos A, Peschos D, Batistatou A, Charalabopoulos K. Genetic models of human cancer as a multistep process. Paradigm models of colorectal cancer, breast cancer, and chronic myelogenous and acute lymphoblastic leukaemia. J Exp Clin Cancer Res (2005) 24(4):505–14.
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
4. Clydesdale GJ, Dandie GW, Muller HK. Ultraviolet light induced injury: immunological and inflammatory effects. Immunol Cell Biol (2001) 79(6):547–68. doi:10.1046/j.1440-1711.2001.01047.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, et al. Sunburn and p53 in the onset of skin cancer. Nature (1994) 372(6508):773–6. doi:10.1038/372773a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol (1994) 65(1):27–33. doi:10.1080/09553009414550041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell (2010) 40(2):294–309. doi:10.1016/j.molcel.2010.09.022
9. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature (1991) 350(6317):429–31. doi:10.1038/350429a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature (1991) 350(6317):427–8. doi:10.1038/350427a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Itoh M, Tsuji T, Nakamura H, Yamaguchi K, Fuchikami J, Takahashi M, et al. Systemic effects of acute cigarette smoke exposure in mice. Inhal Toxicol (2014) 26(8):464–73. doi:10.3109/08958378.2014.917346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair (Amst) (2004) 3(8–9):1109–15. doi:10.1016/j.dnarep.2004.03.002
13. Rotblat B, Grunewald TG, Leprivier G, Melino G, Knight RA. Anti-oxidative stress response genes: bioinformatic analysis of their expression and relevance in multiple cancers. Oncotarget (2013) 4(12):2577–90.
14. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ (2014) 21(1):26–38. doi:10.1038/cdd.2013.48
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Haigis KM, Sweet-Cordero A. New insights into oncogenic stress. Nat Genet (2011) 43(3):177–8. doi:10.1038/ng0311-177
16. Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, et al. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol (2010) 2(6):a001198. doi:10.1101/cshperspect.a001198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta (2009) 1787(5):414–20. doi:10.1016/j.bbabio.2008.10.005
18. Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol (2010) 20(1):14–24. doi:10.1016/j.tcb.2009.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol (2005) 17(6):631–6. doi:10.1016/j.ceb.2005.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet (2012) 28(3):128–36. doi:10.1016/j.tig.2011.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res (2013) 41(18):8403–20. doi:10.1093/nar/gkt635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell (1992) 71(4):587–97. doi:10.1016/0092-8674(92)90593-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ (2006) 13(6):941–50. doi:10.1038/sj.cdd.4401925
24. Murray-Zmijewski F, Lane DP, Bourdon J. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ (2006) 13(6):962–72. doi:10.1038/sj.cdd.4401914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer (2009) 9(10):749–58. doi:10.1038/nrc2723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Müller M, Schleithoff ES, Stremmel W, Melino G, Krammer PH, Schilling T. One, two, three – p53, p63, p73 and chemosensitivity. Drug Resist Updat (2006) 9(6):288–306. doi:10.1016/j.drup.2007.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, et al. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J (2005) 24(13):2458–71. doi:10.1038/sj.emboj.7600708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Shimada A, Kato S, Enjo K, Osada M, Ikawa Y, Kohno K, et al. The transcriptional activities of p53 and its homologue p51/p63: similarities and differences. Cancer Res (1999) 59(12):2781–6.
29. Müller M, Schilling T, Sayan AE, Kairat A, Lorenz K, Schulze-Bergkamen H, et al. TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ (2005) 12(12):1564–77. doi:10.1038/sj.cdd.4401774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Dötsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol (2010) 2(9):a004887. doi:10.1101/cshperspect.a004887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Schilling T, Schleithoff ES, Kairat A, Melino G, Stremmel W, Oren M, et al. Active transcription of the human FAS/CD95/TNFRSF6 gene involves the p53 family. Biochem Biophys Res Commun (2009) 387(2):399–404. doi:10.1016/j.bbrc.2009.07.063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Candi E, Agostini M, Melino G, Bernassola F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: regulators and effectors. Hum Mutat (2014) 35(6):702–14. doi:10.1002/humu.22523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature (1999) 398(6729):714–8. doi:10.1038/19539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature (2000) 404(6773):99–103. doi:10.1038/35003607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Vilgelm A, El-Rifai W, Zaika A. Therapeutic prospects for p73 and p63: rising from the shadow of p53. Drug Resist Updat (2008) 11(4–5):152–63. doi:10.1016/j.drup.2008.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity (1995) 3(6):673–82. doi:10.1016/1074-7613(95)90057-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell (1993) 75(6):1169–78. doi:10.1016/0092-8674(93)90326-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol (1988) 141(8):2629–34.
