 Tomás Gutiérrez
Tomás Gutiérrez Thomas Simmen*
Thomas Simmen*- Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
Endoplasmic reticulum (ER) chaperones and oxidoreductases are abundant enzymes that mediate the production of fully folded secretory and transmembrane proteins. Resisting the Golgi and plasma membrane-directed “bulk flow,” ER chaperones and oxidoreductases enter retrograde trafficking whenever they are pulled outside of the ER by their substrates. Solid tumors are characterized by the increased production of reactive oxygen species (ROS), combined with reduced blood flow that leads to low oxygen supply and ER stress. Under these conditions, hypoxia and the unfolded protein response upregulate their target genes. When this occurs, ER oxidoreductases and chaperones become important regulators of tumor growth. However, under these conditions, these proteins not only promote the folding of proteins, but also alter the properties of the plasma membrane and hence modulate tumor immune recognition. For instance, high levels of calreticulin serve as an “eat-me” signal on the surface of tumor cells. Conversely, both intracellular and surface BiP/GRP78 promotes tumor growth. Other ER folding assistants able to modulate the properties of tumor tissue include protein disulfide isomerase (PDI), Ero1α and GRP94. Understanding the roles and mechanisms of ER chaperones in regulating tumor cell functions and immunorecognition will lead to important insight for the development of novel cancer therapies.
Introduction
The endoplasmic reticulum (ER) is the location of oxidative protein folding, a mechanism that enzymatically manufactures fully folded secretory and transmembrane proteins. These two groups of proteins make up about 10 and 20% of a typical mammalian proteome, respectively (1). Ribosomally produced polypeptides for these two groups of proteins are first targeted to the ER membrane, where they interact with the Sec61 protein translocation channel (translocon) using their signal peptide (2). At this location, polypeptides undergo cytosolic folding that continues during translocation to the ER lumen (3, 4). Subsequently, the interaction with immunoglobulin binding protein (BiP/GRP78), a major ER-lumenal chaperone, initiates the production of secretory and transmembrane proteins (5, 6). If polypeptides are glycosylated they subsequently interact with the lectin chaperones calnexin and calreticulin (7), as well as oxidoreductases including protein disulfide isomerase (PDI) and related family members such as ERp57 (8). The oxidizing activity of these proteins is kept intact by oxygen- or hydrogen-peroxide consuming oxidoreductases such as Ero1α (9). Thus, ER chaperones and oxidoreductases cyclically interact with the ongoing flow of polypeptides emerging from the translocon. The flow of these proteins is massive. Using a vesicular stomatitis virus G protein (VSVG) fusion with green fluorescent protein (GFP), it has been estimated to amount to 7,000 molecules per second for this model protein alone (10, 11).
This observation raises the question as to how cells handle this quantity of export and how they ensure that exported proteins are segregated from resident ER chaperones and oxidoreductases. Early research using glycosylated short peptides had indicated that ER–Golgi trafficking occurs via non-specific “bulk flow” (12). However, this intuitive model may not be correct, since positive signals are not only needed for export from the ER (13, 14), but also for transit toward the Golgi complex (15). Moreover, secretory proteins are actually actively excluded from retrograde trafficking, which describes the trafficking route from the Golgi complex back to the ER (15). Conversely, most ER oxidoreductases and chaperones are equipped with a C-terminal KDEL motif that serves to interact with the KDEL receptor, a retrieval receptor that re-establishes ER localization for proteins with such a motif (16, 17). Therefore, given these efficient retention mechanisms, it makes little sense that tumor immunorecognition should be influenced by ER-restricted chaperones and oxidoreductases, when this mechanism is dictated by the properties of the cell surface. Nevertheless, in a cancer setting, proteins of this group can become localized to the plasma membrane or even secreted (18). When this occurs, ER oxidoreductases and chaperones become important regulators of tumor growth, but also of tumor immune recognition. For instance, the escape of calreticulin from the ER leads to the generation of an “eat-me” signal on the surface of tumor cells (19). Surface BiP/GRP78 is a target for antibody-based experimental therapies as well (20, 21). Understanding how these proteins target to the plasma membrane could therefore lead to important insight for the development of immune-based cancer therapies.
ER Retrieval of Chaperones and Oxidoreductases
To ensure their residence to the ER and their availability for further work on newly synthesized polypeptides, chaperones and oxidoreductases are continually recycled back to the ER (22). Lumenal ER chaperones and oxidoreductases use the lysine-based C-terminal KDEL sequence for this purpose to interact with the KDEL receptor, a sorting receptor that cycles between the Golgi complex and the ER, first discovered by the Pelham lab in 1990 (16, 17). The KDEL receptor is part of a group of transmembrane proteins that all retrieve luminal ER proteins (23). These transmembrane receptor proteins typically use cytosolic, C-terminal lysine-based motifs (KKXX) to travel from the Golgi complex to the ER on a retrograde trafficking route (24). One example is the KKFF motif in the case of the lectin ERGIC53 (25).
KKXX motifs retrieve ER transmembrane proteins by mediating interaction with the coatomer protein complex, also called COPI (26). This is a multi-subunit protein complex composed of seven distinct proteins termed coatomer whose subunits are termed α, β, β′, γ, δ, ε, and ζ COPs (27). The binding of dilysine-bearing cargo molecules on α and β′ subunits nucleates the formation of COPI coats (28, 29). This step also requires the activation of ADP-ribosylation factor 1 (Arf1) (30). Upon pinching off from within the Golgi complex or the ER–Golgi intermediate compartment (ERGIC), retrograde vesicles are uncoated, following GTP hydrolysis on Arf1 mediated by Arf GTPase activating proteins (ArfGAPs) 1, 2, and 3 (31). These vesicles then migrate into the proximity of the ER. Here, a Soluble NSF Attachment Protein Receptor (SNARE) complex becomes important for retrograde trafficking. This trans-SNARE complex forms via Dsl1p (yeast) or Zw10 (mammals) with incoming COPI-derived vesicles (32, 33). These vesicles then fuse with the ER membrane at a site termed ER import sites, whose existence so far has only been demonstrated in plants (34). Trafficking from the Golgi complex to the ER is also under the control of Ras-related GTPases, members of a large regulatory protein family that serve as address tags for intracellular trafficking (35). Rab6 and Rab2 likely work in sequence to facilitate retrograde transport mediated by coatomer and directed to the ER (36–38), whereas Rab18 might regulate coatomer-independent trafficking from the Golgi to the ER (39). Together, the retention of ER chaperones and oxidoreductases clearly requires a large set of proteins, whose identity and mechanisms are now fairly well understood, despite some important outstanding questions (40).
In addition to COPI-mediated retrieval, some ER chaperones and oxidoreductases are retained in this organelle by other retention mechanisms (41). One type of mechanism requires the interaction of ER-resident proteins with COPI adaptors or helper proteins, exemplified by the interaction of a calnexin cytosolic acidic cluster motif with the sorting adaptor PACS-2 that dictates the extent of calnexin ER retention (42). This is particularly important, as calnexin does not have a canonical KKXX motif, but rather a di-arginine-based C-terminal motif involved in its retention (43).
Another way how ER transmembrane proteins are excluded from ER export is by the length of their transmembrane domains. This is demonstrated with artificial 17 transmembrane residue constructs that are unable to enter ER exit sites (ERES), whereas 22 residue long transmembrane domains allow for inclusion into Golgi-destined vesicles (44). The length of these transmembrane domains might facilitate inclusion into specific ER membrane domains (44). Some ER proteins use their transmembrane domains to enter a retrieval cycle similar to KDEL-tagged ER lumenal proteins. This is the case with sarcoendoplasmic reticulum calcium transport ATPase (SERCA) (45). Some of these proteins use the retrieval receptor Rer1 for their localization to the ER, as is the case for rhodopsin or components of the γ-secretase complex (46–48).