39. Lavrik IN. Systems biology of death receptor networks: live and let die. Cell Death Dis (2014) 5:e1259. doi:10.1038/cddis.2014.160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ (2014). doi:10.1038/cdd.2014.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol (2013) 5(4):a008656. doi:10.1101/cshperspect.a008656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol (2014) 30:337–56. doi:10.1146/annurev-cellbio-100913-013226
43. Kumar S. Caspase function in programmed cell death. Cell Death Differ (2007) 14(1):32–43. doi:10.1038/sj.cdd.4402060
44. Elkholi R, Floros KV, Chipuk JE. The role of BH3-only proteins in tumor cell development, signaling, and treatment. Genes Cancer (2011) 2(5):523–37. doi:10.1177/1947601911417177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Ding J, Mooers BH, Zhang Z, Kale J, Falcone D, McNichol J, et al. After embedding in membranes antiapoptotic Bcl-XL protein binds both Bcl-2 homology region 3 and helix 1 of proapoptotic Bax protein to inhibit apoptotic mitochondrial permeabilization. J Biol Chem (2014) 289(17):11873–96. doi:10.1074/jbc.M114.552562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell (2010) 37(3):299–310. doi:10.1016/j.molcel.2010.01.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bourdon J, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev (2005) 19(18):2122–37. doi:10.1101/gad.1339905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Marcel V, Perrier S, Aoubala M, Ageorges S, Groves MJ, Diot A, et al. Δ160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett (2010) 584(21):4463–8. doi:10.1016/j.febslet.2010.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Surget S, Khoury MP, Bourdon J. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther (2013) 7:57–68. doi:10.2147/OTT.S53876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Aoubala M, Murray-Zmijewski F, Khoury MP, Fernandes K, Perrier S, Bernard H, et al. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ (2011) 18(2):248–58. doi:10.1038/cdd.2010.91
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Ungewitter E, Scrable H. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev (2010) 24(21):2408–19. doi:10.1101/gad.1987810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell (2014) 25(3):304–17. doi:10.1016/j.ccr.2014.01.021
53. Mavinahalli JN, Madhumalar A, Beuerman RW, Lane DP, Verma C. Differences in the transactivation domains of p53 family members: a computational study. BMC Genomics (2010) 11(Suppl 1):S5. doi:10.1186/1471-2164-11-S1-S5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Sauer M, Bretz AC, Beinoraviciute-Kellner R, Beitzinger M, Burek C, Rosenwald A, et al. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res (2008) 36(6):1900–12. doi:10.1093/nar/gkn044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Li FP, Fraumeni JF. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med (1969) 71(4):747–52. doi:10.7326/0003-4819-71-4-747
56. Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat (2003) 21(3):313–20. doi:10.1002/humu.10185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature (1992) 356(6366):215–21. doi:10.1038/356215a0
58. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer (2009) 9(6):400–14. doi:10.1038/nrc2657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell (1992) 69(7):1237–45. doi:10.1016/0092-8674(92)90644-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science (1996) 274(5289):948–53. doi:10.1126/science.274.5289.948
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387(6630):296–9. doi:10.1038/387296a0
62. Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol (2000) 20(22):8458–67. doi:10.1128/MCB.20.22.8458-8467.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Barak Y, Gottlieb E, Juven-Gershon T, Oren M. Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev (1994) 8(15):1739–49. doi:10.1101/gad.8.15.1739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol (2003) 13(1):49–58. doi:10.1016/S1044-579X(02)00099-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Perry ME. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb Perspect Biol (2010) 2(1):a000968. doi:10.1101/cshperspect.a000968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell (1995) 83(6):993–1000. doi:10.1016/0092-8674(95)90214-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J (1998) 17(17):5001–14. doi:10.1093/emboj/17.17.5001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Sherr CJ. An Arf(GFP/GFP) reporter mouse reveals that the Arf tumor suppressor monitors latent oncogenic signals in vivo. Cell Cycle (2004) 3(3):239–40. doi:10.4161/cc.3.3.744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci U S A (2001) 98(9):5025–30. doi:10.1073/pnas.091100298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Gregory MA, Qi Y, Hann SR. The ARF tumor suppressor: keeping Myc on a leash. Cell Cycle (2005) 4(2):249–52. doi:10.4161/cc.4.2.1491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Cong F, Zou X, Hinrichs K, Calame K, Goff SP. Inhibition of v-Abl transformation by p53 and p19ARF. Oncogene (1999) 18(54):7731–9. doi:10.1038/sj.onc.1203290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Evangelou K, Bartkova J, Kotsinas A, Pateras IS, Liontos M, Velimezi G, et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ (2013) 20(11):1485–97. doi:10.1038/cdd.2013.76
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol (1999) 1(1):20–6. doi:10.1038/8991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res (1999) 59(9):2217–22.
76. Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS. E2F transcriptional repressor complexes are critical downstream targets of p19(ARF)/p53-induced proliferative arrest. Cancer Cell (2002) 2(1):55–65. doi:10.1016/S1535-6108(02)00085-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Robertson KD, Jones PA. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol Cell Biol (1998) 18(11):6457–73.
78. Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol (2010) 2(8):a000935. doi:10.1101/cshperspect.a000935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell (2003) 11(3):577–90. doi:10.1016/S1097-2765(03)00050-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem (2006) 281(13):8600–6. doi:10.1074/jbc.M507611200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science (2004) 303(5660):1010–4. doi:10.1126/science.1092734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol (2004) 6(5):443–50. doi:10.1038/ncb1123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol (2005) 7(2):165–71. doi:10.1038/ncb1211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Aloni-Grinstein R, Shetzer Y, Kaufman T, Rotter V. p53: the barrier to cancer stem cell formation. FEBS Lett (2014) 588(16):2580–9. doi:10.1016/j.febslet.2014.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med (2011) 17(3):313–9. doi:10.1038/nm.2304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med (1998) 4(7):839–43. doi:10.1038/nm0798-839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Schmale H, Bamberger C. A novel protein with strong homology to the tumor suppressor p53. Oncogene (1997) 15(11):1363–7. doi:10.1038/sj.onc.1201500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell (1998) 2(3):305–16. doi:10.1016/S1097-2765(00)80275-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell (1997) 90(4):809–19. doi:10.1016/S0092-8674(00)80540-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Harms KL, Chen X. The C terminus of p53 family proteins is a cell fate determinant. Mol Cell Biol (2005) 25(5):2014–30. doi:10.1128/MCB.25.5.2014-2030.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Beyer U, Moll-Rocek J, Moll UM, Dobbelstein M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc Natl Acad Sci U S A (2011) 108(9):3624–9. doi:10.1073/pnas.1016201108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Beyer U, Dobbelstein M. Non-hominid TP63 lacks retroviral LTRs but contains a novel conserved upstream exon. Cell Cycle (2011) 10(12):1905–11. doi:10.4161/cc.10.12.15838
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Allocati N, Di Ilio C, de Laurenzi V. p63/p73 in the control of cell cycle and cell death. Exp Cell Res (2012) 318(11):1285–90. doi:10.1016/j.yexcr.2012.01.023
94. Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisà E, Pesole G, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res (2009) 37(18):6092–104. doi:10.1093/nar/gkp674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Suh K, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol (2007) 29(4):374–7. doi:10.1097/DAD.0b013e31812f52bd
96. Helton ES, Zhu J, Chen X. The unique NH2-terminally deleted (DeltaN) residues, the PXXP motif, and the PPXY motif are required for the transcriptional activity of the DeltaN variant of p63. J Biol Chem (2006) 281(5):2533–42. doi:10.1074/jbc.M507964200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Duijf PH, Vanmolkot KR, Propping P, Friedl W, Krieger E, McKeon F, et al. Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum Mol Genet (2002) 11(7):799–804. doi:10.1093/hmg/11.7.799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Ghioni P, Bolognese F, Duijf PH, van Bokhoven H, Mantovani R, Guerrini L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol Cell Biol (2002) 22(24):8659–68. doi:10.1128/MCB.22.24.8659-8668.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ (2011) 18(9):1487–99. doi:10.1038/cdd.2011.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Koster MI, Marinari B, Payne AS, Kantaputra PN, Costanzo A, Roop DR. DeltaNp63 knockdown mice: a mouse model for AEC syndrome. Am J Med Genet A (2009) 149A(9):1942–7. doi:10.1002/ajmg.a.32794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Mundt HM, Stremmel W, Melino G, Krammer PH, Schilling T, Müller M. Dominant negative (DeltaN) p63alpha induces drug resistance in hepatocellular carcinoma by interference with apoptosis signaling pathways. Biochem Biophys Res Commun (2010) 396(2):335–41. doi:10.1016/j.bbrc.2010.04.093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Zangen R, Ratovitski E, Sidransky D. DeltaNp63alpha levels correlate with clinical tumor response to cisplatin. Cell Cycle (2005) 4(10):1313–5. doi:10.4161/cc.4.10.2066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Dohn M, Zhang S, Chen X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene (2001) 20(25):3193–205. doi:10.1038/sj.onc.1204427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Di Como CJ, Urist MJ, Babayan I, Drobnjak M, Hedvat CV, Teruya-Feldstein J, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res (2002) 8(2):494–501.
105. Pruneri G, Fabris S, Dell’Orto P, Biasi MO, Valentini S, Del Curto B, et al. The transactivating isoforms of p63 are overexpressed in high-grade follicular lymphomas independent of the occurrence of p63 gene amplification. J Pathol (2005) 206(3):337–45. doi:10.1002/path.1787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Jung Y, Qian Y, Yan W, Chen X. Pirh2 E3 ubiquitin ligase modulates keratinocyte differentiation through p63. J Invest Dermatol (2013) 133(5):1178–87. doi:10.1038/jid.2012.466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Zhang J, Jun Cho S, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci U S A (2010) 107(21):9614–9. doi:10.1073/pnas.0912594107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Yan W, Zhang Y, Zhang J, Cho S, Chen X. HuR is necessary for mammary epithelial cell proliferation and polarity at least in part via ΔNp63. PLoS One (2012) 7(9):e45336. doi:10.1371/journal.pone.0045336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Cho S, Jung Y, Chen X. Poly (C)-binding protein 1 regulates p63 expression through mRNA stability. PLoS One (2013) 8(8):e71724. doi:10.1371/journal.pone.0071724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Martynova E, Pozzi S, Basile V, Dolfini D, Zambelli F, Imbriano C, et al. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget (2012) 3(2):132–43.
111. Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol (2009) 11(12):1451–7. doi:10.1038/ncb1988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Suh E, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, et al. p63 protects the female germ line during meiotic arrest. Nature (2006) 444(7119):624–8. doi:10.1038/nature05337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med (2009) 15(10):1179–85. doi:10.1038/nm.2033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Vanbokhoven H, Melino G, Candi E, Declercq W. p63, a story of mice and men. J Invest Dermatol (2011) 131(6):1196–207. doi:10.1038/jid.2011.84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Holembowski L, Schulz R, Talos F, Scheel A, Wolff S, Dobbelstein M, et al. While p73 is essential, p63 is completely dispensable for the development of the central nervous system. Cell Cycle (2011) 10(4):680–9. doi:10.4161/cc.10.4.14859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. D’Aguanno S, Barcaroli D, Rossi C, Zucchelli M, Ciavardelli D, Cortese C, et al. p63 isoforms regulate metabolism of cancer stem cells. J Proteome Res (2014) 13(4):2120–36. doi:10.1021/pr4012574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Giacobbe A, Bongiorno-Borbone L, Bernassola F, Terrinoni A, Markert EK, Levine AJ, et al. p63 regulates glutaminase 2 expression. Cell Cycle (2013) 12(9):1395–405. doi:10.4161/cc.24478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell (2005) 7(4):363–73. doi:10.1016/j.ccr.2005.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Keyes WM, Vogel H, Koster MI, Guo X, Qi Y, Petherbridge KM, et al. p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc Natl Acad Sci U S A (2006) 103(22):8435–40. doi:10.1073/pnas.0602477103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Koster MI, Dai D, Roop DR. Conflicting roles for p63 in skin development and carcinogenesis. Cell Cycle (2007) 6(3):269–73. doi:10.4161/cc.6.3.3792
121. Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer (2013) 13(2):136–43. doi:10.1038/nrc3446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Lang GA, Iwakuma T, Suh Y, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell (2004) 119(6):861–72. doi:10.1016/j.cell.2004.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell (2009) 137(1):87–98. doi:10.1016/j.cell.2009.01.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell (2009) 139(7):1327–41. doi:10.1016/j.cell.2009.11.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Muller PA, Trinidad AG, Caswell PT, Norman JC, Vousden KH. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J Biol Chem (2014) 289(1):122–32. doi:10.1074/jbc.M113.502138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Knouf EC, Garg K, Arroyo JD, Correa Y, Sarkar D, Parkin RK, et al. An integrative genomic approach identifies p73 and p63 as activators of miR-200 microRNA family transcription. Nucleic Acids Res (2012) 40(2):499–510. doi:10.1093/nar/gkr731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Tran MN, Choi W, Wszolek MF, Navai N, Lee IC, Nitti G, et al. The p63 protein isoform ΔNp63α inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem (2013) 288(5):3275–88. doi:10.1074/jbc.M112.408104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Bailey SG, Cragg MS, Townsend PA. Family friction as ΔNp73 antagonises p73 and p53. Int J Biochem Cell Biol (2011) 43(4):482–6. doi:10.1016/j.biocel.2010.12.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Ethayathulla AS, Nguyen HT, Viadiu H. Crystal structures of the DNA-binding domain tetramer of the p53 tumor suppressor family member p73 bound to different full-site response elements. J Biol Chem (2013) 288(7):4744–54. doi:10.1074/jbc.M112.408039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Rufini A, Agostini M, Grespi F, Tomasini R, Sayan BS, Niklison-Chirou MV, et al. p73 in cancer. Genes Cancer (2011) 2(4):491–502. doi:10.1177/1947601911408890
131. Tschan MP, Grob TJ, Peters UR, Laurenzi VD, Huegli B, Kreuzer KA, et al. Enhanced p73 expression during differentiation and complex p73 isoforms in myeloid leukemia. Biochem Biophys Res Commun (2000) 277(1):62–5. doi:10.1006/bbrc.2000.3627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Burge S, Teufel DP, Townsley FM, Freund SM, Bycroft M, Fersht AR. Molecular basis of the interactions between the p73 N terminus and p300: effects on transactivation and modulation by phosphorylation. Proc Natl Acad Sci U S A (2009) 106(9):3142–7. doi:10.1073/pnas.0900383106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Ozaki T, Okoshi R, Ono S, Kubo N, Nakagawara A. Deregulated expression of E2F1 promotes proteolytic degradation of tumor suppressor p73 and inhibits its transcriptional activity. Biochem Biophys Res Commun (2009) 387(1):143–8. doi:10.1016/j.bbrc.2009.06.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Zeng X, Li X, Miller A, Yuan Z, Yuan W, Kwok RP, et al. The N-terminal domain of p73 interacts with the CH1 domain of p300/CREB binding protein and mediates transcriptional activation and apoptosis. Mol Cell Biol (2000) 20(4):1299–310. doi:10.1128/MCB.20.4.1299-1310.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Downward J, Basu S. YAP and p73: a complex affair. Mol Cell (2008) 32(6):749–50. doi:10.1016/j.molcel.2008.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Watanabe K, Ozaki T, Nakagawa T, Miyazaki K, Takahashi M, Hosoda M, et al. Physical interaction of p73 with c-Myc and MM1, a c-Myc-binding protein, and modulation of the p73 function. J Biol Chem (2002) 277(17):15113–23. doi:10.1074/jbc.M111281200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Bálint E, Bates S, Vousden KH. Mdm2 binds p73 alpha without targeting degradation. Oncogene (1999) 18(27):3923–9. doi:10.1038/sj.onc.1202781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Munarriz E, Barcaroli D, Stephanou A, Townsend PA, Maisse C, Terrinoni A, et al. PIAS-1 is a checkpoint regulator which affects exit from G1 and G2 by sumoylation of p73. Mol Cell Biol (2004) 24(24):10593–610. doi:10.1128/MCB.24.24.10593-10610.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Olmos Y, Brosens JJ, Lam EW. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat (2011) 14(1):35–44. doi:10.1016/j.drup.2010.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Gaiddon C, Lokshin M, Gross I, Levasseur D, Taya Y, Loeffler J, et al. Cyclin-dependent kinases phosphorylate p73 at threonine 86 in a cell cycle-dependent manner and negatively regulate p73. J Biol Chem (2003) 278(30):27421–31. doi:10.1074/jbc.M300251200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Watson IR, Blanch A, Lin DC, Ohh M, Irwin MS. Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J Biol Chem (2006) 281(45):34096–103. doi:10.1074/jbc.M603654200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Rossi M, Laurenzi V, de Munarriz E, Green DR, Liu Y, Vousden KH, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J (2005) 24(4):836–48. doi:10.1038/sj.emboj.7600444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Zhao LY, Liu Y, Bertos NR, Yang X, Liao D. PCAF is a coactivator for p73-mediated transactivation. Oncogene (2003) 22(51):8316–29. doi:10.1038/sj.onc.1206916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature (1999) 399(6738):814–7. doi:10.1038/21704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Ren J, Datta R, Shioya H, Li Y, Oki E, Biedermann V, et al. p73beta is regulated by protein kinase Cdelta catalytic fragment generated in the apoptotic response to DNA damage. J Biol Chem (2002) 277(37):33758–65. doi:10.1074/jbc.M110667200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Sayan BS, Yang AL, Conforti F, Tucci P, Piro MC, Browne GJ, et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc Natl Acad Sci U S A (2010) 107(29):12877–82. doi:10.1073/pnas.0911828107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Bisso A, Collavin L, Del Sal G. p73 as a pharmaceutical target for cancer therapy. Curr Pharm Des (2011) 17(6):578–90. doi:10.2174/138161211795222667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Balint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57KIP2 expression by p73. Proc Natl Acad Sci U S A (2002) 99(6):3529–34. doi:10.1073/pnas.062491899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Scian MJ, Carchman EH, Mohanraj L, Stagliano KE, Anderson MA, Deb D, et al. Wild-type p53 and p73 negatively regulate expression of proliferation related genes. Oncogene (2007) 27(18):2583–93. doi:10.1038/sj.onc.1210898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Innocente SA, Lee JM. p73 is a p53-independent, Sp1-dependent repressor of cyclin B1 transcription. Biochem Biophys Res Commun (2005) 329(2):713–8. doi:10.1016/j.bbrc.2005.02.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. De Cola A, Bongiorno-Borbone L, Bianchi E, Barcaroli D, Carletti E, Knight RA, et al. FLASH is essential during early embryogenesis and cooperates with p73 to regulate histone gene transcription. Oncogene (2012) 31(5):573–82. doi:10.1038/onc.2011.274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Terrinoni A, Ranalli M, Cadot B, Leta A, Bagetta G, Vousden KH, et al. p73-alpha is capable of inducing scotin and ER stress. Oncogene (2004) 23(20):3721–5. doi:10.1038/sj.onc.1207342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Wetzel MK, Naska S, Laliberté CL, Rymar VV, Fujitani M, Biernaskie JA, et al. p73 Regulates neurodegeneration and phospho-tau accumulation during aging and Alzheimer’s disease. Neuron (2008) 59(5):708–21. doi:10.1016/j.neuron.2008.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Cancino GI, Miller FD, Kaplan DR. p73 haploinsufficiency causes tau hyperphosphorylation and tau kinase dysregulation in mouse models of aging and Alzheimer’s disease. Neurobiol Aging (2013) 34(2):387–99. doi:10.1016/j.neurobiolaging.2012.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev (2008) 22(19):2677–91. doi:10.1101/gad.1695308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Tissir F, Ravni A, Achouri Y, Riethmacher D, Meyer G, Goffinet AM. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci U S A (2009) 106(39):16871–6. doi:10.1073/pnas.0903191106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev (2010) 24(6):549–60. doi:10.1101/gad.1873910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Niklison-Chirou MV, Steinert JR, Agostini M, Knight RA, Dinsdale D, Cattaneo A, et al. TAp73 knockout mice show morphological and functional nervous system defects associated with loss of p75 neurotrophin receptor. Proc Natl Acad Sci U S A (2013) 110(47):18952–7. doi:10.1073/pnas.1221172110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol (2013) 15(8):991–1000. doi:10.1038/ncb2789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev (2012) 26(18):2009–14. doi:10.1101/gad.197640.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Seitz SJ, Schleithoff ES, Koch A, Schuster A, Teufel A, Staib F, et al. Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. Int J Cancer (2010) 126(9):2049–66. doi:10.1002/ijc.24861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Schuster A, Schilling T, De Laurenzi V, Koch AF, Seitz S, Staib F, et al. ΔNp73β is oncogenic in hepatocellular carcinoma by blocking apoptosis signaling via death receptors and mitochondria. Cell Cycle (2010) 9(13):2629–39. doi:10.4161/cc.9.13.12110