Endoplasmic reticulum lumenal chaperones and oxidoreductases further depend on the nature of the ER environment to achieve their typical distributions (49). This phenomenon is best understood for the ER oxidoreductase Ero1α (50). This lumenal ER protein lacks a KDEL motif, but uses interactions with other ER oxidoreductases (PDI and ERp44) to stay within the ER, but only under oxidizing conditions (51, 52). A similar mechanism is used by peroxiredoxin 4 (53). Less is known about the ability of Ca2+ binding domains to assist to ER retention, as is known to occur in the case of the ER chaperone calreticulin (54). While the depletion of ER lumenal Ca2+ is a known inducer of ER stress, the disruption of calreticulin ER localization is uniquely dependent on Ca2+. Calreticulin is not secreted upon induction of an ER stress with, for instance, tunicamycin (55). Potentially, this finding could indicate a requirement of Ca2+ binding to achieve a retrievable conformation of ER chaperones and oxidoreductases and specifically calreticulin. Such a hypothesis would be consistent with known alterations of protein conformation upon the loss of bound Ca2+ (56) and a general loss of chaperone–protein interactions within the ER upon the loss of free Ca2+ (57, 58). Either consequence could then lead to a loss of KDEL retrieval, either via masking of the KDEL sequence or via saturation of KDEL receptors (59). A similar Ca2+-dependent mechanism appears to determine the retention of BiP/GRP78 in the ER (60, 61). Together, ER localization of chaperones and oxidoreductases is lost or reduced upon the interference with retrieval receptors, upon modulation of the oxidative conditions of the ER and upon loss of ER Ca2+.
ER Chaperones and Oxidoreductases on the Plasma Membrane of Tumor Cells
At first glance, the ER retention of chaperones and oxidoreductases appears like an abstract problem of interest only to very specialized cell biologists. Although cell types such as thyrocytes and immature thymocytes retain ER chaperones and oxidoreductases less efficiently, it is not known what the exact biological significance of this finding is (62, 63). However, over the past few years, information has emerged that ER chaperone and oxidoreductase retention in the ER is a critical sentinel mechanism that signals ER stress to the immune system (64). This is not unexpected, since ER chaperones such as calreticulin are functionally linked to the immune system and mediate the folding of major histocompatibility complex (MHC) class I (64). Through this function, ER chaperones and oxidoreductases already exhibit a tight link to the immune system via the regulation of intracellular peptide presentation by MHC class I on the plasma membrane (65). Accordingly, lost retention of ER chaperones and oxidoreductases upon ER stress impairs MHC class I expression on the surface (66). Surprisingly, however, this is not the only consequence. Calreticulin is normally enriched on the rough ER (rER) (67, 68). However, in cells undergoing ER stress, in particular following the depletion of ER Ca2+, calreticulin, PDI, BiP/GRP78, and GRP94 escape ER retention and retrieval (69). These cell surface-exposed chaperones and oxidoreductases can present antigens to the immune system (calreticulin, GRP94), serve as anchors for leukocytes (PDI), but can also activate pro-survival signaling pathways (BiP/GRP78) (18). In addition, ER stress generated from lost ER localization of chaperones and oxidoreductases leads to mitochondrial dysfunction and also triggers the activation of the NLRP3 inflammasome (70). In an organism, these metabolic and chemical changes lead to increased blood flow and leukocyte delivery to cells, where ER stress occurs (71). Therefore, the mechanisms that retain ER chaperones and oxidoreductases communicate ER stress to the immune system via multiple readouts: they determine MHC class I surface exposure, they influence the activation of inflammation, but they also signal the intracellular stress status to the immune system when found on the plasma membrane.
Calreticulin: A DAMP on the Plasma Membrane of Tumor Cells
The appearance of ER chaperones and oxidoreductases on the plasma membrane corresponds to a danger-associated molecular pattern (DAMP) (72). DAMPs are molecules that are normally intracellular, but become exposed on the plasma membrane in stressed, damaged, or dying cells, as well as in tumor cells (73). Their presence on the cell surface leads to the recruitment of innate inflammatory cells, following the interaction of surface DAMPs with pattern-recognition receptors (PRRs) (74). An example for this is CD91, found on the surface of dendritic cells (DC) and other antigen-presenting cells (APC), which interacts with the ER-derived DAMPs calreticulin and GRP94 on stressed or dying cells (75). Upon formation of a complex between these proteins, a potent “eat-me” signal is generated and phagocytosis of calreticulin or GRP94-bearing stressed cells is initiated (19, 76). In contrast, CD47 acts as an inhibitor of this activity of calreticulin by interfering with the calreticulin–CD91 complex formation (64, 76).
This mechanism is particularly important in the cancer scenario (Figure 1), where calreticulin is today one of the most extensively studied DAMPs that dictates the immunogenicity of cancer cells (19, 77). Importantly, calreticulin exposure on the plasma membrane is triggered upon treatment with different chemotherapeutic stimuli, including cisplatin and the anthracyclines doxorubicin, idarubicin, and mitoxantrone (19, 78). However, it is not clear whether calreticulin remains in the membrane of the stressed cell or is transferred over to immune cells (79). Regardless of the exact location of this extracellular calreticulin, stressed and apoptotic cells are subsequently engulfed and eliminated (80). This activation of the immune system can be exploited via the injection of calreticulin-coated cancer cells. Once in the blood stream, these abnormal cells can trigger a tumor-specific immune-response that eventually may activate an anti-tumor immune-response in vivo (81).
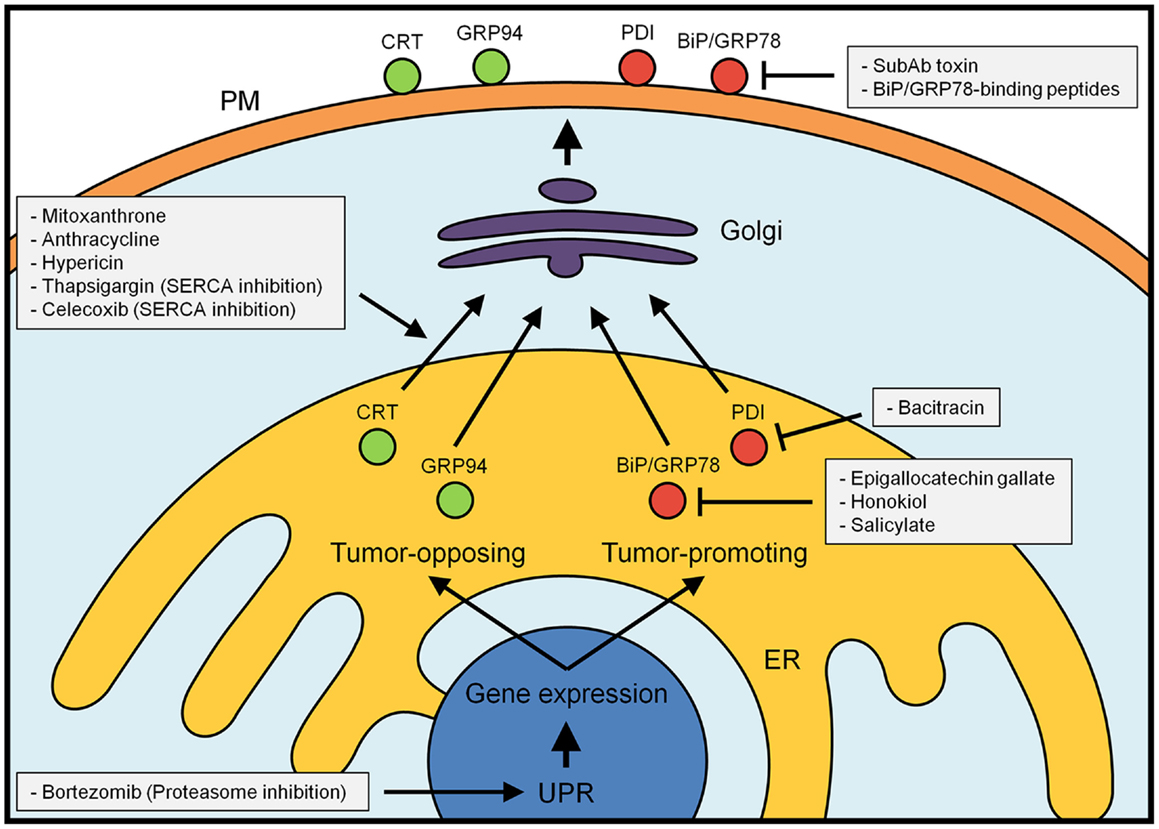
Figure 1. Major tumor-promoting (red) and tumor-inhibitory (green) ER chaperones and oxidoreductases. Their localization and function within the ER can be favorably influenced by a number of currently known drugs (for details see text). At the plasma membrane, inhibitory binding peptides can stop the tumor-promoting activity of this class of proteins. In contrast, simple modulation of the unfolded protein response (UPR) is expected to induce both tumor-promoting and tumor-blocking responses.