163. Norris MD, Gilbert J, Smith SA, Marshall GM, Salwen H, Cohn SL, et al. Expression of the putative tumour suppressor gene, p73, in neuroblastoma and other childhood tumours. Med Pediatr Oncol (2001) 36(1):48–51. doi:10.1002/1096-911X(20010101)36:1<48::AID-MPO1013>3.0.CO;2-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Lo Iacono M, Monica V, Saviozzi S, Ceppi P, Bracco E, Papotti M, et al. p63 and p73 isoform expression in non-small cell lung cancer and corresponding morphological normal lung tissue. J Thorac Oncol (2011) 6(3):473–81. doi:10.1097/JTO.0b013e31820b86b0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Carastro LM, Lin H, Park HY, Kim D, Radlein S, Hampton KK, et al. Role of p73 dinucleotide polymorphism in prostate cancer and p73 protein isoform balance. Prostate Cancer (2014) 2014(3):1–9. doi:10.1155/2014/129582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Arvanitis DA, Lianos E, Soulitzis N, Delakas D, Spandidos DA. Deregulation of p73 isoform equilibrium in benign prostate hyperplasia and prostate cancer. Oncol Rep (2004) 12(5):1131–7. doi:10.3892/or.12.5.1131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Yokomizo A, Mai M, Tindall DJ, Cheng L, Bostwick DG, Naito S, et al. Overexpression of the wild type p73 gene in human bladder cancer. Oncogene (1999) 18(8):1629–33. doi:10.1038/sj.onc.1202474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Guan M, Peng H, Yu B, Lu Y. p73 overexpression and angiogenesis in human colorectal carcinoma. Jpn J Clin Oncol (2003) 33(5):215–20. doi:10.1093/jjco/hyg045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Dominguez G, Silva JM, Silva J, Garcia JM, Sanchez A, Navarro A, et al. Wild type p73 overexpression and high-grade malignancy in breast cancer. Breast Cancer Res Treat (2001) 66(3):183–90. doi:10.1023/A:1010624717311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Diaz R, Gonzalez-Sancho JM, Soldevilla B, Silva J, Garcia JM, Garcia V, et al. Differential regulation of TP73 isoforms by 1alpha,25-dihydroxyvitamin D3 and survivin in human colon and breast carcinomas. Genes Chromosomes Cancer (2010) 49(12):1135–42. doi:10.1002/gcc.20821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Leupin N, Luthi A, Novak U, Grob TJ, Hugli B, Graber H, et al. P73 status in B-cell chronic lymphocytic leukaemia. Leuk Lymphoma (2004) 45(6):1205–7. doi:10.1080/10298190310001623829
172. Ito Y, Takeda T, Wakasa K, Tsujimoto M, Sakon M, Matsuura N. Expression of p73 and p63 proteins in pancreatic adenocarcinoma: p73 overexpression is inversely correlated with biological aggressiveness. Int J Mol Med (2001) 8(1):67–71. doi:10.3892/ijmm.8.1.67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Soussi T, Ishioka C, Claustres M, Béroud C. Locus-specific mutation databases: pitfalls and good practice based on the p53 experience. Nat Rev Cancer (2006) 6(1):83–90. doi:10.1038/nrc1783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol (2010) 2(2):a001107. doi:10.1101/cshperspect.a001107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Solomon H, Madar S, Rotter V. Mutant p53 gain of function is interwoven into the hallmarks of cancer. J Pathol (2011) 225(4):475–8. doi:10.1002/path.2988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Piccinin S, Tonin E, Sessa S, Demontis S, Rossi S, Pecciarini L, et al. A “twist box” code of p53 inactivation: twist box: p53 interaction promotes p53 degradation. Cancer Cell (2012) 22(3):404–15. doi:10.1016/j.ccr.2012.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature (2007) 445(7128):661–5. doi:10.1038/nature05541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature (2007) 445(7128):656–60. doi:10.1038/nature05529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (2004) 303(5659):844–8. doi:10.1126/science.1092472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene (2008) 27(7):997–1003. doi:10.1038/sj.onc.1210707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene (2007) 26(24):3473–81. doi:10.1038/sj.onc.1210136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Wang J, Zheng T, Chen X, Song X, Meng X, Bhatta N, et al. MDM2 antagonist can inhibit tumor growth in hepatocellular carcinoma with different types of p53 in vitro. J Gastroenterol Hepatol (2011) 26(2):371–7. doi:10.1111/j.1440-1746.2010.06440.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Zauli G, Celeghini C, Melloni E, Voltan R, Ongari M, Tiribelli M, et al. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica (2012) 97(11):1722–30. doi:10.3324/haematol.2012.062083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood (2005) 106(9):3150–9. doi:10.1182/blood-2005-02-0553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia (2008) 22(4):730–9. doi:10.1038/leu.2008.11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Secchiero P, Melloni E, Tiribelli M, Gonelli A, Zauli G. Combined treatment of CpG-oligodeoxynucleotide with nutlin-3 induces strong immune stimulation coupled to cytotoxicity in B-chronic lymphocytic leukemic (B-CLL) cells. J Leukoc Biol (2008) 83(2):434–7. doi:10.1189/jlb.0707459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Zauli G, Voltan R, Bosco R, Melloni E, Marmiroli S, Rigolin GM, et al. Dasatinib plus nutlin-3 shows synergistic antileukemic activity in both p53 wild-type and p53 mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clin Cancer Res (2011) 17(4):762–70. doi:10.1158/1078-0432.CCR-10-2572
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Mir R, Tortosa A, Martinez-Soler F, Vidal A, Condom E, Pérez-Perarnau A, et al. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. Int J Cancer (2013) 132(7):1525–36. doi:10.1002/ijc.27832
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Ohnstad HO, Paulsen EB, Noordhuis P, Berg M, Lothe RA, Vassilev LT, et al. MDM2 antagonist nutlin-3a potentiates antitumour activity of cytotoxic drugs in sarcoma cell lines. BMC Cancer (2011) 11(211):1–11. doi:10.1186/1471-2407-11-211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Pishas KI, Neuhaus SJ, Clayer MT, Schreiber AW, Lawrence DM, Perugini M, et al. Nutlin-3a efficacy in sarcoma predicted by transcriptomic and epigenetic profiling. Cancer Res (2014) 74(3):921–31. doi:10.1158/0008-5472.CAN-13-2424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Villalonga-Planells R, Coll-Mulet L, Martínez-Soler F, Castaño E, Acebes J, Giménez-Bonafé P, et al. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS One (2011) 6(4):e18588. doi:10.1371/journal.pone.0018588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Cinatl J, Speidel D, Hardcastle I, Michaelis M. Resistance acquisition to MDM2 inhibitors. Biochem Soc Trans (2014) 42(4):752–7. doi:10.1042/BST20140035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Michaelis M, Rothweiler F, Barth S, Cinatl J, van Rikxoort M, Löschmann N, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis (2011) 2:e243. doi:10.1038/cddis.2011.129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator nutlin-3. Oncogene (2011) 30(46):4678–86. doi:10.1038/onc.2011.185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med (2004) 10(12):1321–8. doi:10.1038/nm1146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Enge M, Bao W, Hedström E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell (2009) 15(3):171–83. doi:10.1016/j.ccr.2009.01.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Roh J, Ko JH, Moon SJ, Ryu CH, Choi JY, Koch WM. The p53-reactivating small-molecule RITA enhances cisplatin-induced cytotoxicity and apoptosis in head and neck cancer. Cancer Lett (2012) 325(1):35–41. doi:10.1016/j.canlet.2012.05.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Burmakin M, Shi Y, Hedström E, Kogner P, Selivanova G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin Cancer Res (2013) 19(18):5092–103. doi:10.1158/1078-0432.CCR-12-2211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A (2008) 105(10):3933–8. doi:10.1073/pnas.0708917105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Sosin AM, Burger AM, Siddiqi A, Abrams J, Mohammad RM, Al-Katib AM. HDM2 antagonist MI-219 (spiro-oxindole), but not nutlin-3 (cis-imidazoline), regulates p53 through enhanced HDM2 autoubiquitination and degradation in human malignant B-cell lymphomas. J Hematol Oncol (2012) 5:57. doi:10.1186/1756-8722-5-57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Zou P, Zheng N, Yu Y, Yu S, Sun W, McEachem D, et al. Preclinical pharmacokinetics of MI-219, a novel human double minute 2 (HDM2) inhibitor and prediction of human pharmacokinetics. J Pharm Pharm Sci (2012) 15(2):265–80.
202. Landré V, Rotblat B, Melino S, Bernassola F, Melino G. Screening for E3-ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget (2014).
203. Wilson JM, Henderson G, Black F, Sutherland A, Ludwig RL, Vousden KH, et al. Synthesis of 5-deazaflavin derivatives and their activation of p53 in cells. Bioorg Med Chem (2007) 15(1):77–86. doi:10.1016/j.bmc.2006.10.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Devine T, Dai M. Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr Pharm Des (2013) 19(18):3248–62. doi:10.2174/1381612811319180009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell (2005) 7(6):547–59. doi:10.1016/j.ccr.2005.04.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Roxburgh P, Hock AK, Dickens MP, Mezna M, Fischer PM, Vousden KH. Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis (2012) 33(4):791–8. doi:10.1093/carcin/bgs092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Kojima K, Burks JK, Arts J, Andreeff M. The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther (2010) 9(9):2545–57. doi:10.1158/1535-7163.MCT-10-0337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Tabernero J, Dirix L, Schöffski P, Cervantes A, Lopez-Martin JA, Capdevila J, et al. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin Cancer Res (2011) 17(19):6313–21. doi:10.1158/1078-0432.CCR-11-1101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med (2011) 3(64):64rv1. doi:10.1126/scitranslmed.3001366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Atkins KM, Thomas LL, Barroso-González J, Thomas L, Auclair S, Yin J, et al. The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep (2014) 8(5):1545–57. doi:10.1016/j.celrep.2014.07.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Botta G, De Santis LP, Saladino R. Current advances in the synthesis and antitumoral activity of SIRT1-2 inhibitors by modulation of p53 and pro-apoptotic proteins. Curr Med Chem (2012) 19(34):5871–84. doi:10.2174/092986712804143303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell (2008) 13(5):454–63. doi:10.1016/j.ccr.2008.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Nihal M, Ahmad N, Wood GS. SIRT1 is upregulated in cutaneous T-cell lymphoma, and its inhibition induces growth arrest and apoptosis. Cell Cycle (2014) 13(4):632–40. doi:10.4161/cc.27523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Ueno T, Endo S, Saito R, Hirose M, Hirai S, Suzuki H, et al. The sirtuin inhibitor tenovin-6 upregulates death receptor 5 and enhances cytotoxic effects of 5-fluorouracil and oxaliplatin in colon cancer cells. Oncol Res (2014) 21(3):155–64. doi:10.3727/096504013X13854886566598
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. MacCallum SF, Groves MJ, James J, Murray K, Appleyard V, Prescott AR, et al. Dysregulation of autophagy in chronic lymphocytic leukemia with the small-molecule sirtuin inhibitor tenovin-6. Sci Rep (2013) 3:1275. doi:10.1038/srep01275
216. Kato S, Han S, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A (2003) 100(14):8424–9. doi:10.1073/pnas.1431692100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene (2007) 26(15):2243–54. doi:10.1038/sj.onc.1210295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8(3):282–8. doi:10.1038/nm0302-282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Bykov VJ, Issaeva N, Zache N, Shilov A, Hultcrantz M, Bergman J, et al. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem (2005) 280(34):30384–91. doi:10.1074/jbc.M501664200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene (2010) 29(30):4245–52. doi:10.1038/onc.2010.188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Piantino CB, Reis ST, Viana NI, Silva IA, Morais DR, Antunes AA, et al. Prima-1 induces apoptosis in bladder cancer cell lines by activating p53. Clinics (Sao Paulo) (2013) 68(3):297–303. doi:10.6061/clinics/2013(03)OA03
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene (2005) 24(21):3484–91. doi:10.1038/sj.onc.1208419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Rökaeus N, Shen J, Eckhardt I, Bykov VJ, Wiman KG, Wilhelm MT. PRIMA-1MET/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene (2010) 29(49):6442–51. doi:10.1038/onc.2010.382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Shalom-Feuerstein R, Serror L, Aberdam E, Müller F, van Bokhoven H, Wiman KG, et al. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc Natl Acad Sci U S A (2013) 110(6):2152–6. doi:10.1073/pnas.1201753109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther (2013) 12(11):2331–41. doi:10.1158/1535-7163.MCT-12-1166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Saha MN, Chen Y, Chen M, Chen G, Chang H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br J Cancer (2014) 110(9):2224–31. doi:10.1038/bjc.2014.164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A (2008) 105(30):10360–5. doi:10.1073/pnas.0805326105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science (1999) 286(5449):2507–10. doi:10.1126/science.286.5449.2507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
229. Wang W, Takimoto R, Rastinejad F, El-Deiry WS. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol Cell Biol (2003) 23(6):2171–81. doi:10.1128/MCB.23.6.2171-2181.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Wischhusen J, Naumann U, Ohgaki H, Rastinejad F, Weller M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene (2003) 22(51):8233–45. doi:10.1038/sj.onc.1207198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Madka V, Zhang Y, Li Q, Mohammed A, Sindhwani P, Lightfoot S, et al. p53-stabilizing agent CP-31398 prevents growth and invasion of urothelial cancer of the bladder in transgenic UPII-SV40T mice. Neoplasia (2013) 15(8):966–74. doi:10.1593/neo.13704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Kravchenko JE, Ilyinskaya GV, Komarov PG, Agapova LS, Kochetkov DV, Strom E, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A (2008) 105(17):6302–7. doi:10.1073/pnas.0802091105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis (2009) 14(4):597–606. doi:10.1007/s10495-009-0330-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Schilling T, Kairat A, Melino G, Krammer PH, Stremmel W, Oren M, et al. Interference with the p53 family network contributes to the gain of oncogenic function of mutant p53 in hepatocellular carcinoma. Biochem Biophys Res Commun (2010) 394(3):817–23. doi:10.1016/j.bbrc.2010.03.082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Zalcenstein A, Stambolsky P, Weisz L, Müller M, Wallach D, Goncharov TM, et al. Mutant p53 gain of function: repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants. Oncogene (2003) 22(36):5667–76. doi:10.1038/sj.onc.1206724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Jackson JG, Pant V, Li Q, Chang LL, Quintás-Cardama A, Garza D, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell (2012) 21(6):793–806. doi:10.1016/j.ccr.2012.04.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: p53, p63, p73, cellular stress, cancer, chemosensitivity, apoptosis
Citation: Pflaum J, Schlosser S and Müller M (2014) p53 family and cellular stress responses in cancer. Front. Oncol. 4:285. doi: 10.3389/fonc.2014.00285
Received: 22 April 2014; Accepted: 03 October 2014;
Published online: 21 October 2014.
Edited by:
Daniel Speidel, Children’s Medical Research Institute, AustraliaReviewed by:
Ella L. Kim, Johannes Gutenberg University Medical Centre, GermanyJean-Christophe Bourdon, University of Dundee, UK
Copyright: © 2014 Pflaum, Schlosser and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martina Müller, Department of Internal Medicine I, University Hospital Regensburg, Franz-Josef-Strauß-Allee 11, Regensburg 93053, Germany e-mail:bWFydGluYS5tdWVsbGVyLXNjaGlsbGluZ0B1a3IuZGU=
†These authors share first authorship