An additional important prerequisite for the immuno-elimination of tumor cells using the calreticulin “eat-me” signal is autophagy: the inhibition of autophagy significantly increases the amounts of calreticulin on the surface of stressed tumor cells, suggesting that autophagy-competent cancer tissue may be less susceptible to calreticulin-mediated immunorecognition of tumor cells (82, 83). In contrast, autophagy promotes the secretion of ATP upon ER stress, another tumor DAMP (84, 85). Interestingly, not only calreticulin on the plasma membrane, but also its overall expression is frequently enhanced in tumor tissue, potentially indicating that this chaperone could indeed provide an avenue for future cancer immunotherapy (86). Specifically, calreticulin over-expression is associated with the development and progression of pancreatic cancer (87). However, studies on infiltrating ductal breast carcinomas (IDCAs) were not able to detect an involvement of calreticulin in the development of a humoral immune-response (88). In defense of the calreticulin role as a protective mechanism against cancer, none of these studies have investigated the intracellular distribution of calreticulin in the respective tumor scenario. Consistent with this caveat, hepatocellular carcinoma has been found associated with high levels of circulating anti-calreticulin antibodies (89). In addition, serum IgG levels of anti-calreticulin autoantibodies have been found to be significantly higher in bladder cancer patients than in normal controls, leading to the proposal of anti-calreticulin antibodies as a novel biomarker for bladder cancer progression (90). It is currently unclear whether the injection of a fragment of recombinant calreticulin blocks tumor growth using these or other mechanisms (91, 92).
ERp57, GRP94, Ero1α, and PDI: Functions Beyond Immunorecognition for Tumor Cell Migration
Other ER chaperones and oxidoreductases also show aberrant targeting to the plasma membrane. One example is ERp57, which is critical for the peptide loading complex for MHC class I together with calreticulin (93). Similar to what occurs with calreticulin, ERp57 also appears on the cell surface following anthracycline treatment. Importantly, ERp57 might not act as a DAMP itself, but rather as a prerequisite for calreticulin surface targeting (94, 95). The expression level of ERp57 in cancer does not provide much insight about its role in cancer, since bladder and gastric cancers appear to be characterized by low levels of calreticulin and ERp57 (96, 97).
GRP94 (also called gp96) is another prominent chaperone of the ER that has a much smaller set of client proteins when compared to calreticulin (98). Its substrates include toll-like receptors (TLRs), important sensors of DAMPs (99). This Hsp90 family protein can escape ER retention like calreticulin, and is found secreted from pancreatic cells and hepatocytes (100, 101). In contrast, tumor cells are decorated with surface-bound GRP94 (102, 103). On this localization, GRP94 acts as a DAMP similar to calreticulin (104) and in parallel to surface-exposed Hsp90 (105). In addition, GRP94 also binds HER2 on the surface of breast cancer cells, and regulates its cancer-promoting activity (106). Interestingly, cell surface GRP94 may interact with the CD91 receptor, like calreticulin, albeit with unclear functional significance (107, 108). Breast cancer tissue is characterized by the over-expression of GRP94 that may modulate the ability of tumor cells to migrate (109).
The oxidoreductase PDI is a central enzyme in the formation of disulfide bonds in secreted proteins (110). This protein also localizes in significant amounts to the cell surface of platelets, CHO, and pancreatic cells, as well as thyrocytes (62, 111–113). Here, it modulates surface-exposed thiols (113, 114) and cellular adhesion of immune cells via the association with integrins (115, 116). This mechanism also determines the ability of T helper cells to migrate through the extracellular matrix (117). PDI expression is tied to tumor vascularization that is often low and results in the activation of the hypoxia-dependent transcription factor HIF-1α (118). This transcription factor then promotes the upregulation of the oxidoreductases PDI and Ero1α (119–121). Subsequently, increased PDI and Ero1α expression also induces the production of vascular endothelial growth factor (VEGF), which, in turn, enables hypoxic tumors to improve angiogenesis (120, 121). Similar to GRP94, the levels of PDI and Ero1α have also been found to correlate with the invasiveness of glioma and the metastatic ability of soft tissue sarcoma, due to the role of PDI in mediating the interaction of cells with integrins (122, 123). Although Ero1α is secreted from hypoxic tumor cells, we currently do not known whether this occurs in vivo and what the function of surface or extracellular Ero1α is (67).
BiP/GRP78, an Inhibitor of Tumor Cell Apoptosis and Immunorecognition
Compared to PDI, more is known about the role of BiP/GRP78 for cancer cells, and specifically when found on the plasma membrane. BiP/GRP78 is over-expressed in many cancers, a hallmark that is associated with aggressive growth, invasive properties, and therapeutic resistance (124). This chaperone is a major regulator of ER protein folding and ER stress (125). By binding hydrophobic surfaces on newly synthesized polypeptides, BiP/GRP78 is first in line for ER protein folding, a role that becomes accentuated when misfolded polypeptides accumulate within the ER. Under that condition, also termed ER stress, BiP/GRP78 binds to unfolded proteins in its ATP-bound form, mediates their folding at the expense of ATP and is released when GDP is exchanged with GTP (126, 127). Folding is typically achieved through multiple rounds of binding and release of BiP/GRP78. Interestingly, when BiP/GRP78 acts as a chaperone, it dissociates from the ER transmembrane stress sensor proteins inositol requiring enzyme 1 (Ire1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) that are then able to trigger the unfolded protein response (UPR) (128). This intracellular signaling pathway activates the transcription of numerous ER chaperones and oxidoreductases to protect the cell from accumulated unfolded proteins, but also acts as an activator of apoptosis (129). Notably, BiP/GRP78 itself is a transcriptional target of the UPR via ER stress-responsive elements that can bind to ATF6 (130).
In cancer tissue, the UPR is frequently constitutively active, because solid tumors are poorly vascularized, leading to low oxygen delivery for mitochondria and low glucose delivery for glycolysis, both a cause of low ATP availability for ER protein folding (131). Consistent with this, BiP/GRP78 has been found over-expressed in prostate, head and neck, melanoma, breast, lung, brain, gastric, colon, and hepatocellular carcinomas (132). High levels of BiP/GRP78 act first of all as suppressors of apoptosis (133, 134), based on its role as a suppressor of the UPR (18), but also from its ability to sequester ER-associated pro-apoptotic Bcl2 family proteins such as Bik (135). Over-expression of BiP/GRP78 also inhibits pro-apoptotic Ca2+ transfer from the ER to mitochondria in astrocytes. This likely occurs due to the inhibitory action of BiP/GRP78 on the inositol-1,4,5 trisphosphate receptors (IP3Rs), major Ca2+ release channels of the ER (136, 137). As expected from these tumor-promoting roles of BiP/GRP78, high levels of this ER chaperone lead to poor prognosis in breast cancer (138).
As a side effect, the UPR not only leads to elevated expression of BiP/GRP78 in tumor tissue, but also leads to aberrant localization of this ER chaperone to the cytosol, mitochondria, and the plasma membrane (124). Cell surface BiP/GRP78 is apparently directly tied to its expression level that is under the control of the UPR, suggesting that high expression of this chaperone leads to saturation of the KDEL receptor retrieval mechanism (61). This phenomenon has been found in prostate, ovarian, and gastric cancer, as well as melanoma (21, 139–141). In some tumors, so much BiP/GRP78 escapes from the ER that secretion results, accompanied by the production of autoantibodies (142). These autoantibodies can promote or inhibit proliferation and apoptosis, but also interfere with phosphoinositide 3-kinase (PI3K), Akt, and MAP kinase pathways with the consequence of increased survival in several types of tumors (143, 144). This latter activity depends on the activating, physical interaction of cell surface BiP/GRP78 with PI3K that subsequently results in the activation of its downstream target Akt (145, 146). This activity of surface BiP/GRP78 may depend on α2-macroglobulin (α2M*), since the association between the two proteins triggers Akt phosphorylation in a PI-3 kinase-dependent manner (147, 148). In contrast, low levels of BiP/GRP78 tend to have opposite effects in mice and result in decreased activity of PI3K signaling in prostate and leukemia cancer models (149, 150).
Like calreticulin, BiP/GRP78 also influences the way cancer cells interact with the immune system. However, whereas calreticulin provides an “eat-me” signal, cell surface BiP/GRP78 protects insulinoma and fibrosarcoma cells from cytotoxic T lymphocytes (151, 152). In addition, BiP/GRP78 also interacts with MHC class I on the cell surface, although the functional significance of this observation is currently unclear (153).
Avenues of Interference with ER Chaperones in Cancer
Increased expression and cell surface appearance of ER chaperones and oxidoreductases have emerged as critical hallmarks of cancer cells and as consequences of low tumor vascularization that results in hypoxia. The observations outlined in our review suggest this insight may be used to develop new strategies to treat cancer (Figure 1) (154). In cancer, an approach under consideration consists in triggering the UPR (155). A number of compounds are currently in preclinical studies or Phase II/III trials and typically attempt to prevent the pro-survival readout of the UPR. This approach led to marked decrease of cancer growth in a multiple myeloma xenograft model (156). A promising strategy appears to be the combination of such drugs with bortezomib, a blocker of the proteasome and inducer of ER stress (157). With this combination of drugs, stress-inducing bortezomib primes cancer cells for death that becomes inevitable, once an inhibitor of protective UPR responses is added to the mix. A similar approach aims to target the redox-modulatory role of ER chaperones and oxidoreductases using the PDI inhibitor bacitracin, which acts as a potent booster of the chemotherapeutics fenretinide and bortezomib (158). Due to the toxicity of bacitracin, novel PDI inhibitors are currently under development (159). Another way how ER redox modulation can be used as an adjuvant for cancer chemotherapy is by interfering with the redox-sensitive activity of SERCA to allow for increased cell stress in tumor tissue due to reduced ER Ca2+ content (160, 161). Given the tumor-promoting (e.g., BiP/GRP78, PDI) and tumor-opposing (e.g., calreticulin) activities of ER chaperones and oxidoreductases, current knowledge suggests more pinpointed approaches are needed to increase efficacy of ER-targeted cancer chemotherapeutic strategies.
Interestingly, the SERCA inhibitor thapsigargin and the non-steroidal anti-inflammatory drug celecoxib both make use of the interference with ER Ca2+ content as a weapon against tumor cells (Figure 1) (162–164). Under this condition, calreticulin, PDI, BiP/GRP78, and GRP94 escape ER retention and retrieval (69, 165). This effect is similar to an indiscriminate activation of the UPR, which turns on ER chaperone and oxidoreductase production, and leads to the saturation of KDEL as well as di-lysine-based retrieval to the ER (166, 167). Similar to blanket interference with the UPR, this approach is bound to have tumor-promoting and tumor-opposing effects: while calreticulin and GRP94 will appear as immune system targets on the cell surface of tumor cells, PDI and BiP/GRP78 will have tumor-promoting effects as promoters of cell mobility and blockers of apoptosis.
Ideally, an efficient therapy would aim to generate cell surface calreticulin to serve as an efficient “eat-me” signal on tumor cells, while down-regulating or inactivating BiP/GRP78 on the plasma membrane, which acts as tumor-promoting. To achieve such a goal, it would be helpful to understand the molecular machinery of calreticulin plasma membrane exposure, currently known to require the triggering of PERK, the cleavage of caspase-8, and the functioning of SNARE proteins (168). Mitoxantrone, an anthracycline that robustly influences these mechanisms and leads to calreticulin surface exposure, is currently in clinical trials against lymphoma (Figure 1) (169).
In contrast to calreticulin, the requirements for BiP/GRP78 cell surface exposure are less understood. As a sole factor, the transmembrane protein MTJ-1 has been identified as critical for BiP/GRP78 surface translocation (170), possibly via the catalysis of ATP exchange through its J domain (171). Interestingly, photo-dynamic therapy (PDT) using the ER-localized photosensitizer hypericin may be such a magic bullet: not only does it result in the reduction of SERCA activity (172), but this treatment specifically results in the surface targeting of only calreticulin, and not BiP/GRP78 (84). Promisingly, this treatment causes tumor regression in BALB/c mice inoculated with colon carcinoma (173).
Conversely, inhibitory agents against BiP/GRP78 could create tumor cell specificity of UPR-targeted anti-cancer strategies (Figure 1). Approaches include the selective destruction of BiP/GRP78 on the surface with the bacterial toxin SubAb that cleaves and inactivates this chaperone (174). This strategy delayed the growth of multiple cancer xenografts in mice (175, 176). Cancer cells also respond well to the inhibition of the BiP/GRP78 ATPase activity with epigallocatechin gallate, honokiol, and salicylate (177–179). It is currently unknown whether these effects stem from inhibiting the activity of MTJ-1 that is needed to transport BiP/GRP78 to the plasma membrane (170). Xenograft growth of tumors is also inhibited in the presence of BiP/GRP78-binding peptides that obstruct the chaperone’s folding pocket (180). Importantly, these peptides bind specifically to tumor cells and abrogate their growth in vivo (181, 182). Such a strategy might be particularly important following the surgical removal of tumor tissue or in combination with chemotherapeutic approaches (183, 184).
Conclusion
Endoplasmic reticulum chaperones and oxidoreductases have emerged as unlikely regulators of tumor growth. While neither being directly connected to the regulation of cell division and growth, nor the triggering of apoptosis, they instead frequently acquire new functions unrelated to their classic ER roles in a cancer setting. These new roles coincide with their relocation from the ER to the plasma membrane. In most cases, this occurs because the UPR triggers the production of more chaperones and oxidoreductases that eventually saturate the KDEL retrieval machinery. Once at the plasma membrane, ER chaperones and oxidoreductases serve as DAMPs for the immune system (calreticulin, GRP94) or modulators of tumor hallmarks (BiP/GRP78, PDI). The exploitation of this group of proteins as cancer therapeutic targets will require a detailed understanding of their intracellular and extracellular roles. Our current knowledge has identified chaperones that serve as DAMPs, whereas modulators of tumor hallmarks including cell death and metabolism are typically tumor-promoting. Specific triggers and inhibitors of the functions of ER chaperones and oxidoreductases will help direct cancer therapeutic approaches in the right direction. This insight warrants further investigation on this class of proteins.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in the Simmen lab is supported by the Cancer Research Society (18325) and CIHR (MOP 133541).
References
1. Kanapin A, Batalov S, Davis MJ, Gough J, Grimmond S, Kawaji H, et al. Mouse proteome analysis. Genome Res (2003) 13(6B):1335–44. doi: 10.1101/gr.978703
2. Song W, Raden D, Mandon EC, Gilmore R. Role of Sec61alpha in the regulated transfer of the ribosome-nascent chain complex from the signal recognition particle to the translocation channel. Cell (2000) 100(3):333–43. doi:10.1016/S0092-8674(00)80669-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Conti BJ, Elferich J, Yang Z, Shinde U, Skach WR. Cotranslational folding inhibits translocation from within the ribosome-Sec61 translocon complex. Nat Struct Mol Biol (2014) 21(3):228–35. doi:10.1038/nsmb.2779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Skach WR. Cellular mechanisms of membrane protein folding. Nat Struct Mol Biol (2009) 16(6):606–12. doi:10.1038/nsmb.1600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Hammond C, Helenius A. Folding of VSV G protein: sequential interaction with BiP and calnexin. Science (1994) 266(5184):456–8. doi:10.1126/science.7939687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Wang N, Daniels R, Hebert DN. The cotranslational maturation of the type I membrane glycoprotein tyrosinase: the heat shock protein 70 system hands off to the lectin-based chaperone system. Mol Biol Cell (2005) 16(8):3740–52. doi:10.1091/mbc.E05-05-0381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, et al. Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell (2004) 13(1):125–35. doi:10.1016/S1097-2765(03)00494-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Daniels R, Kurowski B, Johnson AE, Hebert DN. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell (2003) 11(1):79–90. doi:10.1016/S1097-2765(02)00821-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Bulleid NJ, Ellgaard L. Multiple ways to make disulfides. Trends Biochem Sci (2011) 36(9):485–92. doi:10.1016/j.tibs.2011.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Hirschberg K, Lippincott-Schwartz J. Secretory pathway kinetics and in vivo analysis of protein traffic from the Golgi complex to the cell surface. FASEB J (1999) 13(Suppl 2):S251–6.
11. Lippincott-Schwartz J, Roberts TH, Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol (2000) 16:557–89. doi:10.1146/annurev.cellbio.16.1.557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Wieland FT, Gleason ML, Serafini TA, Rothman JE. The rate of bulk flow from the endoplasmic reticulum to the cell surface. Cell (1987) 50(2):289–300. doi:10.1016/0092-8674(87)90224-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Nishimura N, Bannykh S, Slabough S, Matteson J, Altschuler Y, Hahn K, et al. A di-acidic (DXE) code directs concentration of cargo during export from the endoplasmic reticulum. J Biol Chem (1999) 274(22):15937–46. doi:10.1074/jbc.274.22.15937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Sevier CS, Weisz OA, Davis M, Machamer CE. Efficient export of the vesicular stomatitis virus G protein from the endoplasmic reticulum requires a signal in the cytoplasmic tail that includes both tyrosine-based and di-acidic motifs. Mol Biol Cell (2000) 11(1):13–22. doi:10.1091/mbc.11.1.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Fossati M, Colombo SF, Borgese N. A positive signal prevents secretory membrane cargo from recycling between the Golgi and the ER. EMBO J (2014) 33(18):2080–97. doi:10.15252/embj.201488367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Lewis MJ, Pelham HR. A human homologue of the yeast HDEL receptor. Nature (1990) 348(6297):162–3. doi:10.1038/348162a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Semenza JC, Hardwick KG, Dean N, Pelham HR. ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell (1990) 61(7):1349–57. doi:10.1016/0092-8674(90)90698-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene (2013) 32(7):805–18. doi:10.1038/onc.2012.130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi:10.1038/nm1523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Uckun FM, Qazi S, Ozer Z, Garner AL, Pitt J, Ma H, et al. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br J Haematol (2011) 153(6):741–52. doi:10.1111/j.1365-2141.2011.08671.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Rauschert N, Brändlein S, Holzinger E, Hensel F, Müller-Hermelink HK, Vollmers HP. A new tumor-specific variant of GRP78 as target for antibody-based therapy. Lab Invest (2008) 88(4):375–86. doi:10.1038/labinvest.2008.2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Spang A. Retrograde traffic from the Golgi to the endoplasmic reticulum. Cold Spring Harb Perspect Biol (2013) 5(6):a013391. doi:10.1101/cshperspect.a013391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem (2010) 79:777–802. doi:10.1146/annurev-biochem-061608-091319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Jackson LP, Lewis M, Kent HM, Edeling MA, Evans PR, Duden R, et al. Molecular basis for recognition of dilysine trafficking motifs by COPI. Dev Cell (2012) 23(6):1255–62. doi:10.1016/j.devcel.2012.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Itin C, Schindler R, Hauri HP. Targeting of protein ERGIC-53 to the ER/ERGIC/cis-Golgi recycling pathway. J Cell Biol (1995) 131(1):57–67. doi:10.1083/jcb.131.1.57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science (1994) 263(5153):1629–31. doi:10.1126/science.8128252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Jackson LP, Kümmel D, Reinisch KM, Owen DJ. Structures and mechanisms of vesicle coat components and multisubunit tethering complexes. Curr Opin Cell Biol (2012) 24(4):475–83. doi:10.1016/j.ceb.2012.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Bremser M, Nickel W, Schweikert M, Ravazzola M, Amherdt M, Hughes CA, et al. Coupling of coat assembly and vesicle budding to packaging of putative cargo receptors. Cell (1999) 96(4):495–506. doi:10.1016/S0092-8674(00)80654-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Ma W, Goldberg J. Rules for the recognition of dilysine retrieval motifs by coatomer. EMBO J (2013) 32(7):926–37. doi:10.1038/emboj.2013.41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem (1994) 269(2):1437–48.
31. Cukierman E, Huber I, Rotman M, Cassel D. The ARF1 GTPase-activating protein: zinc finger motif and Golgi complex localization. Science (1995) 270(5244):1999–2002. doi:10.1126/science.270.5244.1999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hirose H, Arasaki K, Dohmae N, Takio K, Hatsuzawa K, Nagahama M, et al. Implication of ZW10 in membrane trafficking between the endoplasmic reticulum and Golgi. EMBO J (2004) 23(6):1267–78. doi:10.1038/sj.emboj.7600135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Zink S, Wenzel D, Wurm CA, Schmitt HD. A link between ER tethering and COP-I vesicle uncoating. Dev Cell (2009) 17(3):403–16. doi:10.1016/j.devcel.2009.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Lerich A, Hillmer S, Langhans M, Scheuring D, van Bentum P, Robinson DG. ER import sites and their relationship to ER exit sites: a new model for bidirectional ER-Golgi transport in higher plants. Front Plant Sci (2012) 3:143. doi:10.3389/fpls.2012.00143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Sandoval CO, Simmen T. Rab proteins of the endoplasmic reticulum: functions and interactors. Biochem Soc Trans (2012) 40(6):1426–32. doi:10.1042/BST20120158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Tisdale EJ, Balch WE. Rab2 is essential for the maturation of pre-Golgi intermediates. J Biol Chem (1996) 271(46):29372–9. doi:10.1074/jbc.271.46.29372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Del Nery E, Miserey-Lenkei S, Falguières T, Nizak C, Johannes L, Perez F, et al. Rab6A and Rab6A’ GTPases play non-overlapping roles in membrane trafficking. Traffic (2006) 7(4):394–407. doi:10.1111/j.1600-0854.2006.00395.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Girod A, Storrie B, Simpson JC, Johannes L, Goud B, Roberts LM, et al. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat Cell Biol (1999) 1(7):423–30. doi:10.1038/15658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Dejgaard SY, Murshid A, Erman A, Kizilay O, Verbich D, Lodge R, et al. Rab18 and Rab43 have key roles in ER-Golgi trafficking. J Cell Sci (2008) 121(Pt 16):2768–81. doi:10.1242/jcs.021808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Spang A. The DSL1 complex: the smallest but not the least CATCHR. Traffic (2012) 13(7):908–13. doi:10.1111/j.1600-0854.2012.01362.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Gao C, Cai Y, Wang Y, Kang BH, Aniento F, Robinson DG, et al. Retention mechanisms for ER and Golgi membrane proteins. Trends Plant Sci (2014) 19(8):508–15. doi:10.1016/j.tplants.2014.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, et al. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell (2008) 19(7):2777–88. doi:10.1091/mbc.E07-10-0995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Schutze MP, Peterson PA, Jackson MR. An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J (1994) 13(7):1696–705.
44. Ronchi P, Colombo S, Francolini M, Borgese N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J Cell Biol (2008) 181(1):105–18. doi:10.1083/jcb.200710093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Watson HR, Butler J, Schuppe HJ, Lee AG, East JM. The localization of the ER retrieval sequence for the calcium pump SERCA1. Mol Membr Biol (2011) 28(4):216–26. doi:10.3109/09687688.2011.572566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Park HJ, Shabashvili D, Nekorchuk MD, Shyqyriu E, Jung JI, Ladd TB, et al. Retention in endoplasmic reticulum 1 (RER1) modulates amyloid-beta (Abeta) production by altering trafficking of gamma-secretase and amyloid precursor protein (APP). J Biol Chem (2012) 287(48):40629–40. doi:10.1074/jbc.M112.418442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Sato K, Nishikawa S, Nakano A. Membrane protein retrieval from the Golgi apparatus to the endoplasmic reticulum (ER): characterization of the RER1 gene product as a component involved in ER localization of Sec12p. Mol Biol Cell (1995) 6(11):1459–77. doi:10.1091/mbc.6.11.1459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Yamasaki A, Hara T, Maejima I, Sato M, Sato K, Sato K. Rer1p regulates the ER retention of immature rhodopsin and modulates its intracellular trafficking. Sci Rep (2014) 4:5973. doi:10.1038/srep05973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Benyair R, Ron E, Lederkremer GZ. Protein quality control, retention, and degradation at the endoplasmic reticulum. Int Rev Cell Mol Biol (2011) 292:197–280. doi:10.1016/B978-0-12-386033-0.00005-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Ramming T, Appenzeller-Herzog C. The physiological functions of mammalian endoplasmic oxidoreductin 1: on disulfides and more. Antioxid Redox Signal (2012) 16(10):1109–18. doi:10.1089/ars.2011.4475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Anelli T, Alessio M, Bachi A, Bergamelli L, Bertoli G, Camerini S, et al. Thiol-mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J (2003) 22(19):5015–22. doi:10.1093/emboj/cdg491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Otsu M, Bertoli G, Fagioli C, Guerini-Rocco E, Nerini-Molteni S, Ruffato E, et al. Dynamic retention of Ero1alpha and Ero1beta in the endoplasmic reticulum by interactions with PDI and ERp44. Antioxid Redox Signal (2006) 8(3–4):274–82. doi:10.1089/ars.2006.8.274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Kakihana T, Araki K, Vavassori S, Iemura S, Cortini M, Fagioli C, et al. Dynamic regulation of Ero1alpha and Prx4 localization in the secretory pathway. J Biol Chem (2013) 288(41):29586–94. doi:10.1074/jbc.M113.467845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Sönnichsen B, Füllekrug J, Nguyen Van P, Diekmann W, Robinson DG, Mieskes G. Retention and retrieval: both mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J Cell Sci (1994) 107(Pt 10):2705–17.
55. Peters LR, Raghavan M. Endoplasmic reticulum calcium depletion impacts chaperone secretion, innate immunity, and phagocytic uptake of cells. J Immunol (2011) 187(2):919–31. doi:10.4049/jimmunol.1100690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Ikura M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem Sci (1996) 21(1):14–7. doi:10.1016/0968-0004(96)80879-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Corbett EF, Michalak KM, Oikawa K, Johnson S, Campbell ID, Eggleton P, et al. The conformation of calreticulin is influenced by the endoplasmic reticulum luminal environment. J Biol Chem (2000) 275(35):27177–85.
58. Corbett EF, Michalak M. Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem Sci (2000) 25(7):307–11. doi:10.1016/S0968-0004(00)01588-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Crofts AJ, Leborgne-Castel N, Hillmer S, Robinson DG, Phillipson B, Carlsson LE, et al. Saturation of the endoplasmic reticulum retention machinery reveals anterograde bulk flow. Plant Cell (1999) 11(11):2233–48. doi:10.2307/3871021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Drummond IA, Lee AS, Resendez E Jr, Steinhardt RA. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J Biol Chem (1987) 262(26):12801–5.
61. Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem (2010) 285(20):15065–75. doi:10.1074/jbc.M109.087445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Mezghrani A, Courageot J, Mani JC, Pugniere M, Bastiani P, Miquelis R. Protein-disulfide isomerase (PDI) in FRTL5 cells. pH-dependent thyroglobulin/PDI interactions determine a novel PDI function in the post-endoplasmic reticulum of thyrocytes. J Biol Chem (2000) 275(3):1920–9. doi:10.1074/jbc.275.3.1920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Wiest DL, Bhandoola A, Punt J, Kreibich G, McKean D, Singer A. Incomplete endoplasmic reticulum (ER) retention in immature thymocytes as revealed by surface expression of “ER-resident” molecular chaperones. Proc Natl Acad Sci U S A (1997) 94(5):1884–9. doi:10.1073/pnas.94.5.1884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Raghavan M, Wijeyesakere SJ, Peters LR, Del Cid N. Calreticulin in the immune system: ins and outs. Trends Immunol (2013) 34(1):13–21. doi:10.1016/j.it.2012.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Li XC, Raghavan M. Structure and function of major histocompatibility complex class I antigens. Curr Opin Organ Transplant (2010) 15(4):499–504. doi:10.1097/MOT.0b013e32833bfb33
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Ulianich L, Terrazzano G, Annunziatella M, Ruggiero G, Beguinot F, Di Jeso B. ER stress impairs MHC class I surface expression and increases susceptibility of thyroid cells to NK-mediated cytotoxicity. Biochim Biophys Acta (2011) 1812(4):431–8. doi:10.1016/j.bbadis.2010.12.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, et al. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones (2010) 15(5):619–29. doi:10.1007/s12192-010-0174-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Opas M, Dziak E, Fliegel L, Michalak M. Regulation of expression and intracellular distribution of calreticulin, a major calcium binding protein of nonmuscle cells. J Cell Physiol (1991) 149(1):160–71. doi:10.1002/jcp.1041490120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A (1990) 87(7):2466–70. doi:10.1073/pnas.87.7.2466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis (2012) 3:e261. doi:10.1038/cddis.2011.132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol (2008) 3:99–126. doi:10.1146/annurev.pathmechdis.3.121806.151456
72. Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol (2007) 28(10):429–36. doi:10.1016/j.it.2007.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ (2014) 21(1):26–38. doi:10.1038/cdd.2013.48
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Escamilla-Tilch M, Filio-Rodríguez G, García-Rocha R, Mancilla-Herrera I, Mitchison NA, Ruiz-Pacheco JA, et al. The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol Cell Biol (2013) 91(10):601–10. doi:10.1038/icb.2013.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity (2001) 14(3):303–13. doi:10.1016/S1074-7613(01)00111-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123(2):321–34. doi:10.1016/j.cell.2005.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Kepp O, Menger L, Vacchelli E, Locher C, Adjemian S, Yamazaki T, et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev (2013) 24(4):311–8. doi:10.1016/j.cytogfr.2013.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Yamamura Y, Tsuchikawa T, Miyauchi K, Takeuchi S, Wada M, Kuwatani T, et al. The key role of calreticulin in immunomodulation induced by chemotherapeutic agents. Int J Clin Oncol (2014). doi:10.1007/s10147-014-0719-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Jeannin P, Jaillon S, Delneste Y. Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol (2008) 20(5):530–7. doi:10.1016/j.coi.2008.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194(6):781–95. doi:10.1084/jem.194.6.781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Wu H, Han Y, Qin Y, Cao C, Xia Y, Liu C, et al. Whole-cell vaccine coated with recombinant calreticulin enhances activation of dendritic cells and induces tumour-specific immune responses. Oncol Rep (2013) 29(2):529–34. doi:10.3892/or.2012.2142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Garg AD, Dudek AM, Agostinis P. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology (2013) 2(10):e26260. doi:10.4161/onci.26260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Garg AD, Dudek AM, Agostinis P. Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis (2013) 4:e826. doi:10.1038/cddis.2013.372
84. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J (2012) 31(5):1062–79. doi:10.1038/emboj.2011.497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334(6062):1573–7. doi:10.1126/science.1208347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med (2010) 2(63):63ra94. doi:10.1126/scitranslmed.3001375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Sheng W, Chen C, Dong M, Zhou J, Liu Q, Dong Q, et al. Overexpression of calreticulin contributes to the development and progression of pancreatic cancer. J Cell Physiol (2014) 229(7):887–97. doi:10.1002/jcp.24519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Kabbage M, Trimeche M, Bergaoui S, Hammann P, Kuhn L, Hamrita B, et al. Calreticulin expression in infiltrating ductal breast carcinomas: relationships with disease progression and humoral immune responses. Tumour Biol (2013) 34(2):1177–88. doi:10.1007/s13277-013-0661-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Pekáriková A, Sánchez D, Palová-Jelínková L, Simsová M, Benes Z, Hoffmanová I, et al. Calreticulin is a B cell molecular target in some gastrointestinal malignancies. Clin Exp Immunol (2010) 160(2):215–22. doi:10.1111/j.1365-2249.2009.04085.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Minami S, Nagashio R, Ueda J, Matsumoto K, Goshima N, Hattori M, et al. Detection of tumor-associated antigens in culture supernatants using autoantibodies in sera from patients with bladder cancer. Biomed Res (2014) 35(1):25–35. doi:10.2220/biomedres.35.25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med (1998) 188(12):2349–56. doi:10.1084/jem.188.12.2349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Pike SE, Yao L, Setsuda J, Jones KD, Cherney B, Appella E, et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood (1999) 94(7):2461–8.
93. Wearsch PA, Cresswell P. The quality control of MHC class I peptide loading. Curr Opin Cell Biol (2008) 20(6):624–31. doi:10.1016/j.ceb.2008.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Obeid M. ERP57 membrane translocation dictates the immunogenicity of tumor cell death by controlling the membrane translocation of calreticulin. J Immunol (2008) 181(4):2533–43. doi:10.4049/jimmunol.181.4.2533
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ (2008) 15(9):1499–509. doi:10.1038/cdd.2008.67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Cathro HP, Smolkin ME, Theodorescu D, Jo VY, Ferrone S, Frierson HF Jr. Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol Immunother (2010) 59(3):465–72. doi:10.1007/s00262-009-0765-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Leys CM, Nomura S, LaFleur BJ, Ferrone S, Kaminishi M, Montgomery E, et al. Expression and prognostic significance of prothymosin-alpha and ERp57 in human gastric cancer. Surgery (2007) 141(1):41–50. doi:10.1016/j.surg.2006.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Marzec M, Eletto D, Argon Y. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta (2012) 1823(3):774–87. doi:10.1016/j.bbamcr.2011.10.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. McGettrick AF, O’Neill LA. Localisation and trafficking of toll-like receptors: an important mode of regulation. Curr Opin Immunol (2010) 22(1):20–7. doi:10.1016/j.coi.2009.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Peter F, Nguyen Van P, Soling HD. Different sorting of Lys-Asp-Glu-Leu proteins in rat liver. J Biol Chem (1992) 267(15):10631–7.
101. Takemoto H, Yoshimori T, Yamamoto A, Miyata Y, Yahara I, Inoue K, et al. Heavy chain binding protein (BiP/GRP78) and endoplasmin are exported from the endoplasmic reticulum in rat exocrine pancreatic cells, similar to protein disulfide-isomerase. Arch Biochem Biophys (1992) 296(1):129–36. doi:10.1016/0003-9861(92)90554-A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Altmeyer A, Maki RG, Feldweg AM, Heike M, Protopopov VP, Masur SK, et al. Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int J Cancer (1996) 69(4):340–9. doi:10.1002/(SICI)1097-0215(19960822)69:4<340::AID-IJC18>3.0.CO;2-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Frasson M, Vitadello M, Brunati AM, La Rocca N, Tibaldi E, Pinna LA, et al. Grp94 is tyr-phosphorylated by Fyn in the lumen of the endoplasmic reticulum and translocates to Golgi in differentiating myoblasts. Biochim Biophys Acta (2009) 1793(2):239–52. doi:10.1016/j.bbamcr.2008.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Robert J, Menoret A, Cohen N. Cell surface expression of the endoplasmic reticular heat shock protein gp96 is phylogenetically conserved. J Immunol (1999) 163(8):4133–9.
105. Suzuki S, Kulkarni AB. Extracellular heat shock protein HSP90beta secreted by MG63 osteosarcoma cells inhibits activation of latent TGF-beta1. Biochem Biophys Res Commun (2010) 398(3):525–31. doi:10.1016/j.bbrc.2010.06.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol (2013) 9(11):677–84. doi:10.1038/nchembio.1335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol (2000) 1(2):151–5. doi:10.1038/77835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Jockheck-Clark AR, Bowers EV, Totonchy MB, Neubauer J, Pizzo SV, Nicchitta CV. Re-examination of CD91 function in GRP94 (glycoprotein 96) surface binding, uptake, and peptide cross-presentation. J Immunol (2010) 185(11):6819–30. doi:10.4049/jimmunol.1000448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Dejeans N, Glorieux C, Guenin S, Beck R, Sid B, Rousseau R, et al. Overexpression of GRP94 in breast cancer cells resistant to oxidative stress promotes high levels of cancer cell proliferation and migration: implications for tumor recurrence. Free Radic Biol Med (2012) 52(6):993–1002. doi:10.1016/j.freeradbiomed.2011.12.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta (2008) 1783(4):535–48. doi:10.1016/j.bbamcr.2007.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Akagi S, Yamamoto A, Yoshimori T, Masaki R, Ogawa R, Tashiro Y. Localization of protein disulfide isomerase on plasma membranes of rat exocrine pancreatic cells. J Histochem Cytochem (1988) 36(8):1069–74. doi:10.1177/36.8.3292644
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Essex DW, Chen K, Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood (1995) 86(6):2168–73.
113. Mandel R, Ryser HJ, Ghani F, Wu M, Peak D. Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide-isomerase. Proc Natl Acad Sci U S A (1993) 90(9):4112–6. doi:10.1073/pnas.90.9.4112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Täger M, Kröning H, Thiel U, Ansorge S. Membrane-bound proteindisulfide isomerase (PDI) is involved in regulation of surface expression of thiols and drug sensitivity of B-CLL cells. Exp Hematol (1997) 25(7):601–7.
115. Bennett TA, Edwards BS, Sklar LA, Rogelj S. Sulfhydryl regulation of L-selectin shedding: phenylarsine oxide promotes activation-independent L-selectin shedding from leukocytes. J Immunol (2000) 164(8):4120–9. doi:10.4049/jimmunol.164.8.4120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Lahav J, Jurk K, Hess O, Barnes MJ, Farndale RW, Luboshitz J, et al. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood (2002) 100(7):2472–8. doi:10.1182/blood-2001-12-0339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Bi S, Hong PW, Lee B, Baum LG. Galectin-9 binding to cell surface protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry. Proc Natl Acad Sci U S A (2011) 108(26):10650–5. doi:10.1073/pnas.1017954108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Liu Q, Berchner-Pfannschmidt U, Möller U, Brecht M, Wotzlaw C, Acker H, et al. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc Natl Acad Sci U S A (2004) 101(12):4302–7. doi:10.1073/pnas.0400265101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Tanaka S, Uehara T, Nomura Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J Biol Chem (2000) 275(14):10388–93. doi:10.1074/jbc.275.14.10388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. May D, Itin A, Gal O, Kalinski H, Feinstein E, Keshet E. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene (2005) 24(6):1011–20. doi:10.1038/sj.onc.1208325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Gess B, Hofbauer KH, Wenger RH, Lohaus C, Meyer HE, Kurtz A. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J Biochem (2003) 270(10):2228–35. doi:10.1046/j.1432-1033.2003.03590.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Goplen D, Wang J, Enger PØ, Tysnes BB, Terzis AJ, Laerum OD, et al. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res (2006) 66(20):9895–902. doi:10.1158/0008-5472.CAN-05-4589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Francis P, Namløs HM, Müller C, Edén P, Fernebro J, Berner JM, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics (2007) 8:73. doi:10.1186/1471-2164-8-73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer (2014) 14(4):263–76. doi:10.1038/nrc3701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Gidalevitz T, Stevens F, Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta (2013) 1833(11):2410–24. doi:10.1016/j.bbamcr.2013.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, et al. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell (1993) 75(4):717–28. doi:10.1016/0092-8674(93)90492-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Blond-Elguindi S, Fourie AM, Sambrook JF, Gething MJ. Peptide-dependent stimulation of the ATPase activity of the molecular chaperone BiP is the result of conversion of oligomers to active monomers. J Biol Chem (1993) 268(17):12730–5.
128. Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol (2013) 5(3):a013169. doi:10.1101/cshperspect.a013169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13(2):89–102. doi:10.1038/nrm3270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Baumeister P, Luo S, Skarnes WC, Sui G, Seto E, Shi Y, et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol (2005) 25(11):4529–40. doi:10.1128/MCB.25.11.4529-4540.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer (2008) 8(11):851–64. doi:10.1038/nrc2501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Li Z. Glucose regulated protein 78: a critical link between tumor microenvironment and cancer hallmarks. Biochim Biophys Acta (2012) 1826(1):13–22. doi:10.1016/j.bbcan.2012.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, et al. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett (2002) 514(2–3):122–8. doi:10.1016/S0014-5793(02)02289-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem (2003) 278(23):20915–24. doi:10.1074/jbc.M212328200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Zhou H, Zhang Y, Fu Y, Chan L, Lee AS. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK). J Biol Chem (2011) 286(29):25687–96. doi:10.1074/jbc.M110.212944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell (2007) 131(3):596–610. doi:10.1016/j.cell.2007.08.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Ouyang YB, Xu LJ, Emery JF, Lee AS, Giffard RG. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion (2011) 11(2):279–86. doi:10.1016/j.mito.2010.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Zheng YZ, Cao ZG, Hu X, Shao ZM. The endoplasmic reticulum stress markers GRP78 and CHOP predict disease-free survival and responsiveness to chemotherapy in breast cancer. Breast Cancer Res Treat (2014) 145(2):349–58. doi:10.1007/s10549-014-2967-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Chinni SR, Falchetto R, Gercel-Taylor C, Shabanowitz J, Hunt DF, Taylor DD. Humoral immune responses to cathepsin D and glucose-regulated protein 78 in ovarian cancer patients. Clin Cancer Res (1997) 3(9):1557–64.
140. Gonzalez-Gronow M, Cuchacovich M, Llanos C, Urzua C, Gawdi G, Pizzo SV. Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res (2006) 66(23):11424–31. doi:10.1158/0008-5472.CAN-06-1721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Papalas JA, Vollmer RT, Gonzalez-Gronow M, Pizzo SV, Burchette J, Youens KE, et al. Patterns of GRP78 and MTJ1 expression in primary cutaneous malignant melanoma. Mod Pathol (2010) 23(1):134–43. doi:10.1038/modpathol.2009.152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Misra UK, Pizzo SV. Modulation of the unfolded protein response in prostate cancer cells by antibody-directed against the carboxyl-terminal domain of GRP78. Apoptosis (2010) 15(2):173–82. doi:10.1007/s10495-009-0430-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Shu CW, Sun FC, Cho JH, Lin CC, Liu PF, Chen PY, et al. GRP78 and Raf-1 cooperatively confer resistance to endoplasmic reticulum stress-induced apoptosis. J Cell Physiol (2008) 215(3):627–35. doi:10.1002/jcp.21340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, et al. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood (2009) 114(18):3960–7. doi:10.1182/blood-2009-03-209668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, Lee AS. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS One (2013) 8(11):e80071. doi:10.1371/journal.pone.0080071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin Cancer Res (2013) 19(24):6802–11. doi:10.1158/1078-0432.CCR-13-1106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Misra UK, Mowery Y, Kaczowka S, Pizzo SV. Ligation of cancer cell surface GRP78 with antibodies directed against its COOH-terminal domain up-regulates p53 activity and promotes apoptosis. Mol Cancer Ther (2009) 8(5):1350–62. doi:10.1158/1535-7163.MCT-08-0990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem (2006) 281(19):13694–707. doi:10.1074/jbc.M511694200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Wey S, Luo B, Tseng CC, Ni M, Zhou H, Fu Y, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood (2012) 119(3):817–25. doi:10.1182/blood-2011-06-357384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A (2008) 105(49):19444–9. doi:10.1073/pnas.0807691105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Jamora C, Dennert G, Lee AS. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci U S A (1996) 93(15):7690–4. doi:10.1073/pnas.93.15.7690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Wang M, Wang P, Liu YQ, Peng JL, Zhao XP, Wu S, et al. The immunosuppressive and protective ability of glucose-regulated protein 78 for improvement of alloimmunity in beta cell transplantation. Clin Exp Immunol (2007) 150(3):546–52. doi:10.1111/j.1365-2249.2007.03525.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Triantafilou M, Fradelizi D, Triantafilou K. Major histocompatibility class one molecule associates with glucose regulated protein (GRP) 78 on the cell surface. Hum Immunol (2001) 62(8):764–70. doi:10.1016/S0198-8859(01)00269-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. McLaughlin M, Vandenbroeck K. The endoplasmic reticulum protein folding factory and its chaperones: new targets for drug discovery? Br J Pharmacol (2011) 162(2):328–45. doi:10.1111/j.1476-5381.2010.01064.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov (2013) 12(9):703–19. doi:10.1038/nrd3976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann N Y Acad Sci (2012) 1271:20–32. doi:10.1111/j.1749-6632.2012.06739.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood (2012) 119(24):5772–81. doi:10.1182/blood-2011-07-366633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Lovat PE, Corazzari M, Armstrong JL, Martin S, Pagliarini V, Hill D, et al. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res (2008) 68(13):5363–9. doi:10.1158/0008-5472.CAN-08-0035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today (2014) 19(3):222–40. doi:10.1016/j.drudis.2013.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther (2005) 4(1):14–22. doi:10.4161/cbt.4.1.1505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med (2012) 4(140):140ra86. doi:10.1126/scitranslmed.3003886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Doan NT, Paulsen ES, Sehgal P, Møller JV, Nissen P, Denmeade SR, et al. Targeting thapsigargin towards tumors. Steroids (2014). doi:10.1016/j.steroids.2014.07.009
163. Pyrko P, Kardosh A, Liu YT, Soriano N, Xiong W, Chow RH, et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Mol Cancer Ther (2007) 6(4):1262–75. doi:10.1158/1535-7163.MCT-06-0629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Wang JL, Lin KL, Chou CT, Kuo CC, Cheng JS, Hsu SS, et al. Effect of celecoxib on Ca(2+) handling and viability in human prostate cancer cells (PC3). Drug Chem Toxicol (2012) 35(4):456–62. doi:10.3109/01480545.2011.638927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Tufi R, Panaretakis T, Bianchi K, Criollo A, Fazi B, Di Sano F, et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ (2008) 15(2):274–82. doi:10.1038/sj.cdd.4402275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Kappeler F, Itin C, Schindler R, Hauri HP. A dual role for COOH-terminal lysine residues in pre-Golgi retention and endocytosis of ERGIC-53. J Biol Chem (1994) 269(9):6279–81.
167. Liaudet E, Garcia M, Rochefort H. Cathepsin D maturation and its stimulatory effect on metastasis are prevented by addition of KDEL retention signal. Oncogene (1994) 9(4):1145–54.
168. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J (2009) 28(5):578–90. doi:10.1038/emboj.2009.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Vacchelli E, Galluzzi L, Fridman WH, Aranda F, Eggermont A, Galon J, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology (2012) 1(2):179–88. doi:10.4161/onci.1.2.19026
170. Misra UK, Gonzalez-Gronow M, Gawdi G, Pizzo SV. The role of MTJ-1 in cell surface translocation of GRP78, a receptor for alpha 2-macroglobulin-dependent signaling. J Immunol (2005) 174(4):2092–7. doi:10.4049/jimmunol.174.4.2092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Chevalier M, Rhee H, Elguindi EC, Blond SY. Interaction of murine BiP/GRP78 with the DnaJ homologue MTJ1. J Biol Chem (2000) 275(26):19620–7. doi:10.1074/jbc.M001333200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J (2006) 20(6):756–8.
173. Sanovic R, Verwanger T, Hartl A, Krammer B. Low dose hypericin-PDT induces complete tumor regression in BALB/c mice bearing CT26 colon carcinoma. Photodiagnosis Photodyn Ther (2011) 8(4):291–6. doi:10.1016/j.pdpdt.2011.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature (2006) 443(7111):548–52. doi:10.1038/nature05124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Backer JM, Krivoshein AV, Hamby CV, Pizzonia J, Gilbert KS, Ray YS, et al. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia (2009) 11(11):1165–73.
176. Prabhu A, Sarcar B, Kahali S, Shan Y, Chinnaiyan P. Targeting the unfolded protein response in glioblastoma cells with the fusion protein EGF-SubA. PLoS One (2012) 7(12):e52265. doi:10.1371/journal.pone.0052265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, et al. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res (2006) 66(18):9260–9. doi:10.1158/0008-5472.CAN-06-1586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Martin S, Lamb HK, Brady C, Lefkove B, Bonner MY, Thompson P, et al. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br J Cancer (2013) 109(2):433–43. doi:10.1038/bjc.2013.325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Deng WG, Ruan KH, Du M, Saunders MA, Wu KK. Aspirin and salicylate bind to immunoglobulin heavy chain binding protein (BiP) and inhibit its ATPase activity in human fibroblasts. FASEB J (2001) 15(13):2463–70. doi:10.1096/fj.01-0259com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Maddalo D, Neeb A, Jehle K, Schmitz K, Muhle-Goll C, Shatkina L, et al. A peptidic unconjugated GRP78/BiP ligand modulates the unfolded protein response and induces prostate cancer cell death. PLoS One (2012) 7(10):e45690. doi:10.1371/journal.pone.0045690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell (2004) 6(3):275–84. doi:10.1016/j.ccr.2004.08.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Ozawa MG, Zurita AJ, Dias-Neto E, Nunes DN, Sidman RL, Gelovani JG, et al. Beyond receptor expression levels: the relevance of target accessibility in ligand-directed pharmacodelivery systems. Trends Cardiovasc Med (2008) 18(4):126–32. doi:10.1016/j.tcm.2008.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res (2006) 66(16):7849–53. doi:10.1158/0008-5472.CAN-06-1660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet (2010) 69:97–114. doi:10.1016/S0065-2660(10)69006-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: endoplasmic reticulum, localization, redox, Ca2+ signaling, cancer, immunity
Citation: Gutiérrez T and Simmen T (2014) Endoplasmic reticulum chaperones and oxidoreductases: critical regulators of tumor cell survival and immunorecognition. Front. Oncol. 4:291. doi: 10.3389/fonc.2014.00291
Received: 10 September 2014; Paper pending published: 01 October 2014;
Accepted: 07 October 2014; Published online: 27 October 2014.
Edited by:
Paul Eggleton, Exeter University Medical School, UKReviewed by:
Amedeo Amedei, University of Florence, ItalyPedro Berraondo, Centro de Investigación Médica Aplicada, Spain
Copyright: © 2014 Gutiérrez and Simmen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Simmen, Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta, Medical Sciences Building Room 565, Edmonton, AB T6G 2H7, Canada e-mail:VGhvbWFzLlNpbW1lbkB1YWxiZXJ0YS5jYQ